

Abstract
Tegument protein VP8 encoded by the UL47 gene of bovine herpesvirus type 1 (BHV-1) is the most abundant constituent of mature virions. In the present report, we describe the characterization of UL47 gene-deleted BHV-1 in cultured cells and its natural host. The UL47 deletion mutant exhibited reduced plaque size and more than 100-fold decrease in intracellular and extracellular viral titers in cultured cells. Ultrastructural observations of infected cells showed normal maturation of BHV-1 virions in the absence of VP8. There was no evidence for a change in immediate-early gene activator function of VP16 in the UL47 deletion mutant virus-infected cells, since bovine ICP4 mRNA and protein levels were similar to those in the wild-type and revertant virus-infected cells throughout the course of infection. Whereas VP16, glycoprotein C (gC), gB, and VP5 were expressed to wild-type levels in the UL47 deletion mutant-infected cells, the gD and VP22 protein levels were significantly reduced. The reduction in gD protein was associated with increased turnover of the protein. Furthermore, some of the analyzed early and late proteins were expressed with earlier kinetics in the absence of VP8. Extracellular virions of the UL47 deletion mutant contained reduced amounts of gD, gB, gC, and VP22 but similar amounts of VP16 compared to those of wild-type or revertant virus particles. In addition, the UL47 gene product was indispensable for BHV-1 replication in vivo, since no clinical manifestations or viral shedding were detected in the UL47 deletion mutant-infected calves, and the virus failed to induce significant levels of humoral and cellular immunity.
Bovine herpesvirus type 1 (BHV-1) is an alphaherpesvirus which is an important pathogen of cattle, causing a variety of clinical manifestations in its natural host (46). BHV-1 virions have a typical herpesvirus structure characterized by the presence of a double-stranded DNA genome enclosed in an icosahedral capsid, the tegument surrounding the capsid, and the outer host-derived lipid envelope bearing virus-encoded glycoproteins. While the major constituents of the viral envelope have been extensively studied (reviewed in reference 17), the proteins present in the tegument and nucleocapsid of BHV-1 have been poorly characterized. Compositionally, the tegument is the most complex compartment of the virion, containing more than 15 viral gene products (32). In addition to their structural role, various regulatory functions, including modulation of transcription (34, 47), kinase activity (39), RNase activity (41), and DNA packaging (43), have been assigned to some tegument proteins, suggesting that these virion constituents function at several stages during virus infection, establishing conditions for efficient viral replication and promoting virus assembly and egress.
Although the UL47 gene product, tegument protein VP8, is the most abundant component of mature BHV-1 virions (5), its function is unknown. Like its herpes simplex virus type 1 (HSV-1) homologue (31), VP8 is posttranslationally modified by phosphorylation (5, 23) and by the addition of O-linked carbohydrates (49). Both HSV-1 and BHV-1 UL47 homologues possess nuclear localization and nuclear export signatures (7, 51, 53, 56), enabling them to shuttle between the nucleus and cytoplasm when expressed in transiently transfected cells (51, 56) or during viral infection (52, 53). Furthermore, both proteins exhibit a steady-state nuclear localization at early stages of infection and during transient expression (6, 35, 49, 51, 52, 56), suggesting a functional role for these homologues in the nucleus. Nucleocytoplasmic shuttling of VP8 is sensitive to treatment with a RNA polymerase II inhibitor, actinomycin D (52). This observation coupled with recently demonstrated RNA binding activity of the HSV-1 and BHV-1 UL47 counterparts suggests that like RNA binding proteins encoded by other viruses (12, 44), these UL47 gene products may be involved in promotion of the export of virus-encoded transcripts from the nucleus to the cytoplasm (8). Early genetic studies suggested a modulatory role of the HSV-1 UL47 gene product in stimulation of viral immediate-early gene transcription, which is primarily mediated by VP16 (54, 55). However, these potential functions are not absolutely required for HSV-1 propagation in cell culture, as corresponding UL47 deletion mutants have been produced using noncomplementing cells (2, 55). The UL47 gene products of Marek's disease virus serotype 1 (9), avian infectious laryngotracheitis virus (ILTV) (13), and pseudorabies virus (PrV) (20) were also considered nonessential in cell culture. A common feature of all UL47-null herpesviruses characterized is a comparatively mild growth defect, which is an approximately 10-fold decrease in final titers. UL47-deleted PrV exhibited normal replication in the nasal epithelium and only a slight delay in neuroinvasion in a mouse model (19). An in vivo phenotype was apparent during infection of a natural host with the UL47 gene-deleted ILTV, which was significantly attenuated in chickens but conferred protection against subsequent challenge with a virulent strain of the virus (13). VP8 was also determined to be nonessential for viability of BHV-1 in cell culture (42). However, the phenotype of a UL47-null BHV-1 has not been characterized either in cell culture or in vivo.
In this report, we describe the construction and functional analysis of a BHV-1 mutant defective in expression of the UL47 gene. This is the first phenotypic characterization of UL47 gene-deleted BHV-1 both in vitro in permissive cells and in vivo in its natural host.
MATERIALS AND METHODS
Cells and viruses.
Madin-Darby bovine kidney (MDBK) cells and fetal bovine testicular (FBT) cells were cultured in Eagle's minimum essential medium supplemented with 10 mM HEPES, 0.1 mM nonessential amino acids, 50 μg/ml gentamicin (cell culture medium), and 10% (vol/vol) fetal bovine serum (FBS). All cell culture reagents were purchased from Gibco/Invitrogen (Carlsbad, CA). A highly virulent BHV-1 strain 108 (16) was used as parental virus in this study. All viral stocks were propagated in FBT cells as described previously (23). Cell culture medium supplemented with 2% FBS and 0.8% agarose type VII (Sigma-Aldrich, St. Louis, MO) was used to overlay cells for viral titration in a 24-well plate format or experiments involving viral plaque formation.
Recombinants were constructed by homologous recombination within MDBK cells. To substitute the UL47 open reading frame (ORF) with the enhanced yellow fluorescent protein (EYFP) coding sequence, the pUL48-YFP-UL46 plasmid bearing the EYFP ORF surrounded by UL47 upstream and downstream flanking sequences was constructed. The upstream homology region was amplified from purified BHV-1 DNA by PCR using primer 5′-ACACGGTACCGAGTTGCGTTGTCTTTGGGATG-3′, including a KpnI restriction site, and primer 5′-TCGGTCGACAAATCCCGTGTAGACCCAAG-3′, including a SalI restriction site. The PCR product was inserted as a KpnI/SalI fragment into pEYFP-N1 (Clontech, Mountain View, CA), creating pUL48-YFP. The homology region downstream of UL47 was amplified from BHV-1 DNA with primer 5′-TGGAATTCTAGCCATCACGCTTGAATGC-3′, containing an EcoRI site, and primer 5′-CAACTAGTTCAGCAGCGCCAGCTTAAAC-3′, containing an SpeI site. This fragment was inserted as an EcoRI/SpeI fragment into the pUL48-YFP plasmid, resulting in pUL48-YFP-UL46. To transfer the UL47 deletion into the virus, a fragment encompassing the EYFP ORF surrounded by UL47 flanking regions was excised from pUL48-YFP-UL46 by KpnI/SpeI digestion, and 2 μg of the fragment DNA was cotransfected with 1 μg of infectious wild-type (WT) BHV-1 DNA into FBT cells using Lipofectamine reagent (Invitrogen, Carlsbad, CA). The resulting virus pool was screened for EYFP-positive plaques. Three rounds of plaque purification on MDBK cells resulted in the appearance of small fluorescent plaques that did not spread efficiently through the monolayer. Therefore, the last round of plaque purification and viral stock preparation was carried out on FBT cells, which better supported the spread of the UL47-null virus (BHV1-ΔUL47) in our experiments. The introduced deletion spanned 40 bp of upstream UL47 flanking sequence and 84 bp of downstream UL47 flanking sequence.
To construct a ΔUL47 rescue virus, the UL47 ORF was excised with BamHI and XhoI from pcDNA6-UL47, which was constructed by cloning a BamHI/XhoI-digested UL47 gene-containing PCR fragment, amplified from BHV-1 DNA with primers 5′-CCGGATCCGCCATGGACGCCGCTAGGGATG-3′ and 5′-CCCTCGAGCTAGCGGCCGCCCAGG-3′, into pcDNA6/V5-His (Invitrogen) cut with the same enzymes. The resulting 2.2-kbp fragment blunted by T4 DNA polymerase was inserted into pUL48-YFP-UL46 digested with BamHI/EcoRI and blunted as described above to create pUL48-UL47-UL46. A fragment encompassing the UL47 ORF surrounded by its flanking sequences was released from pUL48-UL47-UL46 by KpnI/SpeI digestion and transfected into FBT cells, followed by infection with 10,000 PFU of BHV1-ΔUL47. The resulting virus pool was used to initiate three rounds of nonfluorescent plaque selection on MDBK cells. In the revertant virus (BHV1-UL47R) genome, the UL47 ORF, but not the flanking intergenic regions, was repaired.
Analysis of viral growth properties.
To examine the replication kinetics of the viruses, MDBK cells cultured in 35-mm cell culture dishes were infected with preparations of extracellular virus at a multiplicity of infection (MOI) of 1. Starting at 5 h after infection, cell culture medium and cells were collected separately every 5 h for 35 h. The cells, collected into freshly added cell culture medium, were subjected to two freeze-thaw cycles to release the virus. All samples were clarified by low-speed centrifugation and stored at −80°C prior to the determination of the infectious titer by plaque assay on MDBK cells.
For plaque size analysis, MDBK cells cultured in six-well plates were infected with ∼100 PFU/well of WT BHV-1, BHV1-ΔUL47, or BHV1-UL47R. At 48 h postinfection, the infected cell monolayers were fixed with 75% ethanol-25% glacial acetic acid, and viral plaques were stained by incubation with a pool of glycoprotein B (gB)-specific monoclonal antibodies (MAbs), followed by incubation with alkaline phosphatase-conjugated goat anti-mouse immunoglobulin G (IgG) (Kirkegaard and Perry Laboratories, Gaithersburg, MD) and development with 5-bromo-4-chloro-3-indolylphosphate and nitroblue tetrazolium (BCIP/NBT) (Sigma Chemical Co., Saint Louis, MO). Images of 50 discrete plaques for each virus were acquired using a Zeiss Axiovert 200M microscope equipped with a digital camera. The average diameter for each plaque was calculated from two perpendicular diameter measurements performed using AxioVision AC 4.5 software.
To prepare gradient-purified BHV-1 virions, supernatants from infected FBT cells displaying complete cytopathic effects were combined, and cell debris was removed by low-speed centrifugation. Virions were pelleted through a 30% (wt/vol) sucrose cushion in TNE buffer (10 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1 mM EDTA) by centrifugation in a Beckman SW32Ti rotor at 25,000 rpm for 2 h at 4°C. The pellet was resuspended in TNE buffer and layered onto a 36-ml linear 10 to 60% (wt/vol) gradient of potassium sodium tartrate in TNE buffer poured from the bottom using a Hoefer SG50 gradient maker (Amersham, Piscataway, NJ). After centrifugation for 2 h at 25,000 rpm (SW32Ti), purified virions were withdrawn by side puncture with a syringe. The virions were concentrated by centrifugation as described above, resuspended in TNE buffer, and stored at −80°C until use.
To analyze the particle-to-PFU ratio, solubilized proteins of gradient-purified WT BHV-1, BHV1-ΔUL47, and BHV1-UL47R virions were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and the VP5 protein concentration was measured by immunoblotting and densitometry. The volumes of the virion preparations were adjusted to normalize the VP5 concentration, and the infectivity of the viral preparations was determined by plaque assay, whereas the viral genome copy numbers were analyzed by quantitative real-time PCR (qPCR).
Analyses of viral DNA and RNA.
For Southern blotting, cell culture supernatants from 150-cm2 flasks with FBT cells infected for 48 h with 106 PFU/flask of WT BHV-1, BHV1-ΔUL47, or BHV1-UL47R were clarified by centrifugation at 5,000 × g for 10 min, and viral particles were collected by centrifugation at 27,000 rpm (SW32Ti rotor) for 2 h at 4°C. DNA was isolated from the pelleted virions using a Qiagen genomic DNA extraction kit. Viral pellets were resuspended in 5 ml of buffer G2, followed by the addition of 95 μl of proteinase K stock solution and incubation at 50°C for 1 h. After the incubation, the lysates were diluted with an equal volume of buffer QBT and loaded into genomic-tip 100/G columns. Subsequent purification steps were performed strictly according to the protocol of the manufacturer. An aliquot of each DNA preparation was digested overnight with HindIII in the presence of RNase A. The DNA fragments were separated by electrophoresis in a 0.8% agarose gel and transferred to Zeta-Probe nylon membrane (Bio-Rad, Hercules, CA) by standard procedures. Probes used in Southern hybridization were generated by PCR using EYFP gene-specific primers 5′-ACGTAAACGGCCACAAGTTC-3′ and 5′-TCACCTTGATGCCGTTCTTC-3′ with the pEYFP-N1 (Clontech) vector as a template, and UL47 gene-specific primers 5′-TGAGGACGAGAACGTGTATG-3′ and 5′-TCGTCTGGCACAAGGTCCTC-3′ or UL49 ORF-specific primers 5′-TACGAGTACAGCGACCTTTG-3′ and 5′-CCAGACGCAACAGCATTAGC-3′ with purified WT BHV-1 DNA as a template. Probes were labeled with [α-32P]dCTP by random priming using Ready-To-Go DNA labeling beads (without dCTP) (Amersham Biosciences, Buckinghamshire, United Kingdom). The hybridizations were performed in ExpressHyb hybridization solution (Clontech) according to standard protocols.
To compare the viral mRNA levels by reverse transcription and qPCR, MDBK cells grown in six-well plates were mock infected or infected with 5 PFU/cell of WT BHV-1, BHV1-ΔUL47, or BHV1-UL47R in duplicate. Total RNA was extracted from the cells at 3, 4, 5, 10, 15, 20, and 25 h postinfection using Trizol reagent (Invitrogen). The isolated RNA samples were additionally cleaned up using a Qiagen RNeasy MinElute cleanup kit (Hilden, Germany), and the concentration and integrity of the RNA samples were determined with the aid of an Agilent 2100 bioanalyzer. Genomic DNA removal and reverse transcription were carried out with 0.5 μg total RNA per reaction mixture using a QuantiTect reverse transcription kit (Qiagen, Mississauga, Ontario, Canada).
Relative gD, bovine ICP4 (bICP4), and UL46 mRNA levels were quantified by qPCR using 18S rRNA internal controls to normalize template input. The amplifications were performed in 96-well PCR plates (Bio-Rad Laboratories, Hercules, CA) with 1 μl cDNA per well in triplicate using Platinum SYBR green qPCR SuperMix-UDG (Invitrogen) and an iQ5 real-time PCR detection system (Bio-Rad Laboratories). The following primers at a concentration of 10 pmol per reaction mixture were used in qPCR: for gD, primers 5′-CGAGCCCAGGAAGCACTTTG-3′ and 5′-ACCGTGCCGTCGATGTACAG-3′; for bICP4, primers 5′-GGCGCGCTTTTTGACACCCGTA-3′ and 5′-CCCCGCGCCTCTTTCTTGTT-3′; for UL46, primers 5′-TGCCGCTTCGCTCACTTTAC-3′ and 5′-TCAGCAGCGCCAGCTTAAAC-3′, and for 18S rRNA, primers 5′-TGTGATGCCCTTAGATGTCC-3′ and 5′-TTATGACCCGCACTTACTGG-3′. SYBR green fluorescence was measured over the course of 40 amplification cycles. Each plate included serial 10-fold dilutions of cDNA synthesized from total RNA isolated from WT BHV-1-infected cells or mock-infected cells to generate standard curves for BHV-1 gene-specific and 18S rRNA-specific primers, respectively. The data were analyzed by the method of Pfaffl (37). Briefly, the amplification efficiencies were calculated from the slopes of generated standard curves using the formula E = 10E(−1/slope). The mean cycle number at which product accumulation entered the linear range, the cycle threshold (CT), was calculated for each template. The relative expression ratio of a target gene was calculated as a sample versus a control in comparison to a reference gene (18S rRNA) using the equation:
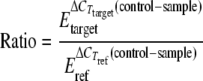 |
where ref stands for reference.
As there were no CT values for BHV-1 gene-specific primers in mock-infected samples, the values for 25 h postinfection samples were used as the control.
The relative copy numbers of WT BHV1, BHV1-ΔUL47, or BHV1-UL47R genomes in gradient-purified virion preparations were determined by qPCR with the gD-specific primers from a standard curve generated by serial dilution of the pSLIAgD plasmid containing the full-length BHV-1 gD gene (3). Viral DNA was extracted using a DNeasy blood and tissue kit (Qiagen, Mississauga, Ontario, Canada). Results were expressed as the number of viral genome copies per milliliter of gradient-purified virion preparation.
Protein analysis.
The following antibodies were used for protein analysis. VP8-specific MAb (49) and VP22-specific polyclonal rabbit antibody (28) have been described previously. Green fluorescent protein (GFP)-specific polyclonal antibody was purchased from Molecular Probes (Eugene, OR). Anti-β-actin MAb clone AC-15 was purchased from Sigma-Aldrich. Polyclonal VP16-specific rabbit serum was a generous gift from Vikram Misra, University of Saskatchewan. VP5-specific serum was raised in rabbits against a synthetic peptide, GVSRSTSDTELQFKQPPGTGEL, representing amino acids 1300 to 1321 of BHV-1 VP5 according to a previously described protocol (18). The same protocol was used to produce polyclonal rabbit serum to synthetic peptide FPAPAGGGRRPRRPRRGPPPRVEVL corresponding to amino acids 1284 to 1308 of BHV-1 bICP4. Mixtures of gB-specific and gC-specific MAbs (48) and gD-specific MAb clone 3D9S (29) have been described elsewhere.
For SDS-PAGE and immunoblotting, whole-cell extracts of virus-infected or mock-infected cells were prepared using radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 1% NP-40, 1% deoxycholate, 0.1% SDS) supplemented with 1 mM phenylmethylsulfonyl fluoride and 1 μg/ml each of aprotinin, leupeptin, and pepstatin A. The solubilized proteins of gradient-purified BHV-1 virions were prepared as previously described (49). Equal amounts of protein preparations were separated by SDS-PAGE, and the gels were either stained with Coomassie blue or transferred to nitrocellulose membranes, which were subjected to incubation with the appropriate primary antibodies. Western blots were further processed using goat IRDye 680-conjugated anti-mouse IgG or IRDye 800CW-conjugated anti-rabbit IgG (LI-COR Biosciences, Lincoln, NE). Antibody-reactive bands were detected using the Odyssey infrared imaging system (LI-COR Biosciences), and the images obtained were processed with the aid of Odyssey 3.0.16 application software (LI-COR Biosciences). In order to reprobe a blot with another antibody when necessary, membrane-bound antibody complexes were removed using the Newblot Nitro stripping buffer (LI-COR Biosciences).
For pulse-chase analysis, MDBK cells cultured in 60-mm cell culture plates were infected with 5 PFU/cell of WT BHV-1, BHV1-ΔUL47, or BHV1-UL47R or mock infected. At 8 h postinfection, the monolayers were washed twice with phosphate-buffered saline (PBS) and starved for 30 min in methionine/cysteine-free Dulbecco's modified Eagle medium supplemented with 2% dialyzed FBS (Gibco/Invitrogen). Subsequently, the cells were pulse-labeled for 40 min by the addition of 100 μCi of [35S]methionine/cysteine (PerkinElmer LAS Canada, Woodbridge, Ontario, Canada) per plate. The cells were washed once with PBS and incubated in Dulbecco's modified Eagle medium supplemented with 2% FBS, 2 mM l-methionine, and 2 mM l-cysteine. At 0, 30, 60, 120, and 180 min of chase, the cells were collected, and total cell lysates were prepared in RIPA buffer. Each lysate was divided into two equal aliquots, and paired aliquots were incubated with either gD-specific or gC-specific MAb for 7 h at 4°C, followed by overnight incubation at 4°C with protein A/G Sepharose CL-4B (GE Healthcare, Uppsala, Sweden). Finally, samples were washed four times with RIPA buffer and analyzed by SDS-8% PAGE and autoradiography.
Transmission electron microscopy.
MDBK cells cultured in 100-mm cell culture plates were mock infected or infected with 5 PFU/cell of WT BHV-1, BHV1-ΔUL47, or BHV1-UL47R. At 14 h postinfection, the cells were collected by trypsinization, washed with PBS by low-speed centrifugation, and fixed with 5% glutaraldehyde in 0.1 M sodium cacodylate buffer (SCB; pH 7.4). After fixation, the cells were washed and postfixed with 1% osmium tetroxide in 0.1 M SCB for 1 h at room temperature. Subsequently, the cells were rinsed with water and dehydrated in 50% ethanol for 5 min, then in 70% ethanol saturated with uranyl acetate for 1 h, followed by a graded ethanol series and three changes of propylene oxide. The samples were embedded in Epon/Araldite, polymerized overnight at 58°C, sectioned, and examined with the aid of a Philips 410 LS transmission electron microscope.
Experimental challenge and virus titration.
Nine- to 10-month-old Angus cross calves were randomly allocated to three groups of five animals each, and the groups were housed in separate isolation pens. Calves from two groups were individually exposed to an aerosol of 4 × 106 PFU of WT BHV-1 or BHV1-ΔUL47 as previously described (40, 50). One group was left as untreated control. After the challenge, the animals were examined for 12 consecutive days for clinical signs, including nasal lesions, conjunctivitis, depression, appetite, and body temperature. At 57 days postinfection, dexamethasone treatment was initiated according to the schedule reported by Inman et al. (14). Nasal swabs and serum samples were collected at the designated times. Nasal swabs collected in serum-free Eagle's minimum essential medium supplemented with 100 U/ml of penicillin and 0.1 mg/ml of streptomycin were snap-frozen in liquid nitrogen and stored at −80°C. All procedures were carried out in accordance with the guidelines of the Canadian Council for Animal Care.
For virus titration, nasal swab samples were thawed quickly in a 37°C water bath, vortexed, and centrifuged at 1,500 × g for 10 min at 4°C. Titrations were performed in a 96-well format with 10-fold serial dilutions, and the diluted samples were plated in three replicate wells. Cell culture medium containing 1% FBS and neutralizing BHV-1-specific bovine serum was used to overlay infected MDBK cells.
IFN-γ ELISPOT assay, virus neutralization assay, and enzyme-linked immunosorbent assay (ELISA).
For the enzyme-linked immunospot (ELISPOT) assay, 96-well Multiscreen-HA filter plates (Millipore, Billerica, MA) were coated overnight at 4°C with a bovine gamma interferon (IFN-γ)-specific MAb (clone 2-2-1) diluted 1:1,500. After the plates were washed with sterile PBS, they were blocked with 1% (wt/vol) bovine serum albumin (Sigma-Aldrich) in PBS. Peripheral blood mononuclear cells (PBMCs) were isolated as described previously (40). Purified PBMCs were resuspended in cell culture medium supplemented with 10% FBS, 2 mM l-glutamine, 50 μM 2-mercaptoethanol, and 2 ng/ml dexamethasone and dispensed at 106 cells per well in triplicate wells in medium or in medium with purified gB (0.1 μg/well), gC (0.1 μg/well), or tgD (0.4 μg/well). After 24 h, the plates were washed, incubated with bovine IFN-γ-specific rabbit antibody (92-131) diluted 1:500, and then incubated with alkaline phosphatase-conjugated goat anti-rabbit IgG (Kirkegaard and Perry Laboratories). IFN-γ spots were developed with BCIP/NBT substrate (Sigma-Aldrich). Finally, the plates were washed with water and air dried. The spots were counted, and the results were expressed as the difference between the number of IFN-γ-secreting cells per 106 cells in the glycoprotein-stimulated wells and the number of IFN-γ-secreting cells per 106 cells in wells with medium.
Virus neutralization titers in sera from experimentally infected animals were determined as described previously (1). The titers were expressed as the reciprocal of the highest dilution that caused a 50% reduction in the number of plaques relative to the virus control. BHV-1-specific IgG titers before and after challenge were determined by ELISA with purified tgD as the antigen according to a procedure described elsewhere (15, 40). ELISA titers were expressed as the reciprocal of the highest dilution resulting in a value of 2 standard deviations above the negative-control serum.
Statistical analyses.
Data were analyzed by using GraphPad Prism version 5.02 software. The differences in plaque sizes between the viruses were analyzed by two-tailed t test. Differences in serum neutralization titers and ELISPOT and ELISA data were investigated using one-way analysis of variance test. Medians between pairs of groups were further compared using the Mann-Whitney U test. Differences were considered significant if P was <0.05.
RESULTS
Construction of UL47 deletion and repair viruses.
To gain more information about the role of VP8 during BHV-1 infection, we constructed a deletion mutant virus carrying a marker gene encoding EYFP in place of the UL47 ORF and the corresponding rescue virus (Fig. 1A). The UL47-null virus demonstrated impaired growth in MDBK cells, which were used for initial rounds of plaque purification. Therefore, the last round of BHV1-ΔUL47 plaque purification and preparation of all viral stocks used in the present study were carried out in FBT cells. The titers of the BHV1-ΔUL47 stocks produced in FBT cells were routinely 2- to 4-fold higher than those obtained in MDBK cells, and these titers were approximately 100-fold lower compared to titers of WT BHV1 or BHV1-UL47R stocks produced in either of these cell lines.
FIG. 1.

Genetic characterization of the UL47 deletion mutant and revertant viruses. (A) Schematic representations of the WT BHV-1, BHV1-ΔUL47, and BHV1-UL47R genomes. The ruler on the top shows DNA size in base pairs. HindIII recognition sites located within the detailed genomic fragment are indicated. The cloning strategy used for the construction of the UL47 deletion and repair viruses led to the incorporation of an additional HindIII site located in the UL48-UL47 intergenic region. IR, inverted repeat; TR, tandem repeat. (B) Southern blot analysis of WT BHV-1, BHV1-ΔUL47, and BHV1-UL47R DNA digested with HindIII. [α-32P]dCTP-labeled UL47, EYFP, or UL49 gene-derived PCR fragments were used as probes. The positions of standard DNA markers (in kilobase pairs) are depicted to the left of the leftmost blot.
The correct genotypes of the recombinant viruses were confirmed by Southern hybridization. Purified viral DNA was cut with HindIII, and the digestion products were separated through a 0.8% agarose gel, transferred to a nylon membrane, and hybridized with [α-35P]dCTP-labeled UL47-, EYFP-, or UL49-specific probes. The UL49 gene is located in the 8,831-bp HindIII H fragment of the BHV-1 genome. As expected, the UL49-specific probe, which was used as the DNA loading control, hybridized to the 8,831-bp fragment present in the HindIII restriction profiles of WT BHV-1, BHV1-ΔUL47, and BHV1-UL47R DNA (Fig. 1B). The cloning strategy used for the construction of the UL47 deletion and repair viruses led to the incorporation of an additional HindIII site located in the UL48-UL47 intergenic region (Fig. 1A). This reduced the size of the UL47 gene-containing HindIII K fragment from 3,737 bp in the WT BHV-1 genome to 2,995 bp in the BHV1-UL47R genomic DNA. As predicted, fragments of approximately 3,737 bp and 2,995 bp were detected by the UL47-specific probe in the HindIII restriction profiles of the WT BHV-1 DNA and BHV1-UL47R DNA, respectively (Fig. 1B). This probe did not show any hybridization with the BHV1-ΔUL47 DNA, confirming deletion of the UL47 gene (Fig. 1B). According to the predicted HindIII profile of the BHV1-ΔUL47 genome, the EYFP ORF is located within the 1,496-bp HindIII L fragment. As anticipated, the restriction pattern of the BHV1-ΔUL47 DNA demonstrated the presence of the 1,496-bp fragment revealed by the EYFP-specific probe, whereas this probe did not hybridize to the WT BHV-1 or BHV1-UL47R restriction fragments (Fig. 1B). Correct recombination was further confirmed by PCR amplification and sequencing of the genetically manipulated regions of the BHV1-ΔUL47 and BHV1-UL47R genomes.
Effect of the UL47 deletion on growth properties of BHV-1.
To further verify the deletion and repair of the UL47 gene and compare the expression kinetics of different viral proteins, a time course experiment of BHV-1 infection was carried out for WT BHV-1, BHV1-ΔUL47, and BHV1-UL47R. MDBK cells were infected at a MOI of 2 and harvested every 5 h up to 25 h postinfection. Equal amounts of whole-cell extracts were subjected to SDS-PAGE and immunoblotting with antibodies specific for VP8, GFP, VP16, bICP4, gD, and β-actin (loading control) (Fig. 2A). VP8 displayed similar expression kinetics for WT BHV-1 and BHV1-UL47R but was absent from lysates of BHV1-ΔUL47-infected cells. EYFP was expressed in BHV1-ΔUL47-infected cells in place of VP8. Importantly, steady-state levels of VP16 were similar in cells infected with WT BHV-1, BHV1-ΔUL47, or BHV1-UL47R, confirming that expression of this protein was not affected by the UL47 deletion. The same applied to an immediate-early gene product, bICP4. In contrast, the expression of gD was significantly reduced in BHV1-ΔUL47-infected cells, while it was expressed with earlier kinetics compared to cells infected with the WT or revertant viruses. To analyze additional viral proteins, infection of cells and preparation of total cell extracts were repeated according to the same protocol, and these samples were subjected to Western blotting. Immunoblotting with gD-specific antibody confirmed the previous results, demonstrating decreased levels of this protein in the BHV1-ΔUL47-infected cells (Fig. 2B). Densitometry data demonstrated that at late stages of infection (20 to 25 h postinfection), the levels of gD protein in the BHV1-ΔUL47-infected cells were about 40% of those detected in the WT and revertant virus-infected cells. Analysis of these samples with gC- and gB-specific antibodies showed that, like gD, gC and gB were expressed with earlier kinetics in the UL47-null virus-infected cells. All glycoproteins tested were readily detectable in the 5-h BHV1-ΔUL47-infected cell extract, whereas in WT BHV-1 and BHV1-UL47R-infected cells, they were not detected until 10 h postinfection. While gC was expressed to WT virus levels by BHV1-ΔUL47 throughout the course of infection, the amount of gB expressed by this virus was slightly reduced at 25 h postinfection. Another protein that showed significantly decreased expression as a result of the UL47 deletion was VP22. Densitometry showed that the VP22 protein content was reduced by ∼54%. Like the three glycoproteins, VP22 was expressed with earlier kinetics. In contrast, the kinetics of expression of the major capsid protein, VP5, were similar for these three viruses. No reactivity with any of the antibodies against viral proteins was detected in mock-infected cell extracts (Fig. 2A and B).
FIG. 2.

Influence of the UL47 gene deletion on viral protein expression. (A) MDBK cells were infected with WT BHV-1, BHV1-ΔUL47, or BHV1-UL47R at a MOI of 2 or mock infected. The cells were harvested at 5-h intervals from 5 h to 25 h postinfection, and equal amounts of total cell lysates were analyzed by SDS-PAGE and immunoblotting with antibodies specific for VP8, GFP, VP16, bICP4, gD, and β-actin. (B) Infections were repeated, and new total cell lysates were immunoblotted with antibodies against gD, gC, gB, VP22, VP5, and β-actin.
During plaque purification, BHV1-ΔUL47 demonstrated a reduced plaque size. To study this phenomenon in more detail, MDBK cells were infected with WT BHV-1, BHV1-ΔUL47, or BHV1-UL47R at a low MOI, and viral plaques were analyzed 48 h postinfection. Relative quantitative analysis of viral plaque diameters showed that compared to that of WT BHV-1, the average diameter of BHV1-ΔUL47-induced plaques was reduced by 43% (P < 0.0001), and the defect caused by the UL47 ORF deletion was corrected in cells infected with the revertant virus (Fig. 3A). Since the titers of BHV1-ΔUL47 stocks were more than 100-fold lower than those of the WT or revertant viruses, the reduced plaque size observed with the UL47 deletion mutant could be primarily due to a defect in virus assembly and egress, rather than impaired cell-to-cell spread. To test the influence of the UL47 deletion on viral assembly, we carried out viral growth curves for WT BHV-1, BHV1-ΔUL47, and BHV1-UL47R. Duplicate monolayers of MDBK cells were infected at a MOI of 1, and cell culture medium and cells were harvested separately at the indicated times postinfection. From 10 h postinfection, the intracellular BHV1-ΔUL47 titers were constantly more than 100-fold lower than the WT BHV-1 titers (Fig. 3B). The difference was even more pronounced when the extracellular virus titers were analyzed. At 15 h and 20 h postinfection, the UL47-null virus demonstrated more than 1,000-fold reduction in the amount of infectious viral particles released into the cell culture medium compared to the WT virus (Fig. 3C). The growth defect exhibited by the UL47 deletion mutant was rescued in cells infected with the revertant virus (Fig. 3B and C), indicating that this defect is specific to the deletion of the UL47 gene.
FIG. 3.

Growth characteristics of WT BHV-1, BHV1-ΔUL47, and BHV1-UL47R in MDBK cells. (A) After 48 h of incubation under a semisolid medium, MDBK cells infected with WT BHV-1, BHV1-ΔUL47, or BHV1-UL47R were fixed with ethanol-acetic acid, and viral plaques were detected using a gB-specific MAb as described in Materials and Methods. The mean diameters of 50 discrete plaques per virus were determined. The data are represented as percentages of the wild-type BHV-1 values. Error bars represent the standard deviations. (B and C) Growth curves of the intracellular virus (B) and extracellular virus (C). Duplicate cultures of MDBK cells were infected with WT BHV-1, BHV1-ΔUL47, or BHV1-UL47R at a MOI of 1, and cell culture medium and cells were harvested separately at the indicated time points. The titer of infectious virus progeny in each sample was determined by plaque assay on MDBK cells.
Taken together, these data demonstrate the following. (i) BHV-1 replication is significantly impaired in the absence of VP8. (ii) Deletion of the UL47 ORF leads to earlier expression of some viral proteins (gD, gC, gB, and VP22), suggesting that VP8 may influence temporal regulation or the level of expression of the corresponding genes. (iii) The gD and VP22 protein levels are reduced at late stages of BHV1-ΔUL47 infection.
The stability of gD is affected by the absence of VP8 in BHV-1-infected cells.
Unlike VP22, which is not required for BHV-1 replication in cell culture (28, 42), the US6 gene product, gD, was shown to be essential (42). Hence, the altered expression of gD could account to some extent for the UL47-null virus growth defect. To test whether the decrease in gD protein levels in the UL47-null virus-infected cells was related to changes at the transcriptional level, we carried out qPCR, comparing the gD mRNA transcript levels over time in MDBK cells infected with 5 PFU/cell of WT BHV-1, BHV1-ΔUL47, or BHV1-UL47R. Since the protein levels of bICP4 were not altered by the UL47 deletion (Fig. 2A), bICP4 mRNA transcripts were included in the analysis for comparison. Furthermore, the UL46 mRNA levels were compared, as this gene is located downstream of the introduced deletion. As shown in Fig. 4A, in line with the earlier kinetics of gD protein expression in infected cells in the absence of VP8, gD mRNA levels were increased 2.6-fold at 3 h postinfection, 6.7-fold at 4 h postinfection, and 6.2-fold at 5 h postinfection in BHV1-ΔUL47-infected cells compared to the WT virus-infected cells. Although the gD mRNA levels appeared to be moderately increased in the revertant virus-infected cells at early stages of infection, these differences were not statistically significant. At 10 h, 15 h, and 20 h postinfection, the gD mRNA levels were very similar among these three viruses. In agreement with the immunoblotting data, the bICP4 mRNA transcript levels in the UL47-null virus-infected cells were comparable to those in the WT and revertant virus-infected cells throughout the course of infection (Fig. 4A). Importantly, the UL46 mRNA levels were also similar for the three viruses (Fig. 4A), indicating that the introduced deletion did not alter the level of UL46 gene transcription. From these results, we draw the following conclusions. (i) The increased gD protein levels detected in the UL47-null virus-infected cells at 5 h postinfection correlate with elevated gD mRNA levels at early times in infection. (ii) Deletion of the UL47 ORF does not affect the expression of gD at the transcriptional level at late stages of infection.
FIG. 4.

Influence of UL47 gene deletion on gD, bICP4, and UL46 mRNA levels and gD and gC protein stability. (A) Analysis of gD, bICP4, and UL46 mRNA levels during the course of infection with WT BHV-1, BHV1-ΔUL47, or BHV1-UL47R. Duplicate monolayers of MDBK cells were infected with the viruses at a MOI of 5 or mock infected, and total RNA was extracted from the cells collected at different time points after infection (from 3 h to 20 h postinfection [hpi]). After genomic DNA removal and reverse transcription, relative gD, bICP4, and UL46 mRNA levels were quantified by qPCR using 18S rRNA internal controls to normalize template input. The statistical significance of the different values is shown as follows: *, P < 0.05; **, P < 0.01; ***, P < 0.001. (B and C) Evaluation of gD and gC stability, respectively, in pulse-chase experiments. MDBK cells were infected for 8 h 30 min with 5 PFU/cell of WT BHV-1, BHV1-ΔUL47, or BHV1-UL47R or mock infected, pulse-labeled for 40 min with [35S]methionine/cysteine, and chased for 0, 30, 60, 120 and 180 min (180′) with medium containing nonradioactive methionine and cysteine. Each total cell lysate was divided into two equal aliquots, and paired aliquots were subjected to immunoprecipitation with either gD-specific or gC-specific MAb. The protein-antibody complexes were analyzed by SDS-PAGE and autoradiography. The values on the right side of the panels are the molecular masses of precursor and mature forms of the proteins. The amount of mature 65-kDa gD and mature 86-kDa gC at each time point was measured by densitometry. Since precursor polypeptides were completely processed into mature forms after 60 min of chase, the obtained values were compared to the corresponding values for 60 min of chase that were set at 100% (bottom of the panels). The values on the left side of the panels are molecular masses in kilodaltons.
To determine whether VP8 expression is required to maintain the stability of gD during infection, MDBK cells were infected for 8 h 30 min with WT BHV-1, BHV1-ΔUL47, or BHV1-UL47R at a MOI of 5 and pulse-labeled for 40 min with [35S]methionine/cysteine. Total cell lysates were prepared from cells, which were either harvested immediately (0 min) or chased for 30, 60, 120, or 180 min. Each lysate was split into two equal aliquots, and the paired aliquots were used to recover gD or gC by immunoprecipitation. Glycoprotein D (Fig. 4B) and gC (Fig. 4C) were expressed as 58-kDa and 65-kDa precursors, respectively. These precursor polypeptides were completely processed into mature forms after 60 min of chase. Therefore, the amount of mature 65-kDa gD and mature 86-kDa gC at each time point was measured by densitometry, and the values obtained were compared to the corresponding values for 60 min of chase, which were set at 100% (baseline). Two hours after the established set point, the amount of gD in WT BHV-1- and BHV1-UL47R-infected cells was close to the baseline level, whereas the UL47 deletion mutant showed a reduction of gD protein level by 28.9% in the same time interval (Fig. 4B). In contrast, the gC protein levels remained stable from 60 min to 180 min of chase in cells infected with any of these three viruses (Fig. 4C). These data indicate that the stability of gD, but not gC, is affected in the absence of VP8.
Characterization of BHV1-ΔUL47 virions.
To ascertain whether the loss of VP8 affected the packaging of other viral proteins into the virion, we purified extracellular viral particles from MDBK cells infected with WT BHV-1, BHV1-ΔUL47, or BHV1-UL47R and analyzed solubilized virion proteins by SDS-PAGE. The Coomassie blue staining results showed that the overall profile of BHV1-ΔUL47 virus particles was similar to those of the WT and revertant viruses, with the exception of the absence of VP8 and increased amounts of proteins with molecular masses of about 44 kDa and 34 kDa (Fig. 5A). To perform relative quantitative analysis, the proteins of purified virions resolved by SDS-PAGE were transferred to nitrocellulose membranes, and each membrane was used to detect a protein of interest and VP5 to normalize the amount of input virus. After densitometry, the normalized values for BHV1-ΔUL47 and BHV1-UL47R virion proteins were compared to the corresponding values for the WT virus, which were set at 100%. The representative images of the blots are illustrated in Fig. 5B. The lower steady-state protein levels of gD in the UL47-null virus-infected cells correlated with a reduction in incorporation of this protein into virions. Specifically, the packaging of gD into BHV1-ΔUL47 virions was reduced by 36%. The gB content in BHV1-ΔUL47 virions was reduced by 24% compared to that in the WT virions. Remarkably, the amount of gC incorporation into the UL47 deletion mutant virions was reduced by 68%, despite the fact that gC was expressed at WT levels in BHV1-ΔUL47-infected cells. The VP22 packaging into the UL47 deletion mutant virions was reduced by 62.5% (Fig. 5B). This agrees with the lower overall expression of this protein in BHV1-ΔUL47-infected cells detected earlier by immunoblotting and the evidence suggesting that the levels of VP22 incorporation into the tegument are primarily regulated by the abundance of the protein in the infected cell (25). Among the proteins analyzed by this method, only VP16 was incorporated into the UL47 deletion mutant virions to WT levels. The recruitment efficiency of the proteins examined into the revertant virus particles was very similar to that in the WT virus, indicating that the packaging defects observed with the UL47-null virus were due to the loss of VP8.
FIG. 5.

Analysis of the influence of the UL47 gene deletion on virion composition and infectivity. (A) Solubilized proteins of purified extracellular virions of WT BHV-1, BHV1-ΔUL47, or BHV1-UL47R were analyzed by SDS-PAGE and Coomassie blue staining. The positions of VP5 and VP8 proteins are indicated to the right of the gel. The bands that are present at larger amounts in the BHV1-ΔUL47 protein profile are indicated by asterisks. The values on the left side of the panel are molecular masses in kilodaltons. (B) The same samples shown in panel A were analyzed by immunoblotting with antibodies against gD, gB, gC, VP16, and VP22. The major nucleocapsid protein, VP5, was simultaneously detected on each membrane to normalize the protein input. Blots were analyzed by densitometry, and average normalized values of two independent experiments are shown as percentages of the corresponding values for the WT virus (below the blots). (C) Relative particle-to-PFU ratios. The volumes of the purified virion preparations were adjusted to normalize the VP5 protein content according to the densitometry results (top panel). The densitometry values expressed as a percentage of WT virus are averages from samples run in duplicate, whereas only one set of bands is shown. The concentration of infectious virus in the obtained preparations was determined by plaque assay (bottom left panel) and compared to the relative number of viral genome copies measured by qPCR (bottom right panel).
The infectivity of BHV1-ΔUL47 virions was analyzed by comparison of the particle-to-PFU ratio. The VP5 protein content in preparations of gradient-purified virions of WT BHV-1, BHV1-ΔUL47, and BHV1-UL47R was determined by Western blotting and densitometry (Fig. 5C, top panel). The volumes of the purified viral stocks were adjusted to the same concentration of VP5, and equal aliquots were withdrawn for titration by plaque assay and quantitation of viral genomes by qPCR. Although there were very similar numbers of viral genome copies in the virus dilutions normalized to VP5 (Fig. 5C, right bottom panel), the BHV1-ΔUL47 titers were approximately 10-fold lower than those of the WT and revertant viruses (Fig. 5C, left bottom panel). Accordingly, there was a ∼13-fold difference in the particle-to-PFU ratios between BHV1-ΔUL47 and the WT and revertant viruses. These data suggest that the infectivity of the BHV1-ΔUL47 virions is slightly lower than that of the WT or BHV1-UL47R virus particles.
Thus, the above data indicate that during BHV-1 infection, the packaging of gD, gB, and particularly gC and VP22, into virions is reduced in the absence of VP8, whereas the virion incorporation of VP16 is not affected. Furthermore, the reduced incorporation of glycoproteins correlated with slightly lower infectivity of BHV1-ΔUL47 virions detected by particle-to-PFU ratio analysis.
Ultrastructural characterization of BHV1-ΔUL47 virion morphogenesis.
The influence of the UL47 ORF deletion on virion morphogenesis was analyzed by electron microscopy of MDBK cells infected for 14 h with 5 PFU/cell of WT BHV-1, BHV1-ΔUL47, or BHV1-UL47R. Analysis of multiple infected cells showed that all major steps of herpesvirus virion maturation were readily detectable in cells infected with any of these three viruses. Although at apparently smaller amounts than in the WT or revertant virus-infected cells, viral capsids were present in the nuclei (Fig. 6A) and in the cytoplasm (Fig. 6B) of UL47-null virus-infected cells, indicating that VP8 is not required for the process of nucleocapsids traversing the nuclear envelope during nuclear egress. The presence of enveloped virions in the cytoplasm of BHV1-ΔUL47-infected cells (Fig. 6C) implied that VP8 is also not critical for the secondary cytoplasmic envelopment step. In agreement with the viral growth characteristics, the UL47 deletion mutant virus-infected cells were surrounded by very few extracellular virions (Fig. 6D) compared to the WT (Fig. 6E) or revertant (Fig. 6F) virus-infected cells where multiple groups of virions were detected across the cell periphery.
FIG. 6.

Transmission electron microscopy of cells infected with BHV1-ΔUL47, WT BHV-1, or BHV1-UL47R. MDBK cells were infected for 14 h with 5 PFU/cell of BHV1-ΔUL47 (A to D), WT BHV-1 (E), or BHV1-UL47R (F). (A and B) Nucleocapsids in the nucleus (N) and the cytoplasm (arrows), respectively, of the BHV1-ΔUL47-infected cells as evidence of unimpaired nuclear egress. (C) Apparently normal secondary envelopment of BHV1-ΔUL47 virions in the cytoplasm. (D to F) Very few extracellular viral particles were detected around the UL47-null virus-infected cells compared to the WT (E) or revertant (F) virus-infected cells, which were surrounded by numerous groups of extracellular virions. Bars, 200 nm.
A comparative analysis of numerous viral particles showed that virions of the WT and revertant viruses had a more pronounced spherical shape, whereas contours of the UL47-null virus particles paralleled the icosahedral capsid shape (Fig. 7). Furthermore, while in the tegument of the WT and revertant virus particles, a more electron-dense layer underlying the viral envelope was distinguished, such layer was not readily seen in the BHV1-ΔUL47 virion tegument. These observations indicate that the loss of VP8, which is the most abundant protein of the BHV-1 tegument, alters the virion morphology to a level detectable by electron microscopy.
FIG. 7.

Representative images of the extracellular WT BHV1, BHV1-ΔUL47, and BHV1-UL47R virions. Compared to the UL47-null virus particles, virions of the WT and revertant viruses were characterized by a more pronounced spherical shape and the presence of a more electron-dense layer of the tegument underlying the viral envelope. Bars, 50 nm.
The UL47-null virus is avirulent in cattle.
To examine the BHV1-ΔUL47 phenotype in vivo, BHV-1-seronegative calves were challenged intranasally with either BHV1-ΔUL47 or WT BHV-1 or left untreated. Calves infected with WT BHV-1, but not the BHV1-ΔUL47-infected or control animals, developed elevated body temperatures, weight loss, and other clinical symptoms of BHV-1 infection, including mild nasal lesions (not shown). Furthermore, WT BHV-1-infected calves began shedding virus on day 2 after challenge and continued to shed for 10 subsequent days, whereas no virus could be detected in the nasal secretions of the animals infected with the UL47-null virus or control animals (Fig. 8A). Similarly, following reactivation from latency, only the WT virus-infected calves shed virus (Fig. 8A). These data showed that the UL47-null virus is avirulent in cattle.
FIG. 8.

Analysis of the responses of calves to challenge with WT BHV-1 or BHV1-ΔUL47. Two groups of five BHV-1-seronegative calves were challenged intranasally with 4 × 106 PFU of either BHV1-ΔUL47 or WT BHV-1. The third group (n = 5) was left uninfected as a negative control. On day 57 after challenge (latency), dexamethasone treatment was initiated to reactivate the latent virus (black arrow pointing down). (A) Nasal virus shedding after the challenge. The swabs were collected at the indicated intervals, and virus titration was performed by plaque assay on MDBK cells. The data are presented as means ± standard errors (error bars). (B) Serum BHV-1 neutralization titers in calves after challenge. Virus-neutralizing titers are expressed as the reciprocal of the highest dilution that caused a 50% reduction in the number of plaques relative to the control. Each data point represents an individual animal, and the line is the median. (C) gD-specific antibody titers in bovine sera collected at indicated time points after challenge. Results are the reciprocal of the highest dilution resulting in a reading of 2 standard deviations above the value of the negative-control serum (pooled serum samples collected before challenge). Data are represented as median with ranges (error bars). (D to F) Numbers of BHV-1-specific IFN-γ-secreting PBMCs in response to in vitro restimulation with tgD, gC, or gB, respectively. Results are expressed as the difference between the number of IFN-γ-secreting cells per 106 cells in the glycoprotein-stimulated wells and the number of IFN-γ-secreting cells per 106 cells in wells with medium. Each data point represents an individual animal, and the median values are indicated by horizontal bars.
From day 12 after challenge and up to day 64, the WT virus-infected calves maintained a mean serum neutralizing antibody titer of 1:54, and as expected, the titer significantly increased upon dexamethasone treatment (P < 0.01) (Fig. 8B). In contrast, the UL47-null virus-infected animals did not develop virus neutralizing antibodies. Similarly, analysis of gD-specific serum antibody titers demonstrated that WT virus-infected animals became seropositive on day 12 after challenge, and a minor reduction of the titers during latency was followed by a significant increase 2 weeks after the first dexamethasone injection (P < 0.01). In contrast, the UL47-null virus did not induce detectable gD-specific antibodies (Fig. 8C). Although there were some differences in the frequency of antigen-induced IFN-γ-secreting cells, the trends were similar for gD, gC, and gB (Fig. 8D, E, and F). In the WT virus-infected group, the frequency of IFN-γ-secreting cells was constantly significantly higher than that in the control group, and the number of IFN-γ-secreting cells increased after reactivation of the latent infection (P < 0.01). In contrast, in the BHV1-ΔUL47-infected group, a significant increase in the number of antigen-induced IFN-γ-secreting cells was registered only on day 14 after challenge (P < 0.05), whereas on other days the numbers of IFN-γ-secreting cells were close to control levels, and the increase after dexamethasone treatment was not statistically significant. Thus, the UL47-null BHV-1 failed to induce significant humoral or lasting cellular immunity in calves.
DISCUSSION
In this study, we constructed a BHV-1 mutant defective in expression of VP8 (BHV1-ΔUL47) to perform a preliminary characterization of the role of this abundant tegument protein in virus infection. Our results have shown that BHV1-ΔUL47 exhibited a severe growth defect in MDBK cells characterized by reduced plaque size and more than 100-fold decrease in extracellular and intracellular viral titers compared to the titer of the WT or revertant viruses. This growth defect was not specific for MDBK cells, as a similar reduction in titers was observed in FBT cells (not shown). Furthermore, the in vivo data presented here demonstrate that a highly virulent BHV-1 challenge strain, strain 108 (27), failed to establish a productive infection in cattle as a result of the UL47 gene deletion. We reported previously that another tegument protein of BHV-1, VP22 encoded by the UL49 gene, is also an important virulence factor, as a corresponding deletion mutant was attenuated in cattle (27). However, infection with this virus elicited low serum neutralizing antibody titers, which were partially protective against WT BHV-1 challenge. In contrast, BHV1-ΔUL47 was unable to induce neutralizing antibody or cell-mediated responses in experimentally infected calves, suggesting that VP8 is indispensable for BHV-1 replication in its natural host.
Intracytoplasmic aggregates of partially tegumented capsids were reported to be present in cells infected with a PrV defective in expression of the UL47 gene product (20). This suggests that the UL47 gene product of PrV plays an essential role in final cytoplasmic envelopment. Our ultrastructural study of BHV1-ΔUL47-infected MDBK cells revealed small amounts of intracellular and extracellular virions, but no apparent defect in the secondary cytoplasmic envelopment. Similar to our data, a UL47 gene-deleted ILTV displayed significantly reduced amounts of intracellular and extracellular virions in the absence of detectable inhibition of any particular virion maturation step (13). These data suggest that the replication deficiency of the UL47 gene-deleted BHV-1 and ILTV is primarily associated with the lack of some unknown nonstructural function(s) of these homologues.
After virus entry, the majority of the tegument proteins are released into the cytoplasm of infected cells, which allows them to play a key role in establishing conditions for efficient viral replication. Essential early functions were assigned to the UL41-encoded virion host shutoff protein (21, 22, 45), and the UL48 gene product, VP16, which initiates viral replication through the recruitment of host factors to the promoters of viral immediate-early genes (4, 34, 36). BHV-1 VP8 is the most abundant component of mature viral particles (5). The large amount of this protein in the tegument suggests an important early function in addition to its structural role. In agreement with this assumption, we and others have shown that VP8, which is a true late protein, accumulates in the nucleus immediately after viral entry (49, 52). The same phenomenon has been demonstrated for the HSV-1 UL47 counterpart (35). Early studies suggested a modulatory role of the HSV-1 UL47 gene product toward the VP16 transcription activator function, as HSV-1 lacking the UL47 gene was dramatically impaired in its ability to induce immediate-early promoter-regulated expression of a reporter gene (54, 55). The results presented here did not confirm such a modulatory role of BHV-1 VP8, as similar levels of immediate-early bICP4 mRNA transcripts were detected in cells infected with BHV1-ΔUL47, WT, or revertant viruses throughout the course of infection, and accordingly, bICP4 protein was expressed with WT virus kinetics in BHV1-ΔUL47-infected cells. Furthermore, equal amounts of VP16 were packaged into extracellular virions of these three viruses and, hence, were available for its early function. These data suggest that the VP16 activator function was not affected by the absence of VP8.
The BHV1-ΔUL47 phenotype in infected cells was also characterized by earlier expression of gD, gC, gB, and VP22 and reduced intracellular protein levels of gD and VP22 at later stages of infection compared to the WT or revertant virus-infected cells. For gD, we have shown that the earlier expression of the protein in BHV1-ΔUL47-infected cells correlated with significantly elevated gD mRNA levels at very early stages of infection. Unlike VP22, gD is essential for the viability of BHV-1 in cell culture (42), suggesting that the low gD protein levels could account at least in part for the UL47 deletion mutant growth defect. We have shown that whereas gD mRNA synthesis and stability in BHV1-ΔUL47-infected cells remained normal, the stability of the protein was reduced. The reason for the increased turnover of gD in the absence of VP8 is not known. However, our preliminary studies have shown that gD is dispersed in the cytoplasm in a speckle-like pattern in UL47-null virus-infected cells at early stages of infection, whereas in the WT or revertant virus-infected cells, the localization of gD in a cytoplasmic area corresponding to the Golgi complex location is more pronounced (not shown). These data raise the possibility that VP8 may alter the localization of gD or the sites of gD mRNA translation in infected cells or protect gD from degradation by some other means.
Although we did not reveal any apparent defect of BHV1-ΔUL47 virion maturation by electron microscopy, comparative biochemical analysis of the mutant, WT, and revertant virus particles showed that the UL47 gene deletion altered the protein composition of the BHV-1 virion. Thus, gD and gB protein levels were moderately decreased in gradient-purified BHV1-ΔUL47 virions compared to the levels in the WT or revertant virus particles, and gC and VP22 virion packaging was significantly reduced in the absence of VP8 (Fig. 5B). While the reduced virion incorporation of gD and VP22 may be primarily associated with the lower steady-state levels of these proteins in the UL47-null virus-infected cells, the defect in gC packaging suggests that the expression of VP8 is important for maintaining the WT levels of gC incorporation into the virion, as gC was expressed normally in BHV1-ΔUL47-infected cells. Given the evidence that gB, gC, and gD mediate virus attachment and penetration into the cell (26), the decrease in packaging of these glycoproteins into BHV1-ΔUL47 virions is consistent with a slightly lower infectivity of BHV1-ΔUL47 viral particles detected by comparative particle-to-PFU ratio analysis. The lower rate of gB and gC packaging into BHV1-ΔUL47 virions is in contrast to ∼50% increase in gB and gC levels detected previously in gradient-purified virions of a HSV-1 UL47 deletion mutant (54). Interestingly, deletion of the UL49 gene from the HSV-1 genome led to a reduction of gD and gE virion packaging (10), whereas our experiments with a UL49 gene-deleted BHV-1 have shown that the lack of the UL49 gene product does not alter the gD virion content (unpublished data). These observations indicate that while deletion of the UL49 gene has a more detrimental effect on HSV-1 virion composition than deletion of the UL47 gene does, the opposite situation exists during BHV-1 infection. Since it is believed that an intricate network of protein-protein interactions with significant redundancy drives tegumentation and secondary envelopment in herpesviruses (reviewed in reference 33), the above difference may be explained in part by the different relative abundances of these proteins in the tegument, as the UL49 gene product is the most abundant protein in the HSV-1 tegument (11). The composition of BHV1-ΔUL47 virions and potential physical interactions of BHV-1 VP8 with viral glycoproteins will be further addressed in future studies.
In this study, we demonstrated that UL47 gene-deleted BHV-1 exhibited a severe growth defect in permissive cells and was avirulent in cattle where it failed to induce clinical disease or significant levels of humoral or cellular immunity. Taken together, our results suggest that the growth defect is predominantly associated with some still unknown nonstructural function of BHV-1 VP8. This function did not appear to be associated with an effect of VP8 on VP16, as our study did not reveal any changes in the VP16 transactivator function in UL47-null BHV-1-infected cells.
Acknowledgments
We are grateful to V. Misra for kindly providing BHV-1 VP16-specific antibody, S. Caldwell for technical assistance with electron microscopy, I. Shirley and J. Cowell for their help in electron microscopy image processing and analysis (the Western College of Veterinary Medicine at the University of Saskatchewan), and the animal support staff at the Vaccine and Infectious Disease Organization for care and handling of the animals.
This work was funded by the Natural Science and Engineering Research Council, and Canadian Institutes of Health Research. L. A. Babiuk is a recipient of a Canada research chair in Vaccinology.
Footnotes
Published ahead of print on 28 October 2009.
Vaccine and Infectious Disease Organization (VIDO) journal series number 546.
REFERENCES
- 1.Babiuk, L. A., R. C. Wardley, and B. T. Rouse. 1975. Defense mechanisms against bovine herpesvirus: relationship of virus-host cell events to susceptibility to antibody-complement cell lysis. Infect. Immun. 12:958-963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barker, D. E., and B. Roizman. 1990. Identification of three genes nonessential for growth in cell culture near the right terminus of the unique sequences of long component of herpes simplex virus 1. Virology 177:684-691. [DOI] [PubMed] [Google Scholar]
- 3.Braun, R., L. A. Babiuk, and H. van Drunen Littel-van den Hurk. 1998. Compatibility of plasmids expressing different antigens in a single DNA vaccine formulation. J. Gen. Virol. 79:2965-2970. [DOI] [PubMed] [Google Scholar]
- 4.Campbell, M. E., J. W. Palfreyman, and C. M. Preston. 1984. Identification of herpes simplex virus DNA sequences which encode a trans-acting polypeptide responsible for stimulation of immediate early transcription. J. Mol. Biol. 180:1-19. [DOI] [PubMed] [Google Scholar]
- 5.Carpenter, D. E., and V. Misra. 1991. The most abundant protein in bovine herpes 1 virions is a homologue of herpes simplex virus type 1 UL47. J. Gen. Virol. 72:3077-3084. [DOI] [PubMed] [Google Scholar]
- 6.Donnelly, M., and G. Elliott. 2001. Fluorescent tagging of herpes simplex virus tegument protein VP13/14 in virus infection. J. Virol. 75:2575-2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Donnelly, M., and G. Elliott. 2001. Nuclear localization and shuttling of herpes simplex virus tegument protein VP13/14. J. Virol. 75:2566-2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Donnelly, M., J. Verhagen, and G. Elliott. 2007. RNA binding by the herpes simplex virus type 1 nucleocytoplasmic shuttling protein UL47 is mediated by an N-terminal arginine-rich domain that also functions as its nuclear localization signal. J. Virol. 81:2283-2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dorange, F., B. K. Tischer, J. F. Vautherot, and N. Osterrieder. 2002. Characterization of Marek's disease virus serotype 1 (MDV-1) deletion mutants that lack UL46 to UL49 genes: MDV-1 UL49, encoding VP22, is indispensable for virus growth. J. Virol. 76:1959-1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duffy, C., J. H. Lavail, A. N. Tauscher, E. G. Wills, J. A. Blaho, and J. D. Baines. 2006. Characterization of a UL49-null mutant: VP22 of herpes simplex virus type 1 facilitates viral spread in cultured cells and the mouse cornea. J. Virol. 80:8664-8675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Elliott, G. D., and D. M. Meredith. 1992. The herpes simplex virus type 1 tegument protein VP22 is encoded by gene UL49. J. Gen. Virol. 73:723-726. [DOI] [PubMed] [Google Scholar]
- 12.Fischer, U., S. Meyer, M. Teufel, C. Heckel, R. Luhrmann, and G. Rautmann. 1994. Evidence that HIV-1 Rev directly promotes the nuclear export of unspliced RNA. EMBO J. 13:4105-4112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Helferich, D., J. Veits, J. P. Teifke, T. C. Mettenleiter, and W. Fuchs. 2007. The UL47 gene of avian infectious laryngotracheitis virus is not essential for in vitro replication but is relevant for virulence in chickens. J. Gen. Virol. 88:732-742. [DOI] [PubMed] [Google Scholar]
- 14.Inman, M., L. Lovato, A. Doster, and C. Jones. 2002. A mutation in the latency-related gene of bovine herpesvirus 1 disrupts the latency reactivation cycle in calves. J. Virol. 76:6771-6779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ioannou, X. P., P. Griebel, R. Hecker, L. A. Babiuk, and S. van Drunen Littel-van den Hurk. 2002. The immunogenicity and protective efficacy of bovine herpesvirus 1 glycoprotein D plus Emulsigen are increased by formulation with CpG oligodeoxynucleotides. J. Virol. 76:9002-9010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jericho, K. W., and C. Q. Darcel. 1978. Response of the respiratory tract of calves kept at controlled climatic conditions to bovine herpesvirus 1 in aerosol. Can. J. Comp. Med. 42:156-167. [PMC free article] [PubMed] [Google Scholar]
- 17.Jones, C., and S. Chowdhury. 2007. A review of the biology of bovine herpesvirus type 1 (BHV-1), its role as a cofactor in the bovine respiratory disease complex and development of improved vaccines. Anim. Health Res. Rev. 8:187-205. [DOI] [PubMed] [Google Scholar]
- 18.Khattar, S. K., S. van Drunen Littel-van den Harke, S. K. Attah-Poku, L. A. Babiuk, and S. K. Tikoo. 1996. Identification and characterization of a bovine herpesvirus-1 (BHV-1) glycoprotein gL which is required for proper antigenicity, processing, and transport of BHV-1 glycoprotein gH. Virology 219:66-76. [DOI] [PubMed] [Google Scholar]
- 19.Klopfleisch, R., J. P. Teifke, W. Fuchs, M. Kopp, B. G. Klupp, and T. C. Mettenleiter. 2004. Influence of tegument proteins of pseudorabies virus on neuroinvasion and transneuronal spread in the nervous system of adult mice after intranasal inoculation. J. Virol. 78:2956-2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kopp, M., B. G. Klupp, H. Granzow, W. Fuchs, and T. C. Mettenleiter. 2002. Identification and characterization of the pseudorabies virus tegument proteins UL46 and UL47: role for UL47 in virion morphogenesis in the cytoplasm. J. Virol. 76:8820-8833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koppers-Lalic, D., F. A. Rijsewijk, S. B. Verschuren, J. A. van Gaans-Van den Brink, A. Neisig, M. E. Ressing, J. Neefjes, and E. J. Wiertz. 2001. The UL41-encoded virion host shutoff (vhs) protein and vhs-independent mechanisms are responsible for down-regulation of MHC class I molecules by bovine herpesvirus 1. J. Gen. Virol. 82:2071-2081. [DOI] [PubMed] [Google Scholar]
- 22.Kwong, A. D., and N. Frenkel. 1989. The herpes simplex virus virion host shutoff function. J. Virol. 63:4834-4839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Labiuk, S. L., L. Babiuk, and S. van Drunen Littel-van den Hurk. 2009. Major tegument protein VP8 of bovine herpesvirus 1 is phosphorylated by viral US3 and cellular CK2 protein kinases. J. Gen. Virol. 90:2829-2839. [DOI] [PubMed] [Google Scholar]
- 24.Reference deleted.
- 25.Leslie, J., F. J. Rixon, and J. McLauchlan. 1996. Overexpression of the herpes simplex virus type 1 tegument protein VP22 increases its incorporation into virus particles. Virology 220:60-68. [DOI] [PubMed] [Google Scholar]
- 26.Li, Y., S. van Drunen Littel-van den Hurk, L. A. Babiuk, and X. Liang. 1995. Characterization of cell-binding properties of bovine herpesvirus 1 glycoproteins B, C, and D: identification of a dual cell-binding function of gB. J. Virol. 69:4758-4768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liang, X., B. Chow, and L. A. Babiuk. 1997. Study of immunogenicity and virulence of bovine herpesvirus 1 mutants deficient in the UL49 homolog, UL49.5 homolog and dUTPase genes in cattle. Vaccine 15:1057-1064. [DOI] [PubMed] [Google Scholar]
- 28.Liang, X., B. Chow, Y. Li, C. Raggo, D. Yoo, S. Attah-Poku, and L. A. Babiuk. 1995. Characterization of bovine herpesvirus 1 UL49 homolog gene and product: bovine herpesvirus 1 UL49 homolog is dispensable for virus growth. J. Virol. 69:3863-3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Manoj, S., P. J. Griebel, L. A. Babiuk, and S. van Drunen Littel-van den Hurk. 2003. Targeting with bovine CD154 enhances humoral immune responses induced by a DNA vaccine in sheep. J. Immunol. 170:989-996. [DOI] [PubMed] [Google Scholar]
- 30.Reference deleted.
- 31.Meredith, D. M., J. A. Lindsay, I. W. Halliburton, and G. R. Whittaker. 1991. Post-translational modification of the tegument proteins (VP13 and VP14) of herpes simplex virus type 1 by glycosylation and phosphorylation. J. Gen. Virol. 72:2771-2775. [DOI] [PubMed] [Google Scholar]
- 32.Mettenleiter, T. C. 2002. Herpesvirus assembly and egress. J. Virol. 76:1537-1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mettenleiter, T. C., B. G. Klupp, and H. Granzow. 2009. Herpesvirus assembly: an update. Virus Res. 143:222-234. [DOI] [PubMed] [Google Scholar]
- 34.Misra, V., S. Walker, S. Hayes, and P. O'Hare. 1995. The bovine herpesvirus alpha gene trans-inducing factor activates transcription by mechanisms different from those of its herpes simplex virus type 1 counterpart VP16. J. Virol. 69:5209-5216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morrison, E. E., A. J. Stevenson, Y. F. Wang, and D. M. Meredith. 1998. Differences in the intracellular localization and fate of herpes simplex virus tegument proteins early in the infection of Vero cells. J. Gen. Virol. 79:2517-2528. [DOI] [PubMed] [Google Scholar]
- 36.O'Hare, P., C. R. Goding, and A. Haigh. 1988. Direct combinatorial interaction between a herpes simplex virus regulatory protein and a cellular octamer-binding factor mediates specific induction of virus immediate-early gene expression. EMBO J. 7:4231-4238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pfaffl, M. W. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29:e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reference deleted.
- 39.Purves, F. C., and B. Roizman. 1992. The UL13 gene of herpes simplex virus 1 encodes the functions for posttranslational processing associated with phosphorylation of the regulatory protein alpha 22. Proc. Natl. Acad. Sci. U. S. A. 89:7310-7314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Raggo, C., M. Habermehl, L. A. Babiuk, and P. Griebel. 2000. The in vivo effects of recombinant bovine herpesvirus-1 expressing bovine interferon-gamma. J. Gen. Virol. 81:2665-2673. [DOI] [PubMed] [Google Scholar]
- 41.Read, G. S., B. M. Karr, and K. Knight. 1993. Isolation of a herpes simplex virus type 1 mutant with a deletion in the virion host shutoff gene and identification of multiple forms of the vhs (UL41) polypeptide. J. Virol. 67:7149-7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Robinson, K. E., J. Meers, J. L. Gravel, F. M. McCarthy, and T. J. Mahony. 2008. The essential and non-essential genes of bovine herpesvirus 1. J. Gen. Virol. 89:2851-2863. [DOI] [PubMed] [Google Scholar]
- 43.Salmon, B., C. Cunningham, A. J. Davison, W. J. Harris, and J. D. Baines. 1998. The herpes simplex virus type 1 U(L)17 gene encodes virion tegument proteins that are required for cleavage and packaging of viral DNA. J. Virol. 72:3779-3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sandri-Goldin, R. M. 1998. ICP27 mediates HSV RNA export by shuttling through a leucine-rich nuclear export signal and binding viral intronless RNAs through an RGG motif. Genes Dev. 12:868-879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Smibert, C. A., D. C. Johnson, and J. R. Smiley. 1992. Identification and characterization of the virion-induced host shutoff product of herpes simplex virus gene UL41. J. Gen. Virol. 73:467-470. [DOI] [PubMed] [Google Scholar]
- 46.Tikoo, S. K., M. Campos, and L. A. Babiuk. 1995. Bovine herpesvirus 1 (BHV-1): biology, pathogenesis, and control. Adv. Virus Res. 45:191-223. [DOI] [PubMed] [Google Scholar]
- 47.Triezenberg, S. J., R. C. Kingsbury, and S. L. McKnight. 1988. Functional dissection of VP16, the trans-activator of herpes simplex virus immediate early gene expression. Genes Dev. 2:718-729. [DOI] [PubMed] [Google Scholar]
- 48.van Drunen Littel-van den Hurk, S., and L. A. Babiuk. 1985. Antigenic and immunogenic characteristics of bovine herpesvirus type-1 glycoproteins GVP 3/9 and GVP 6/11a/16, purified by immunoadsorbent chromatography. Virology 144:204-215. [DOI] [PubMed] [Google Scholar]
- 49.van Drunen Littel-van den Hurk, S., S. Garzon, J. V. van den Hurk, L. A. Babiuk, and P. Tijssen. 1995. The role of the major tegument protein VP8 of bovine herpesvirus-1 in infection and immunity. Virology 206:413-425. [DOI] [PubMed] [Google Scholar]
- 50.van Drunen Littel-van den Hurk, S., M. D. Parker, B. Massie, J. V. van den Hurk, R. Harland, L. A. Babiuk, and T. J. Zamb. 1993. Protection of cattle from BHV-1 infection by immunization with recombinant glycoprotein gIV. Vaccine 11:25-35. [DOI] [PubMed] [Google Scholar]
- 51.Verhagen, J., M. Donnelly, and G. Elliott. 2006. Characterization of a novel transferable CRM-1-independent nuclear export signal in a herpesvirus tegument protein that shuttles between the nucleus and cytoplasm. J. Virol. 80:10021-10035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Verhagen, J., I. Hutchinson, and G. Elliott. 2006. Nucleocytoplasmic shuttling of bovine herpesvirus 1 UL47 protein in infected cells. J. Virol. 80:1059-1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Williams, P., J. Verhagen, and G. Elliott. 2008. Characterization of a CRM1-dependent nuclear export signal in the C terminus of herpes simplex virus type 1 tegument protein UL47. J. Virol. 82:10946-10952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang, Y., and J. L. McKnight. 1993. Herpes simplex virus type 1 UL46 and UL47 deletion mutants lack VP11 and VP12 or VP13 and VP14, respectively, and exhibit altered viral thymidine kinase expression. J. Virol. 67:1482-1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang, Y., D. A. Sirko, and J. L. McKnight. 1991. Role of herpes simplex virus type 1 UL46 and UL47 in alpha TIF-mediated transcriptional induction: characterization of three viral deletion mutants. J. Virol. 65:829-841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zheng, C., R. Brownlie, L. A. Babiuk, and S. van Drunen Littel-van den Hurk. 2004. Characterization of nuclear localization and export signals of the major tegument protein VP8 of bovine herpesvirus-1. Virology 324:327-339. [DOI] [PubMed] [Google Scholar]