

Abstract
Sophisticated models for the regulation of mitotic entry are lacking for human cells. Inactivating human cyclin A/Cdk2 complexes through diverse approaches delays mitotic entry and promotes inhibitory phosphorylation of Cdk1 on tyrosine 15, a modification performed by Wee1. We show here that cyclin A/Cdk2 complexes physically associate with Wee1 in U2OS cells. Mutation of four conserved RXL cyclin A/Cdk binding motifs (RXL1 to RXL4) in Wee1 diminished stable binding. RXL1 resides within a large regulatory region of Wee1 that is predicted to be intrinsically disordered (residues 1 to 292). Near RXL1 is T239, a site of inhibitory Cdk phosphorylation in Xenopus Wee1 proteins. We found that T239 is phosphorylated in human Wee1 and that this phosphorylation was reduced in an RXL1 mutant. RXL1 and T239 mutants each mediated greater Cdk phosphorylation and G2/M inhibition than the wild type, suggesting that cyclin A/Cdk complexes inhibit human Wee1 through these sites. The RXL1 mutant uniquely also displayed increased nuclear localization. RXL1 is embedded within sequences homologous to Crm1-dependent nuclear export signals (NESs). Coimmunoprecipitation showed that Crm1 associated with Wee1. Moreover, treatment with the Crm1 inhibitor leptomycin B or independent mutation of the potential NES (NESm) abolished Wee1 nuclear export. Export was also reduced by Cdk inhibition or cyclin A RNA interference, suggesting that cyclin A/Cdk complexes contribute to Wee1 export. Somewhat surprisingly, NESm did not display increased G2/M inhibition. Thus, nuclear export of Wee1 is not essential for mitotic entry though an important functional role remains likely. These studies identify a novel bifunctional regulatory element in Wee1 that mediates cyclin A/Cdk2 association and nuclear export.
Despite broad progress in studies of cell cycle control in eukaryotes, advanced models are lacking for the regulation of mitotic entry in human cells. This regulation is pivotal in cell cycle control, and a better understanding of it may be crucial to improving cytotoxic cancer chemotherapy, the mainstay of cancer treatment. Models of mitotic entry in higher eukaryotes revolve around activation of the cyclin B/Cdk1 (cyclin-dependent kinase 1 or Cdc2) complex, which drives the major events of mitosis. A rise in the cyclin B level triggers mitotic entry in Xenopus egg extracts but not in mammalian cells (15, 47). Inhibitory phosphorylation of Cdk1 on the ATP-binding site residue tyrosine 15 (Y15) has been recognized as a key constraint throughout eukaryotes (29, 42). Wee1 and Myt kinases perform this phosphorylation in vertebrate cells, where Wee1 appears to be dominant (34). Kim and Ferrell and others have recently developed an elegant model for ultrasensitive, switch-like inactivation of Wee1 by cyclin B/Cdk1 in a positive feedback loop that contributes to mitotic entry in Xenopus egg extracts (27).
Although cyclin A(A2)/Cdk2 is traditionally omitted from models of mitotic entry, accumulating evidence from several different approaches suggests that cyclin A/Cdk complexes play roles. Cyclin A levels rise during S phase and peak in G2 before falling abruptly in prometaphase of mitosis (60). Microinjection of cyclin A/Cdk2 complexes in human G2 phase cells was observed to drive mitotic entry (14). Conversely, microinjection of antibodies directed against cyclin A in S-phase cells inhibited mitotic entry without an apparent effect on bulk DNA synthesis (45). In complementary approaches that supported biochemical analyses, cyclin A RNA interference (RNAi) or induction of a dominant negative mutant of Cdk2 (Cdk2-dn), the major cyclin A binding partner, inhibited mitotic entry (13, 15, 21, 37). In these settings, cyclin B/Cdk1 complexes accumulated in inactive, Y15-phosphorylated forms (13, 21, 37). Cdc25 phosphatases, which can reverse this phosphorylation, show reduced activity in this context (37), but increased Cdc25 activity could not readily overcome the arrest (13). RNAi-mediated knockdown of Wee1 was found capable of overriding the arrest mediated by cyclin A RNAi, suggesting that Wee1 is a key rate-limiting factor (13). However, whether and by what mechanisms cyclin A complexes might regulate Wee1 and drive Cdk1 dephosphorylation and mitotic entry have remained unclear.
Recently, genetic studies in mice have reinforced these observations while providing evidence for some cell type differences (24). Although Cdk2 is not essential, in its absence Cdk1 binds more cyclin A and E and provides redundant functions (4, 25, 44). Deletion of the cyclin A gene is lethal for embryos and adults (24). Gene deletion in fibroblasts in vitro did not completely abrogate their proliferation but caused S and G2/M delays. In this setting cyclin E was upregulated, and combined deletion of cyclin E yielded arrest in G1, S, and G2/M phases. Cyclin A gene deletion was alone sufficient to block proliferation of hematopoietic stem cells, suggesting that cyclin A is essential for their proliferation.
Wee1 is regulated on multiple levels, including inhibitory phosphorylation in the amino-terminal regulatory domain (NRD), residues 1 to 292. This region is predicted to be intrinsically disordered (56), and few functional elements have been identified in it. The cyclin B/Cdk1 complex has been thought to be the principal or exclusive kinase responsible for NRD phosphorylation (18, 27, 28). Two sites in the Xenopus embryonic Wee1 NRD, Thr 104 and Thr 150 (referred to here by the homologous residue, T239, in human somatic Wee1), have been identified as Cdk phosphorylation sites that inhibit Wee1 activity (28). Recent studies of Xenopus somatic Wee1 suggest that T239 phosphorylation may antagonize the function of a surrounding motif, dubbed the Wee box (43). This small, conserved region appears to augment the activity of the carboxy-terminal kinase domain.
We show here that cyclin A/Cdk2 complexes directly bind Wee1 as a substrate in human cells. In particular, a conserved cyclin A/Cdk binding RXL motif in the Wee1 NRD is required for efficient T239 phosphorylation. Further analysis revealed that RXL1 is located within a Crm1 binding site that mediates Wee1 export during S and G2 phases. Cyclin A/Cdk2 activity appears to foster Wee1 export, but this export is not essential for mitotic entry. These findings further define roles of cyclin A/Cdk complexes in regulating Wee1 and mitotic entry in human cells and dissect the mechanisms and consequences of Wee1 redistribution during the run-up to mitosis.
MATERIALS AND METHODS
Plasmids, primers, and mutagenesis.
Human Wee1A coding region cDNA (Origene Inc., Rockville, MD) was amplified by PCR. Primers used for PCR were the following: W5p, 5′-TTAGGATCCATGAGCTTCCTGAGCCGA-3′; w3p, 5′-GGCCTCGAGTCAGTATATAGTAAGGCT-3′. The PCR product was cloned into mammalian expression vector pCMV-3tag-2A (Stratagene, La Jolla, CA) at BamHI/XhoI sites. The constructs M1-4 (RXL1-4 mutation), M1A (RXL1 with RXL and the +5 leucine mutated to alanines) M2AGL (selective mutation of the R alanine in RXL2), qM (quadruple mutant with all four RXL sites mutated), KD (kinase defective), T239A, M1KD, and NESm (mutation of the proline and phenylalanine residues of the nuclear export signal [NES]) were generated by site-directed mutagenesis using a multimutagenesis kit (Stratagene). Primers for mutagenesis were the following: M1, 5′-CCACACAAGACCTTCGCGGCCGCGCGACTCTTCGACAC-3′; M1A, 5′-AAGACCTTCGCGGCAGCGCGAGCCTTCGACACCCCG-3′; M2, 5′-CTTTTGCAAGTTGGCGCGGGCGCCAGGTATATTCATTC-3′; M2AGL, 5′-CTCCTTTTGCAAGTTGGCGCCGGCTTGAGGTATATTC-3′; M3, 5′-GAAGAGGGCGATAGCGCTTTTGCTGCAAATGAAGTTTT-3′; M4, 5′-GCAGCTGAGGAAGCGGCCGCATTCACTGACCGGA-3′; KD, 5′-GTTATGTTTAAAATAGGTAACCTAGGGCATGTAACAAGGATCTCC-3′; T239A, 5′-CAAGTGAATATTAATCCTTTTGCGCCGGATTCTTTGTTGCTTC-3′; NESm, 5′-GGGCACCCCGGCACACAAGACCGCGCGCAAGCTGC-3′.
Cell culture, transfection, induction, synchronization, drug treatment, and extract preparation.
U2-OS cells and HeLa cells were cultured in Dulbecco's modified minimal essential medium supplemented with 10% fetal calf serum, penicillin, and streptomycin and maintained in 5% carbon dioxide and humidified air. Hydroxyurea (HU; Sigma-Aldrich, St. Louis, MO) block and release were described previously (21). For the mimosine (Sigma-Aldrich) block, culture medium and transfection complexes were removed and replaced by medium containing 0.5 mM mimosine for 24 h. Lipofectamine 2000 (Invitrogen, Carlsbad, CA) was used for transfection of small interfering RNA (siRNA) and plasmid according to the manufacturer's instructions. Wee1 siRNAs (sense, GGUAUUGCCUUGUGAAUUUUU; antisense, AAAUUCACAAGGCAAUACCUU) were designed by using the siDESIGN Center from Dharmacon Inc. (Chicago, IL). Cyclin A siRNAs (sense, CUAUGGACAUGUCAAUUGUTT; antisense, ACAAUUGACAUGUCCAUAGTA) are from Applied Biosystems Inc. (Foster City, CA). Cdk2 siRNAs (sense, GCUGAAGAGGGUUGGUAUAUU; antisense, UAUACCAACCCUCUUCAGCUU) are from Dharmacon. The U2-OS cell clones with inducible expression of Cdk2 wild type (Cdk2-wt) and Cdk2-dn have been described previously (21). Induction was achieved by removing cells from the dish with trypsin-EDTA, washing twice with phosphate-buffered saline (PBS), and replating in the absence of tetracycline. In synchronized experiments, induction was begun at the start of HU or mimosine treatment. Cells were released by incubation with standard culture medium for the designated times. Cells were treated with 200 μmol/liter olomoucine (LC Laboratories, Woburn, MA), 20 μmol/liter roscovitine (Sigma-Aldrich), 2.5 ng/ml leptomycin B (LMB; Sigma-Aldrich), or vehicle (dimethyl sulfoxide) alone 6 h after mimosine release. Total cell extracts were prepared in E1A lysis buffer as described previously (21) at the designated time points. Nuclear and cytoplasmic fractions were isolated using a CelLytic NuCLEAR extraction kit (Sigma-Aldrich). For gel electrophoresis, nuclear and cytoplasmic fractions were normalized to cell number.
Antibodies, immunoblotting, and immunoprecipitation.
Immunoblotting and immunoprecipitation were performed as described previously (21). Antibodies directed against cyclin A (H-432 or BF683), cyclin E (C-19 or HE111), cyclin B1 (C-19 or H-433), Cdk2 (M2 or D-12), Cdk1 (C-19 or 17), Crm1 (H-300 or C-1), and Wee1 (C-20 or B-11) were from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-CREB, anti-phospho-Cdk tyrosine 15, and anti-Myc-tag (9B11) antibodies were from Cell Signaling Technology (Danvers, MA). Anti-alpha-tubulin and antiactin antibodies were from Sigma-Aldrich. Anti-glutathione S-transferase (anti-GST) antibody was from GE Healthcare Life Sciences (Piscataway, NJ). The antihemagglutinin (anti-HA) antibody was mouse monoclonal clone 12CA5 (Roche, Indianapolis, IN). Fluorescein isothiocyanate-CD20 antibody was from BD Biosciences (San Jose, CA). Rabbit polyclonal anti-Wee1-phospho-T239 antibody was generated by Covance Immuno Technologies (Denver, PA). It was generated against and purified on a column with the phosphopeptide CKKVNINPFpTPDSL and counterselected against the unphosphorylated peptide. The anti-Wee1 phospho-S121/S123 (S121/123) antibody was kindly provided by Nobumoto Watanabe (Wako, Japan). Secondary antibodies were from GE Healthcare Life Sciences. ImageJ software (National Institutes of Health) was used for quantification of immunoblotting bands. Values for binding or phosphorylation of Wee1 mutants were expressed as ratios to those observed for the wild-type protein expressed in parallel and were calculated following normalization of signal to expression of each Wee1 protein, assayed by immunoblotting.
Flow cytometry and immunofluorescence.
Cell fixation for flow cytometry with propidium iodide and CD20 staining was performed as described previously (21). Data were collected on a Becton Dickinson single laser three-fluorescence FACScan flow analyzer and analyzed with FlowJo and Dean/Jett/Fox (Free Star, Ashland, OR) or ModFit (www.vsh.com/products/mflt) software. Cells were fixed for immunofluorescence as described previously (21) and stained with a Myc-tagged mouse monoclonal antibody (Cell Signaling Technology, Danvers, MA), an Alexa Fluor 568 goat anti-mouse secondary antibody (Invitrogen), and 4′,6′-diamidino-2-phenylindole (DAPI; Sigma-Aldrich). Images were acquired on a Nikon Eclipse E800 microscope with a Nikon DXM1200C camera and using ACT-1C software (Nikon, Medville, NY). In each of two to three independent experiments, cells were scored by a blinded observer.
GST fusion protein expression, purification, and pulldown assays.
Full-length wild-type and mutant (M1, qM, KD, and M1KD) Wee1 coding regions were cloned as BamHI/XhoI fragments into the GST fusion protein expression vector pGEX-4T-1 (GE Healthcare Life Sciences) to generate GST-Wee1 fusion proteins. pGEX-4T-1 and pGEX-Wee1-wt/M1/qM/KD/M1KD were transformed into Escherichia coli strain Rosetta 2 (kindly provided by Yoshihiro Matsumoto, Fox Chase Cancer Center, Philadelphia, PA). Bacterial culture and GST fusion protein purification followed instructions in the GST SpinTrap purification module (GE Healthcare Life Sciences) manual except that an induction temperature of 20°C was used. For pulldown assays, 2 μg of GST or GST-Wee1-wt/M1/qM protein was immobilized with glutathione beads (BD Pharmingen, San Jose, CA) in binding buffer (50 mM HEPES, pH 7.0, 50 mM NaCl, 0.1% NP-40, 1 mM EDTA, 10% glycerol, 1% bovine serum albumin, and protease inhibitors). U2-OS cell extracts were precleared by incubation with GST-glutathione beads at 4°C for 30 min, and then cells were incubated with immobilized GST or GST-Wee1 fusion proteins at 4°C for 2 h. Beads were washed four times with binding buffer and eluted with 2× concentrated sample loading buffer for sodium dodecyl sulfate-gel electrophoresis.
DNA and protein sequence analysis and structure prediction.
DNA and protein sequences were analyzed by Lasergene (DNASTAR Inc., Madison, WI) and Vector NTI (Invitrogen). Predictions of structural order in Wee1 were made using the DIOSPRED server (http://bioinf.cs.ucl.uk.disopred/) (56). Multiple sequence alignments of NES residues from Wee1, cyclin D1, and Snurportin were performed with ClustalW (53). The sequence of Wee1 was aligned to the Snurportin NES templates using the program MolIDE (8), and side chain conformations of the NES peptide were predicted with the program SCWRL (8, 55). This program allowed all Wee1 side chains to move while avoiding clashes with the Crm1 NES binding cleft, whose residues were held rigid during modeling. Structure figures were produced with the program Chimera (University of California, San Francisco).
Statistical analysis.
A paired t test was used to test each hypothesis comparing G2/M fractions obtained following expression of wild-type Wee1 or a specific mutant (or vector). This approach was taken to account for possible subtle differences in conditions among the several independent experiments analyzed for each wild-type-mutant pair.
RESULTS
Cyclin A/Cdk2 complexes physically associate with Wee1.
We chose U2-OS human osteogenic sarcoma cells for these studies because they respond well to synchronization methods and are widely used for human cell cycle studies (12, 33, 36, 54). We first asked whether endogenous Cdk2 and Wee1 are physically associated. Cdk2 is the major Cdk partner of cyclin A in human cells and does not bind appreciably to cyclin B in the presence of Cdk1 (21, 35, 37, 38). Cdk2 was readily detected in somatic Wee1 immunoprecipitates (IPs) derived from asynchronous cells (Fig. 1A, left). Conversely, Wee1 was readily detected in Cdk2 IPs (Fig. 1A, right). Similar results were obtained using extracts from synchronized cultures enriched for cells in late S and G2 phase (Fig. 1B and data not shown; see below for comparable flow cytometry data). RNAi directed against Cdk2 confirmed that the Cdk2 antibody used is specific for Cdk2 and that the band detected in Wee1 IPs was indeed Cdk2 (Fig. 1C). These results suggested that Wee1 and Cdk2 are physically associated in human cells, including cells in late interphase.
FIG. 1.

Wee1 associates with cyclin A/Cdk2 complexes. (A) Wee1 and Cdk2 complexes were immunoprecipitated from lysates of asynchronous U2-OS cells and subjected to immunoblotting (IB) for the opposite protein. Cdk2 was readily detected in Wee1 IPs, and Wee1 was readily detected in Cdk2 IPs. IPs without extract or with nonspecific immunoglobulin G (Ig) served as negative controls. (B) Same experiment as in panel A except that cells were enriched for S and G2 phases, following treatment with HU and release. (C) Cdk2 RNAi confirms association of Wee1 with Cdk2. U2-OS cells were treated with siRNAs directed against Cdk2 or an off-target sequence (control). IB confirmed knockdown of Cdk2 but not Cdk1, thus also confirming the specificity of the respective antibodies (left). Cdk2 RNAi reduced Cdk2 detected in Wee1 IPs (right). Relative band intensity was quantitated, and values are listed below each lane. The numbers below the panels present quantitation of the main bands observed relative to the control (con).
Paucity of Wee1 association with cyclin E or inactive Cdk2 complexes.
We then tested whether Wee1 associates with cyclin A or E complexes, using asynchronous cells. Wee1 was readily detected in cyclin A IPs but not cyclin E IPs, despite comparable precipitation of Cdk2 (Fig. 2A). These results suggest that Wee1 associates preferentially with cyclin A complexes. Because Cdk2 binds most cyclin A present in U2-OS cells during late S and G2 phases (37), most Wee1 bound to cyclin A is likely in complex with Cdk2. Some complexes may also contain Cdk1, which associates with a low but increasing fraction of cyclin A as U2-OS cells approach mitosis (38). Indeed, low-level association of Wee1 with Cdk1 was detected (data not shown).
FIG. 2.

Lack of Wee1 association with cyclin E or inactive Cdk2 complexes. (A) Cyclin A (cyc A) and cyclin E (cyc E) IPs from asynchronous cells were subjected to IB for Wee1, Cdk2, and the respective cyclins (for the last, the blot was divided, probed separately for each cyclin, and rejoined prior to exposure to film). Note similar recovery of Cdk2 (bottom) in IPs of cyclins A (middle left) and E (middle right) but little detectable Wee1 in the cyclin E IP (top). (B) U2-OS cell clones with inducible expression of HA-Cdk2-wt or HA-Cdk2-dn were induced for 40 h. Protein extracts were immunoprecipitated with anti-HA antibody or nonspecific Ig and subjected to IB for Wee1. Note similar recovery of Cdk2 (bottom). Wee1 associated with Cdk2-wt but not Cdk2-dn. (C) The experiment is the same as that in panel B except that the reciprocal immunoprecipitation used a Wee1 antibody and IB with anti-HA antibody.
In principle, Wee1 might bind Cdk complexes as substrates, and/or Cdk complexes might bind Wee1 as substrate. The preferential association of Wee1 with cyclin A/Cdk complexes suggested that the cyclin may dictate association. Recent data have shown that catalytic activity of Cdc28, the major Cdk in Saccharomyces cerevisiae, is required for stable association with the budding yeast Wee1 homologue Swe1 (18). This observation, together with lack of binding of a phosphorylation-resistant Swe1 mutant, suggested the model that phosphorylation of Swe1 by Cdc28 stabilizes the association. We therefore asked whether Cdk2 kinase activity is required for stable association with Wee1. We used U2-OS clones with induction of Cdk2 wild type (Cdk2-wt) or a well-characterized catalytically inactive, dominant negative (Cdk2-dn) mutant (21). The inducible Cdk2 proteins are tagged with HA epitopes to distinguish them from endogenous Cdk2. Reciprocal immunoprecipitations confirmed association of endogenous Wee1 with the induced Cdk2-wt (Fig. 2B and C). In contrast, Wee1 association with Cdk2-dn was not readily detected (Fig. 2B, C), suggesting that Cdk2 kinase activity is required for robust association. These observations further confirm association of Wee1 with Cdk2 complexes and support the notion that Wee1 may be recognized as a substrate of cyclin A/Cdk2 complexes.
Conserved cyclin A binding motifs in Wee1.
To further address whether Wee1 is recognized as a substrate of cyclin A/Cdk2 complexes, we hypothesized that this interaction might be directed by RXL/Cy motifs. Such motifs mediate Cdk2 binding and phosphorylation of pRb, p27, and other substrates via binding to a hydrophobic pocket in cyclin A (1, 7, 52, 59). Most defined RXL interactions have been with cyclin A or, less commonly, cyclin E/Cdk2 complexes but not cyclin B/Cdk1 complexes. Direct evidence has recently been obtained that cyclin A/Cdk2 complexes interact more strongly in vitro with model RXL-containing peptides than do cyclin B/Cdk2 complexes (6). An apparent anomaly is Myt, which contains a C-terminal RXL sequence that mediates binding to cyclin B/Cdk1 complexes (5).
Wee1A contains four RXL sequences (Fig. 3A) that are conserved throughout the known vertebrate somatic Wee1 family members (Fig. 3B). These sites lie, respectively, within the long NRD of Wee1 (RXL1), the kinase domain (RXL2 and RXL3), and the short carboxy-terminal region (RXL4) (Fig. 3A). In the two sites within the catalytic region (RXL2 and RXL3), the R and L residues are not conserved among protein kinases (17), suggesting that they may not be required for catalytic activity. We noted that RXL1 is the most conserved RXL sequence. It is also conserved in flies and, in addition, possesses a leucine in the +5 position relative to the arginine. A leucine residue is preferred in this position in cyclin A recognition motifs (6, 9). The NRD of Wee1 is overall poorly conserved and predicted to be largely disordered by the DISOPRED server (56). This method uses different criteria, judging in part whether the amino acid content is consistent with packing interactions needed for structured domains. In contrast, RXL1 was within a small region of potential order (Fig. 3C). Of note, the Wee box containing T239 occupied a nearby region of potential order (Fig. 3C). Such short regions predicted to be ordered within otherwise largely disordered regions have been identified as potential functional regions by Mohan and colleagues (39).
FIG. 3.

Conserved RXL sequences in Wee1. (A) Primary structure map of human somatic Wee1, with residue numbers at domain boundaries below (black): NRD (light green), kinase domain (dark green), carboxy-terminal domain (light blue), RXL sites (red, with amino acid numbers below), Wee box (single-letter amino acids), and T239 phosphorylation site (aqua). (B) RXL sequences conserved in Wee1 (blue shading). Note that RXL1 and RXL3 are conserved through the fruit fly, and RXL1 is followed by a leucine at position +5, favored in cyclin A binding sequences. The leucine in RXL2 (yellow) is predicted to be involved in packing interactions. (C) Predicted disorder plot of the Wee1 NRD from the DISOPRED server (http://bioinf.cs.ucl.uk/disopred/). The horizontal hatched line marks the 5% default false-positive rate for disordered regions, the solid filter line marks the output from DISOPRED2, and the hatched output line marks the output from a linear support vector machine classifier of lower confidence level. Positions of the RXL1 and Wee box are marked. x axis, Wee1 amino acid sequence; y axis, probability of being disordered.
Reduced cyclin A/Cdk2 binding and T239 phosphorylation of RXL mutants.
To test the potential role of the RXL sites in mediating Wee1 binding and/or phosphorylation by cyclin A/Cdk complexes, we mutated these sites. The RXL residues were mutated to alanines in each site, except the central (X) residues in sites 2 and 3, which are conserved in protein kinases. M1 refers to the RXL1 mutant, M2 to the RXL2 mutant, and other designations use the same form. Amino-terminal Myc epitope tags were added, and the primary structure of each coding region was confirmed by total sequencing. In transient expression studies, each mutant was expressed at levels comparable to the wild-type protein except M2, which displayed lower levels (data not shown). Examination of the crystal structure of the Wee1 kinase domain (46, 51) suggests that the RXL2 leucine is involved in packing interactions. Loss of these interactions might render the protein unstable. We therefore generated an RXL2 mutant with only the arginine replaced, designated M2AGL. This protein was expressed at levels comparable to the level of the wild-type protein (data not shown). We then generated a quadruple mutant (qM) with all four RXL sites altered as in the individual mutants, including M2AGL.
We expressed wild-type Wee1, M1, and qM by transient transfection in U2-OS cells and compared their stable association with cyclin A/Cdk2 complexes in cells enriched in late S and G2 phases (data not shown) (see below). Cdk2 immunoprecipitations recovered modestly and distinctly lower levels of M1 and qM, respectively (Fig. 4A). Cyclin A immunoprecipitations yielded similar results (Fig. 4B). Binding of cyclin A to the RXL mutants was reduced more than that of Cdk2, an observation consistent with the possibility of some reduction in binding of cyclin A/Cdk1 complexes and/or independent binding of cyclin E/Cdk2 complexes. These observations suggest that M1 and the other RXL sites contribute to stable Wee1 association with cyclin A/Cdk2 and possibly cyclin A/Cdk1 complexes. To exclude secondary effects (e.g., cell cycle effects) that might influence associations in vivo, we assayed binding in vitro. We generated amino-terminal GST fusion proteins with full-length wild-type Wee1, M1, and qM. We compared the ability of the respective GST-Wee1 fusion proteins and glutathione beads to pull down cyclin A and Cdk2 from extracts of cells enriched in S and G2 phase. GST fusions of M1 and qM showed modest and distinctly less binding, respectively, than GST-Wee1-wt, providing further evidence that the RXL sites in Wee1 bind cyclin A/Cdk2 complexes (Fig. 4C). It was more difficult to detect binding of cyclin B/Cdk1 complexes to the GST-Wee1 fusion proteins, but no differences were observed between the Wee1 mutants (Fig. 4D), consistent with the established binding preferences of RXL sites for cyclin A complexes.
FIG. 4.

Reduced binding of M1 and qM mutants to cyclin A/Cdk2 complexes. (A) U2-OS cells were transiently transfected with empty vector (V), wild-type (wt) Wee1, M1, or qM and enriched in late S and G2 phases by mimosine treatment and release. Protein extracts were subjected to immunoblotting (IB) for the respective Wee1 proteins (top, Myc tag), or immunoprecipitation (IP) for Cdk2 and IB for the respective Wee1 proteins (Myc tag), cyclin A (cyc A), or Cdk2. M1 and qM showed moderately and markedly reduced binding to Cdk2 complexes, respectively. The mean ratio of Cdk2 bound to M1 versus the wild type was 0.72 ± 0.03 (n = 3), and for qM versus the wild type it was 0.40 ± 0.03 (n = 3) (B) The same extracts used in panel A were subjected to cyclin A IP. Nonspecific antibody (Ig) was used for IP as additional negative controls. M1 and qM showed moderately and markedly reduced binding to cyclin A complexes, respectively. The mean ratio of cyclin A bound to M1 versus the wild type was 0.55 ± 0.04 (n = 3), and for qM versus the wild type it was 0.10 ± 0.01 (range, n = 2). (C) GST alone or GST fusion proteins of wild-type Wee1, M1, or qM were incubated with extracts from U2-OS cells enriched for late-S and G2 phases and retrieved on glutathione beads. GST proteins (GST antibody), cyclin A, and Cdk2 were detected by IB. Note that the qM pull-down was somewhat overloaded. M1 and qM showed reduced binding to Cdk2 and cyclin A complexes. Extr: extract alone. The mean ratio of cyclin A bound to GST-M1 versus the wild type was 0.57 ± 0.09 (n = 4), and for GST-qM versus the wild type it was 0.29 ± 0.08 (n = 3). The mean ratio of Cdk2 bound to GST-M1 versus the wild type was 0.80 ± 0.08 (n = 3), and for GST-qM versus the wild type it was 0.29 ± 0.03 (n = 4). (D) GST alone or GST fusion proteins with wild-type Wee1, M1, or qM were used in pull-down assays to assess binding in vitro to cyclin B (cyc B) or Cdk1, detected by IB. Binding of M1 and qM to cyclin B/Cdk1 complexes was not diminished. The numbers below the panels present quantitation of the main bands observed relative to the control.
RXL motifs have been implicated in directing phosphorylation of particular sites among local alternatives (9, 49). Multiple Cdk phosphorylation sites have been identified in budding yeast Swe1 and Xenopus somatic and embryonic Wee1 proteins. Phosphorylation of T239 stands out in the vertebrate proteins. This site was independently identified as a Cdk1 phosphorylation site in Xenopus somatic and embryonic Wee1 proteins (28, 43). T239 is within the Wee-box, and evidence has been obtained that phosphorylation at this site abrogates Wee-box-mediated stimulation of Wee1 activity, thereby inhibiting Wee1 (28).
To investigate T239 phosphorylation in human somatic Wee1, we generated and purified polyclonal antibodies directed against a T239-phosphorylated peptide (P-T239). This antibody recognized endogenous Wee1 in a manner sensitive to phosphatase treatment (Fig. 5A). The antibody also efficiently recognized wild-type Wee1 expressed by transient transfection and showed decreased reactivity against M1 and qM (Fig. 5B). To confirm the antibody's relative specificity for the P-T239 protein, we generated a T239A mutant. The P-T239 antibody showed only modest reactivity against this protein, likely due to some reactivity against surrounding sequences present in the peptide antigen (Fig. 5C). Relative to T239A, P-T239 reactivity with M1 and qM was slightly greater, suggesting that T239 phosphorylation of M1 and qM, though reduced, was not eliminated. For comparison, we examined phosphorylation of another nearby site suggested to be Cdk dependent and to influence the stability of human somatic Wee1 (58). Reactivity with an antibody demonstrated to recognize this site (57) was unaffected in M1 (Fig. 5D). Less T239 phosphorylation of M1 than the wild type was also seen when both exogenous proteins were expressed at lower levels, approaching those of endogenous Wee1 (data not shown).
FIG. 5.

Reduced phosphorylation of M1 and qM on T239. G1/S-synchronized U2-OS cells were transfected with the indicated Wee1 proteins, synchronized, and used to prepare extracts from cells enriched in late-S and G2 phases. (A) T239 phosphorylation of Wee1. Anti-P-T239 antibody was used for immunoblotting (IB) of endogenous Wee1 IPs, without or with phosphatase treatment. IB with anti-Wee1 antibody demonstrated a shift in mobility with phosphatase treatment and absence of proteolysis. (B) Reduced T239 phosphorylation in M1 and T239A. Cells expressing wild-type (wt) Wee1, T239A, or M1 were subjected to IB for Wee1 (Myc tag), P-T239, or tubulin (loading control). Note the lack of reactivity of T239A with the P-T239 antibody and reduced reactivity of M1. (C) Reduced T239 phosphorylation in M1 and qM. Extracts from cells expressing wild-type Wee1, M1, or qM were subjected to IB for P-T239. The blot was then stripped and reprobed for Wee1 (Myc tag). The ratios (means ± standard deviations) of P-T239 were as follows: M1 to the wild type, 0.48 ± 0.11 (n = 6), T239A to the wild type, 0.11 ± 0.02 (n = 7), qM to the wild type, 0.40 (range, n = 2). (D) Phosphorylation of S121/123 was unaffected in M1. Extracts from cells transfected with empty vector, the wild type, or M1 were subjected to IB with P-S121/123 or Myc antibody. (E) Cyclin A (CycA) knockdown reduces Wee1 T239 phosphorylation. Extracts of cells transfected with wild-type Wee1 and either scrambled (con) or cyclin A siRNA were subjected to IB for Wee1 (Myc tag), P-T239, cyclin A, or tubulin (loading control). The ratio (mean ± standard deviation) of P-T239 for cyclin A RNAi versus control was 0.52 ± 0.07 (n = 4). (F) M1 and T239A mediate greater Cdk phosphorylated at Y-15 (P-Y15) than the wild type in vivo. Synchronized U2-OS cells were transfected with empty vector (V) and wild-type, M1, T239A, and KD proteins, and cell lysates were prepared from late S/G2-enriched populations. Cdk P-Y15 was assayed by IB. The mean (± standard deviation) ratios of Cdk P-Y15 observed following transfection were as follows: wild type to vector, 1.8 ± 0.2 (n = 4), M1 to wild type, 1.7 ± 0.2 (n = 7), T239A to wild type, 2.2 ± 0.2 (n = 6), KD to wild type, 0.93 ± 0.03 (range, n = 2). The numbers below the panels present quantitation of the main bands observed relative to the control. tub, tubulin.
We then tested whether cyclin A knockdown reduced T239 phosphorylation of endogenous Wee1 using synchronized cells harvested as the peaks entered mid-late S phase (data not shown). Cyclin A RNAi reduced T239 phosphorylation (Fig. 5E). Cdk2 knockdown also reduced T239 phosphorylation of endogenous Wee1 (data not shown). We conclude that RXL1 and the other RXL sites contribute to Wee1 binding by cyclin A/Cdk2 complexes and that RXL1 is required for optimal phosphorylation of T239.
As an initial test of the function of M1 and T239A mutants, we assayed their impact on Cdk Y15 phosphorylation in transfected cells. For these studies, we generated a KD derivative. We substituted asparagine for aspartate 145 of the catalytic site, the residue that orients the terminal phosphates of ATP for the phospho-transfer and is mutated in our extensively validated Cdk2-dn (Cdk2 numbering) (21). In vitro auto-phosphorylation confirmed that the KD mutant was inactive (see below). Wild-type Wee1 increased Cdk Y15 phosphorylation compared to empty vector in U2-OS cells (Fig. 5F) and 293 cells (data not shown). M1 was more potent than the wild type (Fig. 5F), and T239A was as potent as M1 (Fig. 5F, right). The KD mutant was less potent than the M1 or T239A mutant. Some Cdk Y15 phosphorylation seen with the KD mutant may be due to protection of the Cdk from phosphatase activity and/or trapping of cells in early S phase (see next section). These findings indicated that M1 and T239A might be more potent Cdk Y15 kinases in vivo.
Increased G2/M inhibition by M1 and T239A.
These results prompted us to examine the ability of M1 and other Wee1 mutants to inhibit progression through G2/M. Asynchronous U2-OS cells were transiently transfected with the respective constructs and analyzed by flow cytometry. Wild-type Wee1 imposed a dose-dependent increase in G2/M fraction (Fig. 6A) (see below). M1 was more potent than the wild type in increasing the G2/M fraction. In contrast, M3 was less potent than either the wild type or M1 (Fig. 6A). M2AGL had a mild phenotype like M3, and M4 had no substantial effect (data not shown). An M1 mutant with the additional mutation of the +5 position leucine to alanine (M1A) showed an increase in the G2/M fraction similar to that of M1 (data not shown).
FIG. 6.

Reduced G2/M inhibition by M3 and increased G2/M inhibition by M1. (A) Asynchronous U2-OS cells were transiently transfected with vector (V), wild-type (wt) Wee1 (with two different amounts of plasmid [μg]), M1, and M3; harvested at 40 h; and subjected to flow cytometry (x axis, DNA content; y axis, relative cell number; profiles normalized to the highest peak) or immunoblotting for exogenous Wee1 (Myc tag). The percentages of cells in each cell cycle phase are listed. Note that the M1 G2/M fraction was nearly as high as that of the wild type expressed at substantially higher levels, and the M1 S phase fraction was lower. (B) Cells transfected with the wild type, M1, or M3 were synchronized by mimosine, released for the designated times, and subjected to flow cytometry.
We explored the effects of Wee1 further using synchronized cells. Cells were transfected, synchronized in G1 (10), released, and harvested at intervals. Sample flow cytometry profiles are shown in Fig. 6B from cells harvested at regular intervals. M3 again showed reduced accumulation of cells at G2/M, the opposite effect of M1, underscoring the specificity of the results. M3 also delayed S-phase progression, an effect phenocopied by the KD mutant (data not shown) (see below). Expression of M1 again resulted in a greater accumulation of cells in G2/M than expression of the wild type (Fig. 6B). These results directly demonstrate that the increased G2/M fraction observed with expression of M1 is due to cell cycle delay, excluding the formal possibility of acceleration of passage through other phases of the cell cycle.
We sought to further characterize the kinase activity of the Wee1 mutants. Immunoprecipitation-kinase assays proved problematic, even with inactive Cdk complexes as substrates. We were, however, able to perform auto-phosphorylation, a well-established assay of intrinsic kinase activity in Wee1 proteins (28), on the GST fusion proteins. GST-Wee1-wt and GST-M1 were similarly active in this assay, whereas GST-KD, GST-M1KD, and GST-qM were inactive (data not shown). Thus, despite preservation of residues conserved in kinases, the kinase domain mutations in qM (most likely M3 and possibly also M2AGL) appear to compromise intrinsic kinase activity (data not shown). We infer that efficient S-phase progression appears to require Wee1 kinase activity, but this issue requires further investigation and is not the focus here.
We extended these studies to other cell types and clarified the stage of the delay. M1 also showed a stronger G2/M delay than the wild type in HeLa and 293 cells (data not shown). Coimmunofluorescent staining for Wee1 (Myc) and DNA (DAPI) in U2-OS and HeLa cells demonstrated that the vast majority of wild-type- and M1-expressing cells in each cell type contained uncondensed DNA and that M1 resulted in a greater fraction (data not shown) (see Fig. 8 below), consistent with a delay in interphase (G2). These results suggest that M1 is a hyperfunctional mutant of Wee1 with enhanced ability to block mitotic entry and that RXL1 fosters inactivation of Wee1.
FIG. 8.

Increased nuclear localization of M1. (A) Asynchronous (asynch) U2-OS cells were transfected with the respective Wee1 proteins, fixed at 40 h, and subjected to indirect IF staining for Wee1 (Myc tag, red; see inserted images) and DAPI (blue). Cells were scored in a range from exclusively nuclear to exclusively cytoplasmic. Note the dramatically increased nuclear localization of M1. (B) U2-OS cells were transfected with the wild type (wt) or M1, synchronized (synch) by mimosine block and release for 10 h, and stained and scored as described for panel A. M1 was again much more nuclear. Dual-phase contrast/fluorescence microscopy confirmed that the vast majority of M1-expressing cells displayed abundant, broadly spread cytoplasm comparable to wild-type-expressing cells; hence, the stronger nuclear staining of M1 was not secondary to cell rounding (data not shown). (C) Quantitation of Wee1 localization in synchronized U2-OS cells from panel B, scored as in panel A. (D) HeLa cells were transfected with the wild type or M1, fixed at 40 h, and stained and scored as in panel A. (E) M1 and M1KD were expressed in U2-OS cells, and their localizations were scored as per panel A. Each result in this figure is representative of at least two independent experiments.
To test this notion further, we performed an extensive series of experiments in both asynchronous and synchronized cells in which pairwise transfections were performed in parallel, using wild-type Wee1 as an internal control. The wild-type protein consistently increased the G2/M fraction compared to the empty vector (Fig. 7A), and M1 was more potent than the wild type (Fig. 7B).
FIG. 7.

Increased G2/M arrest mediated by M1 and T239A. (A) Empty vector (V) and wild-type (wt)-expressing plasmids were expressed by transient transfection in asynchronous cells (Asynch) or cells synchronized with mimosine and released for 18 h (Synch). Exogenous Wee1 levels were assayed by immunoblotting (Myc tag), with a tubulin (tub) loading control. Cell cycle position was assayed by flow cytometry from the same plates of cells (center). The percent G2/M content is quantified above that peak. Results from multiple (N) independent experiments are graphed (right), with paired results from the same experiment connected by lines. The wild type yielded consistently more cells in G2/M than vector (means ± standard deviations, 33 ± 6 versus 20 ± 6 for asynchronous cells [P < 0.0008] and 30 ± 2 versus 20 ± 4 for synchronized cells [P < 0.005]). (B) The same experiment as in panel A with transfection of the wild type and M1. M1 consistently yielded higher G2/M levels (46 ± 3 versus 34 ± 6 for asynchronous cells [P < 0.01] and 37 ± 2 versus 30 ± 1 for synchronized cells [P < 0.009]). (C) The same experiment as in panel B except that the wild type and T239A were compared in synchronized cells. T239A yielded consistently higher G2/M levels (40 ± 5 versus 31 ± 6; P < 0.01).
Exogenous Myt1 has been shown to be capable of inhibiting mitotic entry in a kinase-independent fashion (31). We tested whether the augmented G2/M delay imposed by M1 was dependent on kinase activity. Transient transfection experiments showed that M1KD displayed lower G2/M inhibition than M1 (data not shown). These observations suggest that the M1 phenotype of enhanced G2/M arrest depends, at least in part, on its kinase activity.
We then assessed the cell cycle inhibitory activity of the T239A mutant using synchronized cells. T239A also displayed a stronger G2/M arrest than the wild type (Fig. 7C). The G2/M accumulation observed with T239A was on average modestly less than that of M1, but this difference was not statistically significant in pairwise comparisons (data not shown). These findings confirm that inhibitory phosphorylation of T239 is conserved in human somatic Wee1 and suggest that this modification is a functional target of cyclin A/Cdk2 complexes acting through RXL1.
Nuclear localization of M1.
Wee1 has been described as a nuclear protein that relocates to the cytoplasm around mitosis, a shift expected to facilitate mitotic entry (3, 19, 26). We therefore compared the subcellular localization of exogenously expressed wild-type Wee1 and RXL mutants using indirect immunofluorescence (IF). In asynchronous cultures, wild-type Wee1 displayed a range of localizations, from exclusively nuclear in 13% of stained cells to exclusively cytoplasmic in 18% (Fig. 8A, left). The fraction of cells with partial or exclusive cytoplasmic localization (ca. 70%) was much greater than the mitotic fraction scored by flow cytometry, suggesting that this localization was not restricted to mitotic cells with disrupted nuclear envelopes and, therefore, was not a passive outcome of mitosis. In striking contrast, M1 displayed exclusively nuclear staining in more than 80% of cells (Fig. 8A). This degree of nuclear localization was unique to M1. M2AGL, M3, and a KD derivative of the wild-type protein showed only modestly greater nuclear localization than the wild type (potentially due to their imposing early S phase delays) (Fig. 6 and 8 and data not shown), and M4 and T239A showed no consistent difference from the wild type (Fig. 8A). These observations indicate that T239 phosphorylation is not necessary for export and that reduced T239 phosphorylation is unlikely to account for the reduced export of M1.
We then compared wild-type and M1 localization in cells enriched in mid-late S phase by mimosine block in G1 and release for 10 h. This time point was before a Wee1-mediated G2 arrest in most cells (Fig. 6). The wild type was largely (ca. 80%) cytoplasmic (Fig. 8B and C), out of proportion to the mitotic fraction (ca. 15%) (Fig. 6 and data not shown). Consistent with this notion, IF assays demonstrated that the vast majority of wild-type-expressing cells had an interphase DNA staining pattern (data not shown). Thus, the exogenous wild-type protein appears to be largely exported from the nucleus prior to mitosis. In contrast, M1 was again predominantly (>80%) nuclear (Fig. 8B and C).
We extended these experiments to another cell type, HeLa, with similar results (Fig. 8D).
To test whether the subcellular distribution of M1 might be secondary to increased kinase activity of this mutant, we compared the subcellular distribution of M1 and the KD version (M1KD). The subcellular distribution of the M1KD mutant was very similar to that of M1 itself (Fig. 8E) and showed greater nuclear localization than the KD mutant (with an intact RXL1) (Fig. 8A). These results suggest that the increased nuclear localization of M1 is not dependent on M1 kinase activity or increased inhibition of Cdk proteins.
RXL1 is embedded within a Crm1-dependent NES.
Given the striking nuclear localization of M1, we considered the possibilities that RXL1 may itself regulate or be embedded within an NES or nuclear localization sequence (NLS). The surrounding sequences do not match a consensus for either signal (22, 30, 41), and the M1 mutation, which removes two basic residues, would be predicted to weaken rather than strengthen an NLS. Furthermore, a Wee1 mutant entirely lacking the NRD also localized largely to the nucleus (data not shown). This observation suggested that an NRD NLS, if present, was not essential for Wee1 nuclear localization and that the NRD might harbor an NES. A Crm1 consensus sequence can mediate Crm1 binding so tightly as to prevent dissociation (30). Many functional Crm1 binding sequences only loosely match such a motif. Examination of the Wee1 primary sequence surrounding RXL1 reveals such a loose match to the consensus sequence and the NES of the prototypic export cargo Snurportin (Fig. 9A). These residues are conserved throughout metazoan Wee1 proteins and fill the potentially structured domain surrounding RXL1 (Fig. 3C). Crm1-dependent export of cyclin D1 is directed by a similar loose match to the Crm1 consensus, including proline as the first (somewhat atypical) hydrophobic residue (Fig. 9A) (2).
FIG. 9.

RXL1 is embedded within a Crm1-dependent NES. (A) Amino acid sequence alignment of the RXL1 (bold) domain, the consensus Crm1 NES, and the established Crm1-dependent NES from human cyclin D1 (CycD1). Residues matching the consensus are boxed, with the variant residue in a dashed-line box. (B) LMB treatment blocks nuclear export of Wee1. U2-OS cells were transfected with wild-type (wt) Wee1, synchronized at the G1/S border with mimosine, released from the block, and fixed in mid-late S phase. Localization of the exogenous Wee1 was assessed by IF assay using the Myc-tagged antibody. Subcellular distribution was characterized as described in the legend of Fig. 8. Light purple, nucleus only; dark purple, cytoplasm only. (C) Wee1 is physically associated with Crm1. Reciprocal immunoprecipitations (IPs) from an extract (extr) of U2-OS cells synchronized in S phase and subjected to immunoblotting for Wee1 or Crm1. IgG, immunoglobulin G (nonspecific antibody [Ab] control). (D) Model of the Wee1 NES-Crm1 binding interaction, based on Crm1-Snurportin crystal structures. Space-filling was used for Crm1. Blue marks, positive charge; red, negative charge. The Wee1 backbone is shown in green. (E) Ribbon and stick model for the Wee1 NES-Crm1 interaction, with the NES residues highlighted in magenta and labeled by number. (F) Selective mutation of the potential RXL1 Crm1 NES blocks nuclear export. Wild-type Wee1 and a Wee1 mutant in which P175 and F179 were converted to alanine residues (NESm) were expressed by transient transfection in U2-OS cells synchronized in mid-late S phase. Its subcellular localization was assessed by IF as per the legend of Fig. 8.
As an initial test of whether Wee1 might undergo Crm1-dependent nuclear export, we transfected synchronized cells with wild-type Wee1, allowed these cells to progress into S phase, treated half with the Crm1 inhibitor LMB, and assayed Wee1 localization by IF assay in cells enriched in late S/G2 phases (data not shown). LMB treatment abolished cytoplasmic localization of Wee1 in this assay (Fig. 9B). This finding supports the notion of Crm1-dependent export of Wee1 but does not rule out indirect effects.
We then asked whether endogenous Wee1 and Crm1 are physically associated. Reciprocal coimmunoprecipitation from S-phase-synchronized cells demonstrated such association (Fig. 9C). This observation supports direct Crm1-mediated export of Wee1.
Crystal structures of Crm1 in complex with Snurportin have recently been solved (11, 40). Using the Crm1-Snurportin structure as a template, we aligned the putative Wee1 NES to that of Snurportin. We used a side chain rotamer library (8) to predict the conformation of Wee1 residues that would best fit in the Crm1 NES binding cleft, which was held rigid during the modeling process. The resulting model is shown in Fig. 9D (see Movie S1 in the supplemental material for the rotation of structures). Wee1 residues P175, F179, L182, and L184 readily fit the binding cleft as the hydrophobic residues of the NES (Fig. 9A), while hydrophilic residues were more solvent exposed (Fig. 9E).
As an independent test of the role of the potential NES, we therefore mutated P175 and F179 (together) to alanine residues, leaving RXL1 intact (NESm) (Fig. 9A). After transient expression in U2-OS cells, NESm was almost exclusively nuclear (Fig. 9F). We conclude that RXL1 is embedded within a Crm1-dependent NES. Cyclin A/Cdk2 binding by RXL1 may also contribute to localization (see below).
To confirm that Crm1-dependent export of Wee1 does not depend upon overexpression, we examined the subcellular distribution of M1 following siRNA-mediated knockdown of endogenous Wee1 and low-level expression of exogenous proteins. Immunoblotting confirmed that Wee1 knockdown was effective and reduced Y15 phosphorylation of Cdk proteins (Fig. 10A). We titrated expression of exogenous Myc-tagged proteins down to levels within twofold of the endogenous protein (Fig. 10B). At this level of expression, the distribution of exogenous wild-type Wee1 was more nuclear than after expression at higher levels, consistent with the literature on the endogenous protein (3). This observation suggests that a factor needed for Wee1 nuclear localization may be limiting. However, M1 was still markedly more nuclear than the wild type (Fig. 10C), indicating that this difference is maintained over a wide expression range.
FIG. 10.

Crm1-dependent nuclear export of Wee1 at endogenous levels. (A) U2-OS cells were treated with low and high amounts of Wee1 siRNA for 24 or 48 h. Immunoblotting demonstrated knockdown of endogenous Wee1 and reduction in Cdk Y15 phosphorylation. (B) Expression of exogenous (Exo) proteins at levels similar to endogenous (endog) Wee1, detected by Wee1 immunoblotting. Some signal at the position of endogenous Wee1 in the transfected samples may reflect exogenous protein following proteolysis of the Myc tag. con, control. (C) Nuclear localization of M1 at levels similar to the endogenous protein. The localization of exogenous wild-type (wt) Wee1 and M1 expressed at low levels was assessed by IF and quantified in synchronized cells harvested in mid-to-late S phase, as described in the legend of Fig. 8. (D) LMB treatment inhibits cytoplasmic localization of endogenous Wee1. U2-OS cells were synchronized at the G1/S border, and half of the culture was treated with LMB for the last 4 h. Cells were harvested with the peak in late S/G2. Nuclear (N) and cytoplasmic (C) fractions (Frxn) were subjected to immunoblotting for Wee1. Immunoblotting for tubulin and CREB served as cytoplasmic and nuclear markers, respectively.
The subcellular localization of endogenous Wee1 could not be assessed well by IF assay even though a variety of fixation methods and antibodies were tried (data not shown). We therefore assayed its distribution by biochemical fractionation. LMB treatment of cells enriched in late S phase resulted in increased retention in the nucleus (Fig. 10D). Adjusted for the loading controls, LMB increased the nuclear-to-cytoplasmic ratio from 0.84 to 3.57. Residual Wee1 in the cytoplasm fraction after LMB treatment may reflect contamination of this fraction with nuclear proteins.
The NESm mutant allowed us to test whether nuclear localization per se affected T239 phosphorylation or Wee1-mediated cell cycle inhibition. We first confirmed that this mutation compromised Crm1 binding. When expressed to similar levels, NESm associated much less strongly with Crm1 IPs than wild-type Wee1 (Fig. 11A). Association of M1 with Crm1 was also reduced (Fig. 11A). However, T239 phosphorylation in NESm was unaffected (Fig. 11B). Somewhat surprisingly, we also observed no apparent effect of the NESm mutation on the ability of Wee1 to impose a G2/M delay (Fig. 11C). The simplest interpretation of these results is that the reduced T239 phosphorylation in M1 is due to loss of RXL1-mediated Cdk binding and not reduced nuclear export. Furthermore, the enhanced G2/M inhibition displayed by M1 appears to be derived largely from reduced T239 phosphorylation rather than increased nuclear localization (see Discussion).
FIG. 11.

NESm displays reduced association with Crm1 but unperturbed T239 phosphorylation and cell cycle inhibition. (A) Reduced association of NESm with Crm1. Wild-type (wt), M1, and NESm proteins were expressed by transient transfection. Crm1 IPs were assayed by immunoblotting for Wee1 (Myc tag). (B) No change in P-T239 was seen in immunoblotting of the wild type and NESm. (C) No change in G2/M inhibition in NESm. The wild type and NESm were transfected in synchronized cells (Synch) and subjected to flow cytometry, as described in the legend of Fig. 7 (means ± standard deviations, 28 ± 1 versus 28 ± 2; P = 0.7). The numbers below the panels present quantitation of the main bands observed relative to the control. tub, tubulin.
Cyclin A/Cdk activity fosters cytoplasmic localization of Wee1.
We then tested further whether cyclin A/Cdk2 complexes foster Wee1 cytoplasmic redistribution. We expressed wild-type Wee1 by transient transfection in synchronized cells enriched in mid-S phase, with or without treatment with two well-characterized Cdk1/2 inhibitors, olomoucine and roscovitine (50). The Cdk inhibitors were added 6 h after mimosine release so that progression through S phase was only moderately delayed. In addition, the Cdk inhibitor-treated cells were fixed 4 h later than untreated cells to match cell cycle positions as closely as possible (15% and 18% more S phase cells for olomoucine and roscovitine) (data not shown). Wee1 localization was examined by IF assay. Both Cdk inhibitor treatments substantially increased the nuclear localization of wild-type protein (about 40% more cells with exclusively or largely nuclear staining) (Fig. 12A). These findings provide additional evidence that Cdk1/Cdk2 activity contributes to net nuclear export of Wee1. There is little cyclin B/Cdk1 activity in the cell cycle phases examined, modest cyclin A/Cdk1 activity, and abundant cyclin A/Cdk2 activity (21, 37, 38). The simplest interpretation is that cyclin A/Cdk2 and possibly cyclin A/Cdk1 complexes contribute to nuclear export of Wee1. In complementary assays, partial knockdown of cyclin A by RNAi in synchronized cells also substantially shifted the distribution of cotransfected wild-type Wee1 toward the nucleus (Fig. 12B).
FIG. 12.
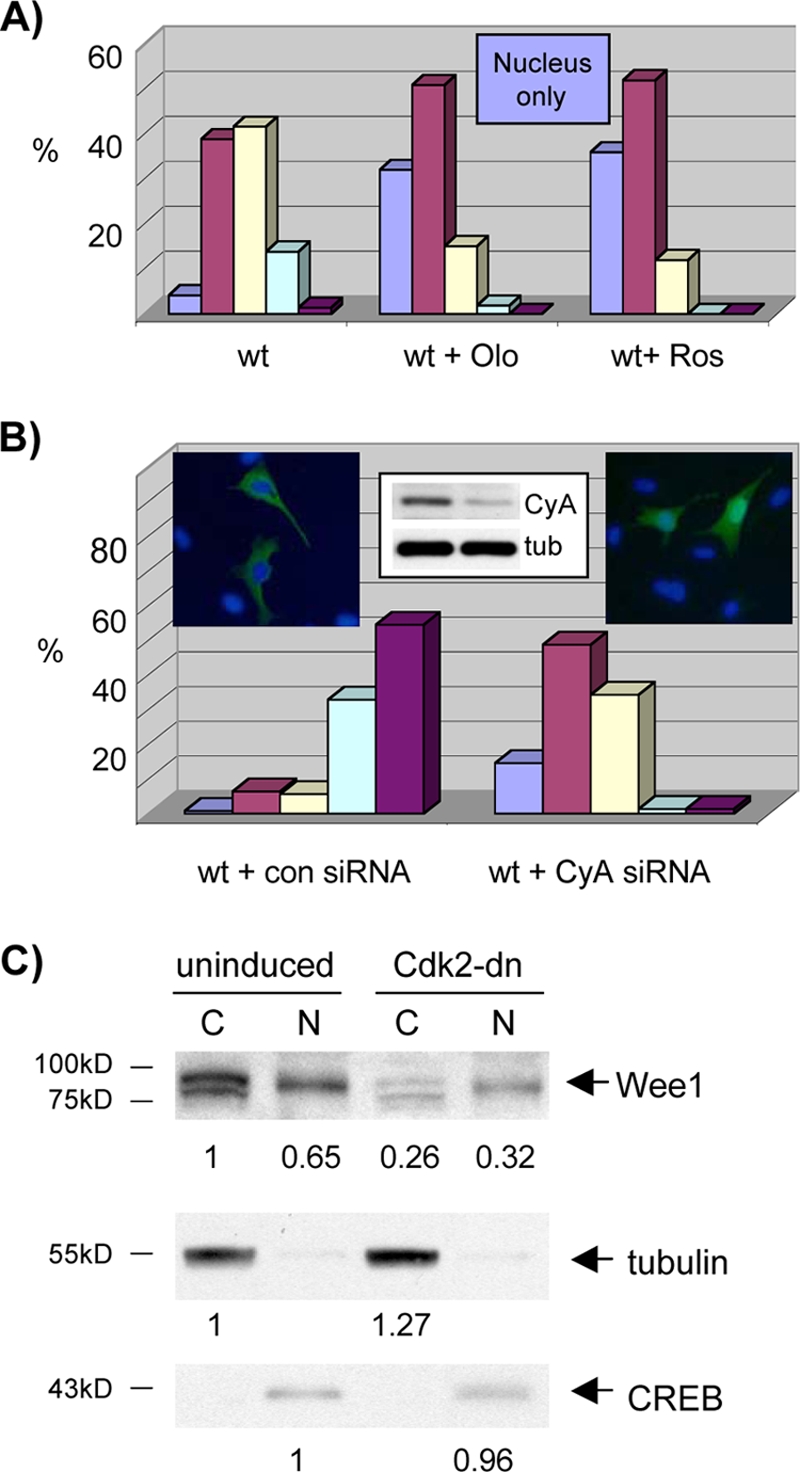
Wee1 nuclear localization is dependent on cyclin A/Cdk activity. (A) Cdk inhibitor treatment reduces nuclear export of Wee1. U2-OS cells were transfected with wild-type (wt) Wee1, synchronized with mimosine, and released. Cells were left untreated or were treated with olomoucine (Olo) or roscovitine (Ros). Cells were harvested with the peak in mid-S phase and subjected to immunofluorescent staining for Wee1 (Myc tag). Results were quantified as described in the legend of Fig. 8A. (B) Cyclin A RNAi reduces nuclear export of Wee1. U2-OS cells were synchronized with mimosine with transfection of control (con) or Wee1 siRNA (see insert for immunoblot of cyclin A and tubulin control). Cytoplasmic and nuclear fractions were analyzed as described in the legend of Fig. 8A. Each result is representative of two independent experiments. CyA, cyclin A; tub, tubulin. (C) Cdk2-dn induction inhibits nuclear export of Wee1. U2-OS cells were synchronized with mimosine with and without induction of Cdk2-dn. Cells were harvested with the peak in late S/G2. Nuclear (N) and cytoplasmic (C) fractions were subjected to immunoblotting for Wee1. Signal intensity was quantified with ImageJ software and normalized to the Cdk2-dn nuclear fraction. Immunoblotting for tubulin and CREB served as cytoplasmic and nuclear markers, respectively. Each result is representative of two independent experiments. The numbers below the panels present quantitation of the main bands observed relative to the control.
We then examined localization of endogenous Wee1. Although the IF signal was weak, increased nuclear localization was seen in synchronized, roscovitine-treated cells (data not shown). As another assay, we performed subcellular fractionation with and without inhibition of cyclin A/Cdk activity by induction of Cdk2-dn. We again used synchronized cells enriched for late S and G2 phases (data not shown). Cdk2-dn induction during S phase was achieved without a dramatic shift in cell cycle profile (data not shown). Wee1 distribution was shifted in favor of the nucleus (Fig. 12C). Adjusted for the loading controls, the nuclear-to-cytoplasmic ratio changed with induction from 0.65 to 1.64. Note that the mobility of cytoplasmic Wee1 is also increased by Cdk2-dn induction, consistent with reduced Wee1 phosphorylation. With roscovitine treatment, the nuclear-to-cytoplasmic ratio changed from 1.0 to 2.13 (data not shown). Although Wee1 levels were reduced by Cdk2-dn induction, they were not reduced by Cdk inhibitor treatment (data not shown), suggesting that the localization shift is not secondary to reduced expression. These results suggest that efficient export of endogenous Wee1 is also dependent on Cdk activity.
DISCUSSION
Wee1 was identified in yeast from mutants that displayed accelerated entry into mitosis, resulting in small cell size (16, 48). These and related studies established that Wee1-mediated Y15 phosphorylation of cyclin B/Cdk1 complexes is a fundamental constraint on mitotic entry in eukaryotes. Evidence that Wee1 can, in turn, be inactivated by Cdk-dependent phosphorylation led to a model in which cyclin B/Cdk1 complexes in higher eukaryotes inactivate Wee1 in a positive feedback loop (47). However, the events that drive the feedback loop past the threshold needed for commitment to mitotic entry have remained obscure in human somatic cells, where a rise in cyclin B levels near mitosis is not routinely observed. Mounting evidence has implicated the dominant Cdk complexes in the preceding cell cycle phases, cyclin A/Cdk2 complexes, and, to a lesser extent, cyclin A/Cdk1 complexes as regulators of mitotic entry and Y15 phosphorylation of Cdk1 (24). These observations have suggested a modified model in which cyclin A/Cdk complexes drive cyclin B/Cdk1 activation past the threshold (13, 14, 15, 21). However, the mechanisms have not been fully defined. Recent data suggest that Wee1 is rate limiting when cyclin A/Cdk activity is compromised (13).
We have reported here that cyclin A/Cdk2 complexes physically associate with Wee1 in S and G2 phase human somatic cells. The preferential association of Wee1 with cyclin A complexes over cyclin E complexes and active over inactive Cdk2 complexes (or inactive Cdk1 complexes [data not shown]) suggests that Wee1 may be recognized as a cyclin A/Cdk2 substrate. The lack of association in vivo with inactive CDKs could reflect either a need for Cdk phosphorylation of Wee1 to stabilize the complex, as observed for Swe1 (18), and/or an inability of inactive Cdk1/Cdk2 complexes to free themselves from competitor substrates (see model below). In Swe1, elimination of many potential phosphorylation sites was required to prevent stable association (18). In any event, these observations are consistent with the notion that Wee1 is recognized as a cyclin A/Cdk substrate.
In further definition of this interaction, we identified an RXL motif, found in many cyclin A substrates, that is conserved in the relatively unstructured amino-terminal regulatory domains of metazoan Wee1 proteins and located in a small region independently predicted to have potential structure. A mutant of this site, M1, demonstrated reduced binding to cyclin A/Cdk2 complexes and reduced phosphorylation of the nearby residue T239. T239 phosphorylation has been implicated in negative regulation of Xenopus Wee1 proteins by Cdk1 complexes and appears to inactivate the Wee box, a domain that augments Wee1 kinase activity (43). RXL1 was the most conserved RXL sequence. Moreover, an RXL1 mutant uniquely displayed enhanced ability to arrest cells in G2. A T239A mutant imposed similarly enhanced G2/M delays, confirming that this phosphorylation is inhibitory in human Wee1 and a functionally significant target of RXL1. Thus, we have identified a molecular interaction through which cyclin A/Cdk complexes can regulate Wee1 and drive mitotic entry.
Evidence has been obtained by others that degradation of human somatic Wee1 is fostered by Cdks (57). Although we did not observe obvious differences in stability between wild-type Wee1 and RXL mutants, this property was not a major focus of our studies, and differences could have been obscured in settings of overexpression.
The reduced cyclin A/Cdk2 binding of the qM mutant compared to M1 suggests that one or more of the other RXL sites may be functional binding sites. T239 phosphorylation does not seem to be further reduced in the qM mutant beyond that of M1. Although expression of M3 (and to a lesser degree M2AGL) delays S phase transit, so does the KD mutant, and initial studies suggest that M3 is catalytically inactive. Therefore, the early S-phase delay observed for M3 may be secondary to misfolding of the kinase domain rather than loss of an RXL3 function that is opposite to that of RXL1. The packing interactions of the RXL2 leucine evident in the crystal structure of the kinase domain (51) argue against its serving as a binding site. M4 has little phenotype.
M1 was also unique in showing dramatically increased nuclear localization in S- and G2-phase cells. This led us to discover that RXL1 is embedded within a Crm1-dependent NES. The function of the NES was supported by the physical association of Wee1 with Crm1, disruption of Crm1 binding in M1 and NESm, and independent blockade of Wee1 nuclear export in M1, NESm, or LMB treatment. Molecular modeling of Crm1-Wee1 NES binding based on the recently defined Crm1-Snurportin crystal structures revealed a good fit within the Crm1 hydrophobic cleft, extending over the predicted structured region around RXL1.
How does cyclin A/Cdk activity contribute to cytoplasmic redistribution of Wee1? Possibilities include activation of nuclear export or cytoplasmic retention signals or inactivation of an NLS. RXL1 may direct additional Wee1 phosphorylation that mediates such an event. Canonical (S/T-P-X-R/K) and noncanonical (S/T-P) Cdk phosphorylation sites have been identified in Swe1 and Xenopus Wee1 NRDs, and a number of such potential sites are present in the human somatic Wee1 NRD. Other kinases may also contribute to Wee1 redistribution. A carboxy-terminal site in Wee1 has been reported to be phosphorylated by Akt, fostering binding to 14-3-3θ and localization in the cytoplasm (26). Cyclin A/Cdk activity might facilitate this mechanism, potentially through the RXL4 site.
It is somewhat surprising that the NESm did not show enhanced G2/M inhibition in our assays. Export of Wee1 is evidently not essential for mitotic entry and may not foster it. One explanation for the lack of an observed delay in mitotic entry of the NESm is that cytoplasmic Wee1 may be needed to prevent early activation of cyclin B/Cdk1 complexes on centrosomes (23). Thus, the role of Wee1 export is currently obscure. However, it is quite possible that such export fosters mitotic entry under more limiting conditions, such as in cell types or conditions (e.g., DNA damage) with less robust Cdk activity. Further work will be needed to define such potential conditions. Alternatively, Wee1 export may render subsequent mitosis more faithful.
A major caveat to models in which cyclin A/Cdk2 complexes drive mitotic entry has been the fact that their activity rises gradually through S and G2 phases. Mitotic entry must be prevented until DNA synthesis and centrosome duplication are complete and then must be fully and promptly executed to avoid genomic instability (20). This conundrum can be resolved if mitotic entry driven by cyclin A/Cdk complexes exhibits an ultrasensitive response, with bistable states separated by a threshold level of activity (27, 61). Kim and Ferrell have provided evidence for ultrasensitivity in inactivation of Xenopus embryonic Wee1 by cyclin B/Cdk1 complexes (27). Competition between Wee1 and alternative cyclin B/Cdk1 substrates contributed to the switch-like inactivation of Wee1. We suggest a revised model for human somatic cells in which cyclin A/Cdk complexes contribute to Wee1 inactivation (Fig. 13). In this model, an ultrasensitive response is based in part on competition between alternative S and G2 RXL-containing substrates and RXL1-mediated cyclin A/Cdk2 phosphorylation of Wee1. Inactivation of Wee1 could be completed by cyclin A/Cdk1 and cyclin B/Cdk1 complexes (data not shown). This model is by no means exclusive. Activation of Cdc25 phosphatases by Cdks (32, 37) and other events are also likely to contribute to mitotic entry.
FIG. 13.

Model of Wee1 regulation by the NES/RXL1 bifunctional regulatory element. Cyclin A/Cdk complexes are freed from RXL-containing S-phase substrates by accumulation of these Cdk complexes and S-phase progression. The Cdk complexes bind RXL1, which directs phosphorylation of T239 and possibly other sites [(S/T)X] that foster nuclear export of Wee1. T239 phosphorylation abrogates stimulation of kinase activity by the Wee box. Crm1 binds the Wee1 NES and directs nuclear export. cycA, cyclin A.
Supplementary Material
Acknowledgments
We thank Tina Chen for contributing to the analysis of Wee1 localization and Sam Litwin for statistical analysis.
This work was supported by NIH grant R01 GM065514 (to G.H.E.) and benefited from NCI institutional support for the Flow Cytometry and Molecular Modeling Facilities at Fox Chase (P30 CA006927).
None of the authors has any conflict of interests to disclose.
Footnotes
Published ahead of print on 26 October 2009.
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Adams, P. D., W. R. Sellers, S. K. Sharma, A. D. Wu, C. M. Nalin, and W. G. Kaelin. 1996. Identification of a cyclin-Cdk2 recognition motif present in substrates and p21-like cyclin-dependent kinase inhibitors. Mol. Cell. Biol. 16:6623-6633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alt, J. R., J. L. Cleveland, M. Hannink, and J. A. Diehl. 2000. Phosphorylation-dependent regulation of cyclin D1 nuclear export and cyclin D1-dependent cellular transformation. Genes Dev. 14:3102-3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baldin, V., and B. Ducommun. 1995. Subcellular localisation of human wee1 kinase is regulated during the cell cycle. J. Cell Sci. 108:2425-2432. [DOI] [PubMed] [Google Scholar]
- 4.Berthet, C., E. Aleem, V. Coppola, L. Tessarollo, and P. Kaldis. 2003. Cdk2 knockout mice are viable. Curr. Biol. 13:1775-1785. [DOI] [PubMed] [Google Scholar]
- 5.Booher, R. N., P. S. Holman, and A. Fattaey. 1997. Human Myt1 is a cell cycle-regulated kinase that inhibits Cdc2 but not Cdk2 activity. J. Biol. Chem. 272:22300-22306. [DOI] [PubMed] [Google Scholar]
- 6.Brown, N. R., E. D. Lowe, E. Petri, V. Skamnaki, R. Antrobus, and L. N. Johnson. 2007. Cyclin B and cyclin A confer different substrate recognition properties on CDK2. Cell Cycle 6:1350-1359. [DOI] [PubMed] [Google Scholar]
- 7.Brown, N. R., M. E. Noble, J. A. Endicott, and L. N. Johnson. 1999. The structural basis for specificity of substrate and recruitment peptides for cyclin-dependent kinases. Nat. Cell Biol. 1:438-443. [DOI] [PubMed] [Google Scholar]
- 8.Canutescu, A. A., A. A. Shelenkov, and R. L. Dunbrack, Jr. 2003. A graph-theory algorithm for rapid protein side-chain prediction. Protein Sci. 12:2001-2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheng, K. Y., M. E. Noble, V. Skamnaki, N. R. Brown, E. D. Lowe, L. Kontogiannis, K. Shen, P. A. Cole, G. Siligardi, and L. N. Johnson. 2006. The role of the phospho-CDK2/cyclin A recruitment site in substrate recognition. J. Biol. Chem. 281:23167-23179. [DOI] [PubMed] [Google Scholar]
- 10.Dalal, S. N., C. M. Schweitzer, J. Gan, and J. A. DeCaprio. 1999. Cytoplasmic localization of human cdc25C during interphase requires an intact 14-3-3 binding site. Mol. Cell. Biol. 19:4465-4479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dong, X., A. Biswas, K. E. Suel, L. K. Jackson, R. Martinez, H. Gu, and Y. M. Chook. 2009. Structural basis for leucine-rich nuclear export signal recognition by CRM1. Nature 458:1136-1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Falck, J., N. Mailand, R. G. Syljuasen, J. Bartek, and J. Lukas. 2001. The ATM-Chk2-Cdc25A checkpoint pathway guards against radioresistant DNA synthesis. Nature 410:842-847. [DOI] [PubMed] [Google Scholar]
- 13.Fung, T. K., H. Tang Ma, and R. Y. Poon. 2007. Specialized roles of the two mitotic cyclins in somatic cells: cyclin as an activator of M phase-promoting factor. Mol. Biol. Cell 18:1861-1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Furuno, N., N. den Elzen, and J. Pines. 1999. Human cyclin A is required for mitosis until mid prophase. J. Cell Biol. 147:295-306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gong, D., J. R. Pomerening, J. W. Myers, C. Gustavsson, J. T. Jones, A. T. Hahn, T. Meyer, and J. E. Ferrell, Jr. 2007. Cyclin A2 regulates nuclear-envelope breakdown and the nuclear accumulation of cyclin B1. Curr. Biol. 17:85-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gould, K. L., and P. Nurse. 1989. Tyrosine phosphorylation of the fission yeast Cdc2+ protein kinase regulates entry into mitosis. Nature 342:39-45. [DOI] [PubMed] [Google Scholar]
- 17.Hanks, S. K., A. M. Quinn, and T. Hunter. 1988. The protein kinase family: conserved features and deduced phylogeny of the catalytic domains. Science 241:42-52. [DOI] [PubMed] [Google Scholar]
- 18.Harvey, S. L., A. Charlet, W. Haas, S. P. Gygi, and D. R. Kellogg. 2005. Cdk1-dependent regulation of the mitotic inhibitor Wee1. Cell 122:407-420. [DOI] [PubMed] [Google Scholar]
- 19.Heald, R., M. McLoughlin, and F. McKeon. 1993. Human wee1 maintains mitotic timing by protecting the nucleus from cytoplasmically activated Cdc2 kinase. Cell 74:463-474. [DOI] [PubMed] [Google Scholar]
- 20.Hernando, E., Z. Nahle, G. Juan, E. Diaz-Rodriguez, M. Alaminos, M. Hemann, L. Michel, V. Mittal, W. Gerald, R. Benezra, S. W. Lowe, and C. Cordon-Cardo. 2004. Rb inactivation promotes genomic instability by uncoupling cell cycle progression from mitotic control. Nature 430:797-802. [DOI] [PubMed] [Google Scholar]
- 21.Hu, B., J. Mitra, S. van den Heuvel, and G. Enders. 2001. S and G2 phase roles for Cdk2 revealed by inducible expression of a dominant negative mutant in human cells. Mol. Cell. Biol. 21:2755-2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hutten, S., and R. H. Kehlenbach. 2007. CRM1-mediated nuclear export: to the pore and beyond. Trends Cell Biol. 17:193-201. [DOI] [PubMed] [Google Scholar]
- 23.Jackman, M., C. Lindon, E. A. Nigg, and J. Pines. 2003. Active cyclin B1-Cdk1 first appears on centrosomes in prophase. Nat. Cell Biol. 5:143-148. [DOI] [PubMed] [Google Scholar]
- 24.Kalaszczynska, I., Y. Geng, T. Iino, S. Mizuno, Y. Choi, I. Kondratiuk, D. P. Silver, D. J. Wolgemuth, K. Akashi, and P. Sicinski. 2009. Cyclin A is redundant in fibroblasts but essential in hematopoietic and embryonic stem cells. Cell 138:352-365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaldis, P., and E. Aleem. 2005. Cell cycle sibling rivalry: Cdc2 vs. Cdk2. Cell Cycle 4:1491-1494. [DOI] [PubMed] [Google Scholar]
- 26.Katayama, K., N. Fujita, and T. Tsuruo. 2005. Akt/protein kinase B-dependent phosphorylation and inactivation of WEE1Hu promote cell cycle progression at G2/M transition. Mol. Cell. Biol. 25:5725-5737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim, S. Y., and J. E. Ferrell, Jr. 2007. Substrate competition as a source of ultrasensitivity in the inactivation of Wee1. Cell 128:1133-1145. [DOI] [PubMed] [Google Scholar]
- 28.Kim, S. Y., E. J. Song, K. J. Lee, and J. E. Ferrell, Jr. 2005. Multisite M-phase phosphorylation of Xenopus Wee1A. Mol. Cell. Biol. 25:10580-10590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Krek, W., and E. A. Nigg. 1991. Mutations of p34cdc2 phosphorylation sites induce premature mitotic events in HeLa cells: evidence for a double block to p34cdc2 kinase activation in vertebrates. EMBO J. 10:3331-3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kutay, U., and S. Guttinger. 2005. Leucine-rich nuclear-export signals: born to be weak. Trends Cell Biol. 15:121-124. [DOI] [PubMed] [Google Scholar]
- 31.Liu, F., C. Rothblum-Oviatt, C. E. Ryan, and H. Piwnica-Worms. 1999. Overproduction of human Myt1 kinase induces a G2 cell cycle delay by interfering with the intracellular trafficking of Cdc2-cyclin B1 complexes. Mol. Cell. Biol. 19:5113-5123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Margolis, S. S., J. A. Perry, C. M. Forester, L. K. Nutt, Y. Guo, M. J. Jardim, M. J. Thomenius, C. D. Freel, R. Darbandi, J. H. Ahn, J. D. Arroyo, X. F. Wang, S. Shenolikar, A. C. Nairn, W. G. Dunphy, W. C. Hahn, D. M. Virshup, and S. Kornbluth. 2006. Role for the PP2A/B56delta phosphatase in regulating 14-3-3 release from Cdc25 to control mitosis. Cell 127:759-773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McConnell, B. B., F. J. Gregory, F. J. Stott, E. Hara, and G. Peters. 1999. Induced expression of p16INK4a inhibits both CDK4- and CDK2- associated kinase activity by reassortment of cyclin-CDK-inhibitor complexes. Mol. Cell. Biol. 19:1981-1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McGowan, C. H., and P. Russell. 1995. Cell cycle regulation of human WEE1. EMBO J. 14:2166-2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Merrick, K. A., S. Larochelle, C. Zhang, J. J. Allen, K. M. Shokat, and R. P. Fisher. 2008. Distinct activation pathways confer cyclin-binding specificity on Cdk1 and Cdk2 in human cells. Mol. Cell 32:662-672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mitra, J., C. Y. Dai, K. Somasundaram, W. S. El-Deiry, K. Satyamoorthy, M. Herlyn, and G. H. Enders. 1999. Induction of p21WAF1/CIP1 and inhibition of Cdk2 mediated by the tumor suppressor p16INK4a. Mol. Cell. Biol. 19:3916-3928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mitra, J., and G. H. Enders. 2004. Cyclin A/Cdk2 complexes regulate activation of Cdk1 and Cdc25 phosphatases in human cells. Oncogene 23:3361-3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mitra, J., G. H. Enders, J. Azizkhan-Clifford, and K. L. Lengel. 2006. Dual regulation of the anaphase promoting complex in human cells by cyclin A-Cdk2 and cyclin A-Cdk1 complexes. Cell Cycle 5:661-666. [PubMed] [Google Scholar]
- 39.Mohan, A., C. J. Oldfield, P. Radivojac, V. Vacic, M. S. Cortese, A. K. Dunker, and V. N. Uversky. 2006. Analysis of molecular recognition features (MoRFs). J. Mol. Biol. 362:1043-1059. [DOI] [PubMed] [Google Scholar]
- 40.Monecke, T., T. Guttler, P. Neumann, A. Dickmanns, D. Gorlich, and R. Ficner. 2009. Crystal structure of the nuclear export receptor CRM1 in complex with Snurportin1 and RanGTP. Science 324:1087-1091. [DOI] [PubMed] [Google Scholar]
- 41.Moore, J. D., J. Yang, R. Truant, and S. Kornbluth. 1999. Nuclear import of Cdk/cyclin complexes: identification of distinct mechanisms for import of Cdk2/cyclin E and Cdc2/cyclin B1. J. Cell Biol. 144:213-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morgan, D. O. 1995. Principles of CDK regulation. Nature 374:131-134. [DOI] [PubMed] [Google Scholar]
- 43.Okamoto, K., and N. Sagata. 2007. Mechanism for inactivation of the mitotic inhibitory kinase Wee1 at M phase. Proc. Natl. Acad. Sci. U. S. A. 104:3753-3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ortega, S., I. Prieto, J. Odajima, A. Martin, P. Dubus, R. Sotillo, J. L. Barbero, M. Malumbres, and M. Barbacid. 2003. Cyclin-dependent kinase 2 is essential for meiosis but not for mitotic cell division in mice. Nat. Genet. 35:25-31. [DOI] [PubMed] [Google Scholar]
- 45.Pagano, M., R. Pepperkok, F. Verde, W. Ansorge, and G. Draetta. 1992. Cyclin A is required at two points in the human cell cycle. EMBO J. 11:961-971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Palmer, B. D., A. M. Thompson, R. J. Booth, E. M. Dobrusin, A. J. Kraker, H. H. Lee, E. A. Lunney, L. H. Mitchell, D. F. Ortwine, J. B. Smaill, L. M. Swan, and W. A. Denny. 2006. 4-Phenylpyrrolo[3,4-c]carbazole-1,3(2H,6H)-dione inhibitors of the checkpoint kinase Wee1. Structure-activity relationships for chromophore modification and phenyl ring substitution. J. Med. Chem. 49:4896-4911. [DOI] [PubMed] [Google Scholar]
- 47.Pomerening, J. R., E. D. Sontag, and J. E. Ferrell, Jr. 2003. Building a cell cycle oscillator: hysteresis and bistability in the activation of Cdc2. Nat. Cell Biol. 5:346-351. [DOI] [PubMed] [Google Scholar]
- 48.Russell, P., and P. Nurse. 1987. Negative regulation of mitosis by wee1+, a gene encoding a protein kinase homolog. Cell 49:559-567. [DOI] [PubMed] [Google Scholar]
- 49.Schulman, B. A., D. L. Lindstrom, and E. Harlow. 1998. Substrate recruitment to cyclin-dependent kinase 2 by a multipurpose docking site on cyclin A. Proc. Natl. Acad. Sci. U. S. A. 95:10453-10458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Senderowicz, A. M. 2003. Small-molecule cyclin-dependent kinase modulators. Oncogene 22:6609-6620. [DOI] [PubMed] [Google Scholar]
- 51.Squire, C. J., J. M. Dickson, I. Ivanovic, and E. N. Baker. 2005. Structure and inhibition of the human cell cycle checkpoint kinase, Wee1A kinase: an atypical tyrosine kinase with a key role in CDK1 regulation. Structure 13:541-550. [DOI] [PubMed] [Google Scholar]
- 52.Takeda, D. Y., J. A. Wohlschlegel, and A. Dutta. 2001. A bipartite substrate recognition motif for cyclin-dependent kinases. J. Biol. Chem. 276:1993-1997. [DOI] [PubMed] [Google Scholar]
- 53.Thompson, J. D., D. G. Higgins, and T. J. Gibson. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673-4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.van Vugt, M. A., A. Bras, and R. H. Medema. 2004. Polo-like kinase-1 controls recovery from a G2 DNA damage-induced arrest in mammalian cells. Mol. Cell 15:799-811. [DOI] [PubMed] [Google Scholar]
- 55.Wang, Q., A. A. Canutescu, and R. L. Dunbrack, Jr. 2008. SCWRL and MolIDE: computer programs for side-chain conformation prediction and homology modeling. Nat. Protoc. 3:1832-1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ward, J. J., L. J. McGuffin, K. Bryson, B. F. Buxton, and D. T. Jones. 2004. The DISOPRED server for the prediction of protein disorder. Bioinformatics 20:2138-2139. [DOI] [PubMed] [Google Scholar]
- 57.Watanabe, N., H. Arai, J. Iwasaki, M. Shiina, K. Ogata, T. Hunter, and H. Osada. 2005. Cyclin-dependent kinase (CDK) phosphorylation destabilizes somatic Wee1 via multiple pathways. Proc. Natl. Acad. Sci. U. S. A. 102:11663-11668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Watanabe, N., H. Arai, Y. Nishihara, M. Taniguchi, N. Watanabe, T. Hunter, and H. Osada. 2004. M-phase kinases induce phospho-dependent ubiquitination of somatic Wee1 by SCFbeta-TrCP. Proc. Natl. Acad. Sci. U. S. A. 101:4419-4424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wilmes, G. M., V. Archambault, R. J. Austin, M. D. Jacobson, S. P. Bell, and F. R. Cross. 2004. Interaction of the S-phase cyclin Clb5 with an “RXL” docking sequence in the initiator protein Orc6 provides an origin-localized replication control switch. Genes Dev. 18:981-991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wolthuis, R., L. Clay-Farrace, W. van Zon, M. Yekezare, L. Koop, J. Ogink, R. Medema, and J. Pines. 2008. Cdc20 and Cks direct the spindle checkpoint-independent destruction of cyclin A. Mol. Cell 30:290-302. [DOI] [PubMed] [Google Scholar]
- 61.Yang, L., W. R. MacLellan, Z. Han, J. N. Weiss, and Z. Qu. 2004. Multisite phosphorylation and network dynamics of cyclin-dependent kinase signaling in the eukaryotic cell cycle. Biophys. J. 86:3432-3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.