

Abstract
Helicobacter pylori colonizes the normal human stomach by maintaining both periplasmic and cytoplasmic pH close to neutral in the presence of gastric acidity. Urease activity, urea flux through the pH-gated urea channel, UreI, and periplasmic α-carbonic anhydrase are essential for colonization. Exposure to pH 4.5 for up to 180 min activates total bacterial urease threefold. Within 30 min at pH 4.5, the urease structural subunits, UreA and UreB, and the Ni2+ insertion protein, UreE, are recruited to UreI at the inner membrane. Formation of this complex and urease activation depend on expression of the cytoplasmic sensor histidine kinase, HP0244. Its deletion abolishes urease activation and assembly, impairs cytoplasmic and periplasmic pH homeostasis, and depolarizes the cells, with an ∼7-log loss of survival at pH 2.5, even in 10 mM urea. Associated with this assembly, UreI is able to transport NH3, NH4+, and CO2, as shown by changes in cytoplasmic pH following exposure to NH4Cl or CO2. To be able to colonize cells in the presence of the highly variable pH of the stomach, the organism expresses two pH-sensor histidine kinases, one, HP0165, responding to a moderate fall in periplasmic pH and the other, HP0244, responding to cytoplasmic acidification at a more acidic medium pH. Assembly of a pH-regulatory complex of active urease with UreI provides an advantage for periplasmic buffering.
The neutralophilic gastric pathogen Helicobacter pylori is able to colonize the normal human stomach by mechanisms we have termed acid acclimation (25). This term contrasts its acid stress responses to the more widespread acid tolerance or resistance responses of other neutralophiles, such as Escherichia coli and Vibrio cholerae. In these bacteria, cytoplasmic but not periplasmic pH is maintained between pH 4 and pH 5, using either amino acid/amine counter transporters or proton export by the F1F0 ATPase, allowing survival but not growth (10). In contrast, H. pylori is able to maintain its periplasmic pH at about 6.1 and its cytoplasmic pH relatively neutral in the presence of external pHs at least as low as ∼2.5, thereby also maintaining an inner membrane potential of about −100 mV (30). This allows both survival and growth in the stomach, enabling gastric colonization. Therefore, although H. pylori may have many acid resistance responses similar to those of other neutralophiles, it also expresses specific acid acclimation responses that may also be regulated differentially depending on the pH of its environment.
Crucial for acid acclimation is the presence of high levels of urease, accounting for as much as 10% of the protein expressed by the organism (8). Urease generates ammonia and carbonic acid, which is converted to the relatively membrane-permeant gas CO2 by a cytoplasmic β-carbonic anhydrase. Urease activity is regulated in several ways. Unique to gastric Helicobacter spp. is UreI, a H+-gated urea channel that has very low open probability at neutral pH and increased open probability as pH falls, with 50% open probability at pH 5.9 (27, 40). In acid, this channel accelerates urea entry, activating intrabacterial urease activity about 20-fold in acidic media (40). UreA and UreB form an apoenzyme complex that is activated by nickel insertion, aided by accessory protein pairs UreE/UreG and UreF/UreH, allowing for control of the level of active urease (37). Interestingly, there is compelling evidence for pH-dependent upregulation of nikR, a nickel-responsive activator of urease expression, implying pH sensitivity of a cascade providing nickel uptake regulation under nickel-deficient conditions (35).
The urease structural subunits (UreA and UreB) and their accessory nickel insertion proteins associate with UreI at neutral medium pH, as shown by native gel analysis and immunoprecipitation (28). Furthermore, postsectioning immuno-electron microscopy found that within 15 min at pH 5.5, there was a UreI dependent increase of both UreA and UreB at the inner membrane (12). The basis for these electron microscopy data has not been investigated, nor is it known whether this association would improve acid acclimation.
Prokaryotes utilize two-component systems (TCSs) to transduce environmental stimuli to produce cellular responses. These are minimally composed of a histidine kinase sensor protein, usually located in the inner membrane, and a response regulator located in the cytoplasm. The H. pylori TCS HP0244/HP0703 (FlgRS) regulates intermediate flagellar gene transcription. The sensor kinase, HP0244, lacks a membrane domain and therefore probably responds only to cytoplasmic stimuli. Recently, it was shown that expression of HP0244 is required for survival at pH 2.5 in the presence of 10 mM urea and that HP0703, its cognate response regulator, is not implicated in this response (42). Transcriptomal analysis showed that deletion of HP0244 at pH 4.5 or 2.5 downregulates ureA, ureB, ureI, ureF, rocF, ansB (asparaginase), and amiE among the pH homeostatic genes and an additional 101 genes outside the HP0703 regulon. Hence, these are part of an HP0244 pH-dependent regulon (42). Thus, HP0244 appears to regulate not only a general acid stress response but also a response specific to periplasmic or cytoplasmic pH changes.
UreI is permeable to urea but not thiourea (40). It is unknown whether UreI is also able to transport the products of urea hydrolysis, i.e., ammonia, ammonium, and carbon dioxide. If UreI were able to transport one or more of these molecules outward, the buffering capacity of the periplasm would be increased, improving growth potential in acid. This provides a rationale for association of urease activity with UreI so as to present these products directly to the channel when bacteria are in the stomach, enabling rapid periplasmic buffering and reducing potentially lethal cytoplasmic alkalization (5).
Hence, in this work, we determined whether assembly of the previously identified urease/UreI complex is regulated by HP0244 (14, 29) and whether UreI is permeable to NH3, NH4+, and CO2 to provide physiological relevance to urease assembly with UreI. The requirement of HP0244 for inner membrane assembly of UreA, UreB, and UreE and for urease activity is shown by comparing membrane locations and activities of urease in wild-type and HP0244 deletion mutant strains. HP0244 deletion leads to a loss of periplasmic pH control after exposure to pH 5.0, as shown by changes in fluorescence of the pH-sensitive dye 2′,7′-bis-(2-carboxyethyl)-5-carboxyfluorescein (BCECF) free acid. Membrane potential measurements indicate that the proton motive force is abolished at pH 2.5 in the deletion mutant in the presence of 10 mM urea, in contrast to the wild type. pHin changes in wild-type and ureI deletion mutant strains exposed to additional NH4Cl or CO2 show that UreI is permeable to the products of urease activity, including NH4+. UreI thus provides a pathway for exit of NH4+, perhaps complementing an as yet undefined active NH4+ transporter (32, 33). The pH changes are accompanied by reciprocal changes in inner membrane potential due to the need to maintain a relatively constant proton motive force (19). Hence, acidification of the cytoplasm results in formation of a pH-regulatory complex of active urease coupled to transport of the products of its reaction through the urea channel, UreI.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
H. pylori strains ATCC 43504 and ATCC 26695 were obtained from the American Type Culture Collection. A nonpolar ATCC 26695 flgS/HP0244 deletion mutant and a nonpolar ATCC 43504 ureI/HP0071 deletion mutant were constructed by allelic exchange using a kanamycin resistance gene as described below. Bacteria were grown under microaerobic conditions (5% O2, 10% CO2, 85% N2) either on Trypticase soy agar (TSA) plates supplemented with 5% sheep blood (Gibco BRL-Life Technologies Inc., Gaithersburg, MD) or in brain heart infusion (BHI) medium (Difco Laboratories, Detroit, MI) supplemented with 7% horse serum (Gibco BRL-Life Technologies Inc.) and 0.25% yeast extract (Difco Laboratories, Detroit, MI). All bacteria grown in media were in the presence of Dent selective supplement (Oxoid Limited, Hampshire, United Kingdom), and the ureI deletion mutant and flgS deletion mutant were grown in the presence of 20 μg/ml kanamycin (Sigma Chemical Co., St. Louis, MO).
Construction of mutant H. pylori strain 26695/ΔHP0244::km by allelic exchange mutagenesis.
H. pylori HP0244 deletion mutants were produced by allelic exchange as previously described (42). Briefly, the isogenic HP0244 deletion mutant 26695/ΔHP0244::km was obtained by transforming H. pylori strain 26695 with a pBluescript II vector (Stratagene) carrying the Tn903 kanamycin resistance gene flanked by a 369-bp fragment comprising the 313-bp 3′ region of the HP0245 gene and the 56-bp 5′ region of the HP0244 gene and a 407-bp fragment comprising the 199-bp intergenic region between the HP0244 and HP0243 genes and the 208-bp 5′ region of the HP0243 gene. The DNA fragments were obtained by PCR performed on H. pylori chromosomal DNA, using primer pairs HP0245-5′P(1229-1248)-KpnI/HP0244-3′P(1597-1577)-EcoRI and HP0243-5′P(2681-2702)-BamHI/HP0243-3′P(3087-3064)-SacI, respectively. The resulting mutants had almost the entire HP0244 coding sequence replaced by the kanamycin resistance gene.
Construction of mutant H. pylori strain 43504/ΔHP0071::km by allelic exchange mutagenesis.
H. pylori ureI deletion mutants were produced by allelic exchange as described in detail elsewhere (24). Briefly, a plasmid was constructed for exchange of the ureI open reading frame of the Helicobacter pylori genome with a kanamycin resistance marker gene. The 3′ region of ureB and the 5′ region of ureE were amplified by PCR, using plasmid pHP808 as a template. The two PCR products were then fused via the BamHI recognition sites, which produced a ureB/ureE hybrid. A kanR open reading frame derived from the pUC4K plasmid was inserted into the ureB/ureE hybrid sequence at the BamHI restriction site. The plasmid was digested with EcoRI and HindIII for DNA fragment cloning. To select for the presence of the plasmid, cells were grown in LB medium (Luria broth base [Gibco BRL]) supplemented with 50 μg/ml ampicillin. Plasmids were assayed for the presence and orientation of the inserted DNA, employing restriction analysis. Plasmid DNA of this vector was used for homologous recombination in H. pylori.
Urease activity of intact bacteria.
H. pylori wild-type and HP0244 deletion mutant strains were incubated in BHI, pH 4.5 or 7.4, for 3 h. Aliquots were removed at 30, 60, 90, and 180 min, and urease activity was measured in the presence of a nonionic detergent to bypass UreI in order to measure total urease activity.
Urease activity was measured radiometrically as described before (30). Ten-microliter aliquots removed at various time points were added to 100 mM phosphate buffer (PB), pH 7.0, containing 5 mM [14C]urea with a specific activity of 10 μCi/μmol and 0.1% C12E8 (dodecyl octaethylene glycol ether). Plastic wells containing 0.5 M KOH-soaked filter paper hung from rubber stoppers were used to collect the total 14CO2 that resulted from the hydrolysis of urea by urease. Urease activity was measured for 30 min at 37°C with constant agitation. The reaction was terminated by the addition of 5 N H2SO4, and the reaction mix was incubated for 30 min at 37°C. The wells were placed in scintillation cocktail (HiIonicFluor; Packard Instruments, Meriden, CT), and the radioactivity was measured by scintillation counting (1216 RackBeta; LKB Institute). Urease activity is reported as the % of control activity. The protein concentration was determined by the bicinchoninic acid (BCA) method.
Measurement of protein synthesis.
Bacteria were suspended at an optical density at 600 of 0.1 to 0.2 in 3 ml of RPMI 1640 medium containing 1% minimal essential medium nonessential amino acid solution (100×), 2 mM l-glutamine, 1 mM minimal essential medium sodium pyruvate solution, 100 mM HEPES adjusted to appropriate pH with HCl or NaOH, and 100 μCi of l-[35S]methionine at a specific activity of 555 MBq/ml. The bacteria were incubated at pH 4.5 and 7.4 in the presence or absence of chloramphenicol (20 mg/ml) in the above medium for 4 h at 37°C in a microaerobic atmosphere (5% O2, 10% CO2, and 85% N2). The bacteria were gently centrifuged at 600 × g for 5 min and washed three times in phosphate-buffered saline (PBS). The bacterial suspension was passed through a French press at 20,000 lb/in2 three times. Protein from the cell lysate was precipitated with acetone followed by centrifugation (20,800 × g, 10 min). The protein pellet was resuspended in 3 ml PBS, 10-μl aliquots were added to scintillation cocktail (HiIonicFluor; Packard Instruments, Meriden, CT), and the radioactivity was measured by scintillation counting (1216 RackBeta; LKB Institute).
Membrane association of urease and UreE. (i) Preparation of H. pylori purified membrane and cytosolic fractions.
Bacteria were harvested after overnight growth on TSA plates and suspended in 1 ml of BHI. A total of 500 μl of the cell suspension was added to 10 ml BHI medium at pH 4.5 or pH 7.4 and was incubated for 30 min at 37°C under microaerobic conditions. Subsequently, the cell suspension was pelleted by centrifugation at 3,000 × g for 10 min at 4°C. The pellet was resuspended in 2 ml of 25 mM phosphate buffer containing 5 mM EDTA solution (PBE) at pH 7.4. This suspension was passed through a French press at 20,000 lb/in2 three times and then centrifuged at 3,000 × g for 10 min to pellet any remaining whole cells. The supernatant was removed and centrifuged at 10,000 × g for 10 min to further purify the lysate. The supernatant from this spin was removed, layered onto a 5-ml 20% sucrose step in PBE solution, and centrifuged at 100,000 × g for 1 h. The pellet from this centrifugation was resuspended in 2 ml PBE solution and again layered onto a 5-ml 20% sucrose step. The pellet obtained after three such centrifugations at 100,000 × g was treated as the membrane fraction. This membrane fraction was devoid of cytosolic protein contamination, as previously shown (17). Sucrose was used to prevent soluble cytoplasmic protein contamination of the membrane pellet. EDTA was added to chelate Ca2+ and to minimize loosely associated proteins. To obtain the cytoplasmic fraction, the supernatant obtained after the 10,000 × g spin for 10 min was subjected to a 100,000 × g spin for 1 h, and the supernatant obtained from this spin was collected. Protein concentration was determined by the BCA (Pierce) and Lowry methods.
(ii) Western blotting.
The proteins from the membrane and cytoplasmic fractions were size fractionated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) on 4 to 12% NuPage Bis-Tris gradient gels (Invitrogen) and transferred onto nitrocellulose membranes (Bio-Rad). Western blot analysis was carried out on the membrane and cytoplasmic fractions, using the following antibodies: anti-UreA (1:250 and 1:500; Santa Cruz Biotechnology, Inc., Santa Cruz, CA), anti-UreB (1:2,500 and 1:3,000; Santa Cruz Biotechnology, Inc., Santa Cruz, CA), anti-UreI (1:2,500 and 1:1,500 [40]), anti-UreE (1:500 and 1:1,000 [28]), and anti-UreH (1:500). Immunolabeling was detected with a SuperSignal West Pico enhanced chemiluminescence detection kit (Pierce). Immunoblots were digitized (Microtek Scanmaker i800) at a resolution of 600 dpi. The scanned images were analyzed using Kodak 1D software. The resulting intensities were expressed as the ratio of the intensity at pH 4.5 to the intensity at pH 7.4.
Urease activity of purified membrane and soluble fractions.
Urease activity was measured radiometrically as described before (30). Ten microliters of the membrane or soluble protein fraction was added to 100 mM PB, pH 7.0, containing 5 mM [14C]urea with a specific activity of 10 μCi/μmol. Plastic wells containing 0.5 M KOH-soaked filter paper hung from rubber stoppers were used to collect the total 14CO2 that resulted from the hydrolysis of urea by urease. Urease activity was measured for 30 min at 37°C with constant agitation. The reaction was terminated by the addition of 5 N H2SO4, and the reaction mix was incubated for 30 min at 37°C. The wells were placed in scintillation cocktail (HiIonicFluor; Packard Instruments, Meriden, CT), and the radioactivity was measured by scintillation counting (1216 RackBeta; LKB Institute). Urease activity is reported as μmol urea hydrolyzed/min/mg protein. Protein concentration was determined by the BCA method. The above experiments defined the alterations of the location of urease protein and activity at 30 min due to HP0244 deletion.
Measurement of intrabacterial pH. (i) BCECF loading.
H. pylori wild-type and deletion mutant strains were grown overnight on TSA plates supplemented with 5% sheep blood (BD). The bacteria were removed from the plate and resuspended in 5 ml BHI medium (Difco). The membrane-permeant, pH-sensitive fluorescent probe 2′,7′-bis-(2-carboxyethyl)-5-carboxyfluorescein acetoxymethyl ester (BCECF-AM) was added to the bacterial suspension to a final concentration of 10 μM. The bacterial suspension was then incubated in a microaerobic (5% O2, 10% CO2, 85% N2) environment at 37°C for 30 min with shaking. The bacteria were pelleted by gentle centrifugation (2,000 × g, 5 min) and resuspended in 300 μl of HP medium (140 mM NaCl, 5 mM KCl, 1.3 mM CaCl2, 0.5 mM MgSO4, 10 mM glucose, 1 mM glutamine) buffered with 1 mM phosphate buffer at pH 7.4.
Measurement and calibration of pHin by use of BCECF at pH 2.5 in the presence of 10 mM urea were possible only with the wild-type strain, since under these conditions the drop in pHin of the HP0244 deletion strain exceeded the range of the dye. Measurement of pHin in the HP0244 deletion strain was therefore done using the fluorescein-based pH-sensitive fluorescent dye Oregon Green AM (OG), with a lower pKa (4.0) than the pKa of 7.0 for BCECF. Loading conditions were identical to those for BCECF-AM. To measure pHin, 20 μl of BCECF- or OG-loaded organisms was added to 3 ml 100 mM glycine-buffered HP medium at pH 2.5, and fluorescence was monitored using a dual-excitation (Ex1 = 502 nm, Ex2 = 436 nm), single-emission (Em = 530 nm) fluorimeter for both dyes.
To determine flux of NH3, NH4+, and CO2 through UreI, the change in pHin was monitored by comparing BCECF-loaded wild-type and ureI deletion strains. The bacteria were harvested and loaded with BCECF-AM as described above. Following loading of the dye, the bacteria were suspended in 3 ml of HP medium buffered to pH 7.4 with 50 mM morpholinepropanesulfonic acid (MOPS). The effect of 50 mM NH4Cl and CO2 on BCECF fluorescence was monitored using a single excitation wavelength (Ex = 502 nm, Em = 530 nm) to allow for rapid data collection (50 ms/data point) to find out whether UreI was able to transport either NH3 or CO2.
(ii) Calibration of pHin.
Each experiment was calibrated independently. Once the fluorescence of the internal BCECF or OG had been measured, 150 nM of the protonophore 3,3′,4′,5-tetrachlorosalicyanilide was added to equilibrate the internal pH with that of the medium. HCl was then added to obtain minimum fluorescence of the dye, followed by the addition of NaOH to obtain maximum fluorescence of the dye. The internal pH was calculated using the equation pHin = pKa + log{[(R − RA)/(RB − R)] × [FA(λ2)/FB(λ2)]}, where pKa = pKa of BCECF = 7.0, pKa of OG = 4.7, R = value at 502 nm/value at 436 nm for each data point, RA = ratio at minimum fluorescence, RB = ratio at maximum fluorescence, FA(λ2) = minimum fluorescence at 436 nm, and FB(λ2) = maximum fluorescence at 436 nm. These experiments were performed after the addition of either NH4Cl or CO2 or exposure to pH 2.5.
Visualizing changes of periplasmic pH.
A Zeiss LSM 510 confocal microscope was used to observe pH changes in the periplasm of H. pylori. Experiments were done using a 100× objective. Higher magnifications were obtained by using the electronic zoom feature provided by the software of the LSM 510 microscope.
H. pylori wild-type and HP0244 deletion strains were grown overnight on TSA plates supplemented with 5% sheep blood (Becton Dickinson). The bacteria were removed from the plates and immobilized on Cel-Tak-coated coverslips.
BCECF in its impermeant, free acid form was used at a final concentration of 5 μM in HP medium buffered with 1 mM phosphate buffer to pH 5.0. The dye was excited at 488 nm and detected at 515 to 545 nm. At the start of the experiment, the BCECF-containing medium was added to the bacteria and images were collected every 5 s. After 20 s, 5 mM urea was added and image collection continued at the same rate for 5 more minutes.
Measurement of membrane potential.
Membrane potential was determined as previously described (18). Briefly, the bacteria were harvested from plates into 300 μl HP medium buffered with 1 mM phosphate. The fluorescent membrane potential-sensitive dye diSC3(5) was dissolved in dimethyl sulfoxide (DMSO), and 3 μl was added to 3 ml of appropriate buffer to give a final concentration of 1 μM. The bacterial suspension was then added to 3 ml of the dye solution at different pHout values in a fluorimeter cuvette to reach an optical density at 600 of 0.160 (usually 15 to 20 μl).
Fluorescence quenching due to potential-dependent uptake of the dye was measured in a fluorimeter set at an excitation wavelength of 600 nm and an emission wavelength of 665 nm. Data points were collected every 0.050 s. The dye solution was added 5 min before adding the bacteria to allow temperature equilibration. With addition of the bacteria, the fluorescence was quenched due to dye uptake dependent on the interior negative potential.
The calibration of the membrane potential was carried out, as previously described, by the addition of valinomycin followed by the addition of K+ until no further change in fluorescence was observed (30). This enables calculation of the K+ equilibrium potential found with the addition of the K+-selective ionophore valinomycin, using the Nernst equation: Δψ = 61 × log([K+]in/5), where [K+]in is equal to the external K+ concentration at which the potential difference becomes zero in the presence of valinomycin, i.e., where [K+]out = [K+]in, and 5 mM is the medium concentration when valinomycin is added. The membrane potential in the absence of valinomycin can then be calculated. No change in medium pH was found in these strong buffers before or after the addition of urea over the time course of measurement. Membrane potential is displayed based on the calibration once the dye reaches equilibrium. The initial fluorescence quench depends on accumulation of the dye inside the bacteria, and the fluorescence present prior to equilibrium does not necessarily imply a positive interior potential.
To determine the membrane potential at high acidity, wild-type or HP0244 deletion strains were added to a cuvette containing 100 mM glycine and 10 mM urea in 3 ml of HP medium buffered to pH 2.5. To determine the effect of NH4Cl addition on membrane potential, wild-type or HP0244 deletion strains were added to a cuvette containing 3 ml of HP medium buffered to pH 7.4 with 50 mM MOPS. After achieving a stable baseline, within about 30 s, 50 mM NH4Cl was added and the membrane potential monitored.
RESULTS
Total urease activities of H. pylori wild-type and HP0244 deletion mutant strains incubated at pH 7.4 and 4.5.
Total urease activity was measured to investigate whether the decreased acid survival of the HP0244 deletion mutants corresponded to a loss of the increase in urease activity of the intact organism (41). The urease activity of the wild-type strain increased >3-fold during the first 30 min of incubation at pH 4.5 compared to that at pH 7.4, and that level of activity was maintained for the remainder of the incubation period (Fig. 1). In contrast, there was an ∼2-fold decrease in urease activity in the HP0244 deletion mutants after 30 min of incubation at pH 4.5 (Fig. 1). No change was seen at pH 7.4 in either strain. Hence, HP0244 directly or indirectly regulates urease activity in response to changes in medium pH within 30 min, and this regulation persists for at least 3 h. Since this activation is independent of protein synthesis, this effect of HP0244 is not due to regulation of transcription, but perhaps is due to phosphorylation of as yet unknown regulatory proteins. None of the proteins encoded by the urease gene cluster contain a canonical bacterial phosphorylation site. The urease activation may be due to HP0244-dependent regulation of Ni2+ uptake and insertion into the UreA-UreB apoenzyme or to regulation of membrane recruitment and activation of urease and the accessory proteins (35, 36). However, nickel removal from the medium did not affect urease activation (28).
FIG. 1.

Urease activity of intact H. pylori incubated in acid. H. pylori wild-type (Wt) and HP0244 deletion mutant strains were incubated in BHI, pH 4.5 or 7.4, for 30 to 180 min, and total urease activity was measured in the presence of a nonionic detergent to bypass UreI. In the wild-type strain, there was a >3-fold increase in urease activity at pH 4.5 compared to that at pH 7.4 after 30 min, and it remained elevated for 3 h. The absence of HP0244 had no effect on urease activity at pH 7.4 but reduced urease activity about 50% with incubation at pH 4.5 within 30 min and further reduced the activity at pH 4.5 after 180 min.
Membrane recruitment of UreA, UreB, and UreE.
To determine whether urease is assembled at the membrane in association with UreI and whether this urease-membrane association is regulated by the HP0244 sensor histidine kinase in response to acidic pH, Western blot analysis was performed on different cellular fractions of H. pylori. Antibodies against UreA, UreB, and UreE were used for immunodetection of these proteins from purified membrane fractions of H. pylori wild-type and HP0244 deletion strains exposed to different pH conditions. As shown in Fig. 2A and C, incubation of H. pylori at pH 4.5 for 30 min, compared to that at pH 7.4, resulted in a threefold increase of membrane-associated UreA and UreB as well as UreE. However, HP0244 deletion abolished this pH-induced increase of membrane-associated UreA, UreB, and UreE (Fig. 2B and C). Hence, membrane recruitment of the urease subunits is dependent on the presence of this cytoplasmic sensor kinase. There was no difference in the level of UreI in either the wild type or the deletion mutant incubated at pH 4.5 compared to that at pH 7.4, but as before, UreI expression was required for urease protein recruitment (28). This suggests that the urease proteins are recruited to the vicinity of this urea channel. Hence, the association of UreA, UreB, and UreE is selective for these members of the urease gene cluster and not a general phenomenon of cytoplasmic proteins, since expressed green fluorescent protein (GFP) was not found in the membranes (29). Crude antibodies against UreH also detected association of this subunit with the membrane fraction (data not shown).
FIG. 2.

Western blot analysis and urease activity of purified membrane fractions from wild-type and HP0244 deletion strains of H. pylori. Representative Western blots are shown for UreA, UreB, UreE, and UreI from wild-type (A) and HP0244 deletion (B) strains that were exposed to pH 7.4 and pH 4.5. The intensities of the protein bands were measured and converted to expression ratios of pH 4.5 to pH 7.4. (C) The amounts of UreA, UreB, and UreE but not UreI protein increased after incubation at pH 4.5 for 30 min in wild-type H. pylori, whereas the absence of HP0244 abolished the recruitment of UreA and UreB, but not UreE, to the membrane. (D) Urease activities from both membrane and soluble fractions incubated at pH 4.5 and pH 7.4 from wild-type and HP0244 deletion strains were measured. The urease activity of the membrane fraction increased ∼2-fold after incubation at pH 4.5 for 30 min in the wild-type strain (black bars) but not in the HP0244 deletion strain (gray bars). Urease activity is expressed as a percentage of that in the pH 7.4 control.
Membrane-associated urease activity.
Incubation of H. pylori at pH 4.5 compared to that at pH 7.4 for 30 min resulted in a >2-fold increase in urease activity of the membrane fraction (Fig. 2D). Deletion of HP0244 prevented the acidic pH-dependent increase of urease activity in the membrane fraction. Acidic medium pH compared to pH 7.4 resulted in a slight reduction of the relative level of urease activity in the cytoplasmic protein fraction in the wild type, but there was no change in the HP0244 deletion strain. Hence, the loss of recruitment and activation of urease at UreI in the inner membrane may account for the loss of urease activation at pH 4.5 in the HP0244 deletion mutants.
Effect of chloramphenicol.
At 10 mM chloramphenicol, there was no incorporation of [35S]methionine into bacterial protein, showing that protein synthesis was absent. Thus, in the absence of chloramphenicol, incorporated protein counts were >7,500 cpm, and in the presence of the inhibitor, incorporated counts were <500 cpm. Nevertheless, there was no inhibition of increased urease activity or membrane recruitment, suggesting that activation of HP0244 occurs by a mechanism distinct from transcription/translation.
Periplasmic pH regulation by HP0244-dependent membrane-associated urease.
To determine the role of HP0244 in periplasmic pH homeostasis, changes in periplasmic pH were visualized using the inner membrane-nonpermeant, pH-sensitive, fluorescent BCECF free acid. Wild-type and HP0244 deletion strains incubated at pH 5.0 were observed by confocal microscopy following the addition of 5 mM urea. There was an increase in fluorescence of the periplasm of the wild-type organisms following urea addition, indicating an increase in pH of this compartment (Fig. 3A). No increase in fluorescence was observed in the HP0244 deletion mutants (Fig. 3B). These results are similar to those found with deletion of both ureI and the periplasmic α-carbonic anhydrase (2, 16). Hence, the HP0244-dependent urease trafficking and activation of urease at the membrane play a role in neutralizing protons entering the periplasm.
FIG. 3.

Deletion of HP0244 abolishes periplasmic buffering in H. pylori. Wild-type (A) and HP0244 deletion (B) strains were incubated at pH 5.0 in the presence of BCECF free acid to monitor periplasmic pH changes. Representative images show that the addition of 5 mM urea resulted in increased peripheral fluorescence due to increased periplasmic pH in the wild type but not in the HP0244 deletion mutant. Arrows indicate periplasmic fluorescence.
Permeation of CO2 through UreI.
CO2 permeation through the inner membrane of H. pylori was measured using BCECF-AM in wild-type and ureI deletion mutant strains as a probe of cytoplasmic pH following the addition of CO2. When pHin was calibrated in the dual-beam mode (Ex = 439 nm and 502 nm), the values obtained were 7.5 ± 0.1 and 7.4 ± 0.3 (n = 3) for the wild-type and ureI deletion mutant strains, respectively. Upon the addition of CO2 to the wild-type strain, pHin fell rapidly, to 6.2 ± 0.1 (n = 3). In contrast, CO2 addition to the UreI deletion strain resulted in a much slower decline in pHin, to 6.5 ± 0.2 (n = 3). These data show the presence of CO2 permeability in the wild-type strain that is absent in the ureI deletion strain (Fig. 4A). To determine more accurately the rate of pHin fall with the addition of CO2, bacterial cytoplasmic pH changes again were measured using BCECF-AM, with single-wavelength excitation (502 nm) to allow collection of data every 50 ms. The addition of CO2 resulted in rapid acidification of the cytoplasm, as shown by the fall in fluorescence (Fig. 4B). The decrease in fluorescence was much more rapid in the wild type than in the mutant. The fall in fluorescence is due to rapid reaction of the penetrating gas (CO2) with OH− to form HCO3− and H+, likely catalyzed by the cytoplasmic β-carbonic anhydrase. In the absence of UreI, the acidification was about fourfold slower, suggesting that UreI is able to transport CO2. Hence, this channel that transports urea is also able to transport CO2, as has been found with several aquaporins (6, 20). These properties of UreI likely have significance for acid acclimation of H. pylori if there is activation and association of urease with UreI.
FIG. 4.

UreI is permeable to CO2, NH3, and NH4+. (A) In the H. pylori wild-type strain (black line), CO2 addition rapidly decreased pHin, followed by a slower decrease in pHin. Deletion of ureI (gray line) greatly reduced the initial rapid fall in pHin but had no effect on the slow phase of pHin acidification. The initial rapid acidification phase is due to CO2 entry through UreI, and the secondary, slow phase of cytoplasmic acidification is due to permeation of CO2 through the lipid bilayer, reflecting continued CO2 addition to the medium. (B) Single-wavelength (502 nm) excitation of BCECF was used for rapid measurement (data capture every 50 ms) of the rate of change of fluorescence. The rate of cytoplasmic fluorescence decrease (alkalization) following CO2 addition was significantly higher in the wild-type organism (black line) than in the ureI deletion mutant (gray line), showing that UreI is able to accelerate CO2 movement across the inner membrane of H. pylori (dF/dt ± standard error of the mean [SEM]) (n = 3; P ≤ 0.05). (C) NH4Cl addition rapidly increases pHin due to NH3 entry, followed by fall in pHin, in the H. pylori wild-type strain (black line). Deletion of ureI (gray line) abolished the secondary fall in pHin. (D) The rate of cytoplasmic fluorescence increase (alkalization) following NH4Cl addition was significantly higher in the wild-type organism (black line) than in the ureI deletion mutant (gray line), showing that UreI is able to accelerate NH4+ movement across the inner membrane of H. pylori (dF/dt ± SEM) (n = 3; P ≤ 0.05). Therefore, the secondary fall in pHin is due to NH4+ entry through UreI, with generation of H+ in the bacterial cytoplasm (dF/dt ± SEM) (n = 3; P ≤ 0.05).
Permeation of NH3 and NH4+ through UreI.
The effect of ureI deletion on NH3 permeability of the inner membrane of H. pylori was measured using BCECF-AM in wild-type and ureI deletion mutants as a probe of cytoplasmic pH following the addition of NH4Cl, as has been done with various eukaryotic cells (11, 20, 26). When pHin was calibrated in the dual-beam mode (Ex = 436 nm and 502 nm), the value was 7.7 ± 0.1 for both wild-type and ureI deletion mutant strains. Upon the addition of NH4Cl, pHin rose rapidly, to 8.3 ± 0.1, in both the wild-type and ureI deletion mutant strains. In the wild-type organisms, the pH then returned to the original pHin within 100 s, whereas the deletion mutants maintained a pHin between 8.3 and 8.2. These data show the presence of NH4+ permeability in the wild-type strain that is absent in the ureI deletion strain (Fig. 4C and Table 1). These properties of UreI likely have significance for acid acclimation of H. pylori if there is activation of urease associated with UreI.
TABLE 1.
Cytoplasmic pH and membrane potential following NH4Cl addition
| Strain | Parameter | Baseline value | Value with 50 mM NH4Cl |
|
|---|---|---|---|---|
| Immediate | After 200 s | |||
| HP43504 | pHin | 7.7 ± 0.1 | 8.3 ± 0.2 | 7.40 ± 0.06 |
| Δψ (mV) | −178.8 ± 12.8 | −157.6 ± 10 | −175.71 ± 13.08 | |
| HP43504 | pHin | 7.7 ± 0.1 | 8.3 ± 0.1 | 8.1 ± 0.1 |
| ΔureI | Δψ (mV) | −184.8 ± 7.8 | −168.8 ± 7.9 | −124.8 ± 11.6 |
The response time in the dual-beam mode is too long to detect the effect of ureI deletion on NH3 penetration. Hence, single-wavelength analysis of BCECF (502 nm) was used as described above to detect the rate of alkalization following NH4Cl addition due to NH3 penetration into the cytoplasm. There was an ∼3-fold increase in the rate of NH3 penetration in the wild-type organism compared to the rate in the ureI deletion mutant (Fig. 4D). The overall rate of NH3 entry appeared to be higher than that of CO2 in terms of the rate of change of fluorescence. Furthermore, the decline of fluorescence and, hence, reacidification occurred only in the wild-type organism, not in the ureI deletion mutant, and this reacidification was likely due to entry of NH4+ (or NH3 + H+) releasing H+ inside the organisms. These results suggest that UreI transports both NH3 and NH4+.
Regulation of membrane potential.
The survival of bacteria in acid depends on maintenance of a proton electrochemical gradient (Δμ̄H+, i.e., proton motive force [PMF]) across the cell membrane, determined as the algebraic sum of the ΔpH and inner membrane potential:
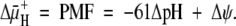 |
The PMF is kept relatively constant by reciprocal changes of ΔpH and Δψ (14). Hence, with NH3 entry, as the cytoplasmic pH increases, Δψ should decrease, and with NH4+ entry, cytoplasmic pH should decrease and Δψ should increase if ΔψμH+ is maintained constant. For growth in acidic media, the pH of the periplasm cannot be allowed to fall to that of the medium, even at pH 4.5, since at a relatively neutral cytoplasmic pH, the inward pH gradient would drive the membrane potential close to 0 mV, impairing solute transport and membrane protein folding, limiting growth if not survival (14, 38).
The cationic dye diSC3(5) was used to measure the Δψ of H. pylori wild-type and ureI deletion mutant strains under the same conditions as those for measurements of pHin. This dye accumulates on hyperpolarized membranes and is translocated into the lipid bilayer. Aggregation within the confined membrane interior results in decreased fluorescence. Thus, a decrease in fluorescence corresponds to an increase in −Δψ.
Following addition of 50 mM NH4Cl, there was a decline of Δψ by about 30 mV that compensated for the increase in ΔpH in both wild-type and mutant bacteria, as predicted by the PMF equation. In wild-type organisms, there was repolarization by about 20 mV due to internal reacidification, whereas the ureI deletion mutants continued to depolarize (Fig. 5 and Table 1). These data corroborate the pH findings, showing that bacterial bioenergetics maintain the PMF in response to changes of ΔpH across the inner membrane. The repolarization observed in the wild type but not in the ureI deletion mutants following NH4Cl addition also suggests, as with pH measurements, that UreI allows NH4+ transport, be it the cation or NH3 + H+, as discussed below.
FIG. 5.

Changes of membrane potential following addition of NH4Cl. In the wild-type and mutant organisms, there was an immediate fall in potential due to alkalization of the interior, as shown in Fig. 4C, with an increase in ΔpH. In the wild-type strain, repolarization is due to a fall in pHin that is absent in the HP0244 deletion mutant, arguing for NH4+ permeation of UreI.
Cytoplasmic pH and Δψ at different medium pHs.
The effects of medium pH on pHin and Δψ for the wild-type strain and the HP0244 deletion mutant were determined in order to identify the role of HP0244 in survival at high acidity. OG-AM was used for pHin measurement in the HP0244 deletion mutant because of its maintained response to a lower pH. There was an immediate fall in cytoplasmic pH, to ≤4.9 ± 0.1, in the HP0244 deletion mutant upon exposure to pH 2.5 in the presence of 10 mM urea. In contrast, the pH was maintained at 7.3 ± 0.2 (n = 3) in the wild type. The wild-type Δψ was maintained at −90.1 ± 7.4 mV (n = 3) at pH 2.5 with 10 mM urea, but in the mutants, Δψ fell to −5.2 ± 0.7 mV (n = 3) within 50 s. This accounts for the decreased survival of HP0244 deletion mutants in vitro, by ∼7 log units after 30 min of incubation (42). At neutral pH and at pH 4.5, there was no difference in pHin or Δψ between wild-type and HP0244 deletion mutant bacteria. Hence, HP0244 is required for maintenance of the PMF only at the highly acidic pH that can occur in its habitat in the stomach (29).
DISCUSSION
Work over the last decade has uncovered some aspects of the mechanism of periplasmic pH regulation by H. pylori necessary for acid survival and gastric colonization (25). At pH 6.0 and below, entry of urea through UreI allows an increase in intrabacterial urease activity. Urea hydrolysis produces 2 NH3 and H2CO3. The NH3 can prevent cytoplasmic acidification by absorbing entering protons, forming NH4+, and can also buffer protons entering the periplasm after exiting through the inner membrane. Since the pKa of NH3/NH4+ is 9.2, ammonia production does not generate a buffer effective at physiological pH. Generation of effective buffer at close-to-neutral pH is performed in the case of H. pylori by a pair of carbonic anhydrases, the cytoplasmic β-carbonic anhydrase and the membrane-bound periplasmic α-carbonic anhydrase. The β-carbonic anhydrase facilitates formation of permeant CO2 from H2CO3 generated by urease activity. This CO2 then enters the periplasm, where the periplasmic α-carbonic anhydrase generates the HCO3− from periplasmic CO2 to buffer the periplasm to a pH of ∼6.1, the pH determined for the periplasm in the presence of physiological concentrations of urea over a range of medium pHs from 2.5 to 6.2 (30).
H. pylori expresses four complete TCSs and two orphan response regulators (1, 3, 34). One two-component system, CheA-CheY, directly regulates chemotaxis, while the remaining two-component systems are probably involved in transcriptional regulation. Of the three remaining TCSs, HP1365-HP1364 (CrdRS) was demonstrated to positively regulate the expression of the copper resistance determinant CrdAB-CzcAB in response to increasing concentrations of copper ions (39). Another TCS of H. pylori, ArsRS (HP0165-HP0166), is an acid-responsive system. HP0165 is an integral membrane protein with two putative transmembrane helices and a relatively large periplasmic domain. HP0165 can mediate a response to small changes in periplasmic pH, and it has been suggested that this sensor kinase responds directly to changes in the hydrogen ion concentration of the periplasm (21-23). This TCS in acid upregulates expression of the majority of the pH homeostatic genes, namely, the urease gene cluster, hypA, rocF (encoding arginase), the α-carbonic anhydrase gene (HP1186), amiE, and amiF, as well as about 75 other genes (22, 43).
The known response regulator for HP0244 is HP0703. HP0703 plays no role in the function of HP0244 in acid-responsive gene regulation, since its deletion has no effect on acid survival (42). The properties of the acid-dependent response of HP0244 are distinct from those of the previously described acid response of the periplasmic pH sensor, HP0165. The HP0165 sensor kinase responds to changes in periplasmic pH, resulting in the regulation of about 100 genes at a medium pH of 5.0, a pH where deletion of HP0244 has no effect (21, 41). There is some overlap of the genes regulated by these two kinases, but HP0165 regulates expression of most of the genes that are potentially pH homeostatic, in contrast to HP0244, which regulates only a few. Furthermore, deletion of HP0165 delays but does not abolish urease activation after incubation at pH 4.5 (data not shown). H. pylori therefore uses two sensor kinases to respond to different degrees of medium acidity, using HP0165 for mild acidity and HP0244 for high acidity. This likely reflects the need for the organism to retain growth potential at the different acidities it is exposed to in the presence or absence of food.
The evidence for a role of HP0244 in membrane assembly of active urease is shown by the loss of recruitment of urease subunits and the loss of urease activation either at the membrane or in the intact organism when this kinase is deleted. This is independent of protein synthesis and therefore suggests either direct interaction of some signaling protein with HP0244 or phosphorylation of such a protein. Without the kinase, the organism is not able to elevate its periplasmic pH above that of the medium and loses >7 log units of survival along with its proton motive force (42). Measurement of cytoplasmic pH and membrane potential at pH 2.5 in the presence of 10 mM urea, where deletion of HP0244 severely impairs survival, shows that both cytoplasmic pH and membrane potential are drastically reduced. In the open reading frame (ORF), HP0244 is the last gene in the operon, consisting of 3 genes (HP0246, HP0245, and HP0244), and is followed by a stretch of ∼200 bp of intergenic sequence preceding HP0243, which is expressed in a separate operon downstream. Therefore, the phenotypes observed in the HP0244 deletion strain are not due to polar effects. Furthermore, we verified that the observed defect in acid survival is due specifically to mutation of HP0244 by replacing the mutated locus with an intact copy of the HP0244 ORF (42). Since HP0244 is cytoplasmic, a fall in pH is probably the cytoplasmic change that is sensed by this histidine kinase, unless there is a response to a small change in inner membrane potential.
There have been several studies evaluating the permeability of the water channel, aquaporin, to CO2, NH3, and NH4+. In general, permeation of CO2 is not described by the solubility-diffusion model, probably requiring the presence of a protein to account for its transport across certain membranes (31). Experiments on AQP1, a member of the MIP family expressed in Xenopus oocytes, which measured internal acidification, showed that this channel was permeable to CO2 (6). By measurement of the external pH of oocytes, it was also shown that AQP1 transported CO2 and NH3 but that AQP4 and AQP5 transported only CO2 (20). There was an approximately fourfold increase in the rate of CO2 penetration in oocytes expressing AQP1. The physiological relevance of this property for mammals is controversial (7).
AQP3, -8, and -9 also transport both NH3 and NH4+ (11). The ammonium transporter, AmtB, has 11 transmembrane segments and is required for nitrogen accumulation by bacteria and yeast. Based on the crystal structure, it appears that the channel is able to bind NH4+ and to lower its pKa to allow deprotonation and generation of NH3 that can pass through the hydrophobic channel while the H+ leaves by another route (13, 15). A similar concept has been suggested for the simpler water channel aquaporin 8 (26). However, in contrast, there are data using methylamine indicating that an ion is actually transported by AmtB (9).
The above data showing the transport properties of UreI in H. pylori were interpreted based on the in vitro results where inward transport of NH3, NH4+, and CO2 was shown. In vivo, transport is likely in the opposite, outward direction. In vivo, protons will enter both the periplasm and cytoplasm, requiring rapid and effective neutralization for gastric survival and colonization.
H. pylori expresses a cytoplasmic β-carbonic anhydrase that accelerates the production of cytoplasmic CO2 from the H2CO3 resulting from urease activity. If the CO2 exit is facilitated by UreI, then the periplasmic α-carbonic anhydrase can more rapidly generate the HCO3− necessary to buffer the periplasm at pH 6.1. The relevance of this to acid acclimation by the organism is shown by the absence of rapid periplasmic pH elevation in the absence of assembly of urease with UreI in the HP0244 deletion mutants. Elimination of α-carbonic anhydrase impairs acid survival, and deletion of both carbonic anhydrases reduces infectivity of the X47 mutant strain in the mouse (4, 16). In this work, deletion of ureI significantly reduced bacterial cytoplasmic acidification due to CO2 addition. This is most simply interpreted as showing that UreI is able to transport CO2. This property of UreI would be predicted to improve periplasmic pH regulation under acidic conditions.
At a medium pH of 2.5, the measured cytoplasmic pH in the presence of urea and UreI is 7.4. With a periplasmic pH of 6.1, there is an inward pH gradient of 1.3 units, and thus a 20-fold outward gradient of NH3 from the cytoplasm to the periplasm and a much larger gradient into the gastric lumen. The gastric juice NH4+ concentration is ∼2 to 3 mM with H. pylori infection, similar to that of gastric juice urea. Urease activity is ∼5 μmol/mg bacterial protein/min, and urea supply from the blood is essentially unlimited, which would allow a rapid and large relative increase of intrabacterial NH3 + NH4+ unless there was an exit path for one or both. Although the concentration of NH3 is only about 1/100 that of NH4+ at cytoplasmic pH, the rapid rate of efflux of NH3 from the cytoplasm via the phospholipid bilayer and UreI and thence the periplasm through the outer membrane is likely sufficient to restrict a large elevation of NH3 + NH4+. Under some circumstances where the ammonia efflux does not keep pace with urease activity, either ammonium transport by UreI or an active transporter could be required (32, 33).
There is a Δψ of −101 mV for the cells, in principle hindering NH4+ efflux as a cation through UreI. Based on analysis of aquaporins with 6 transmembrane segments, the in vitro observation of NH4+ inward conductance through UreI could be translated in vivo to NH4+ binding to a cytoplasmic vestibule in the channel, followed by deprotonation and transport of the formed NH3 outward through UreI concomitant with a separate efflux of H+, resulting in net export of NH4+ (11). This would result in insensitivity of NH4+ export through UreI to membrane potential.
UreI is a pH-gated urea channel that does not transport thiourea, showing the precision of structural restriction by this channel (40). However, NH3, NH4+, and CO2 are much smaller than urea, and the above data show that although the urea pathway is closed at pH 7.4, there is permeation of these smaller molecules. The availability of a high-resolution crystal should illuminate this issue.
A model incorporating these data is shown in Fig. 6. This model shows that the cytoplasmic histidine kinase, HP0244, is necessary for recruitment of urease to the inner membrane at acidic pH, along with the nickel insertion proteins. This allows activation of urease at the membrane in the vicinity of UreI. The membrane association of urease activity provides a localized increase in NH3, NH4+, and H2CO3. NH3 efflux through both the bilayer and UreI allows rapid neutralization of protons entering the periplasm. UreI may also serve as an exit pathway for NH4+, permitting continuing cytoplasmic pH homeostasis (33). CO2 is produced by the cytoplasmic β-carbonic anhydrase that then enters the periplasm largely via UreI, and the periplasmic α-carbonic anhydrase converts this to HCO3− in the periplasm, leading to buffering of the periplasmic space. The lack of survival of HP0244 deletion mutants at pH 2.5, even in the presence of urea, demonstrates the importance of this complex. There is no illustration of the possible role of the complicated acid-responsive nickel cascade, since this is not addressed in this work (35, 36).
FIG. 6.

Model of the mechanism of action of UreI and HP0244. UreI transports urea at acidic periplasmic pH (pH < 6.2) and increasing urease activity, forming H2CO3 and 2 NH3. Carbonic acid is converted to CO2 in the cytoplasm by β-carbonic anhydrase and enters the periplasm, where it is converted by α-carbonic anhydrase to HCO3+, enabling maintenance of periplasmic pH at ∼6.1. NH3 exits via the bilayer and UreI, and NH4+ is formed from the H+ generated by α-carbonic anhydrase and from protons entering from the medium. The NH4+ generated in the cytoplasm exits via UreI, or perhaps via NH3 + H+ exit. Acidification of the cytoplasm activates HP0244, which in turn allows assembly of the apoenzyme UreA/UreB with the nickel insertion pairs, UreE/UreG and UreF/UreH, activating urease at the membrane, providing local generation of carbonic acid and ammonia.
Acknowledgments
This work was supported in part by National Institute of Diabetes and Digestive and Kidney Diseases grants DK053642, DK058333, and T32 DK07180-34, by National Institute of Allergy and Infectious Diseases grant AI078000, and by the Veterans Administration Merit Review.
Thanks are due to Glenn Nagami and Jeffrey Kraut for careful readings of this paper.
Footnotes
Published ahead of print on 23 October 2009.
REFERENCES
- 1.Alm, R. A., L. S. Ling, D. T. Moir, B. L. King, E. D. Brown, P. C. Doig, D. R. Smith, B. Noonan, B. C. Guild, B. L. deJonge, G. Carmel, P. J. Tummino, A. Caruso, M. Uria-Nickelsen, D. M. Mills, C. Ives, R. Gibson, D. Merberg, S. D. Mills, Q. Jiang, D. E. Taylor, G. F. Vovis, and T. J. Trust. 1999. Genomic-sequence comparison of two unrelated isolates of the human gastric pathogen Helicobacter pylori. Nature 397:176-180. [DOI] [PubMed] [Google Scholar]
- 2.Athmann, C., N. Zeng, T. Kang, E. A. Marcus, D. R. Scott, M. Rektorschek, A. Buhmann, K. Melchers, and G. Sachs. 2000. Local pH elevation mediated by the intrabacterial urease of Helicobacter pylori cocultured with gastric cells. J. Clin. Invest. 106:339-347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beier, D., and R. Frank. 2000. Molecular characterization of two-component systems of Helicobacter pylori. J. Bacteriol. 182:2068-2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bury-Mone, S., G. L. Mendz, G. E. Ball, M. Thibonnier, K. Stingl, C. Ecobichon, P. Ave, M. Huerre, A. Labigne, J. M. Thiberge, and H. De Reuse. 2008. Roles of alpha and beta carbonic anhydrases of Helicobacter pylori in the urease-dependent response to acidity and in colonization of the murine gastric mucosa. Infect. Immun. 76:497-509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clyne, M., A. Labigne, and B. Drumm. 1995. Helicobacter pylori requires an acidic environment to survive in the presence of urea. Infect. Immun. 63:1669-1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cooper, G. J., Y. Zhou, P. Bouyer, I. I. Grichtchenko, and W. F. Boron. 2002. Transport of volatile solutes through AQP1. J. Physiol. 542:17-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fang, X., B. Yang, M. A. Matthay, and A. S. Verkman. 2002. Evidence against aquaporin-1-dependent CO2 permeability in lung and kidney. J. Physiol. 542:63-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ferrero, R. L., V. Cussac, P. Courcoux, and A. Labigne. 1992. Construction of isogenic urease-negative mutants of Helicobacter pylori by allelic exchange. J. Bacteriol. 174:4212-4217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fong, R. N., K. S. Kim, C. Yoshihara, W. B. Inwood, and S. Kustu. 2007. The W148L substitution in the Escherichia coli ammonium channel AmtB increases flux and indicates that the substrate is an ion. Proc. Natl. Acad. Sci. USA 104:18706-18711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Foster, J. W. 2004. Escherichia coli acid resistance: tales of an amateur acidophile. Nat. Rev. Microbiol. 2:898-907. [DOI] [PubMed] [Google Scholar]
- 11.Holm, L. M., T. P. Jahn, A. L. Moller, J. K. Schjoerring, D. Ferri, D. A. Klaerke, and T. Zeuthen. 2005. NH3 and NH4+ permeability in aquaporin-expressing Xenopus oocytes. Pflugers Arch. 450:415-428. [DOI] [PubMed] [Google Scholar]
- 12.Hong, W., K. Sano, S. Morimatsu, D. R. Scott, D. L. Weeks, G. Sachs, T. Goto, S. Mohan, F. Harada, N. Nakajima, and T. Nakano. 2003. Medium pH-dependent redistribution of the urease of Helicobacter pylori. J. Med. Microbiol. 52:211-216. [DOI] [PubMed] [Google Scholar]
- 13.Javelle, A., G. Thomas, A. M. Marini, R. Kramer, and M. Merrick. 2005. In vivo functional characterization of the Escherichia coli ammonium channel AmtB: evidence for metabolic coupling of AmtB to glutamine synthetase. Biochem. J. 390:215-222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kashket, E. R., and P. T. Wong. 1969. The intracellular pH of Escherichia coli. Biochim. Biophys. Acta 193:212-214. [DOI] [PubMed] [Google Scholar]
- 15.Khademi, S., and R. M. Stroud. 2006. The Amt/MEP/Rh family: structure of AmtB and the mechanism of ammonia gas conduction. Physiology (Bethesda) 21:419-429. [DOI] [PubMed] [Google Scholar]
- 16.Marcus, E. A., A. P. Moshfegh, G. Sachs, and D. R. Scott. 2005. The periplasmic alpha-carbonic anhydrase activity of Helicobacter pylori is essential for acid acclimation. J. Bacteriol. 187:729-738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marcus, E. A., and D. R. Scott. 2001. Cell lysis is responsible for the appearance of extracellular urease in Helicobacter pylori. Helicobacter 6:93-99. [DOI] [PubMed] [Google Scholar]
- 18.Meyer-Rosberg, K., D. R. Scott, D. Rex, K. Melchers, and G. Sachs. 1996. The effect of environmental pH on the proton motive force of Helicobacter pylori. Gastroenterology 111:886-900. [DOI] [PubMed] [Google Scholar]
- 19.Mitchell, P. 1966. Chemiosmotic coupling in oxidative and photosynthetic phosphorylation. Biol. Rev. Camb. Philos. Soc. 41:445-502. [DOI] [PubMed] [Google Scholar]
- 20.Musa-Aziz, R., L. M. Chen, M. F. Pelletier, and W. F. Boron. 2009. Relative CO2/NH3 selectivities of AQP1, AQP4, AQP5, AmtB, and RhAG. Proc. Natl. Acad. Sci. USA 106:5406-5411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pflock, M., N. Finsterer, B. Joseph, H. Mollenkopf, T. F. Meyer, and D. Beier. 2006. Characterization of the ArsRS regulon of Helicobacter pylori, involved in acid adaptation. J. Bacteriol. 188:3449-3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pflock, M., S. Kennard, I. Delany, V. Scarlato, and D. Beier. 2005. Acid-induced activation of the urease promoters is mediated directly by the ArsRS two-component system of Helicobacter pylori. Infect. Immun. 73:6437-6445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pflock, M., S. Kennard, N. Finsterer, and D. Beier. 2006. Acid-responsive gene regulation in the human pathogen Helicobacter pylori. J. Biotechnol. 126:52-60. [DOI] [PubMed] [Google Scholar]
- 24.Rektorschek, M., A. Buhmann, D. Weeks, D. Schwan, K. W. Bensch, S. Eskandari, D. Scott, G. Sachs, and K. Melchers. 2000. Acid resistance of Helicobacter pylori depends on the UreI membrane protein and an inner membrane proton barrier. Mol. Microbiol. 36:141-152. [DOI] [PubMed] [Google Scholar]
- 25.Sachs, G., J. A. Kraut, Y. Wen, J. Feng, and D. R. Scott. 2006. Urea transport in bacteria: acid acclimation by gastric Helicobacter spp. J. Membr. Biol. 212:71-82. [DOI] [PubMed] [Google Scholar]
- 26.Saparov, S. M., K. Liu, P. Agre, and P. Pohl. 2007. Fast and selective ammonia transport by aquaporin-8. J. Biol. Chem. 282:5296-5301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Scott, D. R., E. A. Marcus, D. L. Weeks, A. Lee, K. Melchers, and G. Sachs. 2000. Expression of the Helicobacter pylori ureI gene is required for acidic pH activation of cytoplasmic urease. Infect. Immun. 68:470-477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scott, D. R., E. A. Marcus, D. L. Weeks, and G. Sachs. 2002. Mechanisms of acid resistance due to the urease system of Helicobacter pylori. Gastroenterology 123:187-195. [DOI] [PubMed] [Google Scholar]
- 29.Scott, D. R., E. A. Marcus, Y. Wen, J. Oh, and G. Sachs. 2007. Gene expression in vivo shows that Helicobacter pylori colonizes an acidic niche on the gastric surface. Proc. Natl. Acad. Sci. USA 104:7235-7240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scott, D. R., D. Weeks, C. Hong, S. Postius, K. Melchers, and G. Sachs. 1998. The role of internal urease in acid resistance of Helicobacter pylori. Gastroenterology 114:58-70. [DOI] [PubMed] [Google Scholar]
- 31.Simon, S. A., and J. Gutknecht. 1980. Solubility of carbon dioxide in lipid bilayer membranes and organic solvents. Biochim. Biophys. Acta 596:352-358. [DOI] [PubMed] [Google Scholar]
- 32.Stingl, K., K. Altendorf, and E. P. Bakker. 2002. Acid survival of Helicobacter pylori: how does urease activity trigger cytoplasmic pH homeostasis? Trends Microbiol. 10:70-74. [DOI] [PubMed] [Google Scholar]
- 33.Stingl, K., E. M. Uhlemann, R. Schmid, K. Altendorf, and E. P. Bakker. 2002. Energetics of Helicobacter pylori and its implications for the mechanism of urease-dependent acid tolerance at pH 1. J. Bacteriol. 184:3053-3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tomb, J. F., O. White, A. R. Kerlavage, R. A. Clayton, G. G. Sutton, R. D. Fleischmann, K. A. Ketchum, H. P. Klenk, S. Gill, B. A. Dougherty, K. Nelson, J. Quackenbush, L. Zhou, E. F. Kirkness, S. Peterson, B. Loftus, D. Richardson, R. Dodson, H. G. Khalak, A. Glodek, K. McKenney, L. M. Fitzegerald, N. Lee, M. D. Adams, E. K. Hickey, D. E. Berg, J. D. Gocayne, T. R. Utterback, J. D. Peterson, J. M. Kelley, M. D. Cotton, J. M. Weidman, C. Fujii, C. Bowman, L. Watthey, E. Wallin, W. S. Hayes, M. Borodovsky, P. D. Karp, H. O. Smith, C. M. Fraser, and J. C. Venter. 1997. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature 388:539-547. [DOI] [PubMed] [Google Scholar]
- 35.van Vliet, A. H., F. D. Ernst, and J. G. Kusters. 2004. NikR-mediated regulation of Helicobacter pylori acid adaptation. Trends Microbiol. 12:489-494. [DOI] [PubMed] [Google Scholar]
- 36.van Vliet, A. H., E. J. Kuipers, J. Stoof, S. W. Poppelaars, and J. G. Kusters. 2004. Acid-responsive gene induction of ammonia-producing enzymes in Helicobacter pylori is mediated via a metal-responsive repressor cascade. Infect. Immun. 72:766-773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Voland, P., D. L. Weeks, E. A. Marcus, C. Prinz, G. Sachs, and D. Scott. 2003. Interactions among the seven Helicobacter pylori proteins encoded by the urease gene cluster. Am. J. Physiol. Gastrointest. Liver Physiol. 284:G96-G106. [DOI] [PubMed] [Google Scholar]
- 38.von Heijne, G. 1995. Membrane protein assembly: rules of the game. Bioessays 17:25-30. [DOI] [PubMed] [Google Scholar]
- 39.Waidner, B., K. Melchers, F. N. Stahler, M. Kist, and S. Bereswill. 2005. The Helicobacter pylori CrdRS two-component regulation system (HP1364/HP1365) is required for copper-mediated induction of the copper resistance determinant CrdA. J. Bacteriol. 187:4683-4688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weeks, D. L., S. Eskandari, D. R. Scott, and G. Sachs. 2000. A H+-gated urea channel: the link between Helicobacter pylori urease and gastric colonization. Science 287:482-485. [DOI] [PubMed] [Google Scholar]
- 41.Wen, Y., J. Feng, D. R. Scott, E. A. Marcus, and G. Sachs. 2006. Involvement of the HP0165-HP0166 two-component system in expression of some acidic-pH-upregulated genes of Helicobacter pylori. J. Bacteriol. 188:1750-1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wen, Y., J. Feng, D. R. Scott, E. A. Marcus, and G. Sachs. 2009. The pH-responsive regulon of HP0244 (FlgS), the cytoplasmic histidine kinase of Helicobacter pylori. J. Bacteriol. 191:449-460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wen, Y., E. A. Marcus, U. Matrubutham, M. A. Gleeson, D. R. Scott, and G. Sachs. 2003. Acid-adaptive genes of Helicobacter pylori. Infect. Immun. 71:5921-5939. [DOI] [PMC free article] [PubMed] [Google Scholar]