

Abstract
Malaria, caused by Plasmodium falciparum and related parasites, is responsible for millions of deaths each year, mainly from complications arising from the blood stages of its life cycle. Macrophage migration inhibitory factor (MIF), a protein expressed by the parasite during these stages, has been characterized in mammals as a cytokine involved in a broad spectrum of immune responses. It also possesses two catalytic activities, a tautomerase and an oxidoreductase, though the physiological significance of neither reaction is known. Here, we have determined the crystal structure of MIF from two malaria parasites, Plasmodium falciparum and Plasmodium berghei at 2.2 Å and 1.8 Å, respectively. The structures have an α/β fold and each reveals a trimer, in agreement with the results of analytical ultracentrifugation. We observed open and closed active sites, these being distinguished by movements of proline-1, the catalytic base in the tautomerase reaction. These states correlate with the covalent modification of cysteine 2 to form a mercaptoethanol adduct, an observation confirmed by mass spectrometry. The Plasmodium MIFs have a different pattern of conserved cysteine residues to the mammalian MIFs and the side chain of Cys58, which is implicated in the oxidoreductase activity, is buried. This observation and the evident redox reactivity of Cys2 suggest quite different oxidoreductase characteristics. Finally, we show in pull-down assays that Plasmodium MIF binds to the cell surface receptor CD74, a known mammalian MIF receptor implying that parasite MIF has the ability to interfere with, or modulate, host MIF activity through a competitive binding mechanism.
Keywords: macrophage migration inhibitory factor (MIF), malaria, crystal structure, tautomerase, oxidoreductase
Introduction
Plasmodium falciparum is responsible for 300–500 million cases and 1–3 million deaths from malaria each year, mainly in sub-Saharan Africa,1 making it one of the main causes of mortality in the world. More than 40% of the world's population are living in areas where malaria is transmitted, meaning around two billion people are at risk of contracting the disease.2 The main causes of mortality are complications due to severe anemia and cerebral malaria. The P. falciparum life cycle is complicated, spanning both human and mosquito hosts, but the clinical manifestations of the disease are a result of the human blood stage cycle in which the parasite invades red blood cells and proliferates asexually.
Patients with severe malarial anemia demonstrate ineffective erythropoiesis.3 It has been proposed that some pathogenic manifestations of severe malaria, such as anemia, could be brought on by the release of proinflammatory cytokines by host macrophages in response to infection by the parasite.4 The proinflammatory cytokine, macrophage migration inhibitory factor (MIF) has been identified as one such host-derived factor that inhibits erythropoiesis3,5 and which therefore could contribute to severe malaria.
MIF was one of the first cytokines to be identified.6,7 Originally described as an immune mediator isolated from T-lymphocytes that inhibit the random migration of macrophages,8 it has over the years, been implicated in many other processes of the innate and adaptive immune responses. For example, MIF is the only cytokine known to counter-regulate the immunosuppressive effects of glucocorticoids.9 It is released by proinflammatory stimuli such as lipopolysaccharides, toxic shock syndrome toxin 1, and malaria parasites.10 The proinflammatory properties of MIF contribute to the pathogenesis of several diseases such as severe sepsis, acute respiratory distress syndrome, asthma, inflammatory bowel disease, and the autoimmune diseases rheumatoid arthritis, and glomerulonephritis.11 Indeed, neutralization of MIF by anti-MIF antibodies has been shown to be therapeutically beneficial to patients with various proinflammatory diseases.12–16 There is also evidence to suggest that MIF is involved in cancer,17,18 diabetes,19 and multiple sclerosis.20 As well as regulating glucocorticoid activity, MIF is involved in a wide range of cellular processes, including transcriptional regulation of inflammatory gene products, cell cycle control, modulation of cell cycle proliferation and differentiation, inactivation of the tumor suppressor factor p53, and signal transduction. A classic cytokine receptor has not yet been identified for MIF and CD74,21 CXCR2,22 the Jun activation domain binding protein (Jab-1),23 and the ribosomal protein S1924 are the only functional MIF-binding proteins to have been described.
MIF is notable among cytokines in having two catalytic activities. The first, a keto-enol tautomerase activity, is mediated by a conserved N-terminal praline.25 The second, an oxidoreductase activity,26 is associated with a thioredoxin-like motif. The physiological substrates for the two enzyme activities remain unknown as does their biological function. In crystals, mammalian MIF is a homotrimer with the tautomerase active site situated at the interface between pairs of subunits.27 The tautomerase active site, lined by residues 1, 33–34, 64–66 (Fig. 1), is highly conserved among MIF homologues. The residues necessary for the protein-thiol oxidoreductase activity, which is associated with a CXXC motif in mammalian MIF, are less conserved with only a single cysteine present in the corresponding region of Plasmodium MIF. However, Plasmodium MIF retains both catalytic activities but at about 20% specific activity compared with recombinant human MIF.29
Figure 1.

Amino acid sequence alignment of parasite and mammalian MIFs. The figure was generated in ESPript28 and adjusted manually to optimize the structure-based alignment. Black shading highlights invariant residues in the alignment whereas boxed residues are conserved. Key catalytic residues for the tautomerase activity are highlighted with an asterisk below the alignment. The conserved CFVR motif (highlighted in gray, first cysteine with a triangle) in the Plasmodium MIFs is replaced by a CALC motif in the mammalian MIFs (also highlighted in gray). GenBank™ accession codes for the sequences are as follows: Human: Q6FHV0, Sheep: Q1ZZU7, Pig: P809028, Cow: P80177, Plasmodium falciparum: Q8I5C5, P. berghei: Q4YQW0, P. yoelii: Q1HEA2, P. chabaudi; Q4Y5M8, P. vivax: A5K093, P. knowlesi; B3LCT3, Toxoplasma gondii: A1XDS9, Brugia malayi: A8PJU3, Leishmania major: 3B64_A. The secondary structure elements of PfMIF and huMIF (1MIF.pdb) are shown above and below the alignment respectively.
The single MIF homologue in Plasmodium was identified through the sequencing of the parasite genome.30 Subsequently, three studies have been published with a more detailed analysis of Plasmodium MIF.29,31,32 It is expressed in all life cycle stages and is localized to both the parasite and the host cytoplasm in the intracellular blood stage forms. Transcription levels peak at the late trophozoite blood stage.29 Like mammalian MIF, which is secreted via nonclassical pathways, Plasmodium MIF lacks a secretion signal. It is likely that it is released from infected red blood cells during schizont rupture and merozoite release.29 Parasite derived MIF has been detected in the serum of patients infected with P. falciparum.32 In vitro experiments showed a decrease in the cell surface markers TLR2, TLR4, and CD86 in response to Plasmodium MIF exposure.31 Although MIF is not essential for parasite survival, there is an increase in the number of circulating reticulocytes in animals infected with PbMIF knockout parasites,29 in agreement with other studies3,5,10,33–35 which found that host MIF inhibits erythropoiesis.
In earlier work, we have determined structures of proteins important in redox regulation in P. falciparum.36,37 To better understand the functional role of macrophage MIF in malaria, including its redox significance, we determined the crystal structures of P. berghei and P. falciparum MIF (PbMIF and PfMIF, respectively). Additionally, as an indication of mechanism of action of Plasmodium MIF, we determined its ability to bind to one receptor for mammalian MIF, the cell surface determinant CD74.
Results
Protein expression and characterization
Following purification of the recombinant Plasmodium MIFs, MALDI mass spectrometry was performed on each sample. The molecular mass of PbMIF was measured as 14,091 Da. This compares with the predicted mass of 14,013 Da, the difference being satisfactorily accounted for by the covalent attachment of a molecule of β-mercaptoethanol (78 g mol−1). The presence of the β-mercaptoethanol species was later confirmed by the crystal structure (see later). The calculated mass of PfMIF is 13,924 Da and so the two observed molecular mass peaks of 13,931 and 14,007 Da correspond to the native protein and a β-mercaptoethanol adduct, respectively.
Analytical ultracentrifugation
The oligomeric state of MIF has been the subject of debate with suggestions that the active species is variously a monomer, a dimer, or a trimer.38 The two Plasmodium MIFs appeared to be trimers in vitro from their gel filtration characteristics. Analytical ultracentrifugation was performed to establish rigorously the oligomerization state of PfMIF. Sedimentation equilibrium analysis showed the average molecular weight to be 40 kDa consistent with a trimer form (calculated molecular weight 41.7 kDa). However, the sample was heterogeneous. Velocity sedimentation analysis revealed a predominant lower molecular weight species with the mass of a trimer, and a three-fold less abundant larger species with a mass five times greater (Fig. 2). The combined results of the equilibrium and velocity sedimentation experiments therefore suggest that Plasmodium MIF consists mainly of trimeric material with about 30% pentamers of the trimer (i.e., 15-mers) in a non-, or very slowly exchanging mixture. The observation of the higher molecular weight species is interesting in the light of an earlier attribution of chaperone-like activities to MIF.39
Figure 2.
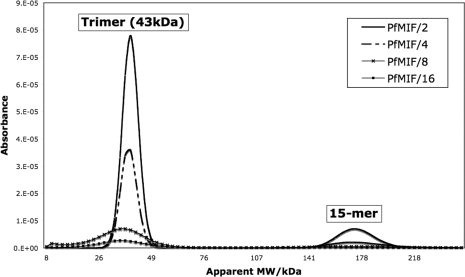
Analytical ultracentrifugation of PfMIF. A solution of PfMIF at a concentration of 2.6 mg mL−1 was serially diluted in 50 mM Tris, 200 mM NaCl, 5% glycerol to give PfMIF/2 (1.3 mg mL−1), PfMIF/4 (0.65 mg mL−1), PfMIF/8 (0.33 mg mL−1), and PfMIF/16 (0.16 mg mL−1) samples. The velocity sedimentation experiment was conducted at 35,000 rpm over 4 h and the absorbance was plotted against the apparent MW. The results show that in solution PfMIF is predominantly trimeric with a small percentage of species at five times this MW.
Although these results show that purified Plasmodium MIF is predominantly a trimer, this is not necessarily the physiologically relevant oligomeric state of the protein. For example, MIF may be stored in the trimeric form but dissociate on release from the cell. Extracellular concentrations of MIF range from 4 ng mL−1 in normal blood plasma to 150 ng mL−1 in patients with septic shock,40 values that are much lower than the concentrations used for these experiments. At these 1000-fold lower concentrations, it is possible that the trimer dissociates and that extracellular Plasmodium MIF is monomeric or dimeric.
Three-dimensional structure of Plasmodium MIF
The crystal structures of PbMIF and PfMIF reveal an identical polypeptide chain topology to human MIF. Each protomer consists of a four-stranded β-sheet (β2-β1-β3-β4) onto one face of which are packed two antiparallel α-helices [Fig. 3(A,B)]. There is a pseudo two-fold axis of symmetry within the subunits of both MIF structures, which relates the secondary structure elements β1 (residues 2–7), β2 (39–44), and α1 (13–31) to β3 (58–63), β4 (96–100), and α2 (72–88). These elements of the structure can be superposed with a root mean squared displacement (rmsΔ) in Cα positions of 1.7 Å. Least squares superposition of the two P. berghei protomers and six P. falciparum MIF subunits gives average pairwise rmsΔ values in the range 0.9–1.2 Å [Fig. 3(C)]. The secondary structure elements superimpose closely with the largest differences occurring in the loop regions, residues 31–37 and 64–70, where the electron density is less well defined.
Figure 3.
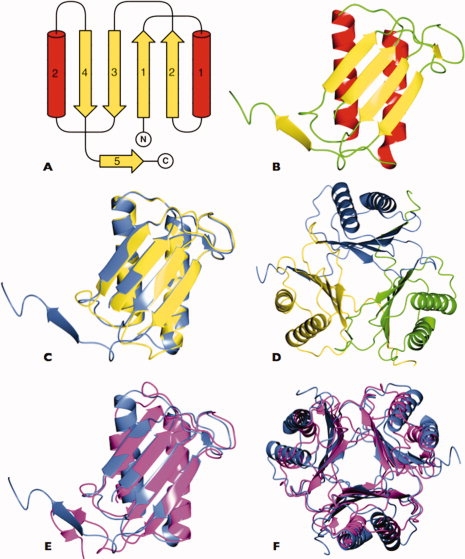
Structure of Plasmodium MIF. A: Topology diagram of the Plasmodium berghei MIF subunit with strands shown as yellow arrows and helices as red cylinders. The secondary structure elements are numbered. The amino (N) and carboxyl (C) termini are indicated. The pseudosymmetry relating β-strands 1 and 2 and α-helix 1 and β-strands 3 and 4 and α-helix 2 is apparent. B: Ribbon diagram of the PbMIF subunit with secondary structure elements colored as in (A). C: Overlay of the P. berghei (blue) and P. falciparum (yellow) MIF subunits showing the high structural similarity between the two proteins. The C-terminal residues of PfMIF are disordered and missing from the model. D: The PbMIF trimer viewed down the molecular three-fold axis with subunits colored individually in yellow, green, and blue. E: Overlay of the MIF subunits from P. berghei (blue) and human (magenta). F: Overlay of the MIF trimers from P. berghei (blue) and human (magenta) showing the close superposition of the β-sheets and the slight variation in the orientation of the α-helices. These and subsequent images were produced in CCP4MG.68
The C-terminal residues (102–114 together with the poly-His tag) are not defined by the electron density maps for either chain of PfMIF and these residues are assumed to be disordered. In PbMIF the chain is defined up to residue 114 and it is seen that the C-terminal residues extend away from the core of the subunit to pack onto an adjacent protomer in the trimer [Fig. 3(C,D)]. Residues 104–107 form a segment of β-strand, β5, which aligns in an antiparallel fashion with β4 of the neighboring subunit so that in the trimer the three β-sheets form a discontinuous β-barrel. A solvent channel coincident with the molecular three-fold axis runs through the center of the assembly. The channel is a feature of all published MIF structures but its biological function, if any, is unknown. Equivalent Cα atoms of the PfMIF and PbMIF trimers can be superimposed with an rmsΔ of 1.1 Å, the principal differences again lying in loop regions. As expected, the Plasmodium MIF trimers are very similar in structure to that of the human homologue25,42–47 with an average rmsΔ between huMIF (1CA725) and PbMIF of 2.6 Å, and between huMIF and PfMIF of 2.2 Å [Fig. 3(E,F)].
The tautomerase active site
The tautomerase active site has been defined in human MIF by the determination of crystal structures of complexes with the substrate p-hydroxyphenylpyruvate (HPP)25 and a competitive inhibitor 2-fluoro p-hydroxycinnamate.48 In each structure, three ligand molecules are bound per trimer each located at a subunit interface in hydrophobic cavities that contain the catalytic base, Pro1. Besides the N-terminal proline, the important substrate binding residues are Lys32, Ile64, Tyr95′, and Asn97′, where the apostrophes signify residues from the neighboring subunit.25 These residues are conserved among the mammalian MIFs (Fig. 1). Comparison of the active site region of unliganded huMIF and huMIF bound to HPP with the corresponding region of PbMIF, following least squares superposition of the respective chains, reveals a noticeable difference in the position of Pro1, the catalytic proline. It is shifted ∼3 Å further into the active site, partially filling the volume that is occupied by the HPP molecule in the huMIF-substrate complex [Fig. 4(A,B)]. Pro1 in PbMIF is in van der Waals contact with Tyr96, a residue whose side chain in huMIF packs over the aromatic ring of the HPP from the opposite face of the cavity. This Pro1-Tyr96 interaction effectively closes off the active site in PbMIF [Fig. 4(A,B)]. Interestingly, the two protomers in the asymmetric unit in the PfMIF crystal exhibit different conformations of Pro1. In chain B, the electron density maps define Pro1 in a position that superimposes with that in PbMIF, whereas in chain-A they reveal the proline in a location reminiscent of that in huMIF [Fig. 4(B)], giving rise to an open active site cavity as observed for huMIF.
Figure 4.
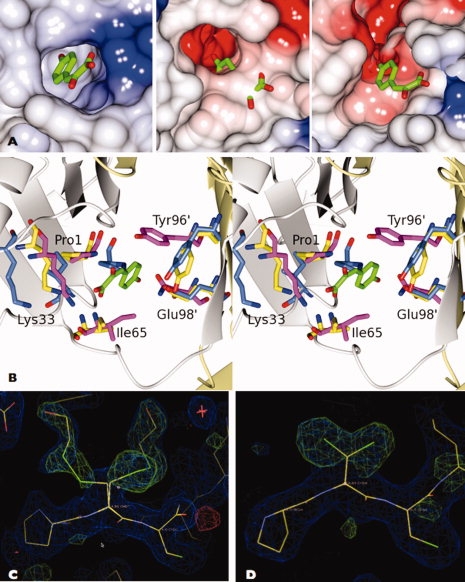
The tautomerase active site. A: Comparison of the human (left), P. berghei (center) and P. falciparum chain-A (right) MIF active site cavities. In the PbMIF and PfMIF images, the HPP substrate is taken from the huMIF coordinate set following superposition with the ligand-bound form of this protein. In huMIF and PfMIF chain-A, the active site is “open,” whereas in PbMIF, it is “closed” and there is a steric clash between the protein and the HPP moiety. The proteins are shown as electrostatic surfaces highlighting the fact that in Plasmodium MIF the active sites are more negatively charged than that of huMIF. B: Stereo view of the active site of MIF. The human (magenta), P. berghei (blue), and P. falciparum chain-A (yellow) MIFs have been superposed and the active site residues are shown in the context of the tautomerase substrate, HPP (colored by atom type C, green; O, red) bound to huMIF. Pro1 of human-MIF and PfMIF-A are in equivalent positions but Pro1 of PbMIF is markedly displaced. In both the Plasmodium MIFs, the aromatic ring of Tyr96 is in a perpendicular plane to that of this tyrosine in the huMIF active site. C, D: Electron density maps displayed in the vicinity of residue 2 of PbMIF chain C and PfMIF chain-A, respectively. The position 2 side chain was truncated to Ala and cycles of REFMAC refinement were carried out prior to the calculation of 2Fo-Fc (blue) and Fo-Fc (green) maps which are displayed at the 1.0 and 3.0 σ levels respectively and displayed on the final refined models. The covalent modification of Cys2 in the PbMIF chain and the dual conformation of the Cys2 side chain in PfMIF chain-A are evident.
The alternative conformations of Pro1 correlate with other changes that distinguish the “closed” active sites of the two PbMIF trimers and trimer B of PfMIF (PfMIF-B) from the “open” active sites of the huMIF trimer and the trimer formed from the PfMIF A chains (PfMIF-A). First, and as discussed in more detail later, Cys2 is covalently modified in PbMIF and PfMIF-B by disulphide linkage to a molecule of β-mercaptoethanol [Fig. 4(C)]. In PfMIF-A, there is no such covalent modification. The distinguishing feature of this protomer is the presence of two molecules of glycerol located at the entrance to the substrate binding cavity.
Second, Ile65 in PbMIF and PfMIF-B is flipped out of the active site with its main chain amide group displaced by 1.2 Å so that it is out of hydrogen bonding distance of the carboxyl group of the HPP moiety observed in huMIF. Third, the aromatic ring of Tyr95 in huMIF lies perpendicular to that of HPP so as to form favorable π-stacking interactions with the substrate. In PbMIF and PfMIF-B, the corresponding ring of tyrosine 96 is displaced from, and has a parallel orientation with respect to, the aromatic ring of HPP [Fig. 4(B)]. This suggests either that HPP has a different mode of binding in Plasmodium MIF compared with huMIF, or more likely, that conformational changes take place in PbMIF and PfMIF-B to generate the catalytically active state.
Receptor interactions
CD74 has been identified as a cell surface receptor for huMIF.49 We used ConSurf50 to determine clusters of conserved residues on the Plasmodium MIF trimer surface that might represent a CD74 binding site. As expected, the highly conserved residues cluster principally around the tautomerase active site (data not shown). To establish whether Plasmodium MIF interacts with CD74 we investigated the protein–protein interaction by GST-pulldown. Since in our hands full length CD74 failed to express in useful amounts, we expressed and purified the mouse CD74 ectodomain (residues 51–279) containing all the elements known to interact with huMIF fused to GST. As shown in Figure 5, the mouse CD74 ectodomain fused to GST (Lane 4) readily pulled down recombinant PbMIF-his6 relative to a GST control (Lane 1). To determine if the stability of this interaction was sensitive to the presence of the tautomerase substrate, we repeated the assay in the presence of 1, 10, and 25 mM HPP. Addition of HPP at concentrations up to 10 times the KM(HPP) of huMIF had a negligible effect on CD74 binding (Lanes 5 and 6). Similar results were obtained for PfMIF (data not shown).
Figure 5.
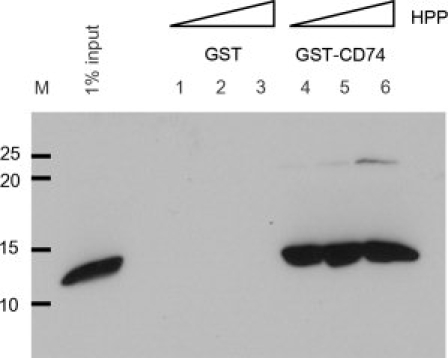
GST-pulldown showing CD74–PbMIF interaction. Left hand lane: 1% input PbMIF; Lanes 1–3: background binding to GST loaded glutathione sepharose beads with 1, 10, and 25 mM HPP, respectively; Lanes 4–6: PbMIF binding to GST-CD74 loaded glutathione sepharose beads with 1, 10, and 25 mM HPP, respectively. The masses in kDa of molecular markers are indicated at the left.
Covalent modification of Cys2
In the six subunits of the asymmetric unit of PbMIF and in one of the two subunits (PfMIF-B) of the asymmetric unit of PfMIF, we observed prominent and unexpected electron density features extending from the side chain of Cys2 [Fig. 4(C)]. In contrast, the electron density at position 2 of PfMIF chain-A (PfMIF-A) defines the expected cysteine residue, albeit one with a dual conformation [Fig. 4(D)]. The size and shape of the electron density associated with the additional feature [Fig. 4(C)] prompted us to infer that there was a straight chain species in a disulphide linkage to Cys2. The presence of β-mercaptoethanol in the protein storage buffer together with the mass spectrometry results, which showed m + 76 and m + 78 peaks, led to the conclusion that Cys2 was covalently linked to an —S—CH2—CH2—OH species. In most of the chains, there was evidence of dual conformations. In the more prominent conformer, the extended position 2 side chain extends into a hydrophobic pocket surrounded by Met39, Asn41 and Glu4 and Ile6′, Leu48′ and Phe59′ from the neighboring subunit. In the lower occupancy conformation, the modified Cys2 residue is stabilized by polar interactions between its OH group and Arg61′ and Glu98′. The covalent modification of Cys2 is linked to a 1.4 Å displacement of the position 2 Cα atom relative to its position in PfMIF-A, which correlates with the movement of the Pro1 residue and the closure of the active site.
The C-terminus
It was recently reported that deletion of residues 105–114 at the C-terminus of MIF results in abolition of the tautomerase activity.51 Although the C-terminal residues are important for tautomerase substrate binding, they form no direct interactions with the HPP moiety and it is logical to conclude that the importance of the C-terminus, for the tautomerase activity, is through its effect on trimer stability. The sequence of this segment is reasonably well conserved (Fig. 1) and in the structure these residues are involved in interactions between subunits in forming the trimer. The crystal structures of human and rat MIF25,46,52 are closely similar except that the C-terminal residues of rat MIF are disordered. This difference recurs in the Plasmodium structures in which the C-terminus is fully ordered in the PbMIF protomers, but disordered in PfMIF beyond residues Asp101 and Asn106 of chains A and B, respectively.
The intersubunit interactions in the Plasmodium MIFs were analyzed using the program PISA.53 In PbMIF, a total of 2674 Å2 of solvent accessible surface becomes buried on formation of the trimer compared with only 1539 Å2 in PfMIF, where the C-terminal residues are missing. Although the PfMIF trimer appears to be less thermodynamically stable than the PbMIF trimer, both molecules are evidently trimeric. In PbMIF, residues 107–112 interact with residues 92–99 of the adjacent subunit. Residues spanning Phe107 to Ser112 of one subunit form main chain hydrogen bonds with β4 of the adjacent protomer to make the additional β-strand [Fig. 6(A)].
Figure 6.
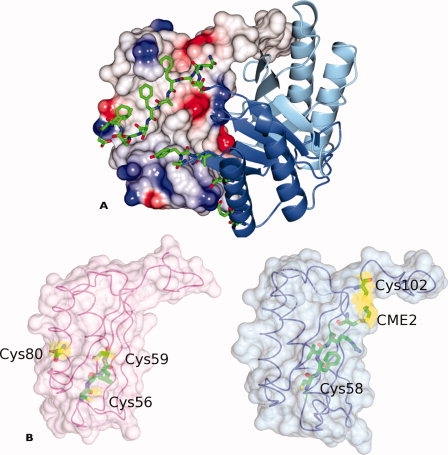
Quaternary interactions in Plasmodium MIFs and distribution of cysteines in huMIF and PbMIF. A: The P. berghei MIF trimer illustrating the extensive intersubunit interactions of the C-terminal residues. Two of the subunits are represented as ribbons (blue and light blue) with the C-terminal amino acid residues (Ser103-Gly114) drawn as bonds and colored by atom type (C, green; N, blue; O, red). The third subunit is shown as a surface whose color indicates the electrostatic potential with red (negative) blue (positive) and white/gray (neutral). The hydrophobic nature of the interface is apparent. B: Surface rendering of single protomers taken from the trimers of huMIF (left) and PbMIF (right). The cysteine and CME residues together with the residues of the CALC and CFVR motifs are shown in cylinder format and colored by atom type (C, green; N, blue; O, red). The Cys/CME residues are shown with a yellow surface. Cys58 of Plasmodium MIF is completely buried within the protomer.
The oxidoreductase active site
In huMIF, the oxidoreductase activity is associated with a conserved motif, C56ALC59. A C56S mutant retains 55 and 68% wild-type activity for insulin and 2-hydroxyethyldisulphide (HED) reduction, respectively, whereas the C59S mutation completely abolishes these activities.26 In the Plasmodium MIFs, the motif is not conserved and the corresponding residues are C58FVR61. In the crystal structures, this motif is quite differently structured [Fig. 6(B)]. Cys58 of Plasmodium MIF is the first residue of strand β3 and its side chain together with that of Val60 are buried in the hydrophobic core of the molecule [Fig. 6(B)]. Phe59 and Arg61 are on the opposite face of this β-strand and contribute to the subunit interface. In huMIF, Cys56 lies in the loop region preceding β3 and it is hypothesized that a disulphide-stabilized β-turn may form in the CALC region on oxidization.26
Discussion
The cytokine macrophage MIF has a widespread distribution with homologues in Plasmodium, Leishmania, Toxoplasma, and parasitic nematodes. The absence of MIF in other protozoans indicates that the expression of MIF in unicellular eukaryotes is confined to parasites that engage in interactions with host cells in the blood. Given the established role of MIF in the immune system, it may be that differences between the parasite and host MIF proteins are important for progression of parasitemia and evasion of the host immune response. MIFs from protozoan parasites have previously been structurally and functionally characterized for the parasitic nematodes Brugia malayi,54 Ancylostoma ceylanicum,55 and the apicomplexan Leishmania major.56
The tertiary and quaternary structures of the P. falciparum and P. berghei MIFs determined here are similar to one another and to the mammalian and parasite MIFs. Electrostatic surface analysis reveals that the tautomerase active site of Plasmodium MIF has more negatively charged character than huMIF [Fig. 4(A)], a phenomenon also observed in the L. major,56 and hookworm55 structures. The extra negative charge is attributable to Glu98; which replaces Asn97 in the human homologue. The differences observed in the active site, in terms of electrostatic potential and the conformation of catalytic residues, may account for why Plasmodium MIF has a lower tautomerase specific activity toward HPP than huMIF.29,32 It may also point to different substrate specificity, given that the physiological substrate has yet to be identified.
Receptor interactions
CD74 has been identified as a cell surface receptor for huMIF49 and our results suggest this interaction is conserved in the Plasmodium MIFs. Although better defined in its intracellular form, it is known that 2–5% of the CD74 protein localizes to the cell surface.57 The NMR structure of the CD74 ectodomain is trimeric,58 sharing the three-fold rotational symmetry of MIF. The programme ConSurf50 shows that highly conserved residues on the Plasmodium MIF trimer surface cluster around the tautomerase active site. A possible coincidence/overlap of the CD74 binding surface and the tautomerase active site has recently been inferred for huMIF following the observation that antagonists of the huMIF-CD74 interaction also inhibit the tautomerase activity of the enzyme.59 This prompted us to investigate the influence of the HPP substrate on Plasmodium MIF—CD74 interaction. Although the mouse CD74 ectodomain was able to pull down recombinant histidine tagged Plasmodium MIF, we found no significant effect of the presence of HPP, even at concentrations up to 10 times the KM for HPP of huMIF, on the interaction with CD74. The fact that saturating concentrations of the tautomerase substrate do not inhibit CD74 binding leads to the tentative suggestion that the tautomerase active site and the CD74 binding epitope are nonoverlapping.
Implications for oxidoreductase mechanism
One of the most interesting aspects of the structures described here is the modification of Cys2, where the presence of disulphide-linked mercaptoethanol adducts is clearly indicated by the electron density maps. The modified residue S,S-(2-hydroxyethyl)thiocysteine (CME) is present in the six crystallographically independent subunits of PbMIF and in one of the two crystallographically independent subunits of PfMIF. There is no evidence of covalent modification of the three other cysteine residues (Cys3, Cys58, or Cys102) in either structure, indicating that Cys2 is uniquely reactive.
Oxidoreductase activity in mammalian MIFs was first considered when the existence of the well-conserved CALC motif was correlated with the presence of CXXC motifs at the catalytic centers of thiol-protein oxidoreductases such as thioredoxin.26 The reaction cycles of thioredoxin-type oxidoreductases feature intermolecular disulphide bonds with substrate proteins and intramolecular disulphide bonds between the adjacent cysteines of the motif.60 HuMIF was subsequently shown to exhibit oxidoreductase activity toward insulin and 2-hydroxyethyl disulphide (HED) substrates.26 In huMIF, site-directed mutagenesis showed that the second cysteine (Cys59) of the CALC motif is essential for oxidoreductase activity, whereas serine substitution of the first cysteine led to quite modest reductions in rate. The location of the CALC motif in the huMIF structure suggests that the oxidoreductase reaction could only plausibly be mediated by the monomeric form. In the trimer, neither of the CALC motif cysteine residues (at positions 56 and 59) nor cysteine 80 is surface accessible. In the monomer, Cys59 and 80 would be on the surface of the protein [Fig. 6(B)].
Since HED reduction produces two molecules of β-mercaptoethanol, the Cys2 adducts observed here could represent intermediates in the Plasmodium MIF-catalyzed reduction of this substrate. This would imply a quite different mechanism of action from that proposed for human MIF consistent with the strikingly different pattern of cysteine residues in the Plasmodium MIF sequences and the absence of the CXXC motif. In the Plasmodium proteins, cysteines are conserved at positions 2, 3, 58, and 102 (Fig. 1), in contrast to the mammalian MIFs where the conserved cysteines occur at 56, 59, and 80. Although, Cys58 in Plasmodium MIF and Cys56 in huMIF are corresponding residues in the sequence alignment (Fig. 1), the structures reveal quite different environments. In contrast to Cys56 in huMIF, which would be surface-exposed in the monomeric form, the Cys58 side chain in Plasmodium MIF is buried in the hydrophobic core of the subunit and clearly incapable of participating in redox reactions in the absence of major conformational changes in the protein [Fig. 6(B)].
The Plasmodium MIF oxidoreductase activity was shown in a previous study to be very low. In this work, we found that the activity of wild-type PfMIF against HED was scarcely above background levels, and much lower than that of recombinant human glutaredoxin 1, which was used as a positive control (data not shown). This very low activity prevented us from evaluating the catalytic contribution of cysteine-2 by site-directed mutagenesis. The uniquely reactive character of Cys2 is unexplained and may be associated with uncharacterized chemistry in MIF possibly involving the adjacent active site proline, Pro1.
The cytokine activity of MIF
The CC motif at the N-terminus of Plasmodium MIF (Cys2-Cys3) is a recognized motif in one of the four chemokine families. As Plasmodium MIF has been characterized as a cytokine, it throws up the possibility that it could mediate some of its activity via this CC motif. Although, there is no evidence of disulphide bonds in the Plasmodium MIF crystal structures, the subunit does exhibit a high degree of structural similarity to the dimeric form of interleukin-8 (IL-8), a CXC chemokine. There is also evidence that MIF is a noncognate ligand of the CXC chemokine receptor CXCR2 that is known to bind IL-8.22 In the same study, it was pointed out that huMIF may qualify as a pseudo-CXC or ΔC chemokine, given its lack of C-terminal cysteines.22 Plasmodium MIF has a C-terminal cysteine (Cys102) that is absent in huMIF, which may indicate that it has added chemokine functionality that could differentiate the two homologues during parasite invasion. An added complexity is that activation of CXCR2 by IL-8 requires an N-terminal ELR motif, of which there is a pseudo motif in huMIF composed of two nonadjacent but adequately spaced Asp and Arg residues. In Plasmodium, MIF Glu55 and Arg93 are surface exposed and spaced such that they could mimic the ELR motif of IL-8.
Conclusion
The role of Plasmodium MIF in the human host is little understood. Human MIF is known to have a role in multiple immune pathways and its function/dysfunction is associated with a multitude of diseases. The malaria parasite releases MIF during the blood stage cycle of infection when it would seem counter-productive to release a protein that could elicit a potentially lethal host immune response. In presenting the crystal structure of MIF from P. berghei and P. falciparum, it is hoped that the important differences, and indeed similarities, between Plasmodium and human MIF may provide an indication of how the parasite-derived protein contributes to the progression of parasitemia. There is a possibility that Plasmodium MIF could be a potential drug target in malaria and it would therefore be important to target the parasite protein selectively relative to the host protein. MIF clearly has potential for inhibitor design with huMIF having been the subject of a virtual screen for MIF-CD74 antagonists,59 and the Plasmodium MIF structures presented here provide a basis for extending this approach.
Materials and Methods
Plasmid preparation
Protein expression constructs were prepared by previously published protocols.29 Briefly, the coding sequences for P. falciparum (PlasmoDB entry: PFL1420w) and P. berghei (GeneDB entry: PB000372.03.0) MIF were amplified from mixed blood stage cDNA using the primers 5′-AAAATTTCCGCATGCCTTGCTGTGAAGTAA TAAC-3′ and 5′-ATTGGATCCGCCGAAAAGAGAACCACTGAAGCG-3′ for P. falciparum and 5′-AAATTTCCGCATGCCGTGCTGTGAATTAATAAC-3′ and 5′-ATTGGATCCACCAAATAGTGAGCCACTAAAAGC-3′ for P. beghei (italicized bases indicate restriction sites). The PCR products were digested with SphI and BamHI and ligated into the similarly digested pQE-70 (QIAGEN) expression vector such that recombinant protein products were expressed with a C-terminal His6-tag. Constructs were sequenced to confirm MIF sequence identity.
Protein expression and purification
PfMIF and PbMIF were expressed in E. coli BL21star (Novagen) cells grown on Luria Broth (10 g tryptone, 5 g yeast extract, 10 g NaCl per liter). Bacteria were grown to an optical density at 600 nm of 0.6 before addition of isopropyl-D-thiogalactoside to 1 mM to induce expression of recombinant protein. Growth was allowed to continue at 37°C for 4 h. Cells were harvested by centrifugation and resuspended and lysed in 50 mM Tris-HCl pH 8.0, 500 mM NaCl, 10% v/v glycerol, 10 mM β-mercaptoethanol, 0.01% Nonidet P-40, and complete cocktail protease inhibitor (Roche). The lysate was cleared by centrifugation at 15, 000 rpm for 40 min at 4°C and loaded onto a 5 mL HisTrap™ HP chelating column (GE Healthcare) equilibrated in buffer A (50 mM Tris-HCl pH 8.0, 200 mM NaCl, 10% v/v glycerol, 10 mM β-mercaptoethanol, 20 mM imidazole). After a washing step, the bound proteins were eluted over 10 column volumes with a 20–500 mM linear imidazole gradient in buffer A. The MIF-containing fractions were identified by SDS-PAGE analysis, concentrated, and loaded onto a Superdex 200 gel filtration column (Amersham Pharmacia) equilibrated in 50 mM Tris pH 8.0, 200 mM NaCl, 5% v/v glycerol, 5 mM β-mercaptoethanol. Following size exclusion chromatography, the MIF proteins were judged to be pure from the Coomassie blue staining pattern following denaturing gel electrophoresis.
GST pulldowns
Pf and PbMIF-His6 were expressed and purified as described previously.29 The ORF of the CD74 ectodomain (residues 51-279) was amplified from mouse cDNA using primers (L2809: 5′-TGGGCAGGCTCATATGGCTTACTTCCTGTACCAGCAACAGG-3′and L2808: 5′-GCCTGGATCCTTACAGGGTGACTTGACCCAGTTCC-3′) and inserted (NdeI, BamHI, italicized in sequence) into pRP265NB (described previously61). The empty vector pRP265NB (for control GST expression) and the GST-CD74 expression plasmid were transformed in BL21 (DE3) pLysS, bacteria were grown at 30°C on Luria Broth (LB, Q-biogene) containing 30 μg/mL chloramphenicol and 0.05 μg/mL ampicillin. Protein expression was induced at OD600 of 0.6 by the addition of isopropyl-d-thiogalactoside to a final concentration of 1 mM. After 5 h, the cells were harvested and lysed in 50 mM Tris (pH 8.0), 500 mM NaCl, 5% (vol/vol) glycerol, 0.01% Tween in the presence of complete protease inhibitor cocktail (Roche) and 1 μg/mL lysozyme. Genomic DNA was fragmented by sonication, after which the lysate was cleared by centrifugation at 30,000 × g for 1 h at 4°C. Approximately 1 μg of GST or GST-CD74 in bacterial lysate was loaded on glutathione sepharose 4b beads (GE Healthcare) by rotating at 4°C for 1 h. After washing with PBS, Pf or PbMIF-His6 was added to a final concentration of roughly 4 μM in 500 μL binding buffer (50 mM KxHyPO4 (pH 7.2), 50 mM NaCl, 5% glycerol, 0.01% Tween) in presence or absence of substrate (p-hydroxyphenylpyruvyc acid, Sigma), which was added dissolved in DMSO. After 1 h binding at 4°C, the glutathione beads were washed three times with binding buffer, supplemented with the appropriate concentration of HPP, before loading on a 15% polyacrylaminde gel. MIF-His6 was detected by western blotting with anti-his and sheep-anti-mouse HRP conjugate (GE Healthcare).
Analytical ultracentrifugation
Sedimentation equilibrium experiments were performed on PfMIF with Dr. Andrew Leech (Molecular Interactions Laboratory, Technology Facility, Department of Biology, University of York) to determine the oligomeric state of the protein. A Beckman Optima XL/A analytical ultracentrifuge with an AN-50Ti rotor was used to conduct the experiment using Beckman cells. PfMIF was dialyzed overnight into 50 mM Tris-HCl pH 8.0, 200 mM NaCl, 5% glycerol. Sample concentrations ranged from 2.6 to 0.16 mg/mL and were prepared by serial two-fold dilutions. 115 μL protein and 120 μL buffer was loaded into sample and reference channels, respectively. Initial absorbance scans were taken at 3,000 rpm to verify that the cell contents were evenly distributed before the speed was increased and sedimentation equilibrium was reached. Absorbance scans were taken at 3 h intervals and all measurements were recorded at 293 K.
The distribution of material between species of different oligomeric state was determined by velocity sedimentation. Experiments were conducted using the same material as for the sedimentation equilibrium analysis. The cell contents were agitated so as to destroy the concentration gradient and ensure even distribution of material. A velocity run was then conducted over 4 h at 35,000 rpm. The data from both experiments were analyzed using HETANAL.
Protein crystallization
PfMIF and PbMIF were concentrated to 5 and 25 mg/mL, respectively, for crystallization experiments. Suitable crystallization conditions were initially screened in 96-well MRC Wilden crystallization plates using a mosquito® (TTP LabTech) liquid handling robot to set up nanoliter drops. Larger crystals were grown via the hanging drop vapor diffusion method, where equal volumes (1 μL) of protein and mother liquor were mixed and allowed to equilibrate at room temperature. PfMIF crystals grew from 0.2M KSCN, 10% PEG 8K, 10% PEG 1K, 100 mM Tris pH 8.0 and belong to space group P213 with unit cell dimensions a = b = c = 83.37 Å. PbMIF crystals grew from 0.2M KSCN, 13% PEG 6K, 100 mM Tris pH 8.5 and belong to space group P21 where a = 78.70 Å, b = 69.17 Å, c = 80.22 Å, and β = 118.20°.
Crystals were soaked in mother liquor containing 25% glycerol as a cryoprotectant and rapidly cooled in liquid N2 (110 K) for data collection.
Data collection and processing
Data for PfMIF and PbMIF were collected on beamlines ID14-2 and ID14-4, respectively, at the European Synchrotron Radiation Facility (ESRF, Grenoble, France) and were processed using the DENZO/SCALEPACK62 and CCP4 packages.63
Determination of the structure of PbMIF
The structure proved difficult to solve by molecular replacement (MR) with the P21 data with calculations producing solutions that were inconclusive. All of the MR programs tested aligned the trimeric three-fold axis correctly but the rotation about this axis was very uncertain (i.e., the translation function was good but the subsequent rotation function was poor). In addition to the three-fold symmetry axis in the trimer, each subunit has pseudo two-fold symmetry. Self-rotation shows the crystallographic two-fold to be parallel to the molecular three-fold, and perpendicular to the pseudo two-fold axes, generating pseudo 222 symmetry. Additionally, the Patterson map showed noncrystallographic translation with a peak that was 43% of the height of the origin peak at 0.50, 0.45, and 0.00. This suggested that there were six molecules in the asymmetric unit correlating with the Matthews coefficient of 2.24 Å3 Da−1 (relating to a solvent content of 45%). All of this may have contributed to the difficulty in finding an MR solution for the P21 data.
A partial solution was found using the automated molecular replacement pipeline Balbes64 following reprocessing of the data in the space group P1. Sixteen monomers were found using chain-A of coordinate set 2OS555 as the MR model. Of these, four protomers, related in pairs by 120°, satisfied the symmetry operations for P21 with the expected pseudo translation. The third chain of each trimer was generated and the subsequent model was good enough to start refinement. ARP/wARP was used to build in the P. berghei sequence, refinement was performed with REFMAC,65 and Coot66 was used to finalize the model.
Determination of the structure of PfMIF
Data collected for PfMIF were processed in the cubic space group P213. An ice ring forced the exclusion of some data during processing. Initial molecular replacement calculations using the human MIF coordinate set (pdb entry 1GD045) as the search model were unsuccessful most likely due to the low-sequence identity between PfMIF and the human homologue (∼29%). A solution was obtained when the PbMIF structure, which had not been available initially, was used as the model for molecular replacement with MolRep.67 Two protomers in the asymmetric unit were sought based on the Matthews coefficient (1.73 Å3 Da−1 with 29% solvent content)68 with the biologically relevant trimers being generated via the crystallographic three-fold axis. Data and refinement statistics for both structures are presented in Table I. PDB accession codes are 2WKB and 2WKF for PbMIF and PfMIF, respectively.
Table I.
Data collection and refinement statistics
| P. berghei | P. falciparum | |
|---|---|---|
| Data collection | ||
| Space group | P21 | P213 |
| Cell dimensions | ||
| a, b, c (Å) | 78.70, 69.17, 80.22 | 83.29, 83.29, 83.29 |
| α, β, γ (°) | 90, 118.20, 90 | 90, 90, 90 |
| Wavelength (Å) | 0.94 | 0.933 |
| Resolution (Å) | 50–1.8 | 50–2.05 |
| No. unique reflections | 72, 962 | 12,045 |
| Rmerge | 0.084 (0.610) | 0.063 (0.393) |
| I/σ(I) | 17.35 (1.36) | 34.6 (6.1) |
| Completeness (%) | 99.6 (99.5) | 99.9 (100) |
| Redundancy | 4.0 (2.7) | 14.2 (12.1) |
| Refinement | ||
| Resolution (Å) | 40.78–1.78 | 58.90–2.05 |
| No. reflections | 68,494 | 12,045 |
| Rwork/Rfree | 0.20/0.25 | 0.25/0.32 |
| No. atoms | ||
| Protein | 5419 (including CME) | 1604 (including CME) |
| Ligand | GOL 48 | GOL 12 |
| Water | 498 | 83 |
| B-factors | ||
| Proteins | 28.30 | 32.43 |
| Ligand | 43.86 | 47.96 |
| Water | 38.05 | 34.51 |
| Rms deviation | ||
| Bond lengths (Å) | 0.025 | 0.020 |
| Bond angles (°) | 2.11 | 1.82 |
Rmerge, I/σ(I), completeness and redundancy are given for all data and for data in the highest resolution shell (numbers in parentheses). Rmerge = Σ|I – (I)|/ΣI, where I is the intensity of an individual observation and (I) is the mean intensity of the same reflection. Rfactor = Σ||Fo| − |Fc||/Σ|Fo|, where Fo are the observed and Fc the calculated structure factors. Rfree was calculated using a test set of 5% of the data. CME is the modified amino acid at position 2, S,S-(2-hydroxyethyl)-thiocysteine, and GOL is glycerol.
Acknowledgments
S.E.D. was the recipient of a BBSRC, UK studentship. The authors are grateful to the ESRF, Grenoble for access to synchrotron radiation for data collection and to Dr. Johan Turkenburg for assistance in data collection and processing.
References
- 1.Miller LH, Good MF, Milon G. Malaria pathogenesis. Science. 1994;264:1878–1883. doi: 10.1126/science.8009217. [DOI] [PubMed] [Google Scholar]
- 2.Tuteja R. Malaria—an overview. Febs J. 2007;274:4670–4679. doi: 10.1111/j.1742-4658.2007.05997.x. [DOI] [PubMed] [Google Scholar]
- 3.Martiney JA, Sherry B, Metz CN, Espinoza M, Ferrer AS, Calandra T, Broxmeyer HE, Bucala R. Macrophage migration inhibitory factor release by macrophages after ingestion of Plasmodium chabaudi-infected erythrocytes: possible role in the pathogenesis of malarial anemia. Infect Immun. 2000;68:2259–2267. doi: 10.1128/iai.68.4.2259-2267.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Clark IA. Cell-mediated immunity in protection and pathology of malaria. Parasitol Today. 1987;3:300–305. doi: 10.1016/0169-4758(87)90187-6. [DOI] [PubMed] [Google Scholar]
- 5.McDevitt MA, Xie J, Shanmugasundaram G, Griffith J, Liu A, McDonald C, Thuma P, Gordeuk VR, Metz CN, Mitchell R, Keefer J, David J, Leng L, Bucala R. A critical role for the host mediator macrophage migration inhibitory factor in the pathogenesis of malarial anemia. J Exp Med. 2006;203:1185–1196. doi: 10.1084/jem.20052398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bloom BR, Bennett B. Mechanism of a reaction in vitro associated with delayed-type hypersensitivity. Science. 1966;153:80–82. doi: 10.1126/science.153.3731.80. [DOI] [PubMed] [Google Scholar]
- 7.David JR. Delayed hypersensitivity in vitro: its mediation by cell-free substances formed by lymphoid cell-antigen interaction. Proc Natl Acad Sci USA. 1966;56:72–77. doi: 10.1073/pnas.56.1.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lue HQ, Kleemann R, Calandra T, Roger T, Bernhagen J. Macrophage migration inhibitory factor (MIF): mechanisms of action and role in disease. Microbes Infect. 2002;4:449–460. doi: 10.1016/s1286-4579(02)01560-5. [DOI] [PubMed] [Google Scholar]
- 9.Calandra T, Bernhagen J, Metz CN, Spiegel LA, Bacher M, Donnelly T, Cerami A, Bucala R. MIF as a glucocorticoid-induced modulator of cytokine production. Nature. 1995;377:68–71. doi: 10.1038/377068a0. [DOI] [PubMed] [Google Scholar]
- 10.Chaisavaneeyakorn S, Lucchi N, Abramowsky C, Othoro C, Chaiyaroj SC, Shi YP, Nahlen BL, Peterson DS, Moore JM, Udhayakumar V. Immunohistological characterization of macrophage migration inhibitory factor expression in Plasmodium falciparum-infected placentas. Infect Immun. 2005;73:3287–3293. doi: 10.1128/IAI.73.6.3287-3293.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Calandra T, Roger T. Macrophage migration inhibitory factor: a regulator of innate immunity. Nat Rev Immunol. 2003;3:791–800. doi: 10.1038/nri1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bernhagen J, Calandra T, Mitchell RA, Martin SB, Tracey KJ, Voelter W, Manogue KR, Cerami A, Bucala R. MIF is a pituitary-derived cytokine that potentiates lethal endotoxaemia. Nature. 1993;365:756–759. doi: 10.1038/365756a0. [DOI] [PubMed] [Google Scholar]
- 13.Calandra T, Spiegel LA, Metz CN, Bucala R. Macrophage migration inhibitory factor is a critical mediator of the activation of immune cells by exotoxins of Gram-positive bacteria. Proc Natl Acad Sci USA. 1998;95:11383–11388. doi: 10.1073/pnas.95.19.11383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Donnelly SC, Bucala R. Macrophage migration inhibitory factor: a regulator of glucocorticoid activity with a critical role in inflammatory disease. Mol Med Today. 1997;3:502–507. doi: 10.1016/S1357-4310(97)01133-7. [DOI] [PubMed] [Google Scholar]
- 15.Lan HY, Bacher M, Yang N, Mu W, Nikolic-Paterson DJ, Metz C, Meinhardt A, Bucala R, Atkins RC. The pathogenic role of macrophage migration inhibitory factor in immunologically induced kidney disease in the rat. J Exp Med. 1997;185:1455–1465. doi: 10.1084/jem.185.8.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mikulowska A, Metz CN, Bucala R, Holmdahl R. Macrophage migration inhibitory factor is involved in the pathogenesis of collagen type II-induced arthritis in mice. J Immunol. 1997;158:5514–5517. [PubMed] [Google Scholar]
- 17.Bucala R, Donnelly SC. Macrophage migration inhibitory factor: a probable link between inflammation and cancer. Immunity. 2007;26:281–285. doi: 10.1016/j.immuni.2007.03.005. [DOI] [PubMed] [Google Scholar]
- 18.Xu X, Wang B, Ye C, Yao C, Lin Y, Huang X, Zhang Y, Wang S. Overexpression of macrophage migration inhibitory factor induces angiogenesis in human breast cancer. Cancer Lett. 2008;261:147–157. doi: 10.1016/j.canlet.2007.11.028. [DOI] [PubMed] [Google Scholar]
- 19.Yabunaka N, Nishihira J, Mizue Y, Tsuji M, Kumagai M, Ohtsuka Y, Imamura M, Asaka M. Elevated serum content of macrophage migration inhibitory factor in patients with type 2 diabetes. Diabetes Care. 2000;23:256–258. doi: 10.2337/diacare.23.2.256. [DOI] [PubMed] [Google Scholar]
- 20.Hoi AY, Iskander MN, Morand EF. Macrophage migration inhibitory factor: a therapeutic target across inflammatory diseases. Inflamm Allergy Drug Targets. 2007;6:183–190. doi: 10.2174/187152807781696455. [DOI] [PubMed] [Google Scholar]
- 21.Le Roch KG, Zhou Y, Blair PL, Grainger M, Moch JK, Haynes JD, De La Vega P, Holder AA, Batalov S, Carucci DJ, Winzeler EA. Discovery of gene function by expression profiling of the malaria parasite life cycle. Science. 2003;301:1503–1508. doi: 10.1126/science.1087025. [DOI] [PubMed] [Google Scholar]
- 22.Bernhagen J, Krohn R, Lue H, Gregory JL, Zernecke A, Koenen RR, Dewor M, Georgiev I, Schober A, Leng L, Kooistra T, Fingerle-Rowson G, Ghezzi P, Kleemann R, McColl SR, Bucala R, Hickey MJ, Weber C. MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat Med. 2007;13:587–596. doi: 10.1038/nm1567. [DOI] [PubMed] [Google Scholar]
- 23.Hoerauf A, Satoguina J, Saeftel M, Specht S. Immunomodulation by filarial nematodes. Parasite Immunol. 2005;27:417–429. doi: 10.1111/j.1365-3024.2005.00792.x. [DOI] [PubMed] [Google Scholar]
- 24.Filip AM, Klug J, Cayli S, Frohlich S, Henke T, Lacher P, Eickhoff R, Bulau P, Linder M, Carlsson-Skwirut C, Leng L, Bucala R, Kraemer S, Bernhagen J, Meinhardt A. Ribosomal protein S19 interacts with macrophage migration inhibitory factor and attenuates its pro-inflammatory function. J Biol Chem. 2009;284:7977–7985. doi: 10.1074/jbc.M808620200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lubetsky JB, Swope M, Dealwis C, Blake P, Lolis E. Pro-1 of macrophage migration inhibitory factor functions as a catalytic base in the phenylpyruvate tautomerase activity. Biochemistry. 1999;38:7346–7354. doi: 10.1021/bi990306m. [DOI] [PubMed] [Google Scholar]
- 26.Kleemann R, Kapurniotu A, Frank RW, Gessner A, Mischke R, Flieger O, Juttner S, Brunner H, Bernhagen J. Disulfide analysis reveals a role for macrophage migration inhibitory factor (MIF) as thiol-protein oxidoreductase. J Mol Biol. 1998;280:85–102. doi: 10.1006/jmbi.1998.1864. [DOI] [PubMed] [Google Scholar]
- 27.Frevert U, Sinnis P, Cerami C, Shreffler W, Takacs B, Nussenzweig V. Malaria circumsporozoite protein binds to heparan sulfate proteoglycans associated with the surface membrane of hepatocytes. J Exp Med. 1993;177:1287–1298. doi: 10.1084/jem.177.5.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Augustijn KD, Kleemann R, Thompson J, Kooistra T, Crawford CE, Reece SE, Pain A, Siebum AH, Janse CJ, Waters AP. Functional characterization of the Plasmodium falciparum and P.berghei homologues of macrophage migration inhibitory factor. Infect Immun. 2007;75:1116–1128. doi: 10.1128/IAI.00902-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gardner MJ, Hall N, Fung E, White O, Berriman M, Hyman RW, Carlton JM, Pain A, Nelson KE, Bowman S, Paulsen IT, James K, Eisen JA, Rutherford K, Salzberg SL, Craig A, Kyes S, Chan MS, Nene V, Shallom SJ, Suh B, Peterson J, Angiuoli S, Pertea M, Allen J, Selengut J, Haft D, Mather MW, Vaidya AB, Martin DM, Fairlamb AH, Fraunholz MJ, Roos DS, Ralph SA, McFadden GI, Cummings LM, Subramanian GM, Mungall C, Venter JC, Carucci DJ, Hoffman SL, Newbold C, Davis RW, Fraser CM, Barrell B. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature. 2002;419:498–511. doi: 10.1038/nature01097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cordery DV, Kishore U, Kyes S, Shafi MJ, Watkins KR, Williams TN, Marsh K, Urban BC. Characterization of a Plasmodium falciparum macrophage-migration inhibitory factor homologue. J Infect Dis. 2007;195:905–912. doi: 10.1086/511309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shao D, Han Z, Lin Y, Zhang L, Zhong X, Feng M, Guo Y, Wang H. Detection of Plasmodium falciparum derived macrophage migration inhibitory factor homologue in the sera of malaria patients. Acta Trop. 2008;106:9–15. doi: 10.1016/j.actatropica.2007.12.008. [DOI] [PubMed] [Google Scholar]
- 32.Awandare GA, Hittner JB, Kremsner PG, Ochiel DO, Keller CC, Weinberg JB, Clark IA, Perkins DJ. Decreased circulating macrophage migration inhibitory factor (MIF) protein and blood mononuclear cell MIF transcripts in children with Plasmodium falciparum malaria. Clin Immunol. 2006;119:219–225. doi: 10.1016/j.clim.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 33.Chaisavaneeyakorn S, Moore JM, Othoro C, Otieno J, Chaiyaroj SC, Shi YP, Nahlen BL, Lal AA, Udhayakumar V. Immunity to placental malaria. IV. Placental malaria is associated with up-regulation of macrophage migration inhibitory factor in intervillous blood. J Infect Dis. 2002;186:1371–1375. doi: 10.1086/344322. [DOI] [PubMed] [Google Scholar]
- 34.Chaiyaroj SC, Rutta ASM, Muenthaisong K, Watkins P, Ubol MN, Looareesuwan S. Reduced levels of transforming growth factor-beta 1, interleukin-12 and increased migration inhibitory factor are associated with severe malaria. Acta Tropica. 2004;89:319–327. doi: 10.1016/j.actatropica.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 35.Boucher IW, Brzozowski AM, Brannigan JA, Schnick C, Smith DJ, Kyes SA, Wilkinson AJ. The crystal structure of superoxide dismutase from Plasmodium falciparum. BMC Struct Biol. 2006;6:20. doi: 10.1186/1472-6807-6-20. DOI: 10.1186/1472-6807-6-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boucher IW, McMillan PJ, Gabrielsen M, Akerman SE, Brannigan JA, Schnick C, Brzozowski AM, Wilkinson AJ, Muller S. Structural and biochemical characterization of a mitochondrial peroxiredoxin from Plasmodium falciparum. Mol Microbiol. 2006;61:948–959. doi: 10.1111/j.1365-2958.2006.05303.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mischke R, Kleemann R, Brunner H, Bernhagen J. Cross-linking and mutational analysis of the oligomerization state of the cytokine macrophage migration inhibitory factor (MIF) FEBS Lett. 1998;427:85–90. doi: 10.1016/s0014-5793(98)00400-1. [DOI] [PubMed] [Google Scholar]
- 38.Cherepkova OA, Lyutova EM, Eronina TB, Gurvits BY. Chaperone-like activity of macrophage migration inhibitory factor. Int J Biochem Cell Biol. 2006;38:43–55. doi: 10.1016/j.biocel.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 39.Calandra T, Echtenacher B, Roy DL, Pugin J, Metz CN, Hultner L, Heumann D, Mannel D, Bucala R, Glauser MP. Protection from septic shock by neutralization of macrophage migration inhibitory factor. Nat Med. 2000;6:164–170. doi: 10.1038/72262. [DOI] [PubMed] [Google Scholar]
- 40.Crichlow GV, Cheng KF, Dabideen D, Ochani M, Aljabari B, Pavlov VA, Miller EJ, Lolis E, Al-Abed Y. Alternative chemical modifications reverse the binding orientation of a pharmacophore scaffold in the active site of macrophage migration inhibitory factor. J Biol Chem. 2007;282:23089–23095. doi: 10.1074/jbc.M701825200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Crichlow GV, Lubetsky JB, Leng L, Bucala R, Lolis EJ. Structural and kinetic analyses of macrophage migration inhibitory factor active site interactions. Biochemistry. 2009;48:132–139. doi: 10.1021/bi8014423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lubetsky JB, Dios A, Han JL, Aljabari B, Ruzsicska B, Mitchell R, Lolis E, Al-Abed Y. The tautomerase active site of macrophage migration inhibitory factor is a potential target for discovery of novel anti-inflammatory agents. J Biol Chem. 2002;277:24976–24982. doi: 10.1074/jbc.M203220200. [DOI] [PubMed] [Google Scholar]
- 43.Orita M, Yamamoto S, Katayama N, Aoki M, Takayama K, Yamagiwa Y, Seki N, Suzuki H, Kurihara H, Sakashita H, Takeuchi M, Fujita S, Yamada T, Tanaka A. Coumarin and chromen-4-one analogues as tautomerase inhibitors of macrophage migration inhibitory factor: discovery and X-ray crystallography. J Med Chem. 2001;44:540–547. doi: 10.1021/jm000386o. [DOI] [PubMed] [Google Scholar]
- 44.Sun HW, Bernhagen J, Bucala R, Lolis E. Crystal structure at 2.6-angstrom resolution of human macrophage migration inhibitory factor. Proc Natl Acad Sci USA. 1996;93:5191–5196. doi: 10.1073/pnas.93.11.5191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Winner M, Meier J, Zierow S, Rendon BE, Crichlow GV, Riggs R, Bucala R, Leng L, Smith N, Lolis E, Trent JO, Mitchell RA. A novel, macrophage migration inhibitory factor suicide substrate inhibits motility and growth of lung cancer cells. Cancer Res. 2008;68:7253–7257. doi: 10.1158/0008-5472.CAN-07-6227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Taylor AB, Johnson WH, Czerwinski RM, Li HS, Hackert ML, Whitman CP. Crystal structure of macrophage migration inhibitory factor-complexed with (E)-2-fluoro-p-bydroxycinnamate at 1.8 angstrom resolution: implications for enzymatic catalysis and inhibition. Biochemistry. 1999;38:7444–7452. doi: 10.1021/bi9904048. [DOI] [PubMed] [Google Scholar]
- 47.Leng L, Metz CN, Fang Y, Xu J, Donnelly S, Baugh J, Delohery T, Chen Y, Mitchell RA, Bucala R. MIF signal transduction initiated by binding to CD74. J Exp Med. 2003;197:1467–1476. doi: 10.1084/jem.20030286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Glaser F, Pupko T, Paz I, Bell RE, Bechor-Shental D, Martz E, Ben-Tal N. ConSurf: identification of functional regions in proteins by surface-mapping of phylogenetic information. Bioinformatics. 2003;19:163–164. doi: 10.1093/bioinformatics/19.1.163. [DOI] [PubMed] [Google Scholar]
- 49.El-Turk F, Cascella M, Ouertatani-Sakouhi H, Narayanan RL, Leng L, Bucala R, Zweckstetter M, Rothlisberger U, Lashuel HA. The conformational flexibility of the carboxy terminal residues 105-114 is a key modulator of the catalytic activity and stability of macrophage migration inhibitory factor. Biochemistry. 2008;47:10740–10756. doi: 10.1021/bi800603x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Suzuki M, Sugimoto H, Nakagawa A, Tanaka I, Nishihira J, Sakai M. Crystal structure of the macrophage migration inhibitory factor from rat liver. Nat Struct Biol. 1996;3:259–266. doi: 10.1038/nsb0396-259. [DOI] [PubMed] [Google Scholar]
- 51.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 52.Zang XX, Taylor P, Wang JM, Meyer DJ, Scott AL, Walkinshaw MD, Maizels RM. Homologues of human macrophage migration inhibitory factor from a parasitic nematode—gene cloning, protein activity, and crystal structure. J Biol Chem. 2002;277:44261–44267. doi: 10.1074/jbc.M204655200. [DOI] [PubMed] [Google Scholar]
- 53.Cho Y, Jones BF, Vermeire JJ, Leng L, DiFedele L, Harrison LM, Xiong H, Kwong YK, Chen Y, Bucala R, Lolis E, Cappello M. Structural and functional characterization of a secreted hookworm macrophage migration inhibitory factor (MIF) that interacts with the human MIF receptor CD74. J Biol Chem. 2007;282:23447–23456. doi: 10.1074/jbc.M702950200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kamir D, Zierow S, Leng L, Cho Y, Diaz Y, Griffith J, McDonald C, Merk M, Mitchell RA, Trent J, Chen Y, Kwong YK, Xiong H, Vermeire J, Cappello M, McMahon-Pratt D, Walker J, Bernhagen J, Lolis E, Bucala R. A Leishmania ortholog of macrophage migration inhibitory factor modulates host macrophage responses. J Immunol. 2008;180:8250–8261. doi: 10.4049/jimmunol.180.12.8250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Leng L, Bucala R. Insight into the biology of macrophage migration inhibitory factor (MIF) revealed by the cloning of its cell surface receptor. Cell Res. 2006;16:162–168. doi: 10.1038/sj.cr.7310022. [DOI] [PubMed] [Google Scholar]
- 56.Jasanoff A, Wagner G, Wiley DC. Structure of a trimeric domain of the MHC class II-associated chaperonin and targeting protein Ii. Embo J. 1998;17:6812–6818. doi: 10.1093/emboj/17.23.6812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cournia Z, Leng L, Gandavadi S, Du X, Bucala R, Jorgensen WL. Discovery of human macrophage migration inhibitory factor (MIF)-CD74 antagonists via virtual screening. J Med Chem. 2009;52:416–424. doi: 10.1021/jm801100v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Berndt C, Lillig CH, Holmgren A. Thioredoxins and glutaredoxins as facilitators of protein folding. Biochim Biophys Acta. 2008;1783:641–650. doi: 10.1016/j.bbamcr.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 59.de Jong RN, Mysiak ME, Meijer LA, van der Linden M, van der Vliet PC. Recruitment of the priming protein pTP and DNA binding occur by overlapping Oct-1 POU homeodomain surfaces. Embo J. 2002;21:725–735. doi: 10.1093/emboj/21.4.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Otwinowski Z, Minor W. Processing of X-ray data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 61.Dodson EJ, Winn M, Ralph A. Collaborative computational project, number 4: providing programs for protein crystallography. Methods Enzymol. 1997;277:620–633. doi: 10.1016/s0076-6879(97)77034-4. [DOI] [PubMed] [Google Scholar]
- 62.Long F, Vagin AA, Young P, Murshudov GN. BALBES: a molecular-replacement pipeline. Acta Crystallogr D Biol Crystallogr. 2008;64:125–132. doi: 10.1107/S0907444907050172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 64.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 65.Vagin A, Teplyakov A. MOLREP: an automated program for molecular replacement. J Appl Crystallogr. 1997;30:1022–1025. [Google Scholar]
- 66.Matthews BW. Solvent content of protein crystals. J Mol Biol. 1968;33:491–497. doi: 10.1016/0022-2836(68)90205-2. [DOI] [PubMed] [Google Scholar]
- 67.Gouet P, Courcelle E, Stuart DI, Metoz F. ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics. 1999;15:305–308. doi: 10.1093/bioinformatics/15.4.305. [DOI] [PubMed] [Google Scholar]
- 68.Potterton L, McNicholas S, Krissinel E, Gruber J, Cowtan K, Emsley P, Murshudov GN, Cohen S, Perrakis A, Noble M. Developments in the CCP4 molecular-graphics project. Acta Crystallogr D Biol Crystallogr. 2004;60:2288–2294. doi: 10.1107/S0907444904023716. [DOI] [PubMed] [Google Scholar]