

Abstract
The measles virus P gene products V and C antagonize the host interferon (IFN) response, blocking both IFN signaling and production. Using Moraten vaccine strain-derived measles virus and isogenic mutants deficient for either V or C protein production (Vko and Cko, respectively), we observed that the Cko virus was a potent inducer of IFN-β, while induction by Vko virus was an order of magnitude lower than that by the Cko virus. The parental recombinant Moraten virus did not significantly induce IFN-β. The enhanced IFN-inducing capacity of the Cko virus correlated with an enhanced activation of IFN regulatory factor 3 (IRF-3), NF-κB, and ATF-2 in Cko-infected compared to Vko or parental virus-infected cells. Furthermore, protein kinase PKR and mitochondrial adapter IPS-1 were required for maximal Cko-mediated IFN-β induction, which correlated with the PKR-mediated enhancement of mitogen-activated protein kinase and NF-κB activation. Our results reveal multiple consequences of C protein expression and document an important function for PKR as an enhancer of IFN-β induction during measles virus infection.
Measles virus (MV), a member of the genus Morbillivirus of the Paramyxoviridae, causes an acute febrile illness. Despite an effective vaccine, measles continues to cause extreme morbidity and mortality worldwide (10), and recently, there has been a resurgence of measles in industrialized countries, where a lack of adherence to vaccine recommendations is an increasing problem (5, 12). The need for improved MV vaccines (11), together with the potential for use of engineered MV vaccine strains with defined mechanisms of attenuation as oncolytic viruses for cancer therapy (4), further justify ongoing efforts to gain an enhanced understanding of the host response to MV infection at the molecular level.
The 15.9-kb negative-stranded RNA genome of MV consists of six genes (N, P/V/C, M, F, H, and L). The P gene is polycistronic, encoding the V and C nonstructural proteins in addition to P, a structural phosphoprotein and essential cofactor for the viral polymerase (2, 3, 10). The V protein shares its N-terminal 231 amino acids with P, but the C-terminal 68 amino acids are unique because of the pseudotemplated G insertion that causes a frameshift in V mRNA, whereas the C protein is synthesized by an alternative translation initiation AUG codon positioned 22 nucleotides downstream of the P/V translation start codon (3, 11). V and C are accessory proteins that serve a variety of functions including the modulation of the host innate immune response to MV infection (8, 9, 27). Isogenic MV mutants that are defective for the expression of either V or C have been generated. Strong adaptive immune responses, but dysregulated innate responses, are seen with these mutants (4, 8, 35).
An important component of the host innate antiviral response is the interferon (IFN) response. IFNs are proinflammatory cytokines that possess antiviral activity (27, 31). IFN action involves IFN binding to cognate receptors and subsequent prototypical JAK-STAT signal transduction that leads to the expression of IFN-stimulated genes, whose products inhibit virus growth. IFN production involves the recognition of pathogen-associated molecular patterns including viral RNAs by retinoic acid-inducible protein (RIG-I)-like cytosolic receptors (RLRs) and membrane-associated Toll-like receptors (TLRs). The RLR and TLR3 sensors signal through cognate adapter proteins, including IPS-1 and TRIF, respectively, to transcriptionally activate IFN expression (36, 40). In the case of the IFN-β gene, IFN regulatory factor 3 (IRF-3) and nuclear factor κB (NF-κB) are activated by RLR or TLR signaling, enter the nucleus, and function together with activating transcription factor 2 (ATF-2)/c-jun to constitute the IFN-β enhanceosome that drives IFN-β transcription (22).
The antagonism of IFN action by MV, which is well documented, occurs through P, V, and, to a lesser extent, C protein-mediated inhibition of STAT phosphorylation and nuclear accumulation, thereby preventing IFN-mediated signal transduction (9, 21, 26, 27, 39). MV also antagonizes IFN production, although the mechanistic basis of this process is largely unresolved. Recent evidence suggests that the V protein inhibits mda-5-triggered RLR signaling (6, 18) and perhaps also IκB kinase α (IKKα)-mediated phosphorylation of IRF-7 mediated by TLRs 7 and 9 (23). In addition, the C protein also inhibits IFN production, presumably indirectly by regulating viral RNA synthesis (18), although the details of the process are not known.
The cellular protein kinase PKR is activated by binding RNA, which leads to the autophosphorylation, dimerization, and subsequent phosphorylation of substrate proteins (16, 30, 34). We showed previously that a C-deficient mutant MV was a potent activator of PKR, whereas the parental wild-type (WT) virus was not (35). In addition, we showed that PKR enhances IFN-β induction mediated by cytoplasmic RLR sensors of double-stranded RNA (dsRNA) and 5′-triphosphate-containing single-stranded RNA (15). In light of these observations, we hypothesized that PKR could be an effector of IFN-β induction by the C-deficient virus (8). To test this possibility, and to directly assess the IFN-inducing capacity of isogenic vaccine-derived virus mutants defective in C or V expression, we measured the IFN-β-inducing capacity of a recombinant MV based on the parental Moraten vaccine strain as well as V-deficient (Vko) and C-deficient (Cko) mutants derived from this attenuated virus (7). We performed infections with these viruses of human HeLa cells stably deficient in PKR expression as a result of stable short hairpin RNA interference-mediated knockdown (PKRkd cells) and with PKR-sufficient control (PKRkd-con) HeLa cells that express levels of PKR comparable to those of PKR-sufficient parental HeLa cells (41, 42). Additionally, we transiently knocked down PKR, IPS-1, and TRIF to further elucidate the signaling pathways that are operative in IFN-β induction by these viruses.
MATERIALS AND METHODS
Cells and viruses.
Parental HeLa (PKR+) and Vero cells were maintained in Dulbecco's modified Eagle's medium supplemented with 5% (vol/vol) fetal bovine serum (HyClone), 100 μg/ml penicillin, and 100 U/ml of streptomycin (Gibco/Invitrogen) as described previously (35, 43). The PKRkd HeLa cell transformant line with a stable knockdown of PKR achieved by the expression of a stably integrated short hairpin silencing RNA targeting PKR utilizing the pSUPER.retro.puro vector with the H1 promoter (41) and the PKR-sufficient drug-resistant PKRkd-con control cell line transfected with the pSUPER vector that lacked the silencing hairpin directed against PKR were both maintained in medium containing 1 μg/ml puromycin (35, 43). The recombinant parental Moraten vaccine strain of measles virus (MVvac), herein designated the WT, includes the gene encoding green fluorescent protein inserted downstream of the H gene (7, 35). This vaccine backbone was used because only vaccine lineage strains can enter cells through CD46, a receptor present on the PKR-sufficient and -deficient HeLa lines (19, 35). For the Vko mutant, V protein expression is selectively abolished by a mutation of the V gene-editing site and the introduction of a stop codon (7, 35). For the Cko mutant, C protein expression is selectively abolished by a mutation of the translation start codon for C and the introduction of a stop codon. The mutations introduced to inactivate V protein expression do not affect the amino acid sequence of P or C, and the mutations that inactivate C protein expression do not affect the amino acid sequence of P or V (7, 35). Virus infections were carried out as previously described (7, 35) by using a multiplicity of infection (MOI) of 5 50% tissue culture infective doses per cell.
IFN-β expression.
The expression of IFN-β was measured by real-time quantitative PCR (qPCR) as previously described (15). Cells were seeded in 12- or 24-well plates and infected with MVvac WT, Vko, or Cko virus or were left uninfected. Total RNA was prepared from uninfected cells or infected cells at 10, 16, and 24 h after infection with an RNeasy minikit (Qiagen) or TRIzol (Invitrogen) according to instructions provided by the manufacturers. Random-primed reverse transcription was carried out by using 500 ng of RNA and SuperScript II (Invitrogen) according to the manufacturer's protocol. The following primer pairs were used: GAPDH (glyceraldehyde-3-phosphate dehydrogenase) forward primer GCCTTCCGTGTCCCCACTG and reverse primer CGCCTGCTTCACCACCTTC and IFN-β forward primer AAACTCATGAGCAGTCTGCA and reverse primer AGGAGATCTTCAGTTTCGGAGG. qPCR reactions were performed in duplicate or triplicate with each reverse transcription template by using IQ SYBR green Supermix (Bio-Rad) and a Bio-Rad MyIQ real-time qPCR instrument (3-min hot start followed by 30 s at 95°C, 45 s at 58°C, and 45 s at 72°C, repeated 40 times). IFN-β values were normalized to GADPH values.
Western immunoblot analysis.
Whole-cell extracts were prepared with extract buffer containing 1 mM phenylmethylsulfonyl fluoride and 1% (vol/vol) protease inhibitor cocktail (Sigma) as previously described (15, 35, 43). Protein concentrations of the extracts were determined by use of the Bradford method. Protein fractionation by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and immunoblot analysis using nitrocellulose membranes blocked in 5% (wt/vol) skim milk in phosphate-buffered saline were described previously (35, 43), and the following primary antibodies were used: PKR (Santa Cruz Biotechnology), phospho-PKR Thr446 (Epitomics), ATF-2 (Santa Cruz Biotechnology), phospho-ATF-2 (Cell Signaling Technology), p38 (Santa Cruz Biotechnology), phospho-p38 (Cell Signaling Technology), Jun N-terminal kinase (JNK) (Santa Cruz Biotechnology), phospho-JNK (Cell Signaling Technology), IRF-3 (Santa Cruz Biotechnology), IPS-1 (Bethyl Laboratories), TRIF (Alexis Biochemicals), α-tubulin (Sigma), and β-actin (Sigma). Antibody against the MV N protein was prepared as previously described (7, 35). Western immunoblot detection was performed with IRDye 800CW-conjugated anti-rabbit immunoglobulin G or IRDye 680-conjugated anti-mouse immunoglobulin G secondary antibody according to the manufacturer's protocols by using an Odyssey infrared imager system (Li-Cor Biosciences).
IPS-1, TRIF, and PKR siRNA transient knockdown.
The sequences targeted in transient knockdown experiments with human cells using validated chemically synthesized short interfering RNAs (siRNAs) prepared by Dharmacon with dTdT overhangs were as follows (15, 43): UAGUUGAUCUCGCGGACGA for IPS-1, GACCAGACGCCACUCCAAC for TRIF, GCAGGGAGUAGUACUUAAAUA for PKR, and, as added controls, either firefly luciferase siRNA (CUUACGCTGAGUACUUCGA) or Dharmacon's negative control, siNEG. For transient knockdown experiments, a double-transfection approach was used to achieve a maximum knockdown of the target proteins (15, 43). Briefly, HeLa cells in 60-mm dishes at ∼50% confluence were transfected with 25 nM siRNA using Lipofectamine 2000 (Invitrogen) on days 1 and 3. On day 2, the cells were reseeded into 60-mm dishes for the second siRNA transfection, and on day 4, the cells were seeded into 12-well plates for MVvac infection.
NF-κB activation assays.
Two assays were used to assess NF-κB activation, a reporter assay and a gel mobility shift assay. For the reporter assay, cells were cotransfected with an NF-κB-dependent firefly luciferase reporter plasmid (generously provided by I. Verma, Salk Institute) and a Renilla luciferase reporter plasmid (Promega). At 6 h after transfection, cells were infected with the indicated MVvac strain for 24 h and then harvested and lysed in passive lysis buffer (Promega). Following centrifugation at 13,400 × g for 10 min, luciferase activities were determined by using the dual-luciferase protocol according to the manufacturer's recommendations (Promega). For the electrophoretic mobility shift assay, cells were infected with wild-type or mutant MVvac as indicated and then harvested after 24 h. Nuclear extracts or high-salt whole-cell extracts as indicated were prepared and analyzed as previously described (15, 37) except that an NF-κB double-stranded oligonucleotide probe with the sense strand sequence AGTTGAGGGGACTTTCCCAGGC was utilized. Incubation was performed for 20 min prior to analysis. The WT probe was 5′ end labeled with IR700 dye (Integrated DNA Technologies), and detection was carried out by using an Odyssey infrared imager system (Li-Cor Biosciences). For competition analysis, a 100-fold excess of unlabeled competitor was used, either the unlabeled WT oligonucleotide or a mutant oligonucleotide, AGTTGAGGCGACTTTCCCAGGC. For supershift analysis, antibody against either the NF-κB p65 subunit or STAT1 (Santa Cruz Biotechnology) was incubated with the extract at 4°C prior to the addition of the probe.
RESULTS AND DISCUSSION
PKR enhances the induction of IFN-β by measles virus V and C mutants.
To determine the capacity of attenuated measles virus vaccine strain isogenic mutants defective in either the V or C protein compared to parental (WT) Moraten virus to induce IFN-β and to test whether PKR plays a role in the induction of IFN-β by V and C mutants, a HeLa cell line in which PKR expression is stably knocked down by RNA interference to less than 5% of the PKR protein expression level found in PKRkd-con cells (41, 42) was examined. As shown in Fig. 1A, the WT virus did not induce detectable IFN-β transcription in either PKR-sufficient PKRkd-con or PKR-deficient PKRkd cells at times up to 24 h postinfection, as measured by real-time quantitative PCR. In contrast, the isogenic Cko virus was a potent inducer of IFN-β, producing more than 100-fold-higher IFN-β transcript levels than WT parental MVvac in PKR-sufficient PKRkd-con cells (Fig. 1A). The isogenic Vko virus also induced IFN in PKRkd-con cells, but the level of induction was ∼10-fold lower than that of the Cko virus (Fig. 1A). However, in PKRkd cells, the inductions of IFN-β by the Cko virus as well as the Vko virus were severely impaired (Fig. 1A). The quantitative difference in IFN-β transcript levels relative to GAPDH transcript levels detected by qPCR was verified by template dilution analysis as illustrated in Fig. 1B for cDNA prepared from uninfected and Cko-infected cells. Regression analyses gave E values of 1.95 for IFN-β and 2.02 for GAPDH, in good agreement with the theoretical value of 2.0. In this experiment (Fig. 1B), the induction of IFN-β by Cko virus was ∼28 or ∼250-fold.
FIG. 1.

PKR is required for maximal induction of IFN-β by measles virus V and C mutants. (A) IFN-β mRNA normalized to GAPDH was measured using quantitative real-time PCR as described previously (15), utilizing reverse-transcribed total cellular RNA prepared from uninfected mock or MV-infected (WT, Vko, or Cko) (MOI of 5) PKRkd-con and PKRkd cells at 10, 16, and 24 h postinfection. Error bars indicate the standard deviations of data from three independent experiments. *, P < 0.01. (B) Template dilution analysis with cDNA prepared from uninfected mock (open symbols) or Cko-infected (24 h) (filled symbols) PKR+ parental cells with primer pairs for IFN-β (triangles) or GAPDH (circles).
These results demonstrate that PKR enhances MV-induced IFN-β transcription and that the expression of the viral accessory proteins impairs this PKR-mediated phenotype. Our results obtained with recombinant MVvac, generated based on the infectious cDNA of the attenuated Moraten vaccine strain, are similar to those obtained with virus generated based on the infectious cDNA of the virulent wild pathogenic strain IC-B, where an upregulation of the IFN-β transcript level was also seen both in cell culture and in monkeys following infection with Cko compared to the parental virus (8, 17, 18).
Activation of mitogen-activated protein (MAP) kinase signaling and ATF-2 phosphorylation is PKR dependent and enhanced in Cko-infected cells.
In PKR-sufficient PKRkd-con cells, IFN-β induction by the Cko virus correlated with the phosphorylation of PKR on threonine 446 (T446), a known activation site, at 24 h postinfection (Fig. 2, lane 4). In contrast, we observed no detectable T446 phosphorylation following the infection of PKR-deficient PKRkd cells with any of the viruses Cko, Vko, or WT (Fig. 2, lanes 6 to 8). This is due to the lower detection threshold, because PKRkd cells express 2 to 5% of the PKR protein compared to PKRkd-con cells or parental HeLa cells (Fig. 2) (41, 42).
FIG. 2.
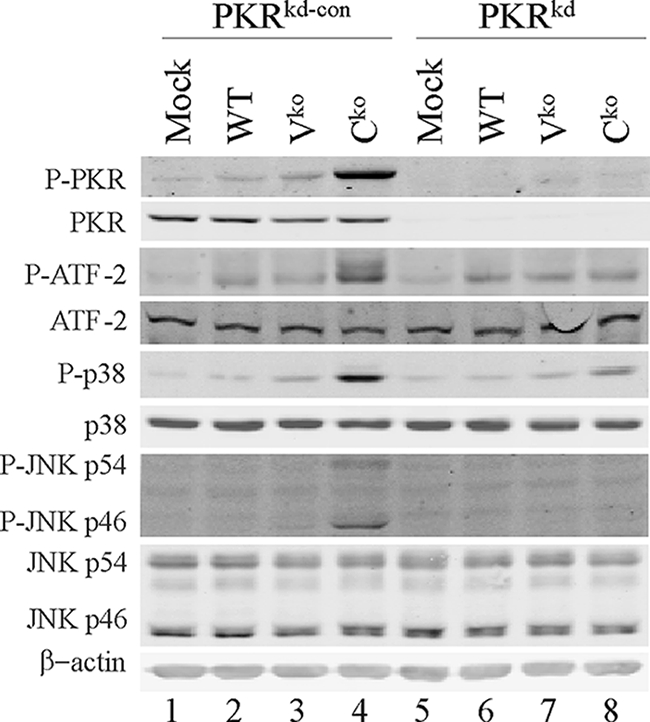
Phosphorylation of p38, JNK, and ATF-2 correlates with PKR activation. Immunoblot analyses of whole-cell extracts prepared from uninfected mock or MV-infected (WT, Vko, or Cko) (MOI of 5) PKRkd-con or PKRkd cells at 24 h postinfection were conducted as previously described (35, 41). Antibodies against PKR, phosphorylated PKR (P-PKR), p38, phosphorylated p38, JNK, phosphorylated JNK, ATF-2, phosphorylated ATF-2, and β-actin were utilized.
To gain insight into the mechanism by which PKR enhances IFN-β induction by the Cko virus, we first considered the p38 and JNK pathways. p38 and JNK are components of two MAP kinase pathways that culminate in the activation of the ATF-2 and c-jun transcription factors, respectively, components of the IFN-β enhanceosome complex (27). The PKR protein has been implicated in p38 and JNK kinase activation in response to cellular stresses including viral infection (27, 29, 34, 44). As shown in Fig. 2, the infection of PKR-sufficient cells with the Cko virus led to the enhanced phosphorylation of ATF-2, p38, and JNK, whereas infection with the WT or Vko virus did not increase (JNK) and only very marginally increased (p38 and ATF-2) phosphorylation. Importantly, the level of phosphorylation of p38, JNK, and ATF-2 was reduced in PKRkd cells, suggesting that PKR-mediated IFN-β induction by the Cko virus occurred, at least in part, through the activation of these MAP kinase pathways (Fig. 2). A similar response was seen with vaccinia virus, where the depletion of PKR by stable knockdown impaired the phosphorylation of both p38 and JNK activated by infection with ΔE3L mutant but not WT vaccinia virus (44).
IRF-3 phosphorylation is enhanced in Cko-infected compared to Vko- or WT-infected cells independently of PKR.
We next examined IRF-3 phosphorylation in PKRkd-con and PKRkd cells infected with the WT, Vko, or Cko virus compared to uninfected cells (Fig. 3). The phosphorylation of IRF-3 was assessed by the reduction in IRF-3 mobility on sodium dodecyl sulfate-polyacrylamide gels, which correlates with C-terminal serine 396 phosphorylation and activation (13, 43). Western analysis using an antibody that recognizes both phosphorylated and nonphosphorylated forms of IRF-3 revealed minimal activation by the WT and Vko MVs (Fig. 3, lanes 2, 3, 6, and 7). One major lower band, corresponding to unphosphorylated IRF-3, was detected in uninfected cells (13, 43). However, infection with the Cko virus led to enhanced IRF-3 activation (Fig. 3, lanes 4 and 8), as revealed by the appearance of IRF-3 forms with reduced gel mobility that corresponded to the virus-induced C-terminal phosphorylation (P-IRF-3) of IRF-3 (13, 43). The enhanced IRF-3 activation observed for Cko virus-infected cells (Fig. 3) provides further explanation for the potent induction of IFN-β observed for Cko-infected cells (Fig. 1). Surprisingly, however, levels of IRF-3 activation for PKRkd-con and PKRkd cells were comparable, suggesting that the activation of IRF-3 induced by the Cko virus likely occurs through a PKR-independent pathway. This is in contrast to the IRF-3 activation seen for vaccinia virus E3L deletion mutant-infected cells, which is PKR dependent (43).
FIG. 3.
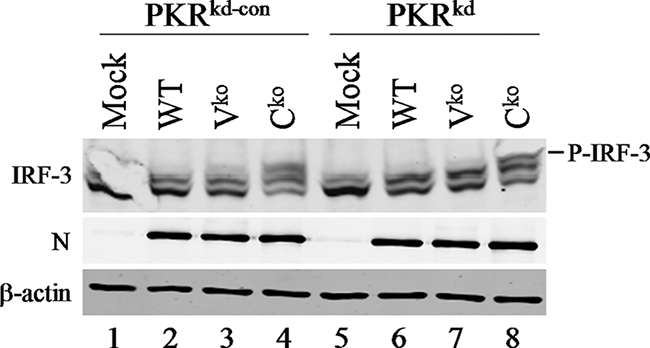
IRF-3 activation by the MV Cko mutant is independent of PKR and the N protein. Shown are immunoblot analyses of whole-cell extracts prepared from uninfected mock and MV-infected (WT, Vko, or Cko) (MOI of 5) PKRkd-con or PKRkd cells at 24 h postinfection using antibodies against IRF-3, the MV N protein, and β-actin. The gel mobility position of C-terminally phosphorylated IRF-3 is indicated.
A previous study suggested that the MV N protein is an activator of IRF-3 (33). We therefore examined the steady-state level of the N protein to determine whether the Cko virus expressed higher levels of N than did the WT and Vko viruses. We found no evidence that the Cko virus expressed higher levels of the N protein than either the WT or Vko virus; all three viruses showed similar infection levels, as measured by determinations of N protein levels (Fig. 3). N protein levels in Cko and WT virus-infected cells, when quantified relative to β-actin, differed by less than 1.5-fold over the time period of 12 to 48 h after infection (data not shown) (35). Additionally, others previously found that none of the MV mRNAs, when tested individually by ectopic plasmid-based expression, were able to activate IFN-β gene transcription, including the N transcript (25). The N protein expression level by itself, therefore, does not likely account for the increased levels of IRF-3 activation and IFN-β transcription seen with the Cko virus compared to the WT or Vko virus. The C protein of MV regulates viral transcription and genome replication (1, 28). The activation of IRF-3 by Cko may happen because of an increased or inaccurate synthesis of RNA that activates PKR-independent IFN induction, which, together with the PKR-dependent enhancement, results in substantially elevated levels of IFN-β transcription.
NF-κB activation in Cko-infected cells is enhanced by PKR.
We also compared the abilities of wild-type and the C and V mutant viruses to activate NF-κB measured by band shift and reporter assays. Selective protein binding to the NF-κB element of the IFN-β promoter was dependent upon MVvac infection as measured by electrophoretic mobility shift assay (Fig. 4A). The infection of PKR-sufficient cells with the Cko mutant virus efficiently activated NF-κB compared to the modest activation level seen following Vko virus infection (Fig. 4A, left). Wild-type MVvac was a poor activator; the band shift was marginally detectable and comparable to that seen with extract prepared from uninfected cells (Fig. 4A). The induced band shift seen with nuclear extracts from Cko-infected cells corresponded to NF-κB as determined by oligonucleotide competition and by antibody supershift analyses (Fig. 4A, right). A 100-fold molar excess of unlabeled wild-type oligonucleotide efficiently competed for the formation of the band shift complex, but a 100-fold excess of mutant oligonucleotide competitor did not. Antibody against the NF-κB p65 subunit abolished complex formation, whereas as a control antibody against STAT1 did not.
FIG. 4.

PKR enhances NF-κB activation in MV Cko mutant-infected cells. (A) Electrophoretic mobility shift analysis of extracts prepared from uninfected mock or MVvac-infected (WT, Vko, or Cko) (MOI of 5) PKRkd-con cells at 24 h postinfection using an NF-κB oligonucleotide probe (15, 37). Nuclear extracts prepared from Cko-infected cells were also analyzed by competition analysis with a 100-fold excess of unlabeled NF-κB oligonucleotide, either the WT or mutant sequence, or following the addition of antibody to either NF-κB p65 or STAT1. (B) Electrophoretic mobility shift assay analysis with high-salt whole-cell extracts prepared from uninfected mock or Cko-infected PKR+ cells following transient knockdown utilizing siRNAs against luciferase as a control (siLUC), IPS-1, PKR, or TRIF or not transfected (NT) with any siRNA. (C) PKR+ parental cells were cotransfected with an NF-κB-dependent firefly luciferase (FF) reporter plasmid and control Renilla luciferase (RL) plasmid following transient knockdown utilizing siRNAs against IPS-1, PKR, or TRIF or the negative control siRNA (siNEG) or were not transfected. Results shown are the means ± standard deviations determined from two to three independent experiments carried out in duplicate.
The adapter IPS-1 but not TRIF mediates NF-κB activation in Cko-infected cells.
To test which of the viral-RNA-sensing pathways may be responsible for the robust activation of NF-κB in Cko virus-infected PKR-sufficient cells (Fig. 4A), we transiently knocked down the adapter proteins IPS-1 for the RIG-like receptors RIG-I and mda-5 (40) and TRIF for TLR3 (36). We then assessed NF-κB activation following Cko infection of parental HeLa cells in which either IPS-1, TRIF, or PKR had been transiently knocked down. As shown in Fig. 4B, the transient knockdown of the IPS-1 mitochondrial adapter abolished the Cko virus-induced activation of NF-κB, whereas TRIF adapter knockdown did not. The transient knockdown of PKR reduced but did not abolish the activation of NF-κB (Fig. 4B). As controls, the Cko virus induced efficient activation in cells either transfected with an siRNA against luciferase or not transfected. Similar results were obtained when an NF-κB-dependent firefly luciferase reporter assay was utilized (Fig. 4C). The transient knockdown of IPS-1 abolished the Cko virus-induced activation of NF-κB-dependent reporter expression, as did the transient knockdown of PKR. In contrast, the TRIF knockdown did not affect reporter expression, with activation comparable to that of cells not transfected with an siRNA or cells treated with an siNEG negative control RNA (Fig. 4C).
The MV C mutant induces IFN-β through the IPS-1 adapter.
MV replicates in the cytoplasm and activates the RIG-I sensor due to the presence of cytosolic 5′-triphosphate-containing viral transcripts (24, 25), but paradoxically, the V protein of MV blocks the mda-5 sensor of the RLR IFN-β induction pathway (6, 18). Furthermore, TLR3 signaling through the adapter TRIF is impaired by V proteins of rubulaviruses but not by the MV V protein (14). Both the cytoplasmic RLRs and membrane-bound TLR3 act as sensors of viral dsRNA to initiate the innate immune response and IFN induction. To ascertain which of these RNA sensors may be involved in the induction of IFN-β following Cko virus infection, we again took advantage of the transient knockdown of the corresponding adapter proteins, IPS-1 for the RLRs and TRIF for TLR3. As shown in Fig. 5A, the knockdown of the mitochondrial IPS-1 protein but not TRIF impaired the induction of IFN-β transcripts following infection with MVvac Cko virus, similar to what was seen for NF-κB activation (Fig. 4B and C). In addition, the transient knockdown of PKR in PKR+ parental cells impaired the induction of IFN-β with the Cko virus (Fig. 5A), consistent with our results obtained with the stable PKR knockdown cell line (Fig. 1). A selective decrease in the steady-state protein level of the targeted transient knockdown was verified by Western immunoblot analysis (Fig. 5B).
FIG. 5.

The MV Cko mutant induces IFN-β through the IPS-1 adapter signal transduction pathway. (A). Total RNA was prepared from parental PKR+ cells, either uninfected or infected with Cko virus, following transient knockdown utilizing siRNAs against luciferase as a control (siLUC), IPS-1, PKR, or TRIF. IFN-β transcript levels normalized to GAPDH were determined by using quantitative real-time PCR at 24 h after infection (MOI of 5) with Cko MVvac compared to uninfected cells. (B) Immunoblot analyses of whole-cell extracts prepared from uninfected mock PKR+ cells following transient knockdown with siRNAs against luciferase as a control (siLUC), IPS-1, PKR, or TRIF. Antibodies against IPS-1, TRIF, PKR, and α-tubulin were utilized.
Although significant amounts of dsRNA, a well-characterized activating trigger of RLRs and TLR3, have been detected in cells infected with positive-strand RNA viruses and DNA viruses, dsRNA was not observed in cells infected with negative-strand RNA viruses including paramyxoviruses (38). However, the IPS-1 dependence of IFN-β induction in cells infected with the Cko virus (Fig. 1 and 5), together with the observed activation of PKR phosphorylation observed for Cko virus-infected cells (Fig. 2), is consistent with an enhanced generation of activator RNA with significant double-stranded character by this mutant virus (32, 35).
Conclusions.
Taken together, our results obtained with the Moraten vaccine strain of MV and derived isogenic mutants establish that the C protein and, to a lesser extent, the V protein are necessary to counteract robust IFN-β transcription. Thus, both an attenuated vaccine strain (Moraten) as shown herein and a pathogenic strain (IC-B) (17, 18) display poor IFN-inducing capacities when C is expressed but enhanced IFN-inducing capacities when deficient of C protein expression. Furthermore, we find that the cellular protein PKR is required for maximal IFN-β induction during infection with an MV vaccine strain. The PKR-mediated enhancement of IFN-β induction by the Cko mutant involves the PKR-dependent activation of ATF-2 and NF-κB. The activation of IRF-3 was maximal in the absence of the C protein, but this activation was PKR independent. Therefore, the C protein of MV functions as a potent antagonist of IFN-β induction, impairing both PKR-dependent (ATF-2 and NF-κB) and PKR-independent (IRF-3) factor activation, leading to IFN-β induction via mitochondrial adapter IPS-1-dependent signaling. The precise mode of action of the MV C protein is unknown: this 186-residue protein can shuttle between the nucleus and cytoplasm (20), and it is conceivable that it modulates the host innate immune response by interacting directly with both cytoplasmic adapter proteins and nuclear transcription factors. The C protein also modulates viral mRNA transcription and genome replication (1, 28), and with the Cko mutant, aberrant RNA synthesis that leads to increased RNA activators of innate response proteins exemplified by RLRs, PKR, and IRF-3 may occur. Our findings establish that PKR functions to enhance IFN-β induction during MV infection with the attenuated Moraten vaccine strain, providing further insight into virus-host interactions and the mechanisms by which the MV C protein modulates the host innate immune response. Since infection with the vaccine strain recapitulates most events occurring during infection with wild viruses, it seems likely that the C proteins of WT MVs have similar mechanisms of action.
Acknowledgments
This work was supported in part by research grants AI-12520 and AI-20611 to C.E.S., AI-63476 to R.C., and AI-76462 to P.D., all from the National Institute of Allergy and Infectious Diseases, NIH, U.S. Public Health Service.
Footnotes
Published ahead of print on 21 October 2009.
REFERENCES
- 1.Bankamp, B., J. Wilson, W. J. Bellini, and P. A. Rota. 2005. Identification of naturally occurring amino acid variations that affect the ability of the measles virus C protein to regulate genome replication and transcription. Virology 336:120-129. [DOI] [PubMed] [Google Scholar]
- 2.Bellini, W. J., G. Englund, S. Rozenblatt, H. Arnheiter, and C. D. Richardson. 1985. Measles virus P gene codes for two proteins. J. Virol. 53:908-919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cattaneo, R., K. Kaelin, K. Baczko, and M. A. Billeter. 1989. Measles virus editing provides an additional cysteine-rich protein. Cell 56:759-764. [DOI] [PubMed] [Google Scholar]
- 4.Cattaneo, R., T. Miest, E. V. Shashkova, and M. A. Barry. 2008. Reprogrammed viruses as cancer therapeutics: targeted, armed and shielded. Nat. Rev. Microbiol. 6:529-540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Centers for Disease Control and Prevention. 2008. Measles—United States, January 1-April 25, 2008. MMWR Morb. Mortal. Wkly. Rep. 57:494-498. [PubMed] [Google Scholar]
- 6.Childs, K., N. Stock, C. Ross, J. Andrejeva, L. Hilton, M. Skinner, R. Randall, and S. Goodbourn. 2007. mda-5, but not RIG-I, is a common target for paramyxovirus V proteins. Virology 359:190-200. [DOI] [PubMed] [Google Scholar]
- 7.Devaux, P., V. von Messling, W. Songsungthong, C. Springfeld, and R. Cattaneo. 2007. Tyrosine 110 in the measles virus phosphoprotein is required to block STAT1 phosphorylation. Virology 360:72-83. [DOI] [PubMed] [Google Scholar]
- 8.Devaux, P., G. Hodge, M. B. McChesney, and R. Cattaneo. 2008. Attenuation of V- or C-defective measles viruses: infection control by the inflammatory and interferon responses of rhesus monkeys. J. Virol. 82:5359-5367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fontana, J. M., B. Bankamp, and P. A. Rota. 2008. Inhibition of interferon induction and signaling by paramyxoviruses. Immunol. Rev. 225:46-67. [DOI] [PubMed] [Google Scholar]
- 10.Griffin, D. E. 2007. Measles virus, p. 1551-1585. In P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus (ed.), Fields virology, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA.
- 11.Griffin, D., C. Pan, and W. Moss. 2008. Measles vaccines. Front. Biosci. 13:1352-1370. [DOI] [PubMed] [Google Scholar]
- 12.Jansen, V. A. A., N. Stollenwerk, H. J. Jensen, M. E. Ramsay, W. J. Edmunds, and C. J. Rhodes. 2003. Measles outbreaks in a population with declining vaccine uptake. Science 301:804. [DOI] [PubMed] [Google Scholar]
- 13.Lin, R., C. Heylbroeck, P. M. Pitha, and J. Hiscott. 1998. Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome-mediated degradation. Mol. Cell. Biol. 18:2986-2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lu, L. L., M. Puri, C. M. Horvath, and G. C. Sen. 2008. Select paramyxoviral V proteins inhibit IRF3 activation by acting as alternative substrates for inhibitor of kappaB kinase epsilon (IKKe)/TBK1. J. Biol. Chem. 283:14269-14276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McAllister, C. S., and C. E. Samuel. 2009. Protein kinase PKR enhances the induction of interferon-beta and apoptosis mediated by cytoplasmic RNA sensors. J. Biol. Chem. 284:1644-1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McCormack, S. J., D. C. Thomis, and C. E. Samuel. 1992. Mechanism of interferon action: identification of a RNA binding domain within the N-terminal region of the human RNA-dependent P1/eIF-2α protein kinase. Virology 188:47-56. [DOI] [PubMed] [Google Scholar]
- 17.Nakatsu, Y., M. Takeda, S. Ohno, R. Koga, and Y. Yanagi. 2006. Translational inhibition and increased interferon induction in cells infected with C protein-deficient measles virus. J. Virol. 80:11861-11867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nakatsu, Y., M. Takeda, S. Ohno, Y. Shirogane, M. Iwasaki, and Y. Yanagi. 2008. Measles virus circumvents the host interferon response by different actions of the C and V proteins. J. Virol. 82:8296-8306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Navaratnarajah, C. K., V. H. L. Leonard, and R. Cattaneo. 2009. Measles virus: glycoprotein complex assembly receptor attachment, and cell entry. Curr. Top. Microbiol. 329:59-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nishie, T., K. Nagata, and K. Takeuchi. 2007. The C protein of wild-type measles virus has the ability to shuttle between the nucleus and the cytoplasm. Microbes Infect. 9:344-354. [DOI] [PubMed] [Google Scholar]
- 21.Palosaari, H., J.-P. Parisien, J. J. Rodriguez, C. M. Ulane, and C. M. Horvath. 2003. STAT protein interference and suppression of cytokine signal transduction by measles virus V protein. J. Virol. 77:7635-7644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Panne, D., T. Maniatis, and S. C. Harrison. 2007. An atomic model of the interferon-β enhanceosome. Cell 129:1111-1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pfaller, C. K., and K.-K. Conzelmann. 2008. Measles virus V protein is a decoy substrate for the kinase IKKα and prevents Toll-like receptor 7/9-mediated interferon induction. J. Virol. 82:12365-12373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pichlmair, A., O. Schulz, C. P. Tan, T. I. Naslund, P. Liljestrom, F. Weber, and C. Reis e Sousa. 2006. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science 314:997-1001. [DOI] [PubMed] [Google Scholar]
- 25.Plumet, S., F. Herschke, J.-M. Bourhis, H. Valentin, S. Longhi, and D. Gerlier. 2007. Cytosolic 5′-triphosphate ended viral leader transcript of measles virus as activator of the RIG I-mediated interferon response. PLoS One 2:e279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ramachandran, A., J.-P. Parisien, and C. M. Horvath. 2008. STAT2 is a primary target for measles virus V protein-mediated alpha/beta interferon signaling inhibition. J. Virol. 82:8330-8338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Randall, R. E., and S. Goodbourn. 2008. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J. Gen. Virol. 89:1-47. [DOI] [PubMed] [Google Scholar]
- 28.Reutter, G. L., C. Cortese-Grogan, J. Wilson, and S. A. Moyer. 2001. Mutations in the measles virus C protein that up regulate viral RNA synthesis. Virology 285:100-109. [DOI] [PubMed] [Google Scholar]
- 29.Sadler, A. J., and B. R. G. Williams. 2007. Structure and function of the protein kinase R. Curr. Top. Microbiol. 316:253-292. [DOI] [PubMed] [Google Scholar]
- 30.Samuel, C. E. 1979. Mechanism of interferon action: phosphorylation of protein synthesis initiation factor eIF-2 in interferon-treated human cells by a ribosome-associated kinase processing site specificity similar to hemin-regulated rabbit reticulocyte kinase. Proc. Natl. Acad. Sci. U. S. A. 76:600-604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Samuel, C. E. 2001. Antiviral actions of interferons. Clin. Microbiol. Rev. 14:778-809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Takeuchi, K., T. Komatsu, Y. Kitagawa, K. Sada, and B. Gotoh. 2008. Sendai virus C protein plays a role in restricting PKR activation by limiting the generation of intracellular double-stranded RNA. J. Virol. 82:10102-10110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.tenOever, B. R., M. J. Servant, N. Grandvaux, R. Lin, and J. Hiscott. 2002. Recognition of the measles virus nucleocapsid as a mechanism of IRF-3 activation. J. Virol. 76:3659-3669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Toth, A. M., P. Zhang, S. Das, C. X. George, and C. E. Samuel. 2006. Interferon action and the double-stranded RNA-dependent enzymes ADAR1 adenosine deaminase and PKR protein kinase. Prog. Nucleic Acid Res. Mol. Biol. 81:369-434. [DOI] [PubMed] [Google Scholar]
- 35.Toth, A. M., P. Devaux, R. Cattaneo, and C. E. Samuel. 2009. Protein kinase PKR mediates the apoptosis induction and growth restriction phenotypes of C protein-deficient measles virus. J. Virol. 83:961-968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Uematsu, S., and S. Akira. 2007. Toll-like receptors and type I interferons. J. Biol. Chem. 282:15319-15323. [DOI] [PubMed] [Google Scholar]
- 37.Ward, S. V., and C. E. Samuel. 2003. The PKR kinase promoter binds both Sp1 and Sp3, but only Sp3 functions as part of the interferon-inducible complex with ISGF-3 proteins. Virology 313:553-566. [DOI] [PubMed] [Google Scholar]
- 38.Weber, F., V. Wagner, S. B. Rasmussen, R. Hartmann, and S. R. Paludan. 2006. Double-stranded RNA is produced by positive-strand RNA viruses and DNA viruses but not in detectable amounts by negative-strand RNA viruses. J. Virol. 80:5059-5064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yokota, S.-I., H. Saito, T. Kubota, N. Yokosawa, K.-I. Amano, and N. Fujii. 2003. Measles virus suppresses interferon-α signaling pathway: suppression of Jak1 phosphorylation and association of viral accessory proteins, C and V, with interferon-α receptor complex. Virology 306:135-146. [DOI] [PubMed] [Google Scholar]
- 40.Yoneyama, M., and T. Fujita. 2007. Function of RIG-I-like receptors in antiviral innate immunity. J. Biol. Chem. 282:15315-15318. [DOI] [PubMed] [Google Scholar]
- 41.Zhang, P., and C. E. Samuel. 2007. Protein kinase PKR plays a stimulus- and virus-dependent role in apoptotic death and virus multiplication in human cells. J. Virol. 81:8192-8200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang, P., B. L. Jacobs, and C. E. Samuel. 2008. Loss of protein kinase PKR expression in human HeLa cells complements the vaccinia virus E3L deletion mutant phenotype by restoration of viral protein synthesis. J. Virol. 82:840-848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang, P., and C. E. Samuel. 2008. Induction of protein kinase PKR-dependent activation of interferon regulatory factor 3 by vaccinia virus occurs through adapter IPS-1 signaling. J. Biol. Chem. 283:34580-34587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang, P., J. O. Langland, B. L. Jacobs, and C. E. Samuel. 2009. Protein kinase PKR-dependent activation of mitogen-activated protein kinases occurs through mitochondrial adapter IPS-1 and is antagonized by vaccinia virus E3L. J. Virol. 83:5718-5725. [DOI] [PMC free article] [PubMed] [Google Scholar]