

Abstract
The lipooligosaccharide (LOS) of Neisseria meningitidis contains heptose (Hep) residues that are modified with phosphoethanolamine (PEtn) at the 3 (3-PEtn) and/or 6 (6-PEtn) position. The lpt3 (NMB2010) and lpt6 (NMA0408) genes of N. meningitidis, which are proposed to encode the required HepII 3- and 6-PEtn transferases, respectively, were cloned and overexpressed as C-terminally polyhistidine-tagged fusion proteins in Escherichia coli and found to localize to the inner membrane, based on sucrose density gradient centrifugation. Lpt3-His6 and Lpt6-His6 were purified from Triton X-100-solubilized membranes by nickel chelation chromatography, and dot blot analysis of enzymatic reactions with 3-PEtn- and 6-PEtn-specific monoclonal antibodies demonstrated conclusively that Lpt3 and Lpt6 are phosphatidylethanolamine-dependent LOS HepII 3- and 6-PEtn transferases, respectively, and that both enzymes are capable of transferring PEtn to both fully acylated LOS and de-O-acylated (de-O-Ac) LOS. Further enzymatic studies using capillary electrophoresis-mass spectrometry (MS) demonstrated that both Lpt3 and Lpt6 are capable of transferring PEtn to de-O-Ac LOS molecules already containing PEtn at the 6 and 3 positions of HepII, respectively, demonstrating that there is no obligate order of PEtn addition in the generation of 3,6-di-PEtn LOS moieties in vitro.
The Gram-negative bacterium Neisseria meningitidis is the causative agent of invasive meningococcal disease, which is a major cause of morbidity and mortality worldwide, especially in children (33). These infections are severe, causing death in 10% of cases and serious long-term effects in 10 to 20% of patients that survive the disease (7). While capsular-polysaccharide-based vaccines against serogroup C are readily available and a combination conjugate vaccine against serogroups A, C, Y, and W-135 has recently been released, there is currently no vaccine that offers protection against serogroup B meningococcal infections. This is primarily due to the poor immunogenicity of the α-2,8-linked N-acetylneuraminic acid capsular polysaccharide produced by serogroup B (44) combined with concerns that structural similarities between the capsule and glycans on host cell surface structures could cause a serogroup B-based capsular conjugate vaccine to elicit an autoimmune response (4, 23). For these reasons, current strategies for vaccine development have focused on alternative surface antigens, including outer membrane proteins and lipooligosaccharides (LOS).
LOS is an important virulence determinant for Neisseria spp., and the severity of the illness caused by neisserial infection correlates with the levels of LOS-containing outer membrane vesicles being released (33). The LOS of Neisseria spp. (Fig. 1) lacks the repeating O-antigen region typically observed in enteric bacteria and contains a 1,3-di-l-glycero-α-d-manno-heptose (di-l,d-Hep)-di-3-deoxy-d-manno-octulosonic acid (di-Kdo) inner-core backbone (15). The heptose proximal to Kdo (HepI) is substituted at the 4 position with β-d-glucose (β-d-Glc), and additional substitutions to this Glc comprise the outer-core region. The distal heptose (HepII) is invariably substituted at the 2 position with α-d-GlcNAc and is also typically substituted with phosphoethanolamine (PEtn) at the 3 (3-PEtn) and/or 6 (6-PEtn) position. The presence of 7-PEtn has also been reported (22, 30); however, recent nuclear magnetic resonance (NMR) studies in our laboratory have confirmed that the LOS from these strains have PEtn substitutions solely at the 6 position (34). Approximately 70% of N. meningitidis strains contain 3-PEtn alone, as demonstrated by reactivity to the 3-PEtn-specific monoclonal antibody (MAb) L3B5 (20), while the majority of the remaining strains contain 6-PEtn alone, with only a few strains containing 3,6-di-PEtn or no PEtn.
FIG. 1.

Neisserial lipooligosaccharides utilized as substrates in this study. (A) Structures of the core regions of the different LOS substrates. (B) Differences in the lipid A region after different chemical modifications. de-O-Ac, LOS after de-O-acylation by hydrazinolysis; HF-de-O-Ac, de-O-Ac LOS after dephosphorylation by incubation with HF; KOH, LOS after complete deacylation by incubation with KOH and re-N-acetylation.
The genes encoding the putative HepII 3- and 6-PEtn transferases in N. meningitidis, lpt3 (NMB2010) and lpt6 (NMA0408), respectively, have previously been identified, and mutants with chromosomal knockouts of these genes were shown to lack PEtn on HepII (20, 43). Interestingly, however, only a small proportion of strains containing both lpt3 and lpt6 were found to exhibit mixtures of 3- and 6- and/or 3,6-di-PEtn-substituted LOS (43). While this could potentially be explained by the presence of a competing 3-Glc transferase gene, lgtG, an N. meningitidis NMB lgtG knockout mutant was generated previously and found not to produce 3-PEtn-containing LOS in spite of the lpt3 gene being present in the chromosome (38), suggesting a more complex scenario. Biochemical characterization of the proteins encoded by the N. meningitidis lpt3 and lpt6 genes is therefore warranted not only to confirm that Lpt3 and Lpt6 exhibit HepII 3- and 6-PEtn transferase activities, respectively, but also to gain insight into the substrate specificities of these enzymes that may explain these observations. The lpt3 gene of N. gonorrhoeae (lpt3Ng) was recently identified by O'Connor et al. (24), based on homology to N. meningitidis lpt3, and they were able to demonstrate that Lpt3Ng is capable of restoring MAb 2-1-L8 reactivity (3-PEtn dependent) to LOS from an N. gonorrhoeae lpt3 mutant. However, they were unable to perform more extensive characterization, due to low activity levels and instability of the purified Lpt3Ng protein (24).
In this study, we describe the overexpression, purification, and functional characterization of both Lpt3 and Lpt6 of N. meningitidis by using a combination of immunological and mass spectrometric techniques.
MATERIALS AND METHODS
Reagents, strains, plasmids, and standard culture conditions.
Unless otherwise stated, all reagents were obtained from Sigma-Aldrich Canada (Oakville, ON, Canada), and all media were obtained from Difco Laboratories (Detroit, MI). All aqueous solutions were prepared using water purified by a Milli-Q Synthesis A10 system (Millipore Canada, Mississauga, ON, Canada). All restriction endonucleases were obtained from New England Biolabs (Mississauga, ON, Canada). The bacterial strains and plasmids used in this study are listed in Table 1. Unless otherwise stated, broth cultures were incubated at 37°C with agitation at 200 rpm, and cultures grown on solid media were incubated at 37°C. Escherichia coli strains were routinely propagated in Luria-Bertani broth (LB) or on Luria-Bertani agar (LA) supplemented with 50 μg ml−1 kanamycin (Km) where appropriate. N. meningitidis strains were routinely propagated on enriched chocolate agar (Oxoid Company, Nepean, ON, Canada). Large-scale 22-liter N. meningitidis fermenter cultures for LOS preparation experiments (below) were grown in Todd-Hewitt-Columbia broth (15 g liter−1 Bacto Todd-Hewitt broth, 17.5 g liter−1 Columbia broth) supplemented with 1% dextrose and 2 ml of the antifoaming agent MAZU DF204 (Mazer Chemicals, Gurnee, IL) in a 30-liter fermenter (newMBR, Zurich, Switzerland) and killed by the addition of phenol (2% [wt/vol] final concentration; 4-h incubation) prior to lyophilization.
TABLE 1.
Strains and plasmids used in this study
| Strain/plasmid | Descriptiona | Source/reference |
|---|---|---|
| Strains | ||
| Neisseria meningitidis | ||
| MC58 | L3 immunotyping reference strain | 20 |
| MC58 galE | L3 galE | 12 |
| MC58 galE lpt3 | L3 galE lpt3::Emr | 19 |
| 89I | L4 immunotyping reference strain | |
| 89I galE | L4 galE::Emr | 39 |
| Escherichia coli | ||
| DH5α | F− φ80 LacZ M15 (lacZY-argF) supE44 thi-1 gyrA96 recA1 endA1 relA1 hsdR17 | |
| BL21(DE3) | F−ompT hsdSB(rB− mB−) gal dcm (DE3) | Novagen |
| CQW50 | Lpt3-His6 expression strain; BL21(DE3) containing pCQJR3; Kmr | This study |
| CQW51 | Lpt6-His6 expression strain; BL21(DE3) containing pCQJR7; Kmr | This study |
| Plasmids | ||
| pET28a | T7 expression vector, N- and C-terminal His6 tags; 5.4 kb; Kmr | Novagen |
| pET30a | T7 expression vector, N- and C-terminal His6 tags; 5.4 kb; Kmr | Novagen |
| pCQJR3 | Lpt3-His6 expression vector; PCR product of CQ-160 and CQ-162, containing lpt3, cloned into the NdeI and XhoI sites of pET30a; 6.8 kb; Kmr | This study |
| pCQJR7 | Lpt6-His6 expression vector; PCR product of CQ-222 and CQ-223, containing lpt6, cloned into the NcoI and XhoI sites of pET28a; 7.0 kb; Kmr | This study |
Em, erythromycin.
DNA manipulation.
Chromosomal DNA was isolated from N. meningitidis according to the method of Goldberg and Ohman (6), and plasmid DNA was isolated using the Qiaprep Spin miniprep kit (Qiagen, Ijamsville, MD) as described by the manufacturer. DNA restriction endonucleases were used as recommended by the manufacturer, and ligation of restriction endonuclease-digested vectors and insert DNA was performed using T4 DNA ligase (Invitrogen Life Technologies, Carlsbad, CA) at 15°C for 16 h. The recipient strain for all cloning experiments was E. coli DH5α, and plasmids were introduced according to the chemical transformation method of Weston et al. (41). PCR amplification reactions were performed with a GeneAmp PCR system 9700 thermocycler (Applied Biosystems Canada, Streetsville, ON, Canada) using PWO polymerase (Roche Diagnostics Corporation, Indianapolis, IN) according to the manufacturer's specifications. The lpt3 gene (NMB2010) was amplified from N. meningitidis MC58 chromosomal DNA using the oligonucleotide primers CQ-160 (forward, 5′-GTGAAATACATATGATGAAAAAATCTTTC-3′) and CQ-162 (reverse, 5′-GTTTTTACTCGAGACATTGCGGATAAAC-3′), which incorporate NdeI and XhoI sites (underlined), respectively, and the lpt6 gene (NMA0408) was amplified from N. meningitidis 89I chromosomal DNA using the primers CQ-222 (forward, 5′-GGGATGTCATGATTGCCTATGTTTTC-3′) and CQ-223 (reverse, 5′-GCGGCAAAGTCGACACGGGCAATTTTC-3′), which incorporate BspHI and SalI sites (underlined), respectively. PCR products were purified using UltraClean 15 (Mo Bio Laboratories, Solana Beach, CA). The lpt3 PCR product and pET30a (Novagen, Madison, WI) were digested with NdeI and XhoI, purified using UltraClean 15 (Mo Bio Laboratories), and ligated together to produce the C-terminally His6-tagged-Lpt3 (Lpt3-His6) expression vector pCQJR3. The lpt6 PCR product was digested with BspHI and SalI and cloned into the compatible NcoI and XhoI sites of pET28a (Novagen) to generate the C-terminally His6-tagged-Lpt6 (Lpt6-His6) expression vector pCQJR7.
Purification and chemical modification of LOS.
LOS samples were purified from bacterial cultures according to the phenol-hot water method of Westphal and Jann (42) using lyophilized cells from fermenter growths that were sequentially washed with ethanol, acetone, and petroleum ether (250 ml/200 g of cells, twice for each solvent) and dried overnight at room temperature. Chemical removal of O-linked acyl chains from purified LOS samples was performed according to the hydrazinolysis method of Holst et al. (8). Chemical removal of phosphate moieties from de-O-Ac LOS was achieved by treatment with 48% hydrofluoric acid (HF; Fisher Scientific, Nepean, ON, Canada) according to the method of Caroff et al. (2). Complete deacylation (removal of both N- and O-linked fatty acids) of LOS (designated KOH LOS) was achieved by the method of Holst et al. (9), except that after incubation with KOH, the samples were neutralized by dropwise addition of acetic anhydride to simultaneously neutralize the reaction and restore the N-acetyl group on the GlcNAc residue of the core. Isolation of core oligosaccharide was performed by mild acid hydrolysis as previously described (8).
Overexpression, subcellular localization, and purification of Lpt3-His6 and Lpt6-His6.
The pCQW3 and pCQW7 expression constructs were each transformed into E. coli BL21(DE3) (Novagen) to generate the Lpt3-His6 and Lpt6-His6 overexpression strains CQW50 and CQW51, respectively. Overexpression and purification conditions were identical for both, proteins with the exception of the pHs of the buffers used (pH 8.5 for Lpt3-His6 and pH 7.5 for Lpt6-His6), due to Lpt6-His6 being inactive in reactions at pH 8.5 (data not shown). The overexpression conditions were adapted from those used by Reynolds et al. (32) for the overexpression of the lipid A-PEtn transferase EptB. Briefly, 100 ml of LB with 50 μg ml−1 Km in a 500-ml flask was inoculated with 0.5 ml of an overnight culture and incubated at 37°C until mid-exponential phase. This culture was used to inoculate 6 liters of LB with 50 μg ml−1 Km (6 volumes of 1 liter in a 4-liter flask) to a starting optical density at 600 nm (OD600) of 0.02, IPTG (isopropyl-β-d-thiogalactopyranoside) was added to 0.1 mM, and the culture was incubated at 37°C for 16 h (final OD600, 2.0 to 2.2). The cells were harvested by centrifugation (4,100 × g, 10 min, 4°C; Sorvall SLA-3000 rotor), resuspended in 100 ml of 100 mM HEPES-150 mM NaCl supplemented with Complete EDTA-free protease inhibitor cocktail (Roche Diagnostics Corporation), and lysed by passage three times through an Emulsiflex C-5 homogenizer (Avestin Inc., Ottawa, ON, Canada). Following removal of cellular debris and inclusion bodies by centrifugation (10,000 × g, 10 min, 4°C; Sorvall SLA-3000 rotor), the resultant soluble fraction was subjected to ultracentrifugation (170,000 × g, 60 min, 4°C; LE-80 60-Ti rotor) to pellet membranes. The membranes were then either separated into inner and outer membrane fractions by sucrose gradient centrifugation according to the method described by Weadge and Clarke (40), by using NADH oxidase activity (26) and the presence/absence of Kdo (16) to assess the purity of the membrane fractions, or solubilized to allow the purification of the overexpressed enzymes. Briefly, the membrane pellets were resuspended in 90 ml of solubilization buffer (100 mM HEPES, 150 mM NaCl, 10% glycerol, 2% Triton X-100) using a Wheaton Potter-Evehjem homogenizer with a PTFE pestle (Fisher Scientific), incubated on ice for 30 min, and then subjected to ultracentrifugation. The supernatant was decanted, combined with 0.5 ml of nickel-nitrilotriacetic acid (Ni-NTA) agarose preequilibrated with solubilization buffer containing 5 mM imidazole, and gently mixed on a nutating mixer for 30 min at 4°C before the buffer-resin slurry was loaded into a column to collect the resin and allow all unbound proteins to flow through. The resin was washed extensively with solubilization buffer containing 20 mM imidazole (50 ml), followed by elution of the target proteins by the addition of 4 bed volumes (2 ml) of solubilization buffer containing 100 mM imidazole to the column and collecting the second bed volume of the eluent. Purified enzyme preparations were adjusted to 25% glycerol and stored at −20°C. Purified proteins were quantitated using the detergent compatible DC protein assay kit (Bio-Rad Laboratories [Canada] Ltd., Mississauga, ON, Canada), and separated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) according to the method of Laemmli (17), except that samples were combined with sample buffer and heated at 37°C for 1 h as described previously (36, 37). Following electrophoresis, samples were either visualized by staining with Coomassie brilliant blue R or transferred electrophoretically to BioTrace nitrocellulose membrane (Gelman Sciences, Ann Arbor, MI) and subjected to Western immunoblot analysis according to the method of Burnette (1), using monoclonal anti-polyhistidine peroxidase conjugate clone His-1 (Sigma Aldrich) according to the manufacturer's instructions with SuperSignal West Pico chemiluminescent substrate (Thermo Scientific, Rockford, IL) as the peroxidase substrate.
Molecular weight determination for purified Lpt3-His6 and Lpt6-His6.
Theoretical molecular weights of Lpt3-His6 and Lpt6-His6 were determined using the ProtParam program (http://www.expasy.org/tools/protparam.html). Molecular weights of the purified proteins were experimentally determined by matrix-assisted laser desorption ionization-time of flight (MALDI-TOF) mass spectrometry (MS). Samples were acidified by the addition of trifluoroacetic acid (TFA; 0.1% final concentration), spotted (1 μl) on a target plate with an equal volume of sinapinic acid solution (12 mg ml−1 sinapinic acid, 0.1% TFA, 33% acetonitrile), and dried. Samples were analyzed using a Voyager DE-STR MALDI-TOF mass spectrometer (Applied Biosystems, Foster City, CA) operated in the linear/positive-ion mode with the following configuration: accelerating voltage, 25,000 V; grid voltage, 92%; guide wire voltage, 0.30%; delay time, 200 ns; mass range, 10,000 to 200,000 Da; low mass gate, 2,000 Da; laser power, 2,174; shots/spectrum: 400.
Functional characterization of Lpt3 and Lpt6.
Enzymatic activity was assayed in 20-μl reaction volumes containing either Lpt3-His6 or Lpt6-His6, 50 mM HEPES, 75 mM NaCl, 1% Triton X-100, 1 mM E. coli phosphatidylethanolamine (PtdEtn), and 1 μg μl−1 of the LOS or chemically modified LOS being evaluated as a substrate acceptor. To prepare the reactions, 2 μl of 10 mg ml−1 of E. coli PtdEtn was added to reaction tubes and the solvent was evaporated by heating at 42°C. After the reaction tubes were allowed to cool at room temperature, the PtdEtn was thoroughly resuspended in 10 μl of 0.5 mg ml−1 purified enzyme by pipetting up and down until the solution was no longer cloudy, followed by the addition of the LOS substrate of interest (10 μl of a 2 mg ml−1 stock). Samples were incubated at 25°C, 30°C, or 37°C overnight and analyzed either by dot blot analysis or by capillary electrophoresis-MS (CE-MS). Dot blot analyses were performed by spotting 2 μl of reaction mixture onto BioTrace nitrocellulose membrane (Gelman Sciences) and using either monoclonal antibody (MAb) L3B5 (specific for N. meningitidis LOS containing HepII-3-PEtn) (28) or MAb L2-16 (specific for N. meningitidis LOS containing HepII-6-PEtn) (5) as the primary antibody and alkaline phosphatase (AP)-conjugated goat anti-mouse F(ab′)2 fragment (Jackson ImmunoResearch Laboratories, West Grove, PA) as the secondary antibody.
CE-MS analysis of Lpt3 and Lpt6 enzymatic reactions.
CE-MS analyses of enzymatic reactions were performed on an Applied Biosystems/MDS Sciex 4000QTRAP (Concord, ON, Canada) coupled to a Prince Technologies CE system (Emmen, The Netherlands), using 10 mM ammonium acetate as the separation buffer. Spectra were acquired using precursor ion scanning for either de-O-Ac lipid A in the negative-ion mode (m/z 951.0) or HexNAc in the positive-ion mode (m/z 204.0) and a collision energy of 65 eV. To maximize detection sensitivity, a narrow scan range (m/z 900 to 1,300) and low-resolution settings were used.
Nucleotide sequence accession number.
The nucleotide sequence of lpt6 from N. meningitidis 89I has been deposited in GenBank (accession number FJ969851).
RESULTS
Lpt3 and Lpt6 are inner membrane proteins.
The cellular locations of Lpt3 and Lpt6 were determined by lysis and fractionation of the expression strains CQW50 and CQW51, respectively, after induction with IPTG (isopropyl-β-d-thiogalactopyranoside). Samples of complete lysate, complete membrane fraction, and inner and outer membrane fractions were analyzed by SDS-PAGE (Fig. 2A [Lpt3-His6] and B [Lpt6-His6]) and Western immunoblotting (Fig. 2C [Lpt3-His6] and D [Lpt6-His6]). Inner membrane fractions exhibited high levels of NADH oxidase activity and did not contain Kdo at detectable levels, while outer membrane fractions contained significant levels of Kdo and exhibited minimal NADH oxidase activity (data not shown). For both enzymes, anti-His MAb-reactive bands were observed in the complete lysate, complete membrane, and inner membrane fractions, demonstrating that both Lpt3 and Lpt6 are membrane proteins, consistent with in silico secondary structure predictions that both enzymes are membrane-anchored proteins (data not shown) and, more specifically, that they localize to the inner membrane.
FIG. 2.
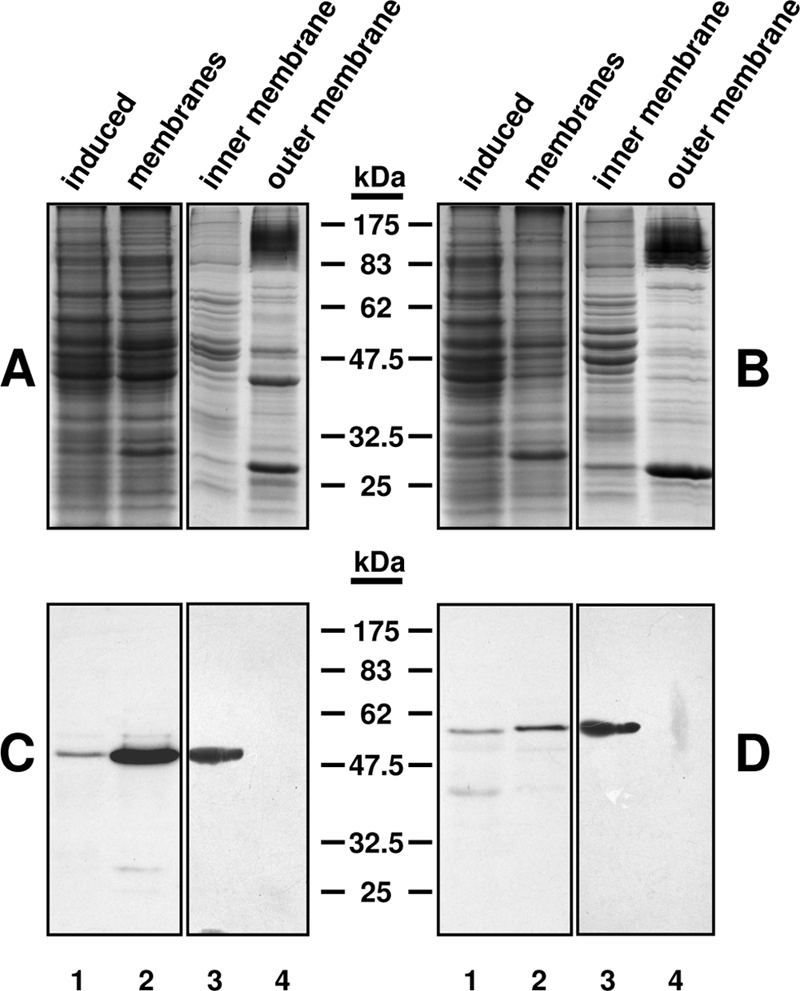
Subcellular localization of Lpt3 and Lpt6. Induced cell culture lysates (lanes 1) were clarified by centrifugation to remove inclusion bodies, followed by isolation of complete membrane fractions by ultracentrifugation (lanes 2), which were further separated into inner membrane (lanes 3) and outer membrane (lanes 4) fractions by sucrose gradient centrifugation. (A and B) Coomassie-stained 12.5% SDS-polyacrylamide gel of fractionation of lysates from induced cultures of the Lpt3-His6 expression strain CQWJR1 (A) and the Lpt6-His6 expression strain CQWJR2 (B). (C and D) Western immunoblot of the samples in panels A and B using monoclonal anti-polyhistidine peroxidase conjugate clone His-1 (Sigma Aldrich). For inner and outer membrane fractions, 10 μg total protein was loaded per lane. Note that both Lpt3-His6 and Lpt6-His6 localize to the inner membrane fraction.
Purified Lpt3 and Lpt6 migrate aberrantly in SDS-PAGE.
Purification of Lpt3-His6 and Lpt6-His6 by metal chelation chromatography purification resulted in 0.5-ml samples with typical yields between 0.7 and 0.9 mg ml−1 (total yield, 0.35 to 0.45 mg of protein) and approximately 85 to 90% purity, based on visual inspection of SDS-PAGE gels (Fig. 3, inset panels). Both Lpt3-His6 and Lpt6-His6 migrated in SDS-PAGE with apparent molecular masses (∼47 kDa and ∼55 kDa, respectively) that were significantly smaller than the corresponding predicted molecular weights for the full-length His6-tagged enzymes (60,277.3 and 63,368.9, respectively), but MALDI-TOF MS spectra of the purified enzyme preparations (Fig. 3A [Lpt3-His6] and B [Lpt6-His6]) each contained a single prominent peak that corresponded closely to the predicted molecular weight for the full-length His6-tagged protein in question (Lpt3-His6, m/z 60,308; Lpt6-His6, m/z 63,249), as well as minor secondary [M + 2H]2+ peaks (Lpt3-His6, m/z 30,177; Lpt6-His6, m/z 31,576).
FIG. 3.

Molecular mass determination of purified Lpt3-His6 and Lpt6-His6. MALDI-TOF MS analysis of purified Lpt3-His6 (A) and Lpt6-His6 (B) and their apparent molecular masses (kDa) as determined by SDS-PAGE (insets of panels A and B, respectively). Note that the MALDI-TOF MS m/z values for Lpt3-His6 and Lpt6-His6 match closely with their predicted molecular weights (60,277.3 and 63,368.9, respectively).
Lpt3 and Lpt6 catalyze phosphatidylethanolamine-dependent HepII-PEtn transferase reactions with distinct regiospecificities.
Initial enzymatic reactions were performed with N. meningitidis MC58 galE lpt3 LOS (Fig. 1A) in both fully acylated (wild-type) and de-O-Ac forms (Fig. 1B). Reaction volumes were initially incubated at 25°C, 30°C, or 37°C overnight to determine the optimal reaction temperatures for Lpt3 and Lpt6 based on relative intensities in dot blot analyses. For both enzymes, reaction volumes incubated at 30°C exhibited the strongest reactivity in dot blot analyses, while reaction volumes incubated at 25°C and 37°C exhibited moderate and weak reactivity levels, respectively (data not shown), and control reactions lacking phosphatidylethanolamine showed no reactivity (Fig. 4A). All subsequent reactions were performed at 30°C and contained 1 mg ml−1 phosphatidylethanolamine. Reactions with Lpt3 restored reactivity to MAb L3B5 (3-PEtn specific) (Fig. 4B) but not to MAb L2-16 (6-PEtn specific) (Fig. 4C), while Lpt6 reactions were recognized by MAb L2-16 (Fig. 4C) but not by MAb L3B5 (Fig. 4B), demonstrating not only that both Lpt3 and Lpt6 are PEtn transferases but that they have distinctly different regiospecificities. These results provided evidence that Lpt3 and Lpt6 are phosphatidylethanolamine-dependent HepII 3- and 6-PEtn transferases, respectively.
FIG. 4.

Lpt3 and Lpt6 exhibit LOS HepII 3- and 6-phosphoethanolamine transferase activities, respectively. (A) Dot blot analyses of Lpt3-His6 and Lpt6-His6 reactions (rxns) with N. meningitidis galE lpt3 de-O-Ac LOS in the absence or presence of 1 mg ml−1 phosphatidylethanolamine, using HepII 3-PEtn-specific MAb L3B5 for Lpt3-His6 reactions and HepII 6-PEtn-specific MAb L2-16 for Lpt6-His6 reactions. (B and C) Dot blot analyses of Lpt3-His6 and Lpt6-His6 reactions with N. meningitidis MC58 galE lpt3 LOS and de-O-Ac LOS, using MAb L3B5 (B) and MAb L2-16 (C), respectively. Negative controls were N. meningitidis MC58 galE lpt3 LOS and de-O-Ac LOS, 3-PEtn controls were N. meningitidis MC58 galE LOS and de-O-Ac LOS, and 6-PEtn controls were N. meningitidis 89I galE LOS and de-O-Ac LOS. Note that MAb L3B5 does not react in Lpt6-His6 reactions and MAb L2-16 does not react in Lpt3-His6 reactions.
Characterization of the substrate specificities of Lpt3 and Lpt6.
To gain insight into the substrate specificities of Lpt3 and Lpt6, enzymatic reactions were performed with each of the enzymes and a range of LOS substrates that varied either in the composition of the core region (Fig. 1A) or by differences in the lipid A region (Fig. 1B). The LOS substrates and their respective PEtn-substituted products were not initially observed in CE-MS analyses under standard conditions. The substrates and their corresponding PEtn-modified reaction products were, however, able to be analyzed directly from reaction mixtures by CE-MS when the mass detection range was limited and precursor scanning for either de-O-Ac lipid A (m/z 951.0 in negative-ion mode) or HexNAc (m/z 204.0 in positive ion mode) was used as appropriate. This optimized CE-MS methodology was essential for the detailed characterization of Lpt3 and Lpt6 reactions with various substrates, as several reactions could not have been effectively analyzed by dot blot analysis. As summarized in Table 2, Lpt3-His6 was found to catalyze the addition of PEtn to N. meningitidis galE lpt3 LOS in de-O-Ac, HF-de-O-Ac, and KOH forms but not core oligosaccharide (data not shown). In contrast, Lpt6 was found to catalyze the addition of PEtn to N. meningitidis galE lpt3 de-O-Ac LOS but was unable to transfer PEtn to the HF-de-O-Ac or KOH form at detectable levels. Furthermore, Lpt3 was able to add PEtn in the presence of 6-PEtn on HepII, as demonstrated in the reaction including N. meningitidis 89I galE de-O-Ac LOS as a substrate acceptor, and Lpt6 was able to add PEtn in the presence of 3-PEtn on HepII, as demonstrated by the reaction including N. meningitidis MC58 galE de-O-Ac LOS. These determinations would not have been possible without CE-MS analysis of the aforementioned reactions, since the resultant spectra clearly indicated the presence of LOS moieties containing two phosphoethanolamine groups, which could not have been determined by immunological means. Representative CE-MS spectra for Lpt3-His6 and Lpt6-His6 reactions with MC58 galE lpt3 de-O-Ac LOS are shown in Fig. 5. Peaks with an m/z of 1,070.5 correspond to the [M − 2H]2− ion of the substrate, and peaks with an m/z of 1,132.0 correspond to the [M − 2H]2− ion of the substrate plus PEtn. The minor peaks observed for the substrate (Fig. 5A) correspond to ions as follows: m/z 1,110.5, [M − 2H]2− of substrate plus PO4; m/z 1,151.5, [M − 2H]2− of substrate plus hexose (Hex); m/z 1,189.5, [M − 2H]2− HEPES adduct of the substrate. Note that the small m/z 1,132.0 peak in Fig. 5A corresponds to a minor form of the substrate that contains a PEtn substitution on the lipid A region described previously (3) similar to the case for the minor forms with PO4 (3) and Hex (18).
TABLE 2.
CE-MS analyses of Lpt3-His6 and Lpt6-His6 reactions with various LOS substrates
| Sample | Observed ion (m/z)a |
Relative intensityd | Molecular mass (Da) |
Proposed compositione | ||
|---|---|---|---|---|---|---|
| [M + 2H]2+ | [M − 2H]2− | Observed | Calculated | |||
| N. meningitidis MC58 galE lpt3 | ||||||
| de-O-Ac LOS | 1,070.5 | 1 | 2,143.0 | 2,143.04 | Hex·HexNAc·Hep2·Kdo2·lipid A (de-O-Ac) | |
| Lpt3-His6 + de-O-Ac LOS | 1,132.0 | 0.39 | 2,266.0 | 2,266.09 | PEtn·Hex·HexNAc·Hep2·Kdo2·lipid A (de-O-Ac) | |
| 1,070.5 | 1 | 2,143.0 | 2,143.04 | Hex·HexNAc·Hep2·Kdo2·lipid A (de-O-Ac) | ||
| Lpt6-His6 + de-O-Ac LOS | 1,132.0 | 0.18 | 2,266.0 | 2,266.09 | PEtn·Hex·HexNAc·Hep2·Kdo2·lipid A (de-O-Ac) | |
| 1,070.5 | 1 | 2,143.0 | 2,143.04 | Hex·HexNAc·Hep2·Kdo2·lipid A (de-O-Ac) | ||
| de-O-Ac HF LOS | 992.5b,c | 1 | 1,983.0 | 1,983.08 | Hex·HexNAc·Hep2·Kdo2·lipid A (de-O-Ac-HF) | |
| Lpt3-His6 + de-O-Ac HF LOS | 1,054.0b,c | 0.31 | 2,106.0 | 2,106.03 | PEtn·Hex·HexNAc·Hep2·Kdo2·lipid A (de-O-Ac-HF) | |
| 992.5b,c | 1 | 1,983.0 | 1,983.08 | Hex·HexNAc·Hep2·Kdo2·lipid A (de-O-Ac-HF) | ||
| Lpt6-His6+ de-O-Ac HF LOS | 992.5b,c | 1 | 1,983.0 | 1,983.08 | Hex·HexNAc·Hep2·Kdo2·lipid A (de-O-Ac-HF) | |
| KOH LOS | 886.0 | 1 | 1,774.0 | 1,774.38 | Hex·HexNAc·Hep2·Kdo2·HexNAc2·(PO4)2 | |
| Lpt3-His6 + KOH LOS | 947.5 | 0.11 | 1,897.0 | 1,897.43 | PEtn·Hex·HexNAc·Hep2·Kdo2·HexNAc2·(PO4)2 | |
| 886.0 | 1 | 1,774.0 | 1,774.38 | Hex·HexNAc·Hep2·Kdo2·HexNAc2·(PO4)2 | ||
| Lpt6-His6 + KOH+NAc LOS | 886.0 | 1 | 1,774.0 | 1,774.38 | Hex·HexNAc·Hep2·Kdo2·HexNAc2·(PO4)2 | |
| N. meningitidis 89I galE | ||||||
| de-O-Ac LOS | 1,132.0c | 1 | 2,266.0 | 2,266.09 | PEtn·Hex·HexNAc·Hep2·Kdo2·lipid A (de-O-Ac) | |
| Lpt3-His6 + de-O-Ac LOS | 1,193.5c | 0.57 | 2,389.0 | 2,389.14 | PEtn2·Hex·HexNAc·Hep2·Kdo2·lipid A (de-O-Ac) | |
| 1,132.0c | 1 | 2,266.0 | 2,266.09 | PEtn·Hex·HexNAc·Hep2·Kdo2·lipid A (de-O-Ac) | ||
| N. meningitidis MC58 galE | ||||||
| de-O-Ac LOS | 1,132.5 | 1 | 2,267.0 | 2,266.09 | PEtn·Hex·HexNAc·Hep2·Kdo2·lipid A (de-O-Ac) | |
| Lpt6-His6 + de-O-Ac LOS | 1,194.0 | 0.12 | 2,390.0 | 2,389.14 | PEtn2·Hex·HexNAc·Hep2·Kdo2·lipid A (de-O-Ac) | |
| 1,132.5 | 1 | 2,267.0 | 2,266.09 | PEtn·Hex·HexNAc·Hep2·Kdo2·lipid A (de-O-Ac) | ||
Obtained using precursor scanning for de-O-Ac lipid A in negative-ion mode or HexNAc in positive-ion mode.
Contained minor loss-of-water ions ([M − H2O + 2H]2+).
Contained minor sodium adducts ([M + H + Na]2+ or [M − 3H + Na]2−).
Values are based on assigning a value of 1 to the largest peak in each spectrum.
Boldface indicates PEtn groups added by Lpt3 or Lpt6, as appropriate.
FIG. 5.

Analysis of Lpt3-His6 and Lpt6 reactions by CE-MS. Shown are CE-MS spectra for N. meningitidis MC58 galE lpt3 de-O-Ac LOS (substrate) (A), the Lpt3-His6 reaction using N. meningitidis MC58 galE lpt3 de-O-Ac LOS as the substrate (B), and the Lpt6-His6 reaction using N. meningitidis MC58 galE lpt3 de-O-Ac LOS as the substrate (C). Spectra were obtained in negative-ion mode, using precursor ion scanning for de-O-Ac lipid A (m/z 951.0). See the text for details regarding the minor peaks observed.
DISCUSSION
The lpt3 and lpt6 genes of N. meningitidis are required for the addition of PEtn to the LOS inner core (20, 43), and in this study, we have demonstrated that Lpt3 and Lpt6 are in fact the HepII 3- and 6-PEtn transferases, respectively. This is the first report of the functional characterization of Lpt6, and while the activity of Lpt3 from N. gonorrhoeae has been demonstrated previously (24), this is the first report of the purification of bacterial PEtn transferases from solubilized membranes in a stable and active form as illustrated by specific MAb reactivities and MS analyses of enzymatic reactions.
The localization of Lpt3 and Lpt6 to the inner membrane was not unexpected, since (i) both proteins were previously predicted to be membrane proteins (24, 27), (ii) the majority of enzymes involved in LOS assembly are located in the inner membrane (29), and (iii) the inner membrane is rich in PtdEtn (25), which is required for the PEtn transferase activities of Lpt3 and Lpt6. This dependence is presumably due to PtdEtn acting as the substrate donor, as is the case for the E. coli Kdo PEtn transferase, EptB (32), although this has not been demonstrated conclusively for either Lpt3 or Lpt6. Furthermore, the localization of Lpt3 and Lpt6 to the inner membrane mimics the localization data for EptB (32), suggesting that this may be a common feature of LOS/lipopolysaccharide (LPS) PEtn transferases. Similarly, Lpt3 and Lpt6 are members of a growing group of membrane proteins that migrate anomalously in SDS-PAGE relative to their predicted molecular mass (36, 37), the cause of which has been investigated recently by Rath et al. (31).
The optimal reaction temperature of ∼30°C for both enzymes is consistent with that used in the characterization of EptB (32), and while this temperature is not unusual for an E. coli enzyme, it is perplexing that this is the case for Lpt3 and Lpt6, given that the only known reservoir for N. meningitidis is the human host. One possible explanation is that this optimal temperature reflects more the stability of the enzyme when isolated from the membrane than the actual optimal temperature in vivo. Alternately, it may simply be that the submaximal rate of catalysis of these enzymes at 37°C is sufficient to fulfill their roles in LOS biosynthesis in vivo.
This is the first study describing in vitro activity of a bacterial PEtn transferase purified in isolation of membrane components, since EptB was characterized by using EptB-containing membrane preparations (32) and reactions with Lpt3Ng utilized enzyme preparations purified from crude cell lysates in the absence of detergent (24). This is an important distinction for several reasons. First, the results of this study indicate that the PEtn transferase activities of Lpt3 and Lpt6 are likely not dependent on accessory membrane proteins or other membrane-associated cofactors, although this cannot be absolutely ruled out, due to the presence of minor contaminating proteins in the enzyme preparations (Fig. 3, inset panels). Second, PtdEtn dependence cannot be shown unequivocally by using enzyme preparations that contain membranes, since PtdEtn is abundant in bacterial membranes. Reynolds et al. (32) were able to successfully demonstrate that the addition of exogenous PtdEtn to reactions with EptB-containing membranes resulted in significant PEtn transferase activity; however, under close inspection of thin-layer chromatography (TLC) results for reactions lacking exogenous PtdEtn (see Fig. 3A, lane 3, in reference 32), a faint band corresponding to the PEtn-modified product is visible. It is interesting to note that PEtn transferase activity was not detected in Lpt3Ng reactions when exogenous PtdEtn was not added, but given that O'Connor et al. (24) reported that the enzyme was unstable and did not function well in solution, it is possible that the level of PEtn transferase activity in the absence of excess PtdEtn was below detection levels and that the excess PtdEtn drove the reaction toward product formation.
The abilities of both Lpt3 and Lpt6 to utilize N. meningitidis MC58 galE lpt3 LOS in the de-O-acylated form proved invaluable for further characterization of these enzymes, since de-O-Ac LOS samples are amenable to CE-MS analyses directly. The conditions developed in this study to allow direct detection and analysis of LOS moieties from Lpt3 and Lpt6 reactions, based on precursor ion scanning for LOS fragments commonly observed in CE-MS/MS analyses, were essential for characterization of the substrate specificities of Lpt3 and Lpt6, since they allowed fairly rapid analysis of reactions and, more importantly, were amenable to the analysis of a broader range of substrates than was possible by MAb recognition. A systematic approach was taken to characterize the substrate specificities of Lpt3 and Lpt6, starting with enzymatic reactions designed to determine the minimum requirements for substrate recognition by each of the enzymes. The genetics of LOS core biosynthesis in N. meningitidis MC58 are well understood (10-15, 35), and previous studies have shown that mutants with a core structure as truncated as Hep2-Kdo2 still contain 3-PEtn (39), indicating that Lpt3 requires no more than Hep2-Kdo2 in the core region for substrate recognition. We postulate that this is also the minimum core structure required for Lpt6, although this has not been demonstrated; the most truncated core structure that has been characterized in a 6-PEtn-containing background to date is the LOS from N. meningitidis 89I galE (43). Therefore, the experiments performed focused on assessing the ability of the enzymes to utilize N. meningitidis galE lpt3 LOS after modification/truncation of the lipid A moiety. Lpt3 was found to be able to transfer PEtn both to substrates that lack the charged phosphate moieties (HF-de-O-Ac LOS) and to fully deacylated (KOH) LOS at low levels but was unable to recognize core oligosaccharide alone (i.e., without lipid A). It is interesting that Lpt3 is better able to utilize dephosphorylated LOS as a substrate acceptor than fully deacylated LOS, given that the former lacks negatively charged phosphate groups while the latter retains them. These results suggest that negative charge plays only a marginal role in substrate binding, whereas the markedly reduced conversion levels with fully deacylated LOS might be due to increased rotational flexibility of the fully deacylated HexN residues, resulting in a significant reduction in the rate of substrate binding. In contrast, we were unable to detect the addition of 6-PEtn in Lpt6 reactions with any of the above-mentioned substrates. While it is possible that Lpt6 has more stringent minimum requirements for substrate recognition than Lpt3, it should be noted that the conversion levels of Lpt6 are significantly lower than those of Lpt3, and therefore, it is possible that Lpt6 is capable of using some or all of the above substrates with low efficiency, thereby forming product at subdetection levels. Regardless, the most truncated LOS structure that we can confirm to be recognized by Lpt6 is that of N. meningitidis MC58 galE lpt3 de-O-Ac LOS.
Although many strains of N. meningitidis contain the genes encoding Lpt3 and Lpt6, only a few strains are known to produce a mixture of 3- and 6-PEtn-containing LOS and/or 3,6-di-PEtn-substituted LOS. This led us to examine if there is a defined or strongly preferred order of addition for 3- and 6-PEtn or if the presence of PEtn at a given position on HepII prevents addition of PEtn to the alternate position. The de-O-Ac LOS of N. meningitidis MC58 galE and N. meningitidis 89I galE were ideal substrates for these experiments, since they have the same structures as the reaction products of the Lpt3 and Lpt6 reactions with N. meningitidis MC58 galE lpt3 de-O-Ac LOS, respectively, thus avoiding the complexities of analyzing sequential enzymatic reactions that could contain a mixture of remaining substrate, 3-, 6-, and 3,6-di-PEtn-containing products. Interestingly, both Lpt3 and Lpt6 were able to add PEtn in the presence of PEtn at the alternate position on HepII, and there was no indication that the presence of PEtn on HepII hindered conversion levels of either enzyme. These results suggest that the absence of di-PEtn-substituted LOS in most N. meningitidis strains containing both lpt3 and lpt6 does not result from the presence of PEtn at one position of HepII precluding the addition of PEtn to the other position, although the possibility that this is not the case in vivo cannot be conclusively ruled out. The reason that di-PEtn modifications are not present on the LOS of the majority of strains with both lpt3 and lpt6, therefore, remains enigmatic.
Acknowledgments
This work was funded in part by research support from Novartis (formerly Chiron Vaccines).
We thank Suzanne Lacelle for supplying MAbs L3B5 and L2-16, Perry Fleming for large-scale fermenter growths of N. meningitidis strains, and Susan Logan for the use of laboratory space.
Footnotes
Published ahead of print on 23 October 2009.
REFERENCES
- 1.Burnette, W. N. 1981. “Western blotting”: electrophoretic transfer of proteins from sodium dodecyl sulfate-polyacrylamide gels to unmodified nitrocellulose and radiographic detection with antibody and radioiodinated protein A. Anal. Biochem. 112:195-203. [DOI] [PubMed] [Google Scholar]
- 2.Caroff, M., S. Lebbar, and L. Szabo. 1987. Do endotoxins devoid of 3-deoxy-D-manno-2-octulosonic acid exist? Biochem. Biophys. Res. Commun. 143:845-847. [DOI] [PubMed] [Google Scholar]
- 3.Cox, A. D., J. C. Wright, J. Li, D. W. Hood, E. R. Moxon, and J. C. Richards. 2003. Phosphorylation of the lipid A region of meningococcal lipopolysaccharide: identification of a family of transferases that add phosphoethanolamine to lipopolysaccharide. J. Bacteriol. 185:3270-3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Finne, J., M. Leinonen, and P. H. Makela. 1983. Antigenic similarities between brain components and bacteria causing meningitis. Implications for vaccine development and pathogenesis. Lancet ii:355-357. [DOI] [PubMed] [Google Scholar]
- 5.Gidney, M. A., J. S. Plested, S. Lacelle, P. A. Coull, J. C. Wright, K. Makepeace, J. R. Brisson, A. D. Cox, E. R. Moxon, and J. C. Richards. 2004. Development, characterization, and functional activity of a panel of specific monoclonal antibodies to inner core lipopolysaccharide epitopes in Neisseria meningitidis. Infect. Immun. 72:559-569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goldberg, J. B., and D. E. Ohman. 1984. Cloning and expression in Pseudomonas aeruginosa of a gene involved in the production of alginate. J. Bacteriol. 158:1115-1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Healy, C. M., and C. J. Baker. 2005. The future of meningococcal vaccines. Pediatr. Infect. Dis. J. 24:175-176. [DOI] [PubMed] [Google Scholar]
- 8.Holst, O., L. Brade, P. Kosma, and H. Brade. 1991. Structure, serological specificity, and synthesis of artificial glycoconjugates representing the genus-specific lipopolysaccharide epitope of Chlamydia spp. J. Bacteriol. 173:1862-1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Holst, O., J. E. Thomas-Oates, and H. Brade. 1994. Preparation and structural analysis of oligosaccharide monophosphates obtained from the lipopolysaccharide of recombinant strains of Salmonella minnesota and Escherichia coli expressing the genus-specific epitope of Chlamydia lipopolysaccharide. Eur. J. Biochem. 222:183-194. [DOI] [PubMed] [Google Scholar]
- 10.Jennings, M. P., M. Bisercic, K. L. Dunn, M. Virji, A. Martin, K. E. Wilks, J. C. Richards, and E. R. Moxon. 1995. Cloning and molecular analysis of the Isi1 (rfaF) gene of Neisseria meningitidis which encodes a heptosyl-2-transferase involved in LPS biosynthesis: evaluation of surface exposed carbohydrates in LPS mediated toxicity for human endothelial cells. Microb. Pathog. 19:391-407. [DOI] [PubMed] [Google Scholar]
- 11.Jennings, M. P., D. W. Hood, I. R. Peak, M. Virji, and E. R. Moxon. 1995. Molecular analysis of a locus for the biosynthesis and phase-variable expression of the lacto-N-neotetraose terminal lipopolysaccharide structure in Neisseria meningitidis. Mol. Microbiol. 18:729-740. [DOI] [PubMed] [Google Scholar]
- 12.Jennings, M. P., P. van der Ley, K. E. Wilks, D. J. Maskell, J. T. Poolman, and E. R. Moxon. 1993. Cloning and molecular analysis of the galE gene of Neisseria meningitidis and its role in lipopolysaccharide biosynthesis. Mol. Microbiol. 10:361-369. [PubMed] [Google Scholar]
- 13.Kahler, C. M., R. W. Carlson, M. M. Rahman, L. E. Martin, and D. S. Stephens. 1996. Inner core biosynthesis of lipooligosaccharide (LOS) in Neisseria meningitidis serogroup B: identification and role in LOS assembly of the α1,2 N-acetylglucosamine transferase (RfaK). J. Bacteriol. 178:1265-1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kahler, C. M., R. W. Carlson, M. M. Rahman, L. E. Martin, and D. S. Stephens. 1996. Two glycosyltransferase genes, lgtF and rfaK, constitute the lipooligosaccharide ice (inner core extension) biosynthesis operon of Neisseria meningitidis. J. Bacteriol. 178:6677-6684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kahler, C. M., and D. S. Stephens. 1998. Genetic basis for biosynthesis, structure, and function of meningococcal lipooligosaccharide (endotoxin). Crit. Rev. Microbiol. 24:281-334. [DOI] [PubMed] [Google Scholar]
- 16.Karkhanis, Y. D., J. Y. Zeltner, J. J. Jackson, and D. J. Carlo. 1978. A new and improved microassay to determine 2-keto-3-deoxyoctonate in lipopolysaccharide of Gram-negative bacteria. Anal. Biochem. 85:595-601. [DOI] [PubMed] [Google Scholar]
- 17.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680-685. [DOI] [PubMed] [Google Scholar]
- 18.Lee, F. K., D. S. Stephens, B. W. Gibson, J. J. Engstrom, D. Zhou, and M. A. Apicella. 1995. Microheterogeneity of Neisseria lipooligosaccharide: analysis of a UDP-glucose 4-epimerase mutant of Neisseria meningitidis NMB. Infect. Immun. 63:2508-2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Logan, S. M., W. Chen, A. Aubry, M. A. Gidney, S. Lacelle, F. St. Michael, R. Kuolee, M. Higgins, S. Neufeld, and A. D. Cox. 2006. Production of a D-glycero-D-manno-heptosyltransferase mutant of Mannheimia haemolytica displaying a veterinary pathogen specific conserved LPS structure; development and functionality of antibodies to this LPS structure. Vet. Microbiol. 116:175-186. [DOI] [PubMed] [Google Scholar]
- 20.Mackinnon, F. G., A. D. Cox, J. S. Plested, C. M. Tang, K. Makepeace, P. A. Coull, J. C. Wright, R. Chalmers, D. W. Hood, J. C. Richards, and E. R. Moxon. 2002. Identification of a gene (lpt-3) required for the addition of phosphoethanolamine to the lipopolysaccharide inner core of Neisseria meningitidis and its role in mediating susceptibility to bactericidal killing and opsonophagocytosis. Mol. Microbiol. 43:931-943. [DOI] [PubMed] [Google Scholar]
- 21.McGuinness, B. T., I. N. Clarke, P. R. Lambden, A. K. Barlow, J. T. Poolman, D. M. Jones, and J. E. Heckels. 1991. Point mutation in meningococcal por A gene associated with increased endemic disease. Lancet 337:514-517. [DOI] [PubMed] [Google Scholar]
- 22.Monteiro, M. A., M. Fortuna-Nevin, J. Farley, and V. Pavliak. 2003. Phase-variation of the truncated lipo-oligosaccharide of Neisseria meningitidis NMB phosphoglucomutase isogenic mutant NMB-R6. Carbohydr. Res. 338:2905-2912. [DOI] [PubMed] [Google Scholar]
- 23.Nedelec, J., J. Boucraut, J. M. Garnier, D. Bernard, and G. Rougon. 1990. Evidence for autoimmune antibodies directed against embryonic neural cell adhesion molecules (N-CAM) in patients with group B meningitis. J. Neuroimmunol. 29:49-56. [DOI] [PubMed] [Google Scholar]
- 24.O'Connor, E. T., A. Piekarowicz, K. V. Swanson, J. M. Griffiss, and D. C. Stein. 2006. Biochemical analysis of Lpt3, a protein responsible for phosphoethanolamine addition to lipooligosaccharide of pathogenic Neisseria. J. Bacteriol. 188:1039-1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Op den Kamp, J. A. 1979. Lipid asymmetry in membranes. Annu. Rev. Biochem. 48:47-71. [DOI] [PubMed] [Google Scholar]
- 26.Osborn, M. J., J. E. Gander, E. Parisi, and J. Carson. 1972. Mechanism of assembly of the outer membrane of Salmonella typhimurium. Isolation and characterization of cytoplasmic and outer membrane. J. Biol. Chem. 247:3962-3972. [PubMed] [Google Scholar]
- 27.Parkhill, J., M. Achtman, K. D. James, S. D. Bentley, C. Churcher, S. R. Klee, G. Morelli, D. Basham, D. Brown, T. Chillingworth, R. M. Davies, P. Davis, K. Devlin, T. Feltwell, N. Hamlin, S. Holroyd, K. Jagels, S. Leather, S. Moule, K. Mungall, M. A. Quail, M. A. Rajandream, K. M. Rutherford, M. Simmonds, J. Skelton, S. Whitehead, B. G. Spratt, and B. G. Barrell. 2000. Complete DNA sequence of a serogroup A strain of Neisseria meningitidis Z2491. Nature 404:502-506. [DOI] [PubMed] [Google Scholar]
- 28.Plested, J. S., K. Makepeace, M. P. Jennings, M. A. Gidney, S. Lacelle, J. Brisson, A. D. Cox, A. Martin, A. G. Bird, C. M. Tang, F. M. Mackinnon, J. C. Richards, and E. R. Moxon. 1999. Conservation and accessibility of an inner core lipopolysaccharide epitope of Neisseria meningitidis. Infect. Immun. 67:5417-5426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Raetz, C. R., and C. Whitfield. 2002. Lipopolysaccharide endotoxins. Annu. Rev. Biochem. 71:635-700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rahman, M. M., D. S. Stephens, C. M. Kahler, J. Glushka, and R. W. Carlson. 1998. The lipooligosaccharide (LOS) of Neisseria meningitidis serogroup B strain NMB contains L2, L3, and novel oligosaccharides, and lacks the lipid-A 4′-phosphate substituent. Carbohydr. Res. 307:311-324. [DOI] [PubMed] [Google Scholar]
- 31.Rath, A., M. Glibowicka, V. G. Nadeau, G. Chen, and C. M. Deber. 2009. Detergent binding explains anomalous SDS-PAGE migration of membrane proteins. Proc. Natl. Acad. Sci. USA 106:1760-1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reynolds, C. M., S. R. Kalb, R. J. Cotter, and C. R. Raetz. 2005. A phosphoethanolamine transferase specific for the outer 3-deoxy-D-manno-octulosonic acid residue of Escherichia coli lipopolysaccharide. Identification of the eptB gene and Ca2+ hypersensitivity of an eptB deletion mutant. J. Biol. Chem. 280:21202-21211. [DOI] [PubMed] [Google Scholar]
- 33.Rosenstein, N. E., B. A. Perkins, D. S. Stephens, T. Popovic, and J. M. Hughes. 2001. Meningococcal disease. N. Engl. J. Med. 344:1378-1388. [DOI] [PubMed] [Google Scholar]
- 34.St. Michael, F., E. Vinogradov, C. Q. Wenzel, B. McIntosh, J. Li, J. C. Hoe, J. C. Richards, and A. D. Cox. 2009. Phosphoethanolamine is located at the 6-position and not the 7-position of the distal heptose residue in the lipopolysaccharide from Neisseria meningitidis. Glycobiology 19:1436-1445. [DOI] [PubMed] [Google Scholar]
- 35.Stojiljkovic, I., V. Hwa, J. Larson, L. Lin, M. So, and X. Nassif. 1997. Cloning and characterization of the Neisseria meningitidis rfaC gene encoding alpha-1,5 heptosyltransferase I. FEMS Microbiol. Lett. 151:41-49. [DOI] [PubMed] [Google Scholar]
- 36.Stoker, N. G., J. M. Pratt, and B. G. Spratt. 1983. Identification of the rodA gene product of Escherichia coli. J. Bacteriol. 155:854-859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Teather, R. M., B. Muller-Hill, U. Abrutsch, G. Aichele, and P. Overath. 1978. Amplification of the lactose carrier protein in Escherichia coli using a plasmid vector. Mol. Gen. Genet. 159:239-248. [DOI] [PubMed] [Google Scholar]
- 38.Tzeng, Y. L., A. Datta, K. Ambrose, M. Lo, J. K. Davies, R. W. Carlson, D. S. Stephens, and C. M. Kahler. 2004. The MisR/MisS two-component regulatory system influences inner core structure and immunotype of lipooligosaccharide in Neisseria meningitidis. J. Biol. Chem. 279:35053-35062. [DOI] [PubMed] [Google Scholar]
- 39.van der Ley, P., M. Kramer, A. Martin, J. C. Richards, and J. T. Poolman. 1997. Analysis of the icsBA locus required for biosynthesis of the inner core region from Neisseria meningitidis lipopolysaccharide. FEMS Microbiol. Lett. 146:247-253. [DOI] [PubMed] [Google Scholar]
- 40.Weadge, J. T., and A. J. Clarke. 2006. Identification and characterization of O-acetylpeptidoglycan esterase: a novel enzyme discovered in Neisseria gonorrhoeae. Biochemistry 45:839-851. [DOI] [PubMed] [Google Scholar]
- 41.Weston, A., M. G. Brown, H. R. Perkins, J. R. Saunders, and G. O. Humphreys. 1981. Transformation of Escherichia coli with plasmid deoxyribonucleic acid: calcium-induced binding of deoxyribonucleic acid to whole cells and to isolated membrane fractions. J. Bacteriol. 145:780-787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Westphal, O., and K. Jann. 1965. Bacterial lipopolysaccharides: extraction with phenol-water and further applications of the procedure. Methods Carbohydr. Chem. 5:81-93. [Google Scholar]
- 43.Wright, J. C., D. W. Hood, G. A. Randle, K. Makepeace, A. D. Cox, J. Li, R. Chalmers, J. C. Richards, and E. R. Moxon. 2004. lpt6, a gene required for addition of phosphoethanolamine to inner-core lipopolysaccharide of Neisseria meningitidis and Haemophilus influenzae. J. Bacteriol. 186:6970-6982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wyle, F. A., M. S. Artenstein, B. L. Brandt, E. C. Tramont, D. L. Kasper, P. L. Altieri, S. L. Berman, and J. P. Lowenthal. 1972. Immunologic response of man to group B meningococcal polysaccharide vaccines. J. Infect. Dis. 126:514-521. [DOI] [PubMed] [Google Scholar]