

Abstract
A stable genome is critical to cell viability and proliferation. During DNA replication, the S-phase checkpoint pathway responds to replication stress. In budding yeast, the chromatin-bound F-box protein Dia2 is required to maintain genomic stability and may help replication complexes overcome sites of damaged DNA and natural fragile regions. SCF (Skp1/Cul1/F-box protein) complexes are modular ubiquitin ligases. We show here that Dia2 is itself targeted for ubiquitin-mediated proteolysis and that activation of the S-phase checkpoint pathway inhibits Dia2 protein degradation. S-phase checkpoint mutants fail to stabilize Dia2 in response to replication stress. Deletion of DIA2 from these checkpoint mutants exacerbates their sensitivity to hydroxyurea, suggesting that stabilization of Dia2 contributes to the replication stress response. Unlike the case for other F-box proteins, deletion of the F-box domain in Dia2 does not stabilize the protein. Rather, an N-terminal domain that is also required for nuclear localization is necessary for degradation. When a strong nuclear localization signal (NLS) is added to dia2 mutants lacking this domain, the Dia2 protein is both stable and nuclear. Together, our results suggest that Dia2 protein turnover does not involve an autocatalytic mechanism and that Dia2 proteolysis is inhibited by activation of the replication stress response.
Maintenance of genomic integrity is required for cellular viability and proliferation. During S phase, problems during DNA synthesis or the presence of genotoxic stress activates conserved checkpoint responses (1, 6, 7, 26, 42). These pathways promote cell viability by mediating a transcriptional response (2), stabilizing replication forks (1, 16, 22, 27), suppressing late-firing origins of replication (33, 35), and slowing S-phase progression (6, 26, 30, 33). In budding yeast, sensing and activation of the S-phase checkpoint response is dependent on many proteins. Sensing of DNA damage or stalled replication forks relies on the Rad24-dependent loading of the heterotrimeric Rad17-Mec3-Ddc1 (9-1-1 complex) sliding clamp onto DNA (14, 23, 45). This leads to Mec1 kinase activation followed by the downstream activation of the primary signaling kinase Rad53 (6, 26, 42). Mec1-dependent activation of the replication checkpoint also requires the adaptor Mrc1, the Saccharomyces cerevisiae homolog of human Claspin that forms a complex to stabilize replication forks at sites of replication stress (1, 16, 22, 27). Additionally, although Chk1 is activated by Mec1 to promote anaphase arrest following DNA damage, it has also been proposed to play a role in response to hydroxyurea (HU)-induced replication blocks (8, 32, 34).
The ubiquitin proteasome system controls a wide variety of cellular processes, including cell division and DNA replication and repair. Members of the highly conserved SCF (Skp1/Cdc53/F-box protein) ubiquitin ligase family are involved in controlling cell proliferation by regulating the ubiquitin-mediated proteolysis of key cell cycle regulators (9, 11, 18, 25, 36, 37, 40, 44, 46). SCF complexes are modular ubiquitin ligases whose specificity is determined by individual F-box proteins, which act as substrate-specific adapters (11, 36). Many F-box proteins have been identified in both humans and model eukaryotic systems, indicating that SCF complexes and their associated roles are highly conserved.
The Saccharomyces cerevisiae F-box protein Dia2 is important for DNA replication and genomic stability. The dia2Δ mutant is hypersensitive to DNA damage, exhibits chromosome loss and rearrangement, and accumulates DNA damage foci (4, 17, 28). Dia2 is a bona fide F-box protein in that it assembles with Skp1, Cdc53, and Rbx1 into a functional SCF ubiquitin ligase complex (15, 17-19). The dia2Δ strain progresses more slowly through S phase than wild-type cells when challenged with the DNA-damaging agent methyl methanesulfonate (MMS), suggesting that Dia2 promotes passage of replication complexes through areas of damaged DNA (4).
F-box proteins are key to the assembly of substrate proteins with a complete SCF ubiquitin ligase complex. There is evidence that the abundance of individual F-box proteins may be regulated to control the ubiquitination of specific target proteins. For example, some F-box proteins have been shown to be targeted for ubiquitin-mediated destruction via an autoubiquitination mechanism (13, 47). Conversely, loss of SCF components appears to negatively affect the stability of some F-box proteins (12, 24, 29, 38). Alternatively, F-box proteins may be targeted for destruction by non-SCF ubiquitin ligases. The human F-box protein Skp2 is a target of the anaphase-promoting complex (APC/C) ubiquitin ligase in a cell cycle-dependent manner (3). In addition, the nuclear localization of yeast Cdc4 has been shown to be important in the turnover of one of its targets, Far1 (5).
The mechanistic role that Dia2 performs in DNA replication is not known. To gain a better understanding of the activity of an SCFDia2 complex throughout the cell cycle, we examined the regulation of the Dia2 protein. Here we demonstrate that Dia2 protein abundance is controlled by ubiquitin-mediated degradation and is independent of an autocatalytic mechanism. Importantly, we find that Dia2 protein turnover is blocked by activation of the S-phase checkpoint, suggesting that SCFDia2 activity is required for the cellular response to replication stress.
MATERIALS AND METHODS
Plasmid and strain construction.
The strains, plasmids, and oligonucleotides used in this study are described in Tables 1 to 3, respectively. Dia2 expression constructs were generated by PCR amplification of the DIA2 coding region with SpeI and BamHI ends using oligonucleotides AK34 and DK96 (full length), AK40 and DK96 (ΔN214), AK41and DK96 (LRR), AK34 and AK43 (TPR), AK51 and DK96 (ΔN 189), AK62 and DK96 (ΔN149), AK72 and DK96 (SVFL), and AK73 and DK96 (SVΔN214). The DIA2 mutant lacking the nuclear localization signal (NLS) region (ΔNLS) was generated using oligonucleotides AK68 and AK69 by the PCR stitching method. The SVΔNLS variant was generated by amplification of the ΔNLS mutant with primers AK72 and DK96. PCR products were digested with SpeI and BamHI and ligated into the p1219 vector (21) to generate galactose-inducible proteins with nine myc epitopes at the N termini. The p1219-myc9 Dia2-ΔF construct was generated by replacing the MluI/NotI fragment from p1219-myc9-Dia2 with a MluI/NotI fragment from pDMK289 (17). To generate the panel of 9MYC-DIA2 integration vectors, a 1-kb KpnI/XhoI fragment of DIA2 5′ untranslated sequence was ligated with an XhoI/NotI fragment from the generated p1219-9MYC-DIA2 plasmids and ligated into the pRS406 vector with KpnI/NotI ends. Constructs were integrated using BclI and transformed into dia2Δ cells using established techniques (31). Integration was monitored by PCR using primers AK44 and AK45 or AK60 and AK61, and protein expression was determined by Western blotting.
TABLE 1.
Strains used in this study
| Strain | Description | Reference |
|---|---|---|
| Y80 | MATacan1-100 ade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-1 | 17 |
| DKY194 | As Y80 but dia2Δ::kanMX | 18 |
| AKY149 | As DKY194 but dia2Δ::kanMX::9MYC-DIA2 URA3 | This study |
| AKY182 | As AKY149 but cdc15-2 | This study |
| AKY184 | As AKY149 but MCM4::MCM4-3HA TRP1 | This study |
| AKY168 | As AKY149 but rpn4Δ::HIS3 pdr5Δ::LEU2 | This study |
| AKY188 | As AKY149 but DIA2-ΔF (bp Δ670-792) | This study |
| AKY189 | As AKY149 but DIA2-ΔN214 (bp 640-2241) | This study |
| AKY190 | As AKY149 but DIA2-LRR (bp 793-2241) | This study |
| AKY192 | As AKY149 but DIA2-TPR (bp 1-669) | This study |
| AKY193 | As AKY149 but DIA2-ΔN189 (bp 565-2241) | This study |
| AKY199 | As AKY149 but DIA2-ΔN149 (bp 447-2241) | This study |
| AKY238 | As DKY194 but dia2Δ::kanMX::9MYC-SV40NLS-DIA2 URA3 | This study |
| AKY239 | As AKY149 but DIA2-ΔNLS (bp Δ580-639) | This study |
| AKY240 | As AKY238 but DIA2-SVΔNLS (bp Δ580-639) | This study |
| AKY241 | As AKY238 but DIA2-ΔN214 (bp 640-2241) | This study |
| DKY404 | As Y80 but dia2Δ::kanMX chk1Δ::HIS3 | 17 |
| DKY405 | As Y80 but dia2Δ::kanMX mrc1Δ::HIS3 | 17 |
| DKY449 | As Y80 but dia2Δ::kanMX rad17Δ::kanMX | 17 |
| DKY450 | As Y80 but dia2Δ::kanMX rad24Δ::kanMX | 17 |
| AKY203 | As DKY404 but dia2Δ::kanMX::9MYC-DIA2 URA3 | 17 |
| AKY204 | As DKY449 but dia2Δ::kanMX::9MYC-DIA2 URA3 | This study |
| AKY205 | As DKY450 but dia2Δ::kanMX::9MYC-DIA2 URA3 | This study |
| AKY206 | rad53-21 but DIA2::9MYC-DIA2 URA3 | This study |
| AKY207 | As DKY405 but dia2Δ::kanMX::9MYC-DIA2 URA3 | This study |
| AKY170 | As AKY149 but skp1-11 | This study |
| AKY174 | As AKY149 but cdc53-1 | This study |
| AKY180 | As AKY149 but cdc34-2 | This study |
TABLE 3.
Oligonucleotides used in this study
| Oligonucleotide | Sequence (5′ → 3′) |
|---|---|
| LM16 | TTAGAAACACTTGTGGTGAACGATAG |
| DK424 | GACTGACTACTTGATGAAGA |
| AK34 | GCGACTAGTATGTCGTATAAATTT |
| DK96 | CGGGATCCCTATGAGTATGAATATGA |
| AK40 | GGACTAGTACCAAGAAAACT |
| AK41 | GGACTAGTTTCAACTTGGCACCA |
| AK43 | CCGGATCCCTAATTGCCAACTAAATC |
| AK44 | AAACGGATTCATCATGAG |
| AK45 | GCCACGCCTGAATTCCCTG |
| AK51 | GGACTAGTGAGACCAAAATAGCA |
| AK60 | TTGTTCACCACTAGCCATGG |
| AK61 | GCAGTACGATATCACCAACGG |
| AK68 | GAGGAGACCAAAATAAGTACCAAGAAAACT |
| AK69 | AGTTTTCTTGGTACTTATTTTGGTCTCCTC |
| AK70 | TGTTCTCGTGGACTGGAGGAGACCAAAATA |
| AK71 | TATTTTGGTCTCCTCCAGTCCACGAGAACA |
| AK72 | GGACTAGTCCGAAGAAGAAACGGAAGGGTATGTCGTATAAATTT |
| AK73 | GGACTAGTCCGAAGAAGAAACGGAAGGGTAGTACCAAGAAAACT |
Yeast cell culture and treatments.
Yeast cells were maintained and cultured according to standard methods (31). For α-factor arrests, cultures were grown in liquid minimal medium or yeast extract-peptone-dextrose (YPD) and pulsed with 4 μg α-factor peptide (αF) (Genscript) every hour for 2 h. Hydroxyurea (HU) (US Biological Corp.)-treated cells were grown in minimal or YPD medium with a final concentration of 200 mM HU for 2 h. Cells arrested with nocodazole (Noc) (Sigma) were incubated in minimal or YPD medium at a final concentration of 15 μg/ml for 2 h. Arrests were monitored by cell morphology and flow cytometry as previously described (41). For proteasome inhibition experiments, the 9MYC-DIA2 rpn4Δ pdr5Δ strain was grown overnight at 30°C in YPD. Cultures were diluted at mid-log phase and incubated for 90 min with a final concentration of 50 μM MG132 (Sigma) or dimethyl sulfoxide (DMSO) (drug vehicle).
Reverse transcription-PCR (RT-PCR).
Cells were grown in 10 ml YPD medium to 2 × 107 cells/ml and collected by centrifugation at 4,000 rpm and 4°C. Cells were processed and total RNA collected using the “PureLink” micro-to-midi kit (Invitrogen). Following RNA elution, DNase I treatment was performed and the RNA was precipitated. Reverse transcription of 2 μg of total RNA was performed using Superscript II (Invitrogen) with oligo(dT)50 primer. The cDNA was used in PCRs with primers DK96 and AK18 or primers LM16 and DK424. Products were analyzed on 2% agarose gels.
Stability assays.
Cells were grown at 30°C in 5 ml YPD overnight, diluted to 1 × 107 cells/ml, and regrown for 90 min. Cell densities of approximately 2 × 107 cells/ml were then used for cell cycle arrest with appropriate chemicals as noted above. Cycloheximide (CHX) (Sigma) was added to log-phase cultures at a final concentration of 100 μg/ml. Cells were collected at the indicated time points. The 9MYC-DIA2 cdc15-2 strain was shifted to 37°C for 2 h prior to addition of CHX. Cells were collected at the indicated time points and processed to extract protein using the trichloroacetic acid (TCA) precipitation method described below. Samples were resolved by SDS-PAGE and immunoblotted with anti-myc (9E10; Covance Research) and anti-Pgk1 (Molecular Probes) primary antibodies, followed by horseradish peroxidase (HRP)-conjugated anti-mouse secondary antibodies. Protein abundance was quantified using Image J software and corrected against the Pgk1 loading control of the same sample. Quantifications were analyzed on images representative of abundance based upon linear film exposure times.
Immunofluorescence.
Cells were prepared as described previously (31) and incubated with 1:500 anti-myc (9E10; Covance Research) primary antibody followed by 1:1,000 fluorescein isothiocyanate (FITC)-conjugated anti-mouse antibodies. Cells were treated with 1:1,000 DAPI (4′,6′-diamidino-2-phenylindole) for nuclear staining. Cells were visualized using a Zeiss Axioscop 2 microscope equipped with a Zeiss Axiocam R2 digital camera and 100× objective with differential interference contrast (DIC), FITC, and DAPI filters. Images were captured using Zeiss Axiovision software release 3.1 (Carl Zeiss, Thornwood, NY).
Immunoprecipitation experiments.
For cells harboring a plasmid under galactose control, the cells were induced for 1 h with 2% galactose at 30°C. Cell pellets were resuspended in NETN lysis buffer (20 mM Tris [pH 8.0], 100 mM NaCl, 1 mM EDTA, 0.5% Igepal,10 mM NaF, 25 mM β-glycerophosphate, 1 mM phenylmethylsulfonyl fluoride [PMSF], and 1 mM pepstatin, plus Complete protease inhibitor cocktail [Roche Applied Science]). Cells were vortexed for 10 min at 4°C with glass beads. Lysates were microcentrifuged at 10,000 rpm at 4°C, and the cleared supernatant was transferred to new tubes. One milligram of lysate was incubated with 1:200 anti-myc or 1:200 anti-Skp1 antibodies (a generous gift from J. Wade Harper, Harvard Medical School) (36). The samples were then incubated with protein A/G-agarose for 2 hours at 4°C (Santa Cruz Biotechnology). Agarose beads were washed three times with NETN lysis buffer and then boiled in 1× Laemmli loading dye prior to SDS-PAGE.
TCA protein precipitation.
Cell pellets were lysed by vortexing in 20% trichloroacetic acid (TCA) for 2 min with glass beads and microcentrifuged at 3,000 rpm for 10 min. The precipitated protein was resuspended in 1× Laemmli loading dye and neutralized with Tris base. Samples were boiled for 5 min and centrifuged at 3,000 rpm for 10 min. Protein concentration was determined using the RCDC kit (Bio-Rad).
RESULTS
Dia2 is an unstable protein.
We investigated Dia2 protein abundance during cell division using a strain that expresses a 9myc-tagged allele of DIA2. The Dia2Myc strain is indistinguishable from a wild-type strain in terms of sensitivity to hydroxyurea, MMS, and growth at room temperature (Fig. 1A). Additionally, Dia2Myc interacts with Skp1 (Fig. 1B) and chromatin (Fig. 1C) as expected (41).
FIG. 1.
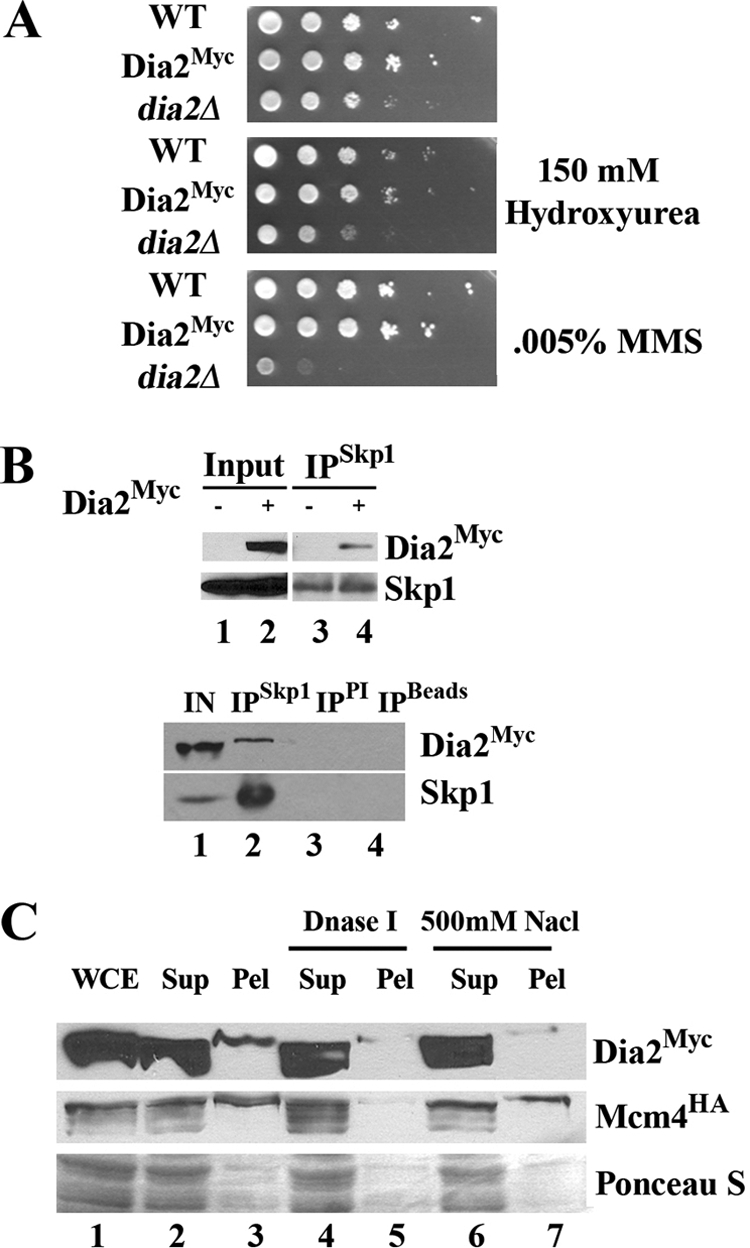
A 9MYC-DIA2 strain behaves like the wild type. (A) Growth of the Dia2Myc strain is indistinguishable from that of the wild type on rich medium with the indicated amounts of HU or methyl methanesulfonate (MMS) at room temperature. Tenfold serial dilutions are shown. (B) Dia2Myc associates with Skp1. Skp1 was immunoprecipitated with anti-Skp1 antibodies (36) from an untagged strain or 9MYC-DIA2 strains (lanes 3 and 4, respectively). Immunoblots were probed with anti-myc or anti-Skp1 antibodies. The bottom panel shows that Dia2Myc is immunoprecipitated (IP) with anti-Skp1 antibodies but not with preimmune serum (PI) or protein A/G beads (Beads) in the absence of antibody. (C) A fraction of Dia2Myc is chromatin bound. Dia2Myc cells were fractionated as described previously (30a). Crude-pellet-associating Dia2Myc is released with DNase I (compare lanes 3 and 5) or high-salt (compare lanes 3 and 7) treatment. Mcm4HA was monitored as a positive control. WCE, whole-cell extract; Su, supernatant; Pel, pellet. Total protein was stained using Ponceau S to visualize overall protein fractionation from equal loaded volumes.
We monitored Dia2 protein levels in cells synchronized with α-factor. Cells were released from an α-factor arrest at 30°C, and then α-factor was added back at 60 min after entry into S phase to capture a single cell division cycle. As shown in Fig. 2A, Dia2 protein abundance is initially low in late G1 but increases at 15 min after release, as cells move into S phase (lanes 1 and 2). The protein levels remain high until the 120-min time point, after cells have returned to G1. We conclude that Dia2 protein abundance is low in late G1 and is higher during the rest of the cell cycle. A similar profile is observed when cells are synchronized using an early G1 arrest, indicating that this is not a specific response to mating pheromone (data not shown). These results are consistent with a requirement for SCFDia2 activity during S phase, and indeed we observe that SCFDia2 complex formation occurs in cells arrested in S phase with hydroxyurea as measured by Skp1 coimmunoprecipitation (Fig. 2B, lane 6). As previously reported, we observe no obvious change in mRNA transcript levels (Fig. 2C) at different points in the cell cycle (10, 39). These results suggest that Dia2 protein levels are modulated through a posttranscriptional mechanism.
FIG. 2.

Dia2Myc protein abundance fluctuates throughout the cell cycle. (A) Dia2 protein levels in synchronized cells. The 9MYC-DIA2 strain was synchronized in G1 with α-factor pheromone and released into rich medium at 30°C. After 60 min, α-factor was added back to the medium for rearrest in G1. Immunoblots were probed with anti-myc or anti-Pgk1 (as a loading control) antibodies. Cell cycle progression was monitored by flow cytometry. (B) Dia2Myc associates with Skp1 during S phase. Skp1 was immunoprecipitated with anti-Skp1 antibodies (36) from Dia2Myc lysates from asynchronous (Asy), α-factor (αF), or HU-arrested cultures. Immunoblots were probed with anti-myc or anti-Skp1 antibodies. WT, wild type. (C) DIA2 transcript abundance does not fluctuate. Total RNA was extracted and RT-PCR was performed as described in Materials and Methods. ACT1 was monitored as a control. Cell cycle stage was monitored by flow cytometry. (D) DIA2 overexpression leads to a growth defect in the presence of MMS. Wild-type cells containing empty vector or DIA2 under the control of the GAL1,10 promoter were spotted in 10-fold serial dilutions to minimal medium with 2% glucose or 2% galactose. Plates were incubated at room temperature. MMS was included at the indicated amount. (E) Dia2 is an unstable protein. Dia2Myc cells were treated with 100 μg/ml CHX and samples taken every 30 min. Samples were immunoblotted with anti-myc antibodies, and anti-Pgk1 antibodies were used to control for loading. Quantitation of three independent experiments is shown on the graph. Error bars indicate standard deviations. (F) Dia2Myc is ubiquitinated. Wild-type or Dia2Myc cells were treated with DMSO (lanes 1 and 2) or MG132 (lanes 3 and 4) for 90 min and immunoprecipitated with anti-myc antibodies (lanes 5 to 8) and immunoblotted with anti-myc or antiubiquitin (Ub) antibodies. Pgk1 was monitored as a loading control.
We hypothesized that if control of Dia2 protein levels was required for normal cellular function, then we would observe a cell growth phenotype when the Dia2 protein is overexpressed. Thus, we transformed wild-type cells with a DIA2 galactose-inducible expression vector. When Dia2 expression is induced on galactose-containing medium, we find that cells exhibit a mild growth defect. Since Dia2 is important for genomic stability, we tested growth of cells overexpressing DIA2 in the presence of the DNA-damaging agent MMS. In this case we find that cells exhibit a strong growth defect (Fig. 2D). These results suggest that regulation of Dia2 protein abundance is required for normal cellular function and are consistent with a proposed role for Dia2 in genomic maintenance.
We tested whether proteolysis controls Dia2 protein levels. An asynchronous culture expressing Dia2Myc was grown to log phase, translation was inhibited by the addition of cycloheximide, and samples were collected every 30 min for 150 min. As shown, Dia2 protein abundance decreased during the time course (Fig. 2E), indicating that the Dia2 protein is unstable, with a half-life of approximately 60 min.
The ubiquitin-dependent proteasome pathway is a major regulatory system for proteolysis within cells. We investigated the possibility that Dia2 was targeted for destruction by the ubiquitin proteasome system by treating cells with the proteasome inhibitor MG132. The abundance of Dia2 protein increased in MG132-treated cells (Fig. 2F, compares lanes 2 and 4), consistent with Dia2 proteolysis via the ubiquitin proteasome system. If Dia2 is targeted for ubiquitin-mediated proteolysis, it should be polyubiquitinated. To test this, 9myc-tagged Dia2 was immunoprecipitated from MG132-inhibited cells and immunoblotted using anti-myc or antiubiquitin antibodies. As shown, higher-molecular-weight species of Dia2, which cross-react with antiubiquitin antibodies, accumulate in the presence of MG132 (Fig. 2F, lane 6 versus 8). No ubiquitin conjugates are observed in an anti-myc immunoprecipitation from an untagged strain, indicating that the conjugates we observe are specific for Dia2. Together, our data provide evidence that the ubiquitin-proteasome pathway controls Dia2 protein turnover.
Dia2 turnover in G1 is not controlled by the SCF pathway.
We next investigated which regions of Dia2 are important for regulating its turnover. A panel of domain and deletion mutants was engineered with an N-terminal 9myc epitope tag and integrated into the endogenous DIA2 locus (Fig. 3A). Protein expression was determined by Western blotting (Fig. 3B). Each mutant protein was tested for competence in forming an SCF complex by coimmunoprecipitation with anti-Skp1 antibodies (Fig. 3C). As expected, full-length Dia2 bound to Skp1, and a strong interaction was seen with the ΔN214 mutant (lanes 6 and 8). We did not observe any coimmunoprecipitation of the ΔF, TPR (tetratricopeptide repeats), or LRR (leucine-rich repeats) domain mutants with Skp1 (lanes 7, 9, and 10); these mutants do not contain the F-box domain required for binding Skp1. None of the Dia2 deletions complement the dia2Δ strain, and mutant cells exhibited similar sensitivity to HU, MMS, and low temperatures (Fig. 3D).
FIG. 3.

Analysis of Dia2 mutants. (A) Diagram of Dia2 domains and mutants used in this study. Relevant amino acid residues or changes are indicated. TPR, tetratricopeptide repeats; F, F-box domain; LRR, leucine-rich repeats; NLS, nuclear localization sequence. (B) Expression of Dia2 deletion mutants. Asynchronous strains expressing Dia2Myc (lane 2) or the various mutants (lanes 3 to 6) were grown at 30°C and collected. TCA precipitates were prepared as described in Materials and Methods and immunoblotted with anti-myc or anti-Pgk1 antibodies. Extract from a wild-type strain was included as an untagged control (lane 1). (C) Mutants lacking the F-box do not associate with Skp1. Dia2 mutants were expressed from plasmids using the GAL1,10 promoter and coimmunoprecipitated with anti-Skp1 antibodies. Immunoblots were probed with anti-myc or anti-Skp1 antibodies. The identity of the protein in each band is indicated. Asterisks indicate breakdown products. (D) Dia2 mutants do not rescue dia2Δ phenotypes. Strains expressing integrated Dia2Myc and variants were spotted to YPD plates containing the indicated amounts of hydroxyurea (HU) or MMS in 10-fold dilution series and grown at room temperature.
To examine the stability of the Dia2 deletion mutants, we performed stability assays with α-factor-arrested cells. Translation was inhibited by the addition of cycloheximide and Dia2 protein abundance followed for 3 hours at 1-hour intervals. Strikingly, full-length Dia2 and the ΔF-box mutant exhibited similar half-lives (Fig. 4A, lanes 1 to 4 and 5 to 8). This suggests that the previously reported in vitro autoubiquitination of SCFDia2 (19) does not control Dia2 protein stability in vivo, at least during G1. When the other mutants were analyzed, it was clear that both the ΔN214 mutant and the LRR domain fragment remained stable during the time course of the experiment, whereas the TPR domain was turned over with a rate similar to that for the wild type (Fig. 4A, lanes 9 to 20). This suggests that Dia2 protein turnover requires the N-terminal domain, deletion of which generates a stabilized form of Dia2.
FIG. 4.

Dia2 protein turnover is not controlled by the SCF pathway. (A) N-terminal deletion mutants of Dia2 are stabilized. Dia2Myc strains were treated with α-factor for 2 h and translation inhibited by CHX. Cells were collected at the indicated time points after inhibition, and protein samples were resolved by SDS-PAGE prior to immunoblotting with anti-myc antibodies. Pgk1 was monitored as a loading control. Quantitation of results from three independent experiments is shown on the graph. Error bars indicate standard deviations. (B) Turnover of Dia2 does not require the SCF pathway. The 9MYC-DIA2 allele was inserted into skp1-11, cdc53-1, and cdc34-2 strains. Cultures were grown in YPD at 25°C and shifted to 37°C for 2 h prior to the addition of CHX. Samples were collected at the indicated times and immunoblotted with anti-myc or anti-Pgk1 antibodies. Quantitation of three independent experiments is shown on the graph. (C) Deletion of the N-terminal region mislocalizes Dia2. Dia2 variants were expressed in dia2Δ cells, and protein was visualized by immunofluorescence as described in Materials and Methods. Scale bar, 5 μm.
To further examine whether Dia2 protein was turned over in an autoubiquitination pathway, we performed stability assays with SCF mutants. We determined the stability of Dia2Myc in temperature-sensitive skp1-11, cdc53-1, and cdc34-2 mutants. Cells were shifted to the nonpermissive temperature of 37°C for 2 h, followed by addition of cycloheximide. Samples were collected at 1-hour intervals. As shown, none of the SCF pathway mutants displayed any detectable stabilization of Dia2 (Fig. 4B). Rather, the skp1-11 and cdc53-1 mutants appeared to increase the turnover rate of Dia2, as has been observed for the Grr1 and Met30 F-box proteins (12, 24, 29, 38). Similar results were observed for the skp1-12 temperature-sensitive strain (data not shown). In contrast, a known SCF ubiquitination target, the CDK inhibitor Sic1, was stabilized in SCF pathway mutants (data not shown). We conclude that Dia2 proteolysis occurs independently of an autocatalytic mechanism.
Dia2 turnover requires a 20-amino-acid N-terminal domain.
We characterized the domain mutants for possible changes in localization (Fig. 4C). Using immunofluorescence, we observed that full-length Dia2 strongly localized to the nucleus at all points in the cell cycle, as expected (4). The F-box domain mutant and the N-terminal TPR region also localized to the nucleus, suggesting that sufficient information for nuclear targeting resides prior to the F-box domain in Dia2. Moreover, Dia2 is not required to interact with Skp1 via the F-box domain for nuclear localization to occur. In contrast, both the ΔN214 mutant and leucine-rich repeat domain were mislocalized throughout the cell. However, these mutants did not appear to be excluded from the nucleus.
We considered whether Dia2 trafficking to the nucleus influenced the rate of protein turnover. To test this possibility, we sought to restore the nuclear localization of the stable, mislocalized DIA2 mutants. Three predicted nuclear localization signals (NLS) exist in Dia2 in a cluster just upstream of the F-box domain (20). The ΔN214 and LRR mutants lack all three and are mislocalized. We therefore generated several mutants to probe the requirement for the NLS-containing region (Fig. 5A). These mutants included two additional N-terminal deletions that begin before the NLS region (ΔN189 and ΔN149) and an in-frame deletion of the entire 20-amino-acid NLS region (ΔNLS). Both the ΔN149 and the ΔN189 mutants localized to nucleus, whereas the ΔNLS mutant was localized throughout the cell, similar to the ΔN214 mutant (Fig. 5B). These results indicate that the 20-amino-acid NLS domain is important for nuclear targeting and encodes at least one functional NLS.
FIG. 5.

Dia2 degradation requires an NLS-containing domain. (A) Diagram of N-terminal Dia2 deletion mutants used in this study. Expression of the mutant proteins is shown in the lower panel. Samples were prepared and blotted as described for Fig. 3B. (B) Dia2 mutants lacking the NLS region are mislocalized. Immunofluorescence was performed as for Fig. 4. Scale bar, 5 μm. (C) Growth phenotypes of Dia2 N-terminal deletions. The indicated strains were spotted to YPD with the indicated amounts of hydroxyurea or MMS in 10-fold dilution series and grown at room temperature. (D) Nucleus-localized Dia2 mutants are susceptible to proteolysis. Dia2 mutants were subjected to stability assays as described for Fig. 4. Quantitation of results from three independent experiments is shown on the graph. Error bars indicate standard deviations.
The N-terminal mutants exhibited various growth phenotypes. Both the ΔN189 and ΔN149 mutants exhibited less sensitivity to HU and MMS than dia2Δ cells, with the ΔN149 mutant approaching near-wild-type levels of sensitivity (Fig. 5C, left panel). In addition, the ΔNLS mutant is indistinguishable from the wild type under these conditions (Fig. 5C, right panel). These results indicate that the residues between 149 and 189 are critical for Dia2 protein function.
We then tested protein stability of these mutants in G1-arrested cells (Fig. 5D). Interestingly, we found that the ΔNLS protein is stable but that the nucleus-localized ΔN189 and ΔΝ149 mutants are unstable, with turnover rates similar to that of wild-type Dia2. These results may be explained by a requirement for the 20-amino-acid domain in both protein turnover and nuclear localization or a requirement for Dia2 to be localized to the nucleus for degradation.
To distinguish between these possibilities, we added the strong NLS sequence from the simian virus 40 (SV40) T antigen (Tag) to stable Dia2 mutants (SVΔN214 and SVΔNLS) and full-length Dia2 as a control (SVFL), as shown in Fig. 5A. As expected, all three of these proteins localized to the nucleus by immunofluorescence (Fig. 6A) and exhibited growth phenotypes nearly identical to those of their counterparts lacking the exogenous SV40 Tag NLS (data not shown). When we performed stability assays with G1-arrested cells and these mutants, we found that the mutants lacking the 20-amino-acid NLS region are still stable, despite being localized to the nucleus via the added SV40 Tag NLS (Fig. 6B). The full-length Dia2 protein with the added SV40 Tag NLS exhibited protein turnover with kinetics similar to those for wild-type Dia2, indicating that the addition of the exogenous NLS does not interfere with normal Dia2 protein turnover. We conclude that the N-terminal 20-amino-acid region contains residues required for Dia2 protein degradation as well as nuclear trafficking.
FIG. 6.

An N-terminal domain required for Dia2 protein turnover overlaps the NLS region. (A) Addition of the SV40 T antigen NLS to Dia2 N-terminal deletions restores nuclear localization. Immunofluorescence was performed as for Fig. 4. Scale bar, 5 μm. (B) Nuclear, N-terminal Dia2 deletions are stable. Dia2 mutants were subjected to stability assays as described for Fig. 4. Quantitation of results from three independent experiments is shown on the graph. Error bars indicate standard deviations. (C) Overexpression of stable, nuclear forms of Dia2 alters cell cycle distribution. Wild-type cells expressing the indicated DIA2 alleles from a galactose-inducible promoter were grown to log phase in galactose-containing medium. DNA content was measured by flow cytometry. Quantitation of results from three independent experiments in shown in the graph.
We investigated whether overexpression of stable, nuclear forms of Dia2 interfered with cell cycle dynamics in wild-type cells. We examined the cell cycle distribution of wild-type cells overexpressing SVFL, SVΔN214, and SVΔNLS from a galactose-inducible promoter using flow cytometry. Cells expressing full-length Dia2 exhibited a slight increase in the percentage of G1 cells (Fig. 6C) coincident with a decrease in cells in G2/M, similar to cells overexpressing full-length Dia2 lacking this NLS (data not shown). Interestingly, cells expressing the stable, nuclear forms of Dia2 (SVΔN214 and SVΔNLS) exacerbated this phenotype, showing significantly increased numbers of cells in G1 and a reduced number in G2/M. These results suggest that excess Dia2 that cannot be efficiently degraded may interfere with either G1 progression or the G1-to-S-phase transition.
Checkpoint activation blocks Dia2 turnover in response to HU treatment.
We compared Dia2 protein stability in G1-arrested cells to that in cells blocked at other stages of the cell cycle. As predicted from the low abundance of Dia2 in G1 cells, we found that Dia2 protein turnover was most rapid in cells arrested with α-factor (Fig. 7A, top). Interestingly, Dia2 was also unstable in cells arrested in late M using the cdc15-2 allele and in cells arrested prior to anaphase using the drug nocodazole (Fig. 7A, lower panels). However, in hydroxyurea-arrested cells we detected very little Dia2 proteolysis under the same conditions (Fig. 7A).
FIG. 7.

Checkpoint proteins inhibit Dia2 turnover in response to replication stress. (A) Dia2 proteolysis is cell cycle regulated. Cells were arrested as described in Materials and Methods and taken at the indicated time points (lanes 1 to 6) after addition of CHX. The 9MYC-DIA2 cdc15-2 strain was shifted to the nonpermissive temperature for 2 h prior to the addition of CHX. The 0-min time point of each arrest was monitored by flow cytometry. Quantitation of results from three independent experiments is shown on the graph. Error bars indicate standard deviations. (B) Checkpoint pathway mutants fail to stabilize Dia2 in response to HU treatment. 9MYC-DIA2 alleles were generated in the indicated checkpoint mutants and arrested with HU as described above. Dia2 protein turnover was monitored for the indicated times after the addition of CHX, and immunoblotting was with anti-myc and anti-Pgk1antibodies (upper panel). Quantitation of results from three independent experiments is shown on the graph. (C) During G1, Dia2 shows turnover rates in checkpoint mutants comparable to those in the wild type. Stability assays were performed as for panel B except that cells were arrested using α-factor. Quantitation of results from a representative experiment is shown on the graph. (D) rad53-21 mutants fail to stabilize Dia2 during replication stress caused by MMS. 9MYC-DIA2 or 9MYC-DIA2 rad53-21 cultures were arrested in G1 with α-factor for 2 h. Cultures were washed and released into fresh medium containing 0.033% MMS. Protein synthesis was halted after 60 min by the addition of CHX, and cells were collected at the indicated time points. Dia2 was monitored with anti-myc antibodies, and anti-Pgk1 antibodies were used as a loading control. (E) dia2Δ and checkpoint mutants show synthetic growth defects in response to HU. The specified strains were spotted in 10-fold serial dilutions on YPD plates containing the indicated amount of HU and grown at room temperature.
Because Dia2 is stable in HU-arrested cells, we considered whether activation of the S-phase checkpoint response inhibits Dia2 protein turnover. To test this, we examined Dia2 protein turnover in S-phase checkpoint mutants. If Dia2 turnover is inhibited by the S-phase checkpoint, we predicted Dia2 to be unstable in mutants that are checkpoint defective. We inserted the 9MYC-DIA2 allele in chk1Δ, rad17Δ, rad24Δ, rad53-21, and mrc1Δ strains. Cells were treated with HU, and Dia2 protein stability was assayed as described above. Cell cycle arrest was monitored by flow cytometry (data not shown). As shown in Fig. 7B, the Dia2 protein was not stabilized in any mutant (Fig. 7B, top, lanes 5 to 24). A requirement for Chk1 was unexpected, as the chk1Δ strain is not sensitive to HU treatment and no defect in the S-phase checkpoint response without the presence of other mutated checkpoint genes has been previously described (32, 34). To rule out an indirect effect of checkpoint mutants on Dia2 protein turnover, we examined Dia2 protein stability in these strains in G1-arrested cells and observed rates of turnover similar to those in wild-type cells (Fig. 7C).
We furthered this analysis by testing Dia2 stability during an intra-S DNA damage checkpoint. α-Factor-arrested cells were released into medium containing MMS to activate the checkpoint, and cycloheximide was added to inhibit translation. In wild-type cells, the Dia2 protein was stabilized under these conditions, whereas in the rad53-21 checkpoint mutant, the Dia2 protein was degraded (Fig. 7D, lanes 7 to 12). These results suggest that inhibition of Dia2 protein turnover is dependent on functional DNA damage and replication checkpoint pathways.
If Dia2 stabilization is required for the cellular response to replication stress, then removal of DIA2 should exacerbate the phenotypes of S-phase checkpoint mutants. We and others have shown that dia2Δ cells exhibit synthetic growth phenotypes on normal media in combination with checkpoint mutants (4, 17, 28, 43). We tested a series of these double mutants for their sensitivity to HU, which induces replication stress. We find that deletion of DIA2 increased sensitivity to HU in chk1Δ, rad17Δ, rad24Δ, and mrc1Δ mutants (Fig. 7E). Thus, we conclude that blocking Dia2 protein turnover contributes to the cellular response to replication stress.
Altogether our results support a model (Fig. 8) in which the ubiquitin-mediated destruction of Dia2 is inhibited by induction of the S-phase checkpoint. Dia2 protein levels are low in G1 due to ubiquitin-mediated turnover. This is not the result of an autoubiquitination or SCF-dependent reaction and is dependent on a 20-amino-acid N-terminal domain in Dia2. These findings are consistent with previous data that suggest a role for Dia2 in DNA and genomic maintenance.
FIG. 8.
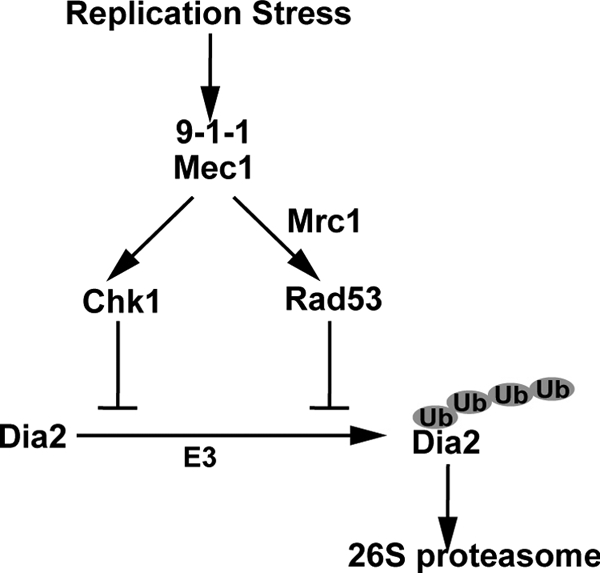
Model for S-phase checkpoint regulation of Dia2. Dia2 is targeted for ubiquitin-mediated destruction by the 26S proteasome and an unknown E3 ligase. Replication stress activates S-phase checkpoints and leads to stabilization of Dia2.
DISCUSSION
In this study, we have found that Dia2 is an unstable protein, that it is targeted for ubiquitin-mediated proteolysis, and that an N-terminal domain is important for both degradation and nuclear localization. The turnover of the Dia2 protein does not seem to be the sole result of an autoubiquitination mechanism, as deletion of the F-box domain does not stabilize the protein, despite the SCFDia2 complex exhibiting autoubiquitination activity in vitro (19). Additionally, other F-box proteins are stabilized in SCF pathway mutants; however, this is not the case for Dia2 (13, 47). Rather, Dia2 is even less stable in skp1-11 and cdc53-1 mutants, consistent with the observation that other F-box proteins are destabilized when core SCF components are defective (12, 24, 29, 38). Some F-box proteins are targets of other ubiquitin ligases, such as the APC/C. We anticipate that Dia2 is also the target of a non-SCF ubiquitin ligase, and we look forward to future studies that identify such a ubiquitin ligase.
The observation that the same 20-amino-acid domain is required for both nuclear localization and protein degradation is interesting. NLS sequences are rich in positively charged residues, and the region in Dia2 contains nine lysine residues but no arginines. It may simply be a coincidence that the NLS sequences in Dia2 could also potentially serve as ubiquitination sites. Alternatively, the regulation of Dia2 localization and ubiquitination may be intertwined. Further studies will be necessary to determine whether nuclear localization and degradation of Dia2 are linked and to dissect critical residues for each in the relevant 20-amino-acid domain.
The inhibition of Dia2 protein turnover by the S-phase checkpoint is consistent with the role of Dia2 in genome maintenance and DNA replication. It has been proposed that Dia2 may regulate the activity of replisome complexes as they move through areas of the genome that are difficult to replicate and prone to DNA damage (4). The accumulation of the Dia2 protein as cells enter S and the stabilization of the Dia2 protein in response to replication stress fit this model nicely. Our results suggest that stabilization of Dia2 occurs downstream of either Rad53 or Chk1 activation. We do not know how the S-phase checkpoint interfaces with the Dia2 protein turnover pathway. Two obvious possibilities exist: (i) Dia2 itself could be a target of the S-phase checkpoint pathway, or (ii) the ubiquitination pathway that controls Dia2 turnover may be regulated by the S-phase checkpoint. Regardless of the mechanism, cells are clearly sensitive to Dia2 protein levels. In the absence of Dia2, cells are hypersensitive to replication block by HU, and in wild-type cells, these same conditions lead to a stabilization of Dia2 protein levels. Moreover, overexpression of Dia2 in wild-type cells leads to increased sensitivity to MMS. Together, these observations suggest that during times of replication stress, SCFDia2 activity is maintained to promote genomic integrity but that there is an optimal level of SCFDia2 activity. One mechanism used to control SCFDia2 activity appears to be regulation of the abundance of the Dia2 protein itself.
In summary, by studying the regulation of the F-box protein Dia2, we have found that it is targeted for proteolysis by a ubiquitin-mediated pathway. The turnover of Dia2 is distinct from that of other F-box proteins in that it is independent of an autoubiquitination mechanism. Dia2 protein turnover is inhibited by the activation of the S-phase checkpoint, and we propose that SCFDia2 activity is required for the cellular response to replication stress.
TABLE 2.
Plasmids used in this study
| Plasmid | Relevant Features | Reference |
|---|---|---|
| p1219 | GAL1,10 promoter, CEN TRP1 Ampr | 21 |
| pACK135 | GAL1,10 promoter, 9MYC-DIA2 CEN TRP1 Ampr | This study |
| pACK136 | GAL1,10 promoter, 9MYC-DIA2-ΔF CEN TRP1 Ampr | This study |
| pACK137 | GAL1,10 promoter, 9MYC-DIA2-ΔN214 CEN TRP1 Ampr | This study |
| pACK138 | GAL1,10 promoter, 9MYC-DIA2-LRR CEN TRP1 Ampr | This study |
| pACK140 | GAL1,10 promoter, 9MYC-DIA2-TPR CEN TRP1 Ampr | This study |
| pACK154 | GAL1,10 promoter, 9MYC-DIA2-ΔN189 CEN TRP1 Ampr | This study |
| pACK171 | GAL1,10 promoter, 9MYC-DIA2-ΔN149 CEN TRP1 Ampr | This study |
| pACK176 | GAL1,10 promoter, 9MYC-SV40NLS-DIA2 CEN TRP1 Ampr | This study |
| pACK177 | GAL1,10 promoter, 9MYC-DIA2-ΔNLS CEN TRP1 Ampr | This study |
| pACK178 | GAL1,10 promoter, 9MYC-SV40NLS-DIA2-ΔNLS CEN TRP1 Ampr | This study |
| pACK179 | GAL1,10 promoter, 9MYC-SV40NLS-DIA2-ΔN214 CEN TRP1 Ampr | This study |
| pACK142 | pRS406 1-kb 5′DIA2 UTR 9MYC-DIA2 URA3 Ampr | This study |
| pACK143 | pRS406 1-kb 5′DIA2 UTR 9MYC-ΔF-DIA2 URA3 Ampr | This study |
| pACK144 | pRS406 1-kb 5′DIA2 UTR 9MYC-ΔN214-DIA2 URA3 Ampr | This study |
| pACK145 | pRS406 1-kb 5′DIA2 UTR 9MYC-DIA2-LRR URA3 Ampr | This study |
| pACK147 | pRS406 1-kb 5′DIA2 UTR 9MYC-DIA2-TPR URA3 Ampr | This study |
| pACK155 | pRS406 1-kb 5′DIA2 UTR 9MYC-DIA2-ΔN189 URA3 Ampr | This study |
| pACK174 | pRS406 1-kb 5′DIA2 UTR 9MYC-DIA2-ΔN149 URA3 Ampr | This study |
| pACK181 | pRS406 1-kb 5′DIA2 UTR 9MYC-SV40NLS-DIA2 URA3 Ampr | This study |
| pACK182 | pRS406 1-kb 5′DIA2 UTR 9MYC-DIA2-ΔNLS URA3 Ampr | This study |
| pACK183 | pRS406 1-kb 5′DIA2 UTR 9MYC-SV40NLS-DIA2-ΔNLS URA3 Ampr | This study |
| pACK184 | pRS406 1-kb 5′DIA2 UTR 9MYC-SV40NLS-DIA2-ΔN214 URA3 Ampr | This study |
Acknowledgments
This work was funded by NIH grant R01GM076663 and awards from the Minnesota Medical Foundation and the University of Minnesota Graduate School Grant-in-Aid to D.M.K.
We thank Yolanda Sanchez (Dartmouth Medical School), Anja-Katrin Bielinsky (University of Minnesota), and J. Wade Harper (Harvard Medical School) for gifts of strains and reagents.
Footnotes
Published ahead of print on 26 October 2009.
REFERENCES
- 1.Alcasabas, A. A., A. J. Osborn, J. Bachant, F. Hu, P. J. Werler, K. Bousset, K. Furuya, J. F. Diffley, A. M. Carr, and S. J. Elledge. 2001. Mrc1 transduces signals of DNA replication stress to activate Rad53. Nat. Cell Biol. 3:958-965. [DOI] [PubMed] [Google Scholar]
- 2.Allen, J. B., Z. Zhou, W. Siede, E. C. Friedberg, and S. J. Elledge. 1994. The SAD1/RAD53 protein kinase controls multiple checkpoints and DNA damage-induced transcription in yeast. Genes Dev. 8:2401-2415. [DOI] [PubMed] [Google Scholar]
- 3.Bashir, T., N. V. Dorrello, V. Amador, D. Guardavaccaro, and M. Pagano. 2004. Control of the SCF(Skp2-Cks1) ubiquitin ligase by the APC/C(Cdh1) ubiquitin ligase. Nature 428:190-193. [DOI] [PubMed] [Google Scholar]
- 4.Blake, D., B. Luke, P. Kanellis, P. Jorgensen, T. Goh, S. Penfold, B. J. Breitkreutz, D. Durocher, M. Peter, and M. Tyers. 2006. The F-box protein Dia2 overcomes replication impedance to promote genome stability in Saccharomyces cerevisiae. Genetics 174:1709-1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blondel, M., J. M. Galan, Y. Chi, C. Lafourcade, C. Longaretti, R. J. Deshaies, and M. Peter. 2000. Nuclear-specific degradation of Far1 is controlled by the localization of the F-box protein Cdc4. EMBO J. 19:6085-6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boddy, M. N., and P. Russell. 2001. DNA replication checkpoint. Curr. Biol. 11:R953-R956. [DOI] [PubMed] [Google Scholar]
- 7.Branzei, D., and M. Foiani. 2005. The DNA damage response during DNA replication. Curr. Opin. Cell Biol. 17:568-575. [DOI] [PubMed] [Google Scholar]
- 8.Caldwell, J. M., Y. Chen, K. L. Schollaert, J. F. Theis, G. F. Babcock, C. S. Newlon, and Y. Sanchez. 2008. Orchestration of the S-phase and DNA damage checkpoint pathways by replication forks from early origins. J. Cell Biol. 180:1073-1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carrano, A. C., E. Eytan, A. Hershko, and M. Pagano. 1999. SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat. Cell Biol. 1:193-199. [DOI] [PubMed] [Google Scholar]
- 10.Cho, R. J., M. J. Campbell, E. A. Winzeler, L. Steinmetz, A. Conway, L. Wodicka, T. G. Wolfsberg, A. E. Gabrielian, D. Landsman, D. J. Lockhart, and R. W. Davis. 1998. A genome-wide transcriptional analysis of the mitotic cell cycle. Mol. Cell 2:65-73. [DOI] [PubMed] [Google Scholar]
- 11.Feldman, R. M., C. C. Correll, K. B. Kaplan, and R. J. Deshaies. 1997. A complex of Cdc4p, Skp1p, and Cdc53p/cullin catalyzes ubiquitination of the phosphorylated CDK inhibitor Sic1p. Cell 91:221-230. [DOI] [PubMed] [Google Scholar]
- 12.Fey, J. P., and S. Lanker. 2007. Delayed accumulation of the yeast G(l) cyclins Clnl and Cln2 and the F-box protein Grrl in response to glucose. Yeast 24:419-429. [DOI] [PubMed] [Google Scholar]
- 13.Galan, J. M., and M. Peter. 1999. Ubiquitin-dependent degradation of multiple F-box proteins by an autocatalytic mechanism. Proc. Natl. Acad. Sci. U.S.A. 96:9124-9129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harrison, J. C., and J. E. Haber. 2006. Surviving the breakup: the DNA damage checkpoint. Annu. Rev. Genet. 40:209-235. [DOI] [PubMed] [Google Scholar]
- 15.Ho, Y., A. Gruhler, A. Heilbut, G. D. Bader, L. Moore, S. L. Adams, A. Millar, P. Taylor, K. Bennett, K. Boutilier, L. Yang, C. Wolting, I. Donaldson, S. Schandorff, J. Shewnarane, M. Vo, J. Taggart, M. Goudreault, B. Muskat, C. Alfarano, D. Dewar, Z. Lin, K. Michalickova, A. R. Willems, H. Sassi, P. A. Nielsen, K. J. Rasmussen, J. R. Andersen, L. E. Johansen, L. H. Hansen, H. Jespersen, A. Podtelejnikov, E. Nielsen, J. Crawford, V. Poulsen, B. D. Sorensen, J. Matthiesen, R. C. Hendrickson, F. Gleeson, T. Pawson, M. F. Moran, D. Durocher, M. Mann, C. W. Hogue, D. Figeys, and M. Tyers. 2002. Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature 415:180-183. [DOI] [PubMed] [Google Scholar]
- 16.Katou, Y., Y. Kanoh, M. Bando, H. Noguchi, H. Tanaka, T. Ashikari, K. Sugimoto, and K. Shirahige. 2003. S-phase checkpoint proteins Tof1 and Mrc1 form a stable replication-pausing complex. Nature 424:1078-1083. [DOI] [PubMed] [Google Scholar]
- 17.Koepp, D. M., A. C. Kile, S. Swaminathan, and V. Rodriguez-Rivera. 2006. The F-box protein Dia2 regulates DNA replication. Mol. Biol. Cell 17:1540-1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koepp, D. M., L. K. Schaefer, X. Ye, K. Keyomarsi, C. Chu, J. W. Harper, and S. J. Elledge. 2001. Phosphorylation-dependent ubiquitination of cyclin E by the SCFFbw7 ubiquitin ligase. Science 294:173-177. [DOI] [PubMed] [Google Scholar]
- 19.Kus, B. M., C. E. Caldon, R. Andorn-Broza, and A. M. Edwards. 2004. Functional interaction of 13 yeast SCF complexes with a set of yeast E2 enzymes in vitro. Proteins 54:455-467. [DOI] [PubMed] [Google Scholar]
- 20.Lange, A., R. E. Mills, C. J. Lange, M. Stewart, S. E. Devine, and A. H. Corbett. 2007. Classical nuclear localization signals: definition, function, and interaction with importin alpha. J. Biol. Chem. 282:5101-5105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu, Q., M. Z. Li, D. Leibham, D. Cortez, and S. J. Elledge. 1998. The univector plasmid-fusion system, a method for rapid construction of recombinant DNA without restriction enzymes. Curr. Biol. 8:1300-1309. [DOI] [PubMed] [Google Scholar]
- 22.Lopes, M., C. Cotta-Ramusino, A. Pellicioli, G. Liberi, P. Plevani, M. Muzi-Falconi, C. S. Newlon, and M. Foiani. 2001. The DNA replication checkpoint response stabilizes stalled replication forks. Nature 412:557-561. [DOI] [PubMed] [Google Scholar]
- 23.Majka, J., and P. M. Burgers. 2003. Yeast Rad17/Mec3/Ddc1: a sliding clamp for the DNA damage checkpoint. Proc. Natl. Acad. Sci. U.S.A. 100:2249-2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mathias, N., S. Johnson, B. Byers, and M. Goebl. 1999. The abundance of cell cycle regulatory protein Cdc4p is controlled by interactions between its F box and Skp1p. Mol. Cell. Biol. 19:1759-1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mathias, N., S. L. Johnson, M. Winey, A. E. Adams, L. Goetsch, J. R. Pringle, B. Byers, and M. G. Goebl. 1996. Cdc53p acts in concert with Cdc4p and Cdc34p to control the G1-to-S-phase transition and identifies a conserved family of proteins. Mol. Cell. Biol. 16:6634-6643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nyberg, K. A., R. J. Michelson, C. W. Putnam, and T. A. Weinert. 2002. Toward maintaining the genome: DNA damage and replication checkpoints. Annu. Rev. Genet. 36:617-656. [DOI] [PubMed] [Google Scholar]
- 27.Osborn, A. J., and S. J. Elledge. 2003. Mrc1 is a replication fork component whose phosphorylation in response to DNA replication stress activates Rad53. Genes Dev. 17:1755-1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pan, X., P. Ye, D. S. Yuan, X. Wang, J. S. Bader, and J. D. Boeke. 2006. A DNA integrity network in the yeast Saccharomyces cerevisiae. Cell 124:1069-1081. [DOI] [PubMed] [Google Scholar]
- 29.Patton, E. E., A. R. Willems, D. Sa, L. Kuras, D. Thomas, K. L. Craig, and M. Tyers. 1998. Cdc53 is a scaffold protein for multiple Cdc34/Skp1/F box protein complexes that regulate cell division and methionine biosynthesis in yeast (vol 12, pg 914, 1998). Genes Dev. 12:3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paulovich, A. G., and L. H. Hartwell. 1995. A checkpoint regulates the rate of progression through S phase in S. cerevisiae in response to DNA damage. Cell 82:841-847. [DOI] [PubMed] [Google Scholar]
- 30a.Ricke, R. M., and A. K. Bielinsky. 2004. Mcm10 regulates the stability and chromatin association of DNA polymerase-alpha. Mol. Cell 16:173-185. [DOI] [PubMed] [Google Scholar]
- 31.Rose, M. D., F. Winston, and P. Hieter. 1990. Methods in yeast genetics: a laboratory course manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 32.Sanchez, Y., J. Bachant, H. Wang, F. Hu, D. Liu, M. Tetzlaff, and S. J. Elledge. 1999. Control of the DNA damage checkpoint by chk1 and rad53 protein kinases through distinct mechanisms. Science 286:1166-1171. [DOI] [PubMed] [Google Scholar]
- 33.Santocanale, C., and J. F. Diffley. 1998. A Mec1- and Rad53-dependent checkpoint controls late-firing origins of DNA replication. Nature 395:615-618. [DOI] [PubMed] [Google Scholar]
- 34.Schollaert, K. L., J. M. Poisson, J. S. Searle, J. A. Schwanekamp, C. R. Tomlinson, and Y. Sanchez. 2004. A role for Saccharomyces cerevisiae Chk1p in the response to replication blocks. Mol. Biol. Cell 15:4051-4063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shirahige, K., Y. Hori, K. Shiraishi, M. Yamashita, K. Takahashi, C. Obuse, T. Tsurimoto, and H. Yoshikawa. 1998. Regulation of DNA-replication origins during cell-cycle progression. Nature 395:618-621. [DOI] [PubMed] [Google Scholar]
- 36.Skowyra, D., K. L. Craig, M. Tyers, S. J. Elledge, and J. W. Harper. 1997. F-box proteins are receptors that recruit phosphorylated substrates to the SCF ubiquitin-ligase complex. Cell 91:209-219. [DOI] [PubMed] [Google Scholar]
- 37.Skowyra, D., D. M. Koepp, T. Kamura, M. N. Conrad, R. C. Conaway, J. W. Conaway, S. J. Elledge, and J. W. Harper. 1999. Reconstitution of G1 cyclin ubiquitination with complexes containing SCFGrr1 and Rbx1. Science 284:662-665. [DOI] [PubMed] [Google Scholar]
- 38.Smothers, D. B., L. Kozubowski, C. Dixon, M. G. Goebl, and N. Mathias. 2000. The abundance of Met30p limits SCFMet30p complex activity and is regulated by methionine availability. Mol. Cell. Biol. 20:7845-7852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Spellman, P. T., G. Sherlock, M. Q. Zhang, V. R. Iyer, K. Anders, M. B. Eisen, P. O. Brown, D. Botstein, and B. Futcher. 1998. Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization. Mol. Biol. Cell 9:3273-3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Strohmaier, H., C. H. Spruck, P. Kaiser, K. A. Won, O. Sangfelt, and S. I. Reed. 2001. Human F-box protein hCdc4 targets cyclin E for proteolysis and is mutated in a breast cancer cell line. Nature 413:316-322. [DOI] [PubMed] [Google Scholar]
- 41.Swaminathan, S., A. C. Kile, E. M. MacDonald, and D. M. Koepp. 2007. Yra1 is required for S phase entry and affects Dia2 binding to replication origins. Mol. Cell. Biol. 27:4674-4684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tercero, J. A., and J. F. Diffley. 2001. Regulation of DNA replication fork progression through damaged DNA by the Mec1/Rad53 checkpoint. Nature 412:553-557. [DOI] [PubMed] [Google Scholar]
- 43.Tong, A. H., G. Lesage, G. D. Bader, H. Ding, H. Xu, X. Xin, J. Young, G. F. Berriz, R. L. Brost, M. Chang, Y. Chen, X. Cheng, G. Chua, H. Friesen, D. S. Goldberg, J. Haynes, C. Humphries, G. He, S. Hussein, L. Ke, N. Krogan, Z. Li, J. N. Levinson, H. Lu, P. Menard, C. Munyana, A. B. Parsons, O. Ryan, R. Tonikian, T. Roberts, A. M. Sdicu, J. Shapiro, B. Sheikh, B. Suter, S. L. Wong, L. V. Zhang, H. Zhu, C. G. Burd, S. Munro, C. Sander, J. Rine, J. Greenblatt, M. Peter, A. Bretscher, G. Bell, F. P. Roth, G. W. Brown, B. Andrews, H. Bussey, and C. Boone. 2004. Global mapping of the yeast genetic interaction network. Science 303:808-813. [DOI] [PubMed] [Google Scholar]
- 44.Tsvetkov, L. M., K. H. Yeh, S. J. Lee, H. Sun, and H. Zhang. 1999. p27(Kip1) ubiquitination and degradation is regulated by the SCF(Skp2) complex through phosphorylated Thr187 in p27. Curr. Biol. 9:661-664. [DOI] [PubMed] [Google Scholar]
- 45.Venclovas, C., and M. P. Thelen. 2000. Structure-based predictions of Rad1, Rad9, Hus1 and Rad17 participation in sliding clamp and clamp-loading complexes. Nucleic Acids Res. 28:2481-2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Willems, A. R., S. Lanker, E. E. Patton, K. L. Craig, T. F. Nason, N. Mathias, R. Kobayashi, C. Wittenberg, and M. Tyers. 1996. Cdc53 targets phosphorylated G1 cyclins for degradation by the ubiquitin proteolytic pathway. Cell 86:453-463. [DOI] [PubMed] [Google Scholar]
- 47.Zhou, P., and P. M. Howley. 1998. Ubiquitination and degradation of the substrate recognition subunits of SCF ubiquitin-protein ligases. Mol. Cell 2:571-580. [DOI] [PubMed] [Google Scholar]