

Abstract
Studies of circulating T (CD3+) lymphocytes have shown that on a population basis T-cell numbers remain stable for many years after HIV-1 infection (blind T-cell homeostasis), but decline rapidly beginning approximately 1.5–2.5 years before the onset of clinical AIDS. We derived a general method for defining the loss of homeostasis on the individual level and for determining the prevalence of homeostasis loss according to HIV status and the occurrence of AIDS in more than 5,000 men enrolled in the Multicenter AIDS Cohort Study. We used a segmented regression model for log10 CD3+ cell counts that included separate T-cell trajectories before and after a time (the T-cell inflection point) where the loss of T-cell homeostasis was most likely to have occurred. The average slope of CD3+ lymphocyte counts before the inflection point was close to zero for HIV− and HIV+ men, consistent with blind T-cell homeostasis. After the inflection point, the HIV+ individuals who developed AIDS generally showed a dramatic decline in CD3+ cell counts relative to HIV− men and HIV+ men not developing AIDS. A CD3+ cell decline of greater than 10 percent per year was present in 77% of HIV+ men developing AIDS but in only 23% of HIV+ men with no onset of AIDS. Our findings at the individual level support the blind T-cell homeostasis hypothesis and provide strong evidence that the loss of homeostasis is an important mechanism in the pathogenesis of the severe immunodeficiency that characterizes the late stages of HIV infection.
Cell-mediated immunity from circulating (CD3+) T lymphocytes is one of the primary defenses against viral infections. Two mechanisms of this antiviral immunity are the destruction of infected cells by CD8+ cytotoxic T lymphocytes and the immune-stimulating activity of CD4+ helper T cells. These latter cells are the primary target of HIV-1 infection, and the depletion of CD4+ cells has long been recognized as a characteristic feature of HIV-1 disease (1). The pattern of CD8+ cell counts is more complicated, with increases early in HIV-1 infection followed by a decline before the onset of AIDS (2). These increases in CD8+ cells occurring concomitantly with a decrease in CD4+ cells suggest that a compensatory mechanism for total T lymphocytes exists in the earlier stages of HIV infection. This compensatory mechanism cannot continue throughout the entire course of HIV infection, however, because people with AIDS have lower numbers of circulating lymphocytes than people with earlier stages of the infection (1).
Longitudinal analyses of T-cell counts from several cohort studies have shown that T-cell numbers in fact remain stable near pre-seroconversion levels without regard to phenotype (CD4+ or CD8+ expression) for many years after HIV-1 infection. Furthermore, this phenomenon, referred to as blind T-cell homeostasis (2, 3), appears to break down an average of 1.5–2.5 years before the onset of clinically defined AIDS. At this time, an inflection occurs in the trajectory of the number of circulating T cells, changing from little or no decline to an average decline of about 30% per year (2, 4). In populations of people with known times of HIV-1 seroconversion and AIDS onset, varying incubation periods of HIV-1 were largely, if not entirely, accounted for by variations in the time from seroconversion to the time of rapid T-cell loss (2). In other words, the interval from the time at which T-cell loss accelerates to the time of AIDS onset was relatively constant, suggesting that a failure of T-cell homeostasis initiated a common final pathway, resulting in opportunistic illnesses after an average of 1.5–2.5 years.
These previous studies of HIV-1 infection addressed the characteristics and failure of T-cell homeostasis at the population level with primary emphasis on the changes in groups of individuals developing AIDS. The present study was undertaken to extend these studies to the level of the individual and to develop a statistical method that could provide, for any longitudinal series of T-cell counts, the best estimate of the time point at which T-cell inflection occurred. We then developed criteria for determining the prevalence of the loss of T-cell homeostasis among well-characterized groups, based on the T-cell slopes before and after the putative inflection points (IPs). For these analyses, we used the extensive longitudinal data from the Multicenter AIDS Cohort Study (MACS) that has followed approximately 5,500 homosexual and bisexual men since 1984.
The utility of our methods for developing IPs on the individual level is to promote targeted retrospective laboratory studies that characterize the factors occurring around the time of T-cell inflection, as specimens can be classified into those before and after T-cell inflection. More generally, however, this paper describes an important integration of laboratory, statistical, and clinical sciences. The methods developed are appropriate for general problems of investigating changes in trajectories. The phenomenon under study, loss of T-cell homeostasis, is one that is naturally defined by the statistical model, and the immunologic changes defined by statistical parameters are related to important clinical outcomes.
METHODS
Study Participants.
The MACS was initiated in 1983 to study the natural history of HIV-1 infection among homosexual and bisexual men in the United States. The study design has been previously described (5), and only aspects pertinent to this analysis are presented here. In 1984–1985, 4,954 men were enrolled in Baltimore/Washington, Chicago, Los Angeles, and Pittsburgh. From 1987 to 1991, recruitment was reopened, and an additional 625 men, mostly nonwhite, were enrolled. Men with pre-existing clinical conditions diagnostic of AIDS and those who were younger than 18 years were excluded from enrollment. At semiannual visits, men returned to the clinics to provide specimens for laboratory analyses, undergo a physical examination, and complete self-administered data forms and an interviewer-administered questionnaire. HIV-1 seropositivity was determined by a positive ELISA confirmed by a Western blot with bands corresponding to at least two of the gag, pol, and env proteins of HIV-1.
T-cell subset levels at each visit were obtained from peripheral blood mononuclear cells stained with mAbs by a whole blood lysing method and analyzed by two-color flow cytometry (6, 7) and mAbs specific for CD3, CD4, and CD8. Absolute numbers of cells per ml of blood were calculated by using the complete blood count with automated 10,000 cell differential.
Methods for Determining Individual CD3+ Lymphocyte IPs.
To characterize whether individual CD3+ lymphocyte trajectories were indicative of homeostasis failure, we developed a method to determine the time at which a change in trajectory, denoted as the T-cell IP, was most likely to have occurred. The method was required to be applicable to any series of CD3+ cell counts, regardless of infection status or disease stage. A (possible) T-cell IP was defined as the midpoint in time between any two semiannual participant visits. At least three CD3+ cell counts were required before and after the midpoint to ensure that slopes around the IP were evaluated by using several observations. Thus, only individuals with at least six CD3+ cell counts were included in the present analysis. Additionally, the CD3+ lymphocyte count was required to decrease between the two visits immediately surrounding the midpoint, because CD3+ cell counts were hypothesized to decline after homeostasis failure. Because this algorithm assigned an IP only between decreasing CD3+ cell counts, there were individuals for whom an IP could not be estimated (e.g., those with monotonically increasing CD3+ cell counts).
Denote Yij as the log10 of the jth of ni CD3+ lymphocyte counts on the ith individual, collected at time tij. Let ti(k) be the time of the kth eligible time of inflection for the ith individual (i.e., ti(k) is a midpoint point between two consecutive tij where CD3+ cell counts decline and there are at least three measurements both before and after ti(k)). Using all available Yij (not just the six required measurements around ti(k)), a segmented regression model was fit with the form:
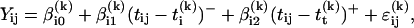 |
where (tij − ti(k))− equals tij − ti(k) if tij < ti(k) and equals zero if tij > ti(k), (tij − ti(k))+ equals tij − ti(k) if tij > ti(k) and equals zero if tij < ti(k), and ɛij(k) represents a normal (Gaussian) error term with mean 0 and SD σi(k). The parameter βi0 can be interpreted as the estimated log10 CD3+ cell count at IPi, and 10βi1 and 10βi2 represent the relative (proportional) rate of change in CD3+ cell counts before and after IPi, respectively.
We obtained one triplet of parameter estimates (β̂i0(k), β̂i1(k), β̂i2(k)) for each eligible ti(k) of the ith individual. The ti(k) that resulted in the smallest residual variability between the data and the fitted line (i.e., mink Σj=1ni [ɛ̂ij(k)]2) was chosen as the estimated IP (IP̂i) for the ith individual. Fig. 1 illustrates the selection method for a hypothetical set of serial CD3+ cell counts on an individual. The eligible times for the IP, denoted with an asterisk along the x-axis, do not include all midpoints because of the requirements of decreasing CD3+ cell counts and the occurrence of at least three CD3+ cell counts before and after the midpoint.
Figure 1.

Graphical illustration of IP model. Post-SC log10 CD3+ cell counts are represented by points (•), and eligible times for inflection are denoted by ∗. The IP for individual i (IPi) is the time for which the segmented regression model (solid lines) gives the smallest residual variability.
After applying the above methods, estimates of the IP (IP̂i) and each of the regression coefficients (β̂i0, β̂i1, β̂i2) were obtained for each individual. The CD3+ lymphocyte trajectories before (βi1) and after (βi2) the IP are key parameters for evaluating the methods relative to the T-cell homeostasis hypothesis. For individuals who are maintaining T-cell homeostasis, we would expect βi1 ≈ βi2 ≈ 0. For those who have lost T-cell homeostasis, we would expect βi1 ≈ 0 and βi1 ≫ βi2, so that either the difference (βi2 − βi1) or the post-IP slope (βi2) would be a measure discriminating those with homeostasis loss from those preserving homeostasis. Below, we describe the methods to address the important question of which of these variables provides better discrimination.
Application of the Methods and Evaluation of IPs.
We primarily were interested in comparing the regression parameters among two groups: (i) HIV+ men who had developed AIDS as of 6/97, and were expected to have lost T-cell homeostasis (referred to as the HIV+/AIDS group), and (ii) HIV+ men who were AIDS-free as of 6/97 and therefore were expected to have maintained T-cell homeostasis (referred to as the HIV+/AIDS-free group). T-cell counts from HIV− men, who obviously were expected to have maintained T-cell homeostasis, were used as controls in the analysis. AIDS surveillance and reporting are conducted continuously in the MACS. Our definition of AIDS included only clinical illnesses diagnostic of AIDS (8) that were confirmed by review of medical records. Individuals with CD4+ cell counts below 200 cells/mm3 without clinical disease were not classified as having AIDS. CD3+ lymphocyte counts from the baseline visit of the seroprevalent (SP) men and measurements before and within 1 year after seroconversion of the seroincident men were not used, to avoid changes occurring around seroconversion.
We restricted AIDS cases to those occurring before 10/94 to obtain an unbiased evaluation of the interval between the IP and AIDS. If AIDS cases after 10/94 had been included in the analysis, IPs would have been forced to occur before onset of AIDS because of insufficient follow-up after AIDS. To ensure that all incipient cases of AIDS were excluded, only CD3+ lymphocyte counts obtained up to 10/94 were included among those AIDS-free through 6/97. Men with AIDS before 10/94 were required to have six CD3+ cell counts before AIDS, although counts after AIDS were used in estimating the IP and trajectories.
Distributions of the three model parameters (CD3+ cell count at IPi, slope before IPi, and slope after IPi) were compared graphically. We also investigated whether larger post-IP declines (βi2) were associated with shorter times between IP and AIDS. The sensitivity and specificity for the development of AIDS was calculated by using varying levels of CD3+ lymphocyte slopes after the IP as cutoffs for identifying those with “true inflection,” with graphical representation facilitated through use of a receiver operating characteristic (ROC) curve (9). ROC curves plot sensitivity versus (1 − specificity) over all possible cutoffs and allow for comparison of classification properties independent of any particular cutoff. ROC curves defined by parameters that better distinguish those with or without the onset of AIDS (our gold standard for the sensitivity and specificity calculations) manifest a larger area under the curve.
The preceding classification scheme does not take into account the variability of an individual’s CD3+ lymphocyte data. Thus, although two individuals might show a decline of 10% per year, this decline may fall within the range of variability for one individual but not the other. As an alternative to classifying individuals based on the ROC curves, we also used a statistical criterion based on the statistical significance of F-statistics. For each individual, suppose
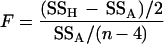 |
is computed for testing whether the segmented regression model is appropriate, where SSH represents the residual sums of squares under the linear regression model (βi1 = βi2), and SSA represents the residual sums of squares under the two-phase regression model. This statistic has an F(n−4)2 distribution when the time of inflection is fixed (or known independently of the data) (10). That is, if the time of inflection were known for each individual, we could compare the F-statistic to an F-distribution cutoff to determine whether the CD3+ cell trajectory after the IP was statistically different from the trajectory before the IP (10). However, our method estimates the IP from each individual’s T-cell data, and the standard F-distribution has been shown to be inappropriate for the segmented regression model when the time of inflection is not fixed but estimated from each individual’s data (11–13). To address this problem, simulation methods were used to approximate the required critical values for the F-statistic as described in the Appendix. Using these values for determining statistical significance, we classified individuals as “true inflectors” and investigated the sensitivity and specificity of true inflection for the onset of AIDS within 3 years.
RESULTS
Fig. 2 depicts the numbers of individuals included after we applied the different restriction criteria. Of the 5,579 men enrolled in the MACS, 2,195 were HIV SP, and 491 of 3,384 seronegative (SN) individuals were observed to seroconvert as of 6/97. The impact of requiring at least six CD3+ cell counts is shown in the second to last line. The final line displays the number of individuals for whom the method estimated an IP.
Figure 2.

MACS study population and final sample. Seroconversion and AIDS events are as reported by June 1997. ∗, 136 SP men and 53 SC men with AIDS after 10/94 were excluded in this analysis to allow for at least three follow-up visits after AIDS.
Fig. 3 displays the distribution of two different estimated parameters from the IP model for individuals in each of five groups resulting from subdividing the AIDS-free and AIDS groups by whether they were SP or seroconverter (SC). There were virtually no differences in these variables between the SP and SC men in either those developing or those not developing AIDS, suggesting that the duration of HIV infection did not affect our conclusions.
Figure 3.

Boxplots (light middle bar denotes median, ends of dark boxes denote 25th and 75th percentiles, brackets indicate 150% of IQR outside the quartiles, outliers are indicated with points) displaying the distribution of estimated parameters by group. (a) CD3+ slope (expressed as change in CD3+ count per year) before IP [(1 − 10β̂i1) × 100]. (b) CD3+ slope (change per year) after IP [(1 − 10β̂i2) × 100].
Fig. 3 a and b describes the slopes in CD3+ cell counts around the IP. Fig. 3a shows the slope of CD3+ cell counts before IP in terms of the percentage change in CD3+ cell counts per year [i.e., (1 − 10β̂i1) × 100]. Although variable, the distributions were essentially symmetric around 0.0 with median (inter-quartile range, IQR) annual change of 0.5% (−7.7% to 7.8%) for the groups developing AIDS, 2.0% (−5.2% to 9.7%) for the groups not developing AIDS, and 1.5% (−6.3% to 8.5%) for the SN group. This finding is consistent with the homeostasis hypothesis, which predicts a stability in CD3+ cell counts before the IP.
Fig. 3b displays the post-IP CD3+ lymphocyte slopes, also expressed in terms of the percentage change of CD3+ cell counts per year [i.e., (1 − 10β̂i2) × 100]. The distribution of this parameter was also very close to symmetric around 0.0 for the HIV− group (median −0.3%, IQR −5.0% to +5.0%), and the HIV+ group remaining AIDS-free (median change −1.9%, IQR −8.7% to +5.5%). However, the distribution for the groups developing AIDS was clearly distinct from these, with a median (IQR) annual decline of 32.0% (−50% to −11.8%). An examination of the exponentiated difference (ratio) of pre-IP and post-IP slopes (10(β̂i2−β̂i1) = 10β̂i2/10β̂i1) also shows similarity between the HIV− group and the HIV+ group without onset of AIDS and much larger declines in the group developing AIDS, although the distributions are not as distinct as in Fig. 3b. Both the discordance in the distributions of post-IP among those with onset of AIDS and the similarity of pre-IP and post-IP slopes for the HIV− and HIV+ men with no onset of AIDS strongly suggest that the methods are identifying a meaningful time point among those with onset of AIDS and a nonmeaningful time point among the other two groups.
The groups developing AIDS had slightly lower CD3+ lymphocyte levels just before the estimated IP (median 1,354, IQR 1,010 to 1,761) than the SP and SC groups not developing AIDS (median 1,572, IQR 1,232 to 1,952) and the SNs (median 1,601, IQR 1,275 to 1,970). The median CD4+ lymphocyte count for the groups developing AIDS (346) was much lower than the median for the groups remaining AIDS-free (619), which was lower than the median for the SNs (1,109). However, the groups developing AIDS had slightly higher CD8+ cell counts (median 1,054) relative to the groups remaining AIDS-free (median 1,015) and higher than the CD8+ cell count at IP for the SNs (median 653).
The distribution of time differences (in years) between the estimated IP and the diagnosis of AIDS were nearly identical for the SP and SC groups. The median (IQR) times from the estimated IPs to AIDS were 1.73 (0.68 to 2.69) and 1.71 (0.81 to 2.99) years for SC and SP groups, respectively. Overall, the vast majority (87%) of IPs were estimated to occur before the onset of AIDS, despite using CD3+ cell counts occurring before and after AIDS. There was a dose–response relationship between post-IP slope and time to AIDS, in which larger post-IP CD3+ lymphocyte declines were associated with shorter times between IP and an onset of AIDS (P < 0.0001, data not shown).
Fig. 4 displays longitudinal CD3+ counts and model estimates for three individuals who had representative slopes for the three subject groups: HIV− (Fig. 4a), HIV+ but without AIDS onset (Fig. 4b), and HIV+ developing AIDS (Fig. 4c). The individuals selected for this figure were among those with preinflection and postinflection slopes closest to the median preinflection and postinflection slopes for each of the three groups. Before the estimated IP, the slopes for all individuals were slightly positive (+1.6%, +2.8%, and +2.0% per year, respectively). After the IP, however, the HIV− and HIV+/AIDS-free individuals showed very modest declines (−0.5% and −2.9% per year, respectively) but the individual who developed AIDS showed a clear change in T-cell trajectory (to −31% per year) occurring 2.3 years before AIDS. Fig. 4 illustrates the need to distinguish true inflection from trajectory changes estimated in the presence of the natural variability of serial T-cell counts.
Figure 4.

Representative T-cell lymphocyte counts (solid lines and points) for three men who were: (a) HIV−, (b) HIV+ without AIDS onset, and (c) HIV+ with AIDS onset. The best-fit segmented regression model is denoted by the dashed line with the ∗ denoting the estimated IP. “A” indicates the date of onset of clinical AIDS. The fitted regression lines are curved because of the linear structure used to model the log10 transformed values, and we have exponentiated the values to plot the data and the regression estimates in the original scale (cells per mm3).
We evaluated the operating characteristics (sensitivity, specificity) that result from classifying individuals with and without onset of AIDS as “inflectors” or “noninflectors” by using only the magnitude of the parameter estimates β̂i1,β̂i2,β̂i−β̂i1 from the model. For example, by using a post-IP slope (β̂i2) cutoff of −20% per year to define inflection, each individual could be classified as an inflector or noninflector. When these data then were combined with our gold standard of disease defined by an AIDS diagnosis, we could compute the sensitivity and specificity associated with this classification rule. In the analysis, shown in Fig. 5, we did not include the 92 individuals with an estimated IP occurring after AIDS, although the analysis was nearly identical with them included. The slope before IP (β̂i1) was a poor parameter to distinguish those developing and not developing AIDS, because it fell very close to the diagonal line. In contrast, the slope after IP better distinguished groups. The change in slopes (β̂i2 − β̂i1) did not give as good characteristics as β̂i2 alone, as indicated by a smaller area under the curve, but performed much better than β̂i1.
Figure 5.

ROC curves (sensitivity vs. 1 − specificity) when distinguishing AIDS and AIDS-free men (SC and SP groups combined) using three different criteria: CD3+ slope before IP (β̂i1), CD3+ slope after IP (β̂i2), and the change in log10(CD3+) slopes from before IP to after IP (β̂i2 − β̂i1). The arrow indicates the point of maximal sensitivity and specificity. ∗ and + denote the characteristics when distinguishing individuals by using the statistical criteria (i.e., classification based on F-statistics) for all individuals and those with 15 or more visits, respectively.
The cutoff of β̂i2, which maximized both sensitivity and specificity (i.e., closest to the upper left corner in Fig. 5) was very close to log10(.90) (Fig. 5, arrow). Thus, by using the criteria that post-IP CD3+ cell counts, which fall by more than 10% per year represent true inflection, 77.5% of the men developing AIDS had true inflection as opposed to only 23.1% of the men remaining AIDS-free. These values were nearly identical when including the 92 men with IP after onset of AIDS (69, or 75%, of whom declined by more than 10% after IP). Among the SNs, 12% (264/2,152) showed a post-IP decline of this magnitude or more. Using the cutoff that maximized the sensitivity and specificity for the difference in slopes, there was slightly less discrimination: 71.8% of the men developing AIDS and 27.9% of the men remaining AIDS-free showed this pattern of decline.
The discrimination using the statistical criterion based on individual F-statistics provided a lower sensitivity (proportion of men developing AIDS who showed inflection) of 38.4% and a correspondingly higher specificity (proportion of men remaining AIDS-free classified without inflection) of 88.8%. These characteristics are plotted as an asterisk in Fig. 5, where we can see the criterion based on the ROC curves performing better in terms of the sensitivity and specificity. Furthermore, approximately 8% of the SNs showed significant CD3+ lymphocyte inflections, which was only slightly higher than the 5% expected as a consequence of how the empirical distribution was simulated to generate the 95th percentile cutoffs.
Because the statistical criterion depended on the number of CD3+ lymphocyte counts available, an immediate concern was the statistical power for detecting a decline in CD3+ cell counts when few data were available. In a separate set of simulations, we estimated the power to detect a 10% drop in CD3+ cell counts per year to be 30% when six semiannual CD3+ cell counts were available but greater than 99% when 15 or more semiannual visits were available. Examining only those individuals with 15 or more visits, we found an increase in the percentage of men developing AIDS with inflection by the statistical criterion (54%) and a slight decrease in the men remaining AIDS-free with significant inflection (13%) (Fig. 5, +). Both the sensitivity and specificity when using the post-IP slope cutoff (β2) among those with 15 or more CD3+ cell counts also increased slightly, to 81%.
DISCUSSION
The data obtained in this study and described by our methods lend strong support to the hypothesis that the loss of T-cell homeostasis is important in the pathogenesis of the severe immunodeficiency that characterizes the late stages of HIV infection. Based on our hypothesis regarding the patterns of CD3+ changes over time, we constructed a segmented regression model with different CD3+ trajectories before and after an IP. Criteria were developed for determining whether an individual could be considered as having a true inflection. We found that the optimal criteria for discriminating men who did and did not develop AIDS was whether their post-IP CD3+ lymphocyte counts declined by more than 10% per year.
Supported by the methods for identifying individuals with true inflection, we applied our model to a variety of groups in the MACS to evaluate the blind T-cell homeostasis hypothesis (2, 3, 14–16). Consonant with this hypothesis, in which CD8+ cells compensate for CD4+ loss to maintain a constant number of circulating CD3+ cells, the trajectories before the IP were closely distributed around zero. Furthermore, there were no differences in the pre-IP trajectories of the HIV− and HIV+ men remaining free of AIDS, who were expected to have maintained homeostasis, and the HIV+ men with onset of AIDS, who were expected to have lost homeostasis, but only after the IP. Independent from whether homeostasis is perfectly maintained (versus a slight decline over time), the individual trajectories after the IP observed in the men who developed AIDS indicated a rapid decline in CD3+ cell counts preceding the onset of AIDS in the vast majority of these men.
With our methods for the estimation of inflection at the individual level, we could investigate issues that cannot be addressed at the population level, particularly the prevalence of inflection among those destined to develop AIDS or not within 3 years. It is not necessary that every person exhibit an IP for the phenomenon to exist in the aggregate of HIV-infected individuals. In the population averages, a few men could have accounted for the observed declines before AIDS. This was not found, however, as we estimated that 77% percent of the men developing AIDS, but only 23% of the men remaining AIDS-free, had T-cell inflection. The use of the statistical criteria qualitatively validated the difference in prevalence among men developing AIDS and men remaining AIDS-free, despite using a more conservative cutoff. Taken together, these data strongly support the idea that failure of T-cell homeostasis is an important milestone in the progression of HIV-1 infection to AIDS.
Nevertheless, even these estimates of the prevalence of CD3+ homeostasis loss before AIDS may be conservative for several reasons. First, the limitations in the data available on each individual may have had an impact. Because the methods required a minimum of six CD3+ cell counts, some individuals with true inflection may have been missed. Furthermore, our analysis did not exclude individuals who supplied the minimum data but who had large gaps between their visits, or those individuals who met the entry criteria with data collected much earlier than AIDS.
Second, our decision criteria used only the magnitude and statistical significance of the trajectories of CD3+ lymphocyte counts. No concomitant immunologic (e.g., CD4+ level, β2-microglobulin, neopterin), virologic (e.g., viral load), or epidemiologic (e.g., antiretroviral therapy) variables were used to assess whether there was evidence of true inflection. Using additional T-cell data might improve the sensitivity and specificity. For example, we could have required both the CD3+ trajectory to be larger than 10% and the CD4+ cell count to be below 500 cells/mm3 to assess men as having evidence of true inflection. Although previous analyses have not demonstrated any influence of therapy on the loss of homeostasis (2), more research in this area is needed, particularly in the new era of highly active antiretroviral therapy.
The occurrence of inflection after AIDS suggests a more complicated relationship between loss of homeostasis and AIDS. However, further investigation of the data from these men suggested that some of the IPts after AIDS might have been consequences of the data structure. The distribution of IPs among these individuals was highly skewed so that 40% of the IPs among these individuals were within 1 year of AIDS. It is likely that many of these AIDS occurrences were within the expected random variation of the IP time from pre-AIDS time points. In addition, we found that these men had a higher median number of CD3+ cell counts (17 versus 12 in those with IP estimated before AIDS) and that AIDS occurred earlier in these men by 1.1 years versus those with an IP estimated before AIDS. Thus, a relatively long follow-up after AIDS might have resulted in an IP estimated after AIDS. Because the method used allowed for only one IP per individual, these individuals also might have had a less dramatic inflection earlier in their disease. In support of this, when the algorithm was rerun on only pre-AIDS data, the prevalence of a pre-AIDS IP characterized by more than a 10% post-IP decline in CD3+ trajectories was nearly identical for these individuals (75%) as for those with an IP estimated before AIDS (77%).
The identification of CD3+ lymphocyte inflection on an individual level provides an approach for investigating immunologic and virologic mechanisms that elucidate the pathogenesis of HIV infection. Using the methods described in this paper, studies now can be designed to examine the occurrence of landmark events (e.g., CD4+ lymphocyte counts less than 50 cells/mm3) relative to the timing of T-cell inflection. This type of design will be critical to resolve factors that are consequences of T-cell inflection from those antecedent factors that may help precipitate CD3+ inflection.
More generally, our approach demonstrates the ability of statistical models to elucidate pathogenesis mechanisms. The methods proposed could be applied to any longitudinal series of outcomes in any time scale. The segmented regression model with random IPs is particularly well-suited for investigating the timing of trajectory changes. Along these lines, they previously have been proven useful in other medical fields (e.g., respiratory medicine, ref. 17) and other scientific disciplines (e.g., allometry, ref. 18). The use of the segmented regression model in HIV research also has additional promise. For example, the application of the IP model to serial viral RNA measurements in plasma would give a formal method to test the hypothesis that a “set-point” exits (19). That is, one could use the methods described in this paper to examine whether longitudinal HIV RNA measurements are constant, consistently rising, or initially constant but rising as a prelude to AIDS. In the latter case, a comparison of T-cell inflection with that of HIV RNA inflection could shed important light on the relative timing of viral events (e.g., the emergence of viral quasi-species (20, 21) and macrophage-tropic variants (22)) and of immunologic events (e.g., immununologic exhaustion (23, 24) and changes in receptor expression (25).
Acknowledgments
We gratefully acknowledge the insightful suggestions from the reviewers of the paper. This work was supported by National Institutes of Health Grants and Contracts UO1-AI-35042, 5-M01-RR-00722 (General Clinical Research Center), UO1-AI-35043, UO1-AI-37984, UO1-AI-35039, UO1-AI-35040, UO1-AI-37613, and UO1-AI-35041.
ABBREVIATIONS
- MACS
Multicenter AIDS Cohort Study
- IP
inflection point
- ROC
receiver operating characteristic
- IQR
inter-quartile range
- SP
seroprevalent
- SC
seroconverter
- SN
seronegative
Appendix
The following methods were used to approximate the critical values used for testing whether the segmented regression model or a linear model was most appropriate for an individual’s CD3+ cell counts. They are approximate because the exact distribution depends on the exact spacing of the observations (12) and simulated critical values used only equally spaced observations. For a fixed value of Ni (the total number of CD3+ cell counts), the algorithm can be described in three steps. Note that our algorithm is similar to that described by Kiuchi et al. (11) except that they generate independent observations in step 1.
Step 1: Generate a series of Ni CD3+ counts under the null hypothesis assumption that βi1 = βi2 = 0.
We assumed the log10 CD3+ responses followed a multivariate normal distribution with mean βi0 and a variance-covariance structure described by a damped-exponential model (26). The damped-exponential regression model is a very flexible and parsimonious method for modeling correlated data and has been specifically used to describe markers of HIV infection (27). The model includes a covariance of responses s years apart described by σ2 γsθ. Estimates for βi0 and the covariance parameters were obtained by applying a regression model of this form to a subset of CD3+ cell counts for 500 HIV− individuals (β̂i0 = 3.24, σ̂2 = 0.0203, γ̂ = 0.643, θ̂ = 0.126.
Step 2: Apply a linear model and a segmented regression model to this series of CD3+ counts.
Standard linear regression (i.e., a model with no IP) and the segmented regression model algorithm described in Methods were applied to the simulated data generated in step 1.
Step 3: Compute the F-statistic for testing the appropriateness of a linear model versus a segmented regression model.
The F-statistic
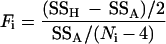 |
was computed, where SSH represents the residual sums of squares under the linear regression model (βi1 = βi2), SSA represents the residual sums of squares under the segmented regression model with the breakpoint estimated by using methods described in Methods.
Step 4: Repeat steps 1–3 to generate an empirical distribution for the F-statistics.
We repeated steps 1–3 a total of 3,000 times (i = 1,… ,3000) to obtain 3,000 F-statistics. We then ordered the F-statistics and computed the 95th percentile. This value is the critical value we need whereby, given the null hypothesis (βi1 = βi2), only 5% of the F-statistics will exceed this critical value.
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
References
- 1.Zolla-Pazner S, Des Jarlais D C, Friedman S R, Spira T J, Marmor M, Holzman R, Mildvan D, Yancovitz S, Mathur-Wagh U, Garber J, et al. Proc Natl Acad Sci USA. 1987;84:5404–5408. doi: 10.1073/pnas.84.15.5404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Margolick J B, Muñoz A, Donnenberg A D, Park L P, Galai N, Giorgi J V, O’Gorman M R G, Ferbas J for the Multicenter AIDS Cohort Study Group. Nat Med. 1995;1:674–680. doi: 10.1038/nm0795-674. [DOI] [PubMed] [Google Scholar]
- 3.Adleman L M, Wofsy D. J AIDS Hum Retrovir. 1993;6:144–152. [PubMed] [Google Scholar]
- 4.Galai N, Margolick J B, Astemborski J, Vlahov D. Clin Immunol Immunopathol. 1996;79:134–141. doi: 10.1006/clin.1996.0060. [DOI] [PubMed] [Google Scholar]
- 5.Kaslow R A, Ostrow D G, Detels R, Phair J P, Polk B F, Rinaldo C R. Am J Epidemiol. 1987;126:14–24. doi: 10.1093/aje/126.2.310. [DOI] [PubMed] [Google Scholar]
- 6.Giorgi J V, Cheng H L, Margolick J B, Bauer J D, Ferbas J, Waxdal M, Schmid I, Hultin L E, Jackson A L, Park L, Taylor J M G for the Multicenter AIDS Cohort Study Group. Clin Immunol Immunopathol. 1990;55:173–186. doi: 10.1016/0090-1229(90)90096-9. [DOI] [PubMed] [Google Scholar]
- 7.Schenker E L, Hultin L E, Bauer K D, Ferbas J, Margolick J B, Giorgi J V. Cytometry. 1993;14:307–317. doi: 10.1002/cyto.990140311. [DOI] [PubMed] [Google Scholar]
- 8.Centers for Disease Control and Prevention. Morbidity Mortality Weekly Report. 1987;36:1s–15s. [Google Scholar]
- 9.Metz C E. Semin Nuclear Med. 1978;8:283–298. doi: 10.1016/s0001-2998(78)80014-2. [DOI] [PubMed] [Google Scholar]
- 10.Draper N, Smith H. Applied Regression Analysis. 2nd Ed. New York: Wiley; 1981. [Google Scholar]
- 11.Kiuchi A S, Hartigan J A, Holford T R, Rubinstein P, Stevens C. Biometrics. 1995;51:236–248. [PubMed] [Google Scholar]
- 12.Hinkley D V. J Am Stat Assoc. 1971;66:736–743. [Google Scholar]
- 13.Feder P I. Ann Stat. 1975;3:84–97. [Google Scholar]
- 14.Adleman L M, Wofsy D. J AIDS Hum Retrovir. 1996;11:334–340. doi: 10.1097/00042560-199604010-00003. [DOI] [PubMed] [Google Scholar]
- 15.Mehr R, Perelson A S. J AIDS Hum Retrovir. 1997;14:387–398. doi: 10.1097/00042560-199704150-00001. [DOI] [PubMed] [Google Scholar]
- 16.Stanley S, Fauci A S. J AIDS Hum Retrovir. 1993;6:142–143. [Google Scholar]
- 17.Kerstjens H A M, Brand P L P, Postma D S. Am J Respir Crit Care Med. 1996;154:S266–S272. doi: 10.1164/ajrccm/154.6_Pt_2.S266. [DOI] [PubMed] [Google Scholar]
- 18.Chappell R. J Theor Biol. 1989;138:235–256. doi: 10.1016/s0022-5193(89)80141-9. [DOI] [PubMed] [Google Scholar]
- 19.Ho D. Science. 1996;272:1124–1125. doi: 10.1126/science.272.5265.1124. [DOI] [PubMed] [Google Scholar]
- 20.Delwart E L, Pan H, Sheppart H W, Wolpert D, Neumann A U, Korber B, Mullins J I. J Virol. 1997;71:7498–7508. doi: 10.1128/jvi.71.10.7498-7508.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McNearney T, Hornickova Z, Markham R, Birdwell A, Arens A, Saah A, Ratner L. Proc Natl Acad Sci USA. 1992;89:10247–10251. doi: 10.1073/pnas.89.21.10247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koot M, Keet I P, Vos A H, deGoede R E, Roos M T, Coutinho R A, Miedema F, Schellekens P T, Tersmette M. Ann Int Med. 1993;118:681–688. doi: 10.7326/0003-4819-118-9-199305010-00004. [DOI] [PubMed] [Google Scholar]
- 23.Miedema F, Klein M R. Science. 1996;272:505–506. doi: 10.1126/science.272.5261.505. [DOI] [PubMed] [Google Scholar]
- 24.Effros R B, Allsopp R, Chiu C P, Hausner M A, Hirji K, Wang L, Harley C B, Villeponteau B, West M D, Giorgi J V. AIDS. 1996;10:F17–F22. doi: 10.1097/00002030-199607000-00001. [DOI] [PubMed] [Google Scholar]
- 25.Flamand L, Crowley R W, Lusso P, Colombini-Hatch S, Margolis D M, Gallo R C. Proc Natl Acad Sci USA. 1998;95:3111–3116. doi: 10.1073/pnas.95.6.3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Muñoz A, Carey V J, Schouten J P, Segal M, Rosner B. Biometrics. 1992;48:733–742. [PubMed] [Google Scholar]
- 27.Galai N, Muñoz A, Chen K, Carey V J, Chmiel J, Zhao S. Stat Med. 1993;12:2133–2145. doi: 10.1002/sim.4780122207. [DOI] [PubMed] [Google Scholar]