

Abstract
We previously reported coreceptor switch in rhesus macaques inoculated intravenously with R5 simian-human immunodeficiency virus SF162P3N (SHIVSF162P3N). Whether R5-to-X4 virus evolution occurs in mucosally infected animals and in which anatomic site the switch occurs, however, were not addressed. We herein report a change in coreceptor preference in macaques infected intrarectally with SHIVSF162P3N. The switch occurred in infected animals with high levels of virus replication and undetectable antiviral antibody response and required sequence changes in the V3 loop of the gp120 envelope protein. X4 virus emergence was associated with an accelerated drop in peripheral CD4+ T-cell count but followed rather than preceded the onset of CD4+ T-cell loss. The conditions, genotypic requirements, and patterns of coreceptor switch in intrarectally infected animals were thus remarkably consistent with those found in macaques infected intravenously. They also overlapped with those reported for humans, suggestive of a common mechanism for coreceptor switch in the two hosts. Furthermore, two independent R5-to-X4 evolutionary pathways were identified in one infected animal, giving rise to dual-tropic and X4 viruses which differed in switch kinetics and tissue localization. The dual-tropic switch event predominated early, and the virus established infection in multiple tissues sites. In contrast, the switch to X4 virus occurred later, initiating and expanding mainly in peripheral lymph nodes. These findings help define R5 SHIVSF162P3N infection of rhesus macaques as a model to study the mechanistic basis, dynamics, and sites of HIV-1 coreceptor switch.
The human immunodeficiency virus (HIV) enters target cells via binding of the viral envelope glycoprotein to the CD4 receptor, triggering envelope conformational changes that allow for interaction with either the CCR5 or CXCR4 chemokine receptor (1, 3, 8, 15, 16, 18). Most HIV type 1 (HIV-1) transmissions are initiated with CCR5-using (R5) viruses (58, 68). With time, CXCR4-tropic (X4) viruses emerge and coexist with R5 viruses in close to 50% of subtype B-infected individuals, and this is accompanied by a rise in viremia, rapid CD4+ T-cell loss, and progression to disease (4, 7, 11, 34, 57, 65). The mechanistic basis and reasons for HIV-1 coreceptor switch, however, are still not well understood. Several factors including high viral load, low CD4+ T-cell numbers, reduced availability of CCR5+ cells, and progressive immune dysfunction have been proposed as playing important roles (48, 54). Since X4 virus emergence is associated with a faster rate of disease progression, insights into the determinants of HIV-1 coreceptor switch are of interest in understanding viral pathogenesis. Furthermore, with the introduction of CCR5 entry inhibitors as anti-HIV therapeutics (19, 23, 24, 38), there is a need not only to identify the presence of X4 variants in patients when treatment options are considered but also to understand the factors that influence X4 virus evolution. Although the majority of individuals failing on short-term CCR5 antagonist monotherapy harbor preexisting minor X4 variants (71), it is conceivable that given the right conditions and selective forces, inhibiting HIV-1 entry via CCR5 may drive the virus to evolve to CXCR4 usage and exacerbate disease. An animal model that faithfully recapitulates the process of coreceptor switch will be highly useful to study and identify the determinants and conditions that facilitate the change in coreceptor preference. In addition, an animal model provides the opportunity to track the kinetics of coreceptor switching at different anatomical sites, which may inform on the mechanisms of X4 virus emergence.
In this regard, we recently reported coreceptor switch in two of nine rhesus macaques (RM) inoculated intravenously with simian-human immunodeficiency virus SF162P3N (SHIVSF162P3N) that bears an HIV-1 CCR5-tropic Env (28, 29). In order to establish a reproducible model for coreceptor switch, however, it was crucial to document additional switching events. Furthermore, since the majority of HIV transmission occurs via mucosal surfaces, it was important to demonstrate coreceptor switch in macaques infected with R5 SHIVSF162P3N by the mucosal route to validate this animal model in studying the in vivo evolution of HIV-1 coreceptor usage. Additionally, the tissue compartment(s) where CXCR4-using viruses evolve and expand is not well characterized. A recent study indicates that the thymus may play an important role in the evolution and/or amplification of coreceptor variants in pediatric HIV infection (56). Since the thymus is the primary source of T lymphopoiesis during early life (45) and since CXCR4 is the predominant coreceptor expressed on thymocytes (33, 64), this organ would seem to provide the ideal milieu for X4 amplification in infants and children. Indeed, we previously showed that whereas X4 SHIV infection of newborn RM resulted in severe thymic involution, R5 SHIV infection induced only a minor disruption in thymic morphology (55), lending support to the idea that the thymus is a preferred site for X4 replication in pediatric HIV infections. Nevertheless, thymopoietic function declines with age (17, 42, 60), and naïve T cells that express high levels of CXCR4 are also enriched in peripheral lymph nodes (5, 27, 36, 66). Thus, the role of the thymus and other lymphoid tissues in HIV-1 coreceptor switch in older individuals remains to be determined. To address these issues, we inoculated adult RM intrarectally (i.r.) with R5 SHIVSF162P3N and performed frequent longitudinal blood and tissue samplings. Our goal was to document changes in coreceptor preference in mucosally infected macaques, as well as to obtain a more detailed picture of the kinetics and site of X4 virus evolution and amplification in vivo.
MATERIALS AND METHODS
Animal inoculation and clinical assessments.
All inoculations were carried out in adult RM (Macaca mulatta) individually housed at the Tulane National Primate Research Center in compliance with its Guide for the Care and use of Laboratory Animals. Animals were confirmed to be serologically negative for simian type D retrovirus, SIV, and simian T-cell lymphotropic virus prior to infection. Animals were inoculated i.r. with 104 50% tissue culture infectious doses of R5 SHIVSF162P3N. This virus was generated through successive rapid transfer in RM of the molecular clone SHIVSF162, recovered from passage 3 macaque T353 at the time of necropsy (25, 28). Whole blood was collected weekly, and an external lymph node was sampled before and at 4, 8, 12, and 16 weeks postinfection (wpi). Furthermore, surgery was performed at peak (2 to 3 wpi) and postacute (13 wpi) infection for collection of tissues including a superficial external (inguinal or axillary) lymph node and one or more internal lymph nodes (iliac, colonic, and mesenteric). Additional tissues collected included sections or wedges of small intestinal jejunum and ileum, bone marrow, thymus, and spleen. Briefly, before sample collection, animals were preanesthetized with ketamine-HCl and then maintained on isoflurane and oxygen. Peripheral lymph nodes were obtained via a small skin incision in the inguinal or axillary region. An open laparotomy was used to obtain 20-cm sections of jejunum, which were collected by resection and anastomosis. Smaller wedge biopsies of ileum and colon were also obtained. Colonic and small intestinal mesenteric lymph nodes were obtained from each animal, and iliac lymph nodes were collected when present. Approximately 2 ml of bone marrow was collected from the femur, and thymic biopsy samples were obtained via a minimally invasive procedure using a rigid thoracoscope and cup biopsy forceps. For the spleen, a mechanical stapling device was used to harvest a small wedge. Analgesics were provided while animals were anesthetized and for several days postsurgery. Plasma viremia was quantified by branched DNA analysis (Siemens Medical Solutions Diagnostic Clinical Lab, Emeryville, CA), and absolute CD4+ and CD8+ cell counts were monitored by TruCount (BD Biosciences, Palo Alto, CA). Animals with clinical signs of AIDS were euthanized by intravenous administration of ketamine-HCl, followed by an overdose of sodium pentobarbital, and tissue from multiple sites was collected. Fresh tissue cells were prepared for immunophenotyping and archiving. A portion of each sample was frozen for DNA extraction, while another was fixed in Z-fix, embedded in paraffin, and stored at room temperature for immunohistological analyses when needed. The percentages of CD4+ T cells in the tissue samples were analyzed by flow cytometry (FACSCalibur) using CD3-fluorescein isothiocyanate (FITC), CD4-phycoerythrin, and CD8-peridinin chlorophyll protein antibodies. Except for CD3-FITC (BioSource, Camarillo, CA), all antibodies were obtained from BD Biosciences.
Cell culture.
Rhesus peripheral blood mononuclear cells (PBMCs) were obtained by Ficoll-Hypaque gradient purification, followed by stimulation with 3 μg/ml Staphylococcus enterotoxin B (SEB; Sigma-Aldrich, St. Louis, MO) in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS), 2 mM glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 20 U/ml of interleukin-2 (Novartis, Emeryville, CA). 293T cells and TZM-bl cells expressing CD4, CCR5, and CXCR4 and containing integrated reporter genes for firefly luciferase and β-galactosidase under the control of the HIV-1 long terminal repeat (70) were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FBS, penicillin, streptomycin, and l-glutamine. U87 cells stably expressing CD4 and one of the chemokine receptors (15) were maintained in DMEM supplemented with 10% FBS, antibiotics, 1 μg/ml puromycin (Sigma-Aldrich), and 300 μg/ml G418 (Geneticin; Invitrogen, Carlsbad, CA).
Virus isolation.
SHIVs were recovered and amplified by culturing plasma or PBMCs from infected macaques with SEB-stimulated PBMCs from uninfected monkeys. p27 Gag antigen content of the recovered virus was quantified according to the manufacturer's instructions (Coulter Corp., Miami, FL).
DNA, RNA extraction, sequencing, and analysis.
Proviral DNA was extracted from 3 × 106 infected RM PBMCs or tissue cells with a DNA extraction kit (Qiagen) while viral RNA was prepared from 300 to 500 μl of plasma or virus supernatant using a commercially available RNA extraction kit (Qiagen, Chatsworth, CA), followed by reverse transcription (RT) with Superscript III RT (Invitrogen) and random hexamer primers (Amersham Pharmacia, Piscataway, NJ). The V1 to V5 region of gp120 was amplified from the viral DNA or RT products by Taq DNA polymerase (Qiagen) with primers ED5 and ED12 or ES7 and ES8 as previously described (14). PCR products were cloned with the TOPO TA cloning kit (Invitrogen) per the manufacturer's instructions, followed by automated sequencing of cloned gp120 amplicons (Genewiz, South Plainfield, NJ). Nucleotide sequences were aligned with ClustalX (39) and edited manually using BioEdit, version 7.0.9. A neighbor-joining tree was constructed using a maximum-composite-likelihood model in the MEGA, version 4, software (62). Gaps were treated as pairwise deletions, and bootstrap values were generated with 1,000 replicates. The substitution rates among sites were set in gamma distribution with gamma parameter at 1.0. The tree was also tested in PhyML using the general time reversible model, with estimated gamma distribution and no invariable sites, and bootstrap was set at 1,000 interactions (22).
Plasmid constructs and pseudotyped virus production.
For expression of envelope glycoproteins, full-length gp160 coding sequences of viruses recovered from macaques at end-stage disease was amplified from RT products with primers SH43 (5′-AAGACAGAATTCATGAGAGTGAAGGGGATCAGGAAG-3′) and SH44 (5′-AGAGAGGGATCCTTATAGCAAAGCCCTTTCAAAGCCCT-3′) and subcloned into the pCAGGS vector. For construction of V3 recombinant plasmids, PCR-based overlapping extension methodology was employed to replace the V3 loop of Env P3N with that of variants from infected macaques, as described previously (27), and the resulting V3 chimeric plasmids were verified by sequencing. To generate luciferase reporter viruses capable of only a single round of replication, an envelope trans-complementation assay was used as previously described (10). Briefly, the Env expression plasmid and the NL4.3LucE-R- vector were cotransfected by polyethylenimine (Polyscience, Warrington, PA) into 2.5 × 106 293T cells plated in a 100-mm plate. Cell culture supernatants were harvested 72 h later, filtered through 0.45-μm-pore-size filters, and stored at −70°C in 1-ml aliquots. Pseudotyped viruses were quantified for p24 Gag content (Beckman Coulter, Fullerton, CA).
Determination of coreceptor usage.
Coreceptor usage of replication-competent SHIVs and pseudotyped reporter viruses was determined by infection of U87.CD4 indicator cell lines. Alternatively, infection of TZM-bl cells was blocked with the CCR5 inhibitor TAK-779 or the CXCR4 antagonist AMD3100. For infection with replication-competent SHIVs, 104 U87.CD4, U87.CD4.CXCR4, and U87.CD4.CCR5 cells in each well of a 12-well plate were infected with 3 ng of p27 Gag antigen equivalent of the indicated virus for 3 h at 37°C. Infected cells were then washed three times and cultured in 2 ml of medium for 7 to 10 days at 37°C, with supernatants collected every 2 to 3 days for p27 Gag antigen content quantification (Coulter Corp., Miami, FL). For assessment of coreceptor usage of reporter viruses, 7 × 103 U87.CD4.CCR5 or U87.CD4.CXCR4 cells were seeded in 96-well plates 24 h before use and infected, in triplicates, with 2 to 3 ng of p24 Gag equivalent of the indicated pseudotyped viruses, followed by incubation for 72 h at 37°C. Infection of U87.CD4 cells served as a negative control. At the end of the incubation period, the cells were harvested, lysed, and processed for luciferase activity according to the manufacturer's instructions (Luciferase Assay System; Promega, Madison, WI). Entry, as quantified by luciferase activity, was measured with an MLX microtiter plate luminometer (Dynex Technologies, Inc., Chantilly, VA). For the entry-blocking experiments, 7 × 103 TZM-bl cells per well of a 96-well plate were inoculated, in triplicates, with 2 to 3 ng of p24 Gag antigen equivalent of the indicated pseudotyped virus in the absence or presence of 1 μM CXCR4 inhibitor AMD3100 or CCR5 inhibitor TAK-779. The cells were lysed after 72 h at 37°C and processed for β-galactosidase activity (Galacto-Star System; Applied Biosystems, Bedford, MA). Relative entry was then determined by calculating the amount of β-galactosidase activity in the presence of inhibitor relative to that in the absence of inhibitor.
Neutralization assay.
Virus neutralization was assessed using TZM-bl cells in 96-well plates. Briefly, equal volumes (50 μl) of SHIV (1 ng of p27 Gag equivalent) and fourfold serial dilutions of CD4-immunoglobulin G2 (IgG2) fusion protein (PRO 542; Progenics Pharmaceuticals, Tarrytown, NY) were incubated for 1 h at 37°C and then added to cells, in triplicate wells, for an additional 2 h at 37°C. A total of 100 μl of medium was then added to each well, and the virus-protein cultures were maintained for 72 h. Control cultures received virus in the absence of CD4-IgG2. At the end of the culture period, the cells were lysed and processed for β-galactosidase activity. A neutralization curve was generated by plotting the percentage of neutralization versus fusion-protein dilution, and 50% inhibitory concentrations (IC50s) were determined using Prism, version 4, software (GraphPad, San Diego, CA).
Sequence-specific PCR.
Detection of HR (histidine and arginine) sequence in envelope amplicons from plasma, PBMCs or tissue cells was performed as previously described with minor modifications (28). Briefly, cellular DNA and plasma cDNA products from various time points postinfection were subjected to PCR using Hot Star Taq DNA polymerase and first-round primers SH70 (5′-AAGAAAAAGTATACGTATACATAG-3′) and V3-OAS (5′-CAGTAGAAAAATTCCCCTCCACA-3′) using the following cycling profile: 95°C for 15 min followed by 25 cycles of 95°C for 15 s, 61.4°C for 30 s, and 72°C for 60 s. Second-round PCR was a seminested reaction with primers SH70 and V3-AS (5′-AATTTCTGGGTCCCCTCCTGA-3′) for 40 cycles using the same cycling conditions. Amplified products were visualized by electrophoresis in ethidium bromide-stained 2% agarose gels. Appropriate mixing and titration experiments with R5 and X4 variant target sequences that differ in the HR region showed that the sensitivity of this detection assay was one X4 variant copy among 105 R5 targets. For detection of the V3 loop sequence carrying a deletion of residues 22 to 25 (Δ22-25), plasma cDNA products were subjected to PCR with primers V3-del (5′-AATTAAAACTGTGCATTACAA-3′) and WR8 (5′-CGGGGAGAGCATTTTACATA-3′) using the following cycling conditions: 95°C for 10 min followed by 35 cycles of 95°C for 30 s, 57.5°C for 20 s, and 72°C for 20 s, with final extension at 72°C for 10 min. The sensitivity of the detection assay for Δ22-25 V3 sequence was one variant copy among 104 R5 targets.
RESULTS
R5 SHIVSF162P3N is mucosally transmissible and replicates to high levels in infected macaques.
The major route of HIV-1 transmission is across mucosal surfaces. To determine whether, similar to HIV-1, R5 SHIVSF162P3N can transmit across mucosal surfaces and evolve to use CXCR4, we inoculated six Indian RM by the i.r. route. All six macaques were infected, with three different patterns of virus replication. Two of the six infected macaques (animals DG08 and DR16) had peak viremia of 7 to 8 log10 RNA copies/ml plasma at 2 wpi and continued to replicate virus at high levels, with progression to disease and euthanasia within 20 wpi (Fig. 1A). A notable spike in viremia (1-log increase) was seen in DG08 at 14 wpi and in DR16 at 16 wpi, near the time of necropsy. Three macaques (EN13, DG88, and DN57) had peak viremia of 7 log10 RNA copies/ml plasma. Virus replication declined thereafter, reaching set points of 5 to 6 log10 RNA copies/ml plasma. One of these three animals (DN57) developed clinical signs consistent with AIDS and was euthanized at 30 wpi. The sixth macaque (DP85) had peak and set point viremias that were 2 to 4 logs lower than in other animals. Major histocompatibility complex genotyping showed that this animal was B*17 positive, an allele known to be associated with efficient control of SIV replication (75). EN13, DG88, DN57, and DP85 seroconverted by 3 to 4 wpi, but DG08 and DR16 failed to develop an anti-SHIV binding antibody response (data not shown). The high levels of virus replication, undetectable antiviral antibody response, and fast disease development in DG08 and DR16 classified them as rapid progressors (RPs).
FIG. 1.

(A) Viral load and absolute CD4+ T-cell count in SHIVSF162P3N i.r. inoculated macaques. +, time of euthanasia. (B) Percentages of CD4+ T cells in the colonic (Col), inguinal (Ing), and mesenteric (Mes) lymph nodes and IELs and LPLs from the jejunum during acute infection (week 2 or 3) and at time necropsy of macaques DN57, DG08, and DR16 are shown. Baseline values generated from three uninfected macaques are used for reference (100% values), with error bars indicating standard deviations.
A drop in peripheral CD4+ T-cell count accompanied peak virus replication in all infected macaques, and this was followed by a progressive CD4+ T-cell decline. The exceptions were DG08 and DR16, the two macaques with high viral set points and rapid progression to disease, who showed accelerated and dramatic loss of CD4+ T cells toward end-stage disease (Fig. 1A). Analysis of tissue cells collected at the time of euthanasia showed depletion of gut as well as lymph node CD4+ T cells in these two animals, with DG08 displaying more severe loss in lymph nodes than DR16 (>95% and 80% CD4+ T-cell loss, respectively) (Fig. 1B). In contrast, whereas CD4+ T cells were depleted in the gut laminar propria of DN57, 30 to 40% of these target cells were preserved in secondary lymphoid tissues such as the colonic, inguinal, and mesenteric lymph nodes. The pattern of high virus replication, precipitous peripheral as well as lymphoid CD4+ T-cell loss, and lack of antiviral antibody response in DG08 and DR16 was reminiscent of conditions associated with X4 emergence in SHIVSF162P3N intravenously (i.v.)-infected macaques, prompting us to examine the gp120 V3 loop sequence and coreceptor usage of viruses present in these animals at the time of overt AIDS.
Emergence of evolutionarily divergent X4 and dual-R variants in SHIVSF162P3N i.r. infected macaques that rapidly progressed to disease.
Indeed, analysis of plasma and PBMC samples demonstrated that V3 loop sequences predictive of CXCR4 usage could be detected in DG08 and DR16 (Fig. 2). For DG08, all plasma-derived clones had sequences similar to the inoculum-dominant sequence (wild-type, [WT]), but insertion of two basic amino acids (histidine and arginine) immediately upstream of the GPGR crown V3 loop sequence, a change that increased the net positive charge of this domain, was present in 2 of 10 envelope clones sequenced from PBMCs. This HR insertion sequence can be found in the Amsterdam cohort patient Ams-32, and was previously shown to be associated with CXCR4 usage in SHIVSF162P3N i.v.-infected monkey BR24 (28). Furthermore, a potential N-linked glycosylation site (PNGS) at the base of the V3 loop has been demonstrated to modify virus tropism and neutralization sensitivity (2, 40, 43, 44, 53). This PNGS, which was lacking in BR24 and Ams-32, was absent in one of the two HR-containing V3 loop sequences from DG08 PBMCs, which should increase neutralization sensitivity of the virus and facilitate coreceptor switch. Of the eight remaining PBMC-derived clones, five were WT, but three carried a 4-amino-acid deletion in the stem of the V3 loop (ATGD; Δ22-25), which increased the net positive charge of this domain to +6, with the N-terminal PNGS intact. For DR16, replacement of the serine residue at position 11 of the V3 loop with the positively charged amino acid arginine, a sequence motif commonly associated with HIV-1 CXCR4 usage (20), could be detected in the majority of envelope sequences amplified from both PBMCs and plasma. In contrast, only V3 sequences similar to that of the R5 SHIV162P3N inoculum were present in DN57.
FIG. 2.

V3 loop sequence comparison of SHIVSF162P3N, SHIVBR24N, reference HIV-1 X4 viruses HXB2 and Ams-32, and PBMC and plasma viruses in macaques DG08, DR16, and DN57 near end-stage disease. Dots indicate gaps; dashes stand for identity in sequences. The net positive charge of this region is shown on the right. Positions 11 and 25 within the V3 loop are indicated by arrows, the PNGS is in bold, and the 4-amino-acid deletion in DG08 is underlined. The numbers in parentheses represent the numbers of clones matching the indicated sequence per total number of clones sequenced. Amino acid residues predicted to confer CXCR4 usage are boxed.
We recovered viruses from macaques DG08, DR16, and DN57 postmortem and tested their coreceptor usage by infecting U87 indicator cell lines. Results showed productive infection of U87.CD4.CCR5 as well as U87.CD4.CXCR4 cells with viruses from macaques DG08 and DR16 but infection of U87.CD4.CCR5 cells only with virus from DN57 (Fig. 3A). These findings indicate that while the replicating virus in macaque DN57 maintained CCR5 usage, as expected, viruses recovered from DG08 and DR16 used CXCR4 instead of or in addition to CCR5. To assess the phenotypic composition of envelope variants present in viruses from macaques DG08 and DR16, full-length gp160 was amplified and used in the generation of pseudotyped reporter viruses. Infectivity studies with pseudotyped viruses in the U87 indicator cell lines demonstrated that envelope variants from DG08 bearing WT V3 loop sequence used only CCR5, but those bearing HR insertions in the V3 loop now used CXCR4 almost exclusively for entry (Fig. 3B). In contrast, virus pseudotyped with envelope bearing the Δ22-25 V3 loop sequence found in DG08 or the S11R V3 loop sequence present in macaque DR16 infected CXCR4- as well as CCR5-expressing cells, indicating that they functioned with both coreceptors. Thus, while viruses in macaques DG08 are comprised of a mixture of R5, X4, and dual-tropic viruses, those in macaque DR16 are predominantly dual-tropic.
FIG. 3.

Coreceptor usage of viruses recovered from macaques DG08, DR16, and DN57 at time of overt AIDS. Coreceptor utilization, determinants, and preference were assessed by infection of U87.CD4.CCR5 (black bars) and U87.CD4.CXCR4 (striped bars) cells with SHIV viruses recovered from macaques DG08, DR16, and DN57 near the time of death (A); luciferase reporter viruses expressing the envelopes with WT or Δ22-25- or HR-bearing V3 loop sequence of macaque DG08 and the S11R V3 loop sequence of DR16 (B); and V3 chimeric pseudotyped viruses (C). (D) Blocking of entry of the indicated pseudotyped viruses into TZM-bl cells with 1 μM CCR5-specific (TAK779) and CXCR4-specific (AMD3100) inhibitors. Data in panels B, C, and D are the means and standard deviations from triplicate wells and are representative of at least two independent experiments. RLU, relative light units.
To determine if sequence changes in the V3 loop alone are sufficient to determine CXCR4 usage, recombinant viruses expressing the V3 domain of DG08 and DR16 Env variants in the backbone of parental SHIVSF162P3N Env were generated and tested for coreceptor preference in single-round infectivity assays. Results showed that the HR and Δ22-25 V3 chimeric viruses infected U87.T4.X4 but not U87.CD4.R5 cells, while the S11R V3 chimeric virus infected both U87.CD4.X4 and U87.CD4.R5 cells but with greater efficiency in the latter (Fig. 3C). These findings demonstrate that the V3 sequence changes alone confer CXCR4 usage to the dual-tropic and X4 viruses in DG08 and DR16. Interestingly, neighbor-joining tree analysis of the V1-V5 region of the HR- and Δ22-25-bearing Env sequences in DG08 showed clustering on different branches of the phylogenetic tree (Fig. 4), suggestive of the coexistence of distinct switch events in this animal. Furthermore, these signature V3 sequences were not present in the original SHIV inoculum when it was probed using sequence-specific PCR assays that are able to detect one Δ22-25 and HR variant copy among 104 and 105 R5 targets, respectively (data not shown), supporting evolution rather than outgrowth of dual-tropic and X4 viruses in DG08.
FIG. 4.
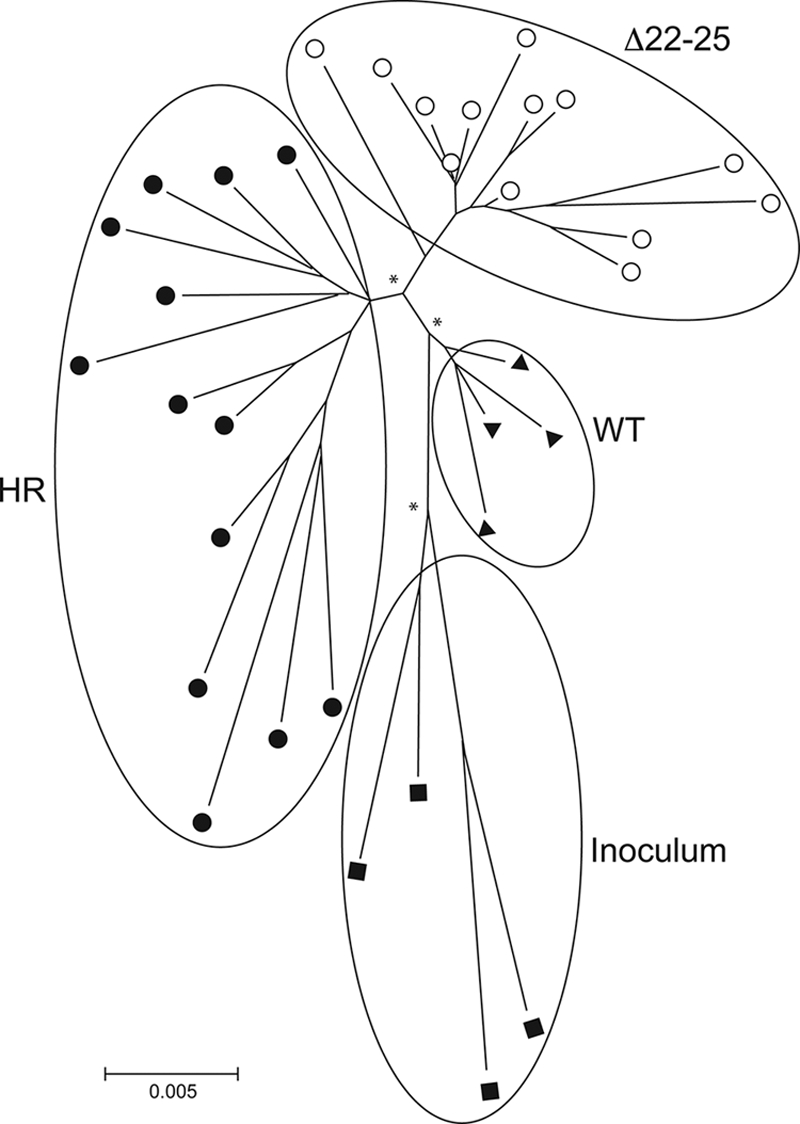
Phylogenetic tree showing the relationship between Env variant sequences (V1 to V5) in macaque DG08. A neighbor-joining tree rooted on the sequences of the inoculating virus SHIVSF162P3N was generated. The scale bar indicates the genetic distance along the branches in nucleotides, and the asterisk along a branch represents a bootstrap value of >70%. ▪, representative clones of the inoculating virus; ▴, WT variants; •, HR-bearing variants; and ○, Δ22-25-bearing variants.
Phenotypically distinct categories of dual-tropic HIV-1 variants have been described, with those that prefer CCR5 over CXCR4 usage being classified as “dual-R” tropic while those that use CXCR4 more efficiently than CCR5 are designated “dual-X” (21, 30). To investigate coreceptor preference of the dual-tropic viruses in DG08 and DR16, blocking experiments with CCR5 and CXCR4 antagonists in TZM-bl cells that express both coreceptors were performed. Pseudotyped viruses bearing WT and HR insertion sequences from DG08 served as R5 and X4 controls, respectively. Results confirmed coreceptor preference of viruses pseudotyped with DG08 WT or HR V3 loop-bearing envelopes (Fig. 3D). Infection with WT V3 pseudotyped virus was blocked with the CCR5 inhibitor TAK-779 but not with the CXCR4 inhibitor AMD3100 while the converse was observed for viruses containing HR insertions in the V3 loop. Infection of TZM-bl cells with virus expressing the Δ22-25 V3 sequence of DG08, however, was partially (50%) blocked by TAK-779 and minimally (<5%) by AMD3100 while infection mediated by virus pseudotyped with the S11R V3 sequence of DR16 was efficiently blocked with the CCR5 but not the CXCR4 inhibitor. Thus, although the Δ22-25 and S11R Env variants could function with CXCR4, they showed a preference for CCR5 usage and thus could be classified as dual-R.
The newly emerging X4 viruses in SHIVSF162P3N i.v. infected macaques were found to be highly sensitive to neutralization with soluble CD4 (CD4-IgG2) (28, 29). To determine whether viruses recovered from i.r. infected macaques with coreceptor switch shared this biological property, their susceptibility to CD4-IgG2 neutralization was examined. Results showed that viruses in DG08 and DR16 at end-stage disease were also highly sensitive to soluble CD4 neutralization (data not shown). Compared to the inoculating SHIVSF162P3N virus, with an IC50 of 2.1 μg/ml of CD4-IgG2, the IC50s for DG08 (0.014 μg/ml) and DR16 (0.08 μg/ml) were 150- and 26-fold lower.
Different switch kinetics and tissue localization of X4 and dual-R viruses in infected macaque DG08.
As an objective of this study was to obtain a more dynamic picture of the tempo and anatomical site(s) of R5-to-X4 virus evolution and amplification, frequent samplings were performed in the SHIVSF162P3N i.r. inoculated macaques, which allowed genotypic and phenotypic analyses when warranted. These included weekly bleeds, longitudinal (4, 8, 12, and 16 wpi) biopsy samples from an axillary lymph node, surgery at acute (2 wpi) and postacute (13 wpi) stages of infection, and full necropsy at the time of euthanasia for collection of tissues from lymphoid and nonlymphoid organs. We first used the highly sensitive sequence-specific PCR assays to track the emergence of X4 HR and dual-R Δ22-25 viruses in the plasma of DG08. Results showed that neither the X4 nor the dual-R signature sequence could be detected in the plasma of macaque DG08 prior to 12 wpi (Fig. 5A). Weak PCR signal was present 1 week later (13 wpi) for the X4 HR sequence but was readily detectable at 14 wpi, a time that coincided with the 1-log rise in plasma viremia (Fig. 5B). Amplification signal for the X4 HR-specific sequence remained strong up to 18 wpi but was weaker at time of euthanasia (20 wpi). For the dual-R Δ22-25 V3 variant, strong and persistent PCR signal was present in the plasma from 13 to 18 wpi but was undetectable at the time of necropsy. Consistent with observations made in the i.v. infected macaques (28, 29), emergence of the X4 and dual-R viruses in DG08 was associated with accelerated peripheral CD4+ T-cell loss but appeared after and not before the onset of CD4+ T-cell decline at 12 wpi.
FIG. 5.

Emergence of X4 and dual-R variants in plasma of macaque DG08 as tracked by the presence of the signature X4 HR insertion and dual-R Δ22-25 motifs in plasma over time using sequence-specific PCR (A) and relationship of X4 and dual-R emergence in plasma to plasma viral load and CD4+ T-cell count (B). +, presence of sequence; −, absence of signature sequence; ±, weak signal; m, marker.
Surgery performed for DG08 at 13 wpi, a time point concomitant with the first detection of X4 HR and dual-R Δ22-25 V3 signals in the plasma by differential PCR, provided us with the unique opportunity to identify the site of R5-to-X4 evolution and amplification in this macaque. Accordingly, V3 sequence analysis of envelope clones amplified from tissue cells was performed to determine the localization as well as the frequency of the different tropism variants. Results showed that the dual-R Δ22-25 variant was present in the lamina propria lymphocyte (LPL) of the gut at high frequency (54%) and at reduced and varying frequencies (3 to 25%) in the spleen, bone marrow, inguinal and mesenteric lymph nodes (LNs), but absent in the intraepithelial lymphocytes (IELs) of the gut and the thymus (Fig. 6A). The X4 variant, however, could not be detected by sequence analysis of over 30 Env clones from each of the tissue sites, suggesting that it was present at frequencies lower than that of the dual-R Δ22-25 virus at this time in infection. At the time of necropsy 7 weeks later, the dual-R Δ22-25 virus could still be found in the mesenteric as well as the inguinal, iliac, and axillary LNs; spleen; and LPLs (3 to 20%) but not in the colonic LN, IELs, bone marrow, or the thymus. In contrast, the X4 HR variant now represented 3 to 31% of the envelope clones sequenced from superficial external (axillary and inguinal) as well as internal (iliac, colonic, and mesenteric) LNs but was not present in the gut, bone marrow, or thymus. Together, these findings in DG08 suggest that although the X4 and dual-R viruses emerged in parallel in DG08 plasma at 13 wpi, the dual-R Δ22-25 mutational event preceded the X4 HR switch pathway temporally, explaining its predominance and presence in multiple tissue sites at this time in infection. That the dual-R Δ22-25 PCR signal was stronger than that of the X4 HR signal seen in the plasma, despite lower sensitivity of the Δ22-25 PCR detection method, supports this scenario. The decreased frequency of the dual-R variant in tissue cells, notably in the gut (from 54% to 4%), bone marrow (from 7% to 0%), and mesenteric LN (26% to 15%) at 20 wpi compared to 13 wpi, however, implied a loss of competitive fitness of this virus over time. Conversely, the increase in tissue representation of the X4 variant over the same period of time implied gain of fitness. Furthermore, the X4 and dual-R viruses differed in anatomical localization, with the former residing mainly within lymph nodes and not in the gut while the latter had a much wider distribution, establishing infection in peripheral as well as mucosal lymphoid tissues.
FIG. 6.

Distribution of V3 variants in tissue compartments of macaques DG08 (A) and DR16 (B) at 13 wpi and time of necropsy as determined by clonal sequence analysis. Numbers in parentheses indicate number of gp120 clones sequenced from each of the tissue sites. Ing, inguinal LN; Mes, mesenteric LN; Spl, spleen; Thy, thymus; BM, bone marrow; axi, axillary LN; ili, iliac LN; Col, colonic LN.
In the case of macaque DR16, the animal was too ill at 13 wpi for full surgery to be performed, with only samples from axillary LN and bone marrow collected. Sequence analysis of amplified Env clones showed the presence of only WT viruses (Fig. 6B). However, a rise in viral load that may be indicative of X4 emergence was seen at 16 wpi. Indeed, sequence analysis of Env clones derived from 15 wpi PBMCs (n = 24) failed to detect the signature dual-R S11R V3 loop motif, but 20/20 clones derived from 16 wpi PBMCs harbored this V3 mutation (data not shown). The switch point in DR16, therefore, also followed, rather than preceded, the onset of precipitous CD4+ T-cell decline at 13 wpi. Furthermore, the dual-R S11R sequence was dominant in all tissue samples examined at the time of necropsy 2 days later, including the LPLs, spleen, and thymus (Fig. 6B), compatible with a generalized switch. This latter finding, together with results seen for the dual-R Δ22-25 V3 variant when it first emerged in DG08, supports systemic spread and amplification of variants that can use both CXCR4 and CCR5.
Peripheral lymph nodes are the site of X4 evolution and amplification.
The inability to detect the X4 HR variant in tissue cells of DG08 at 13 wpi by clonal sequence analysis despite its detection in plasma by differential PCR suggested limited virus spread, prompting us to identify its tissue compartmentalization using the PCR detection method, which is more sensitive. Results showed clear presence of the X4 virus in the inguinal LN but not in PBMCs, mesenteric LN, bone marrow, spleen, thymus, or the LPLs sampled at 13 wpi (Fig. 7A). Consistent with data obtained by clonal sequencing, the X4 variant was found in all lymph nodes and the spleen examined postmortem but remained undetectable in the bone marrow, thymus, and LPLs (Fig. 7B). Similar analyses in mononuclear cells prepared from axillary lymph node biopsies collected at 4, 8, 12, and 16 wpi showed that HR- as well as Δ22-25-bearing sequences were absent prior to 12 wpi but could be detected at 16 and 20 wpi despite the extremely low percentage of CD4+ T cells in this lymph node compartment, indicative of severe depletion (Table 1). The highly restricted tissue localization of the X4 variant at 13 wpi contrasts with the generalized dissemination of the dual-R Δ22-25 virus seen at the same time, reinforcing the notion that the X4 switch event was caught early while the dual-R switch had already disseminated.
FIG. 7.

HR V3 loop-bearing X4 variants in tissues of macaque DG08 at 13 wpi (A) and time of necropsy (20 wpi) (B) as determined by differential PCR analysis. m, marker; + and −, positive and negative controls, respectively; Mes, mesenteric LN; Ing, inguinal LN; BM, bone marrow; Ax, axillary; Col, colonic LN. Results shown are representative of two independent PCR amplifications.
TABLE 1.
Percentage of CD4+ T cells and X4 HR and dual-R Δ22-25 presence in longitudinal axillary lymph node samples as determined by differential PCR
| Characteristic | Value for the characteristic at:a |
|||||
|---|---|---|---|---|---|---|
| Week 0 | Week 4 | Week 8 | Week 12 | Week 16 | Week 20 | |
| % CD4+ T cells | 64 | 30.1 | 31.9 | 29.6 | 0.02 | 1.26 |
| HR insertion | − | − | − | − | + | + |
| Δ22-25 sequence | − | − | − | − | + | + |
HR and Δ22-25 V3-specific amplification signals are designated as absent (−) or present (+).
We previously reported that X4 sequences in the i.v. infected switch macaque BR24 were poorly represented in the gut (IELs and LPLs) but predominated in peripheral and visceral lymph nodes (63). Similar observations were made when tissue localization of X4 variants in DG08 and in the other i.v. infected macaque CA28 at the time of necropsy was analyzed (29), showing lymph node compartmentalization of X4 viruses (Table 2). These data from three switch macaques, coupled with the finding that X4 signature sequence was first detected in the inguinal lymph node of DG08, support lymph nodes as niches where the chances for X4 virus evolution as well as expansion are the greatest.
TABLE 2.
Summary of sites of localization for CXCR4-using variants in R5 SHIVSF162P3N-infected switch macaques
| Animal | Infection route | Tropism variant | Localization of variant in the indicated tissuea |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Primary tissue |
Secondary tissue |
|||||||||||
| LNs |
Spl | GALT |
||||||||||
| Thy | BM | Ax | Ili | Ing | Col | IEL | LPL | |||||
| BR24b | i.v. | X4 | NA | NA | + | + | + | + | + | − | − | − |
| CA28c | i.v. | X4 | − | NA | + | + | + | − | − | + | − | − |
| DG08d | i.r. | X4 | − | − | + | + | + | + | + | − | − | − |
| Dual | − | + | + | + | + | − | + | + | − | + | ||
| DR16d | i.r. | Dual | + | + | + | + | + | + | + | + | + | + |
The presence (+) or absence (−) of the indicated variant was determined by envelope sequence analysis. NA, not available; Thy, thymus; BM, bone marrow; Ax, axillary; Ili, iliac; Ing, inguinal; Col, colonic; Mes, mesenteric; Spl, spleen; GALT, gut-associated lymphoid tissues.
Tasca et al. (63).
Ho et al. (29); also unpublished data.
This study.
DISCUSSION
In this study, we investigated R5 SHIVSF162P3N pathogenesis in mucosally infected macaques and examined the tempo and location of dual-tropic and X4 virus evolution and amplification. We found that the SHIVSF162P3N virus is highly pathogenic in i.r. infected macaques, with development of clinical signs consistent with AIDS within 30 wpi in three of six infected animals. Importantly, change or expansion in coreceptor preference was detected in two of the three diseased animals. The infected macaques in which X4 and dual-tropic viruses emerged were RPs, with a clinical course that was characterized by high levels of virus replication and weak or undetectable antiviral antibody response. Sequence changes in the V3 loop of envelope gp120 that were associated with phenotypic change from R5-to-X4 tropism in infected humans were found to determine CXCR4 usage in i.r. infected macaques (9, 12, 20, 21, 61). Furthermore, the newly emerging CXCR4-using viruses were highly sensitive to neutralization with soluble CD4, supporting the notion that immune failure contributes to emergence of X4 variants at late-stage disease (46, 72). Although X4 and dual-tropic virus emergence was associated with an accelerated drop in peripheral CD4+ T-cell count, it followed rather than preceded the onset of CD4+ T-cell loss, suggesting that limited CD4+ T-cell availability or function provided the selective pressure for coreceptor switch (6, 67). While the numbers of switch events and variants studied are relatively small, the conditions, genotypic requirements and patterns for coreceptor switching in i.r. SHIVSF162P3N-infected macaques are remarkably similar to those previously reported for macaques infected intravenously with the same virus (28, 29), demonstrating that the route of inoculation does not constrain the emergence of X4 and dual-tropic viruses. They also overlapped with those reported for humans, suggestive of a common mechanism for coreceptor switching in the two hosts.
As observed for the switch in SHIVSF162P3N i.v. infected macaque (28), the switch or expansion to CXCR4 usage in both i.r. infected macaques was associated with V3 loop sequence mutations similar to those reported in humans but differed from those required to confer expanded CXCR4 usage to the parental SF162 envelope in tissue culture systems (13, 26, 32). In the case of DR16, the emergence of a positively charged amino acid at position 11 in V3 was shown to be associated with CXCR4 coreceptor phenotype and with increased clinical progression in HIV-1-infected individuals (12, 20). This dual-tropic virus was present in the majority of envelope sequences amplified from the blood and multiple lymphoid organs at end-stage disease (Fig. 2 and 6B). The X4 virus in DG08 harbored two basic amino acid insertions upstream of the GPGR crown of the V3 loop, a sequence motif that is also found in the Amsterdam cohort patient Ams-32 and which is linked to a switch to CXCR4 usage in the SHIVSF162P3N i.v. infected macaque BR24 (28). The dual-R variant, Δ22-25, however, had a 4-amino-acid deletion in the C-terminal stem region of the V3 loop (ATGD) that increased the net positive charge of this domain. V3 deletion Envs with altered neutralization sensitivity and tropism have been described for HIV-1 as well as HIV-2 (37, 41, 49), with the stem region reportedly playing a critical role in CXCR4 utilization (49). Thus, as in humans (20, 31, 47, 59), a positively charged V3 is important for X4 tropism in macaques. And while a coreceptor switch, so far, has been seen only in RP macaques that fail to mount or maintain an antiviral antibody response, the similarity in Env V3 loop sequence changes required for CXCR4 usage in RP macaques and in infected humans who usually have developed neutralizing antibodies argues against a major role of humoral immune selective pressure in driving X4 evolution. Indeed, coreceptor switching was observed in only two of six RP macaques infected intravenously with SHIVSF162P3N (28, 29), and experimental B-cell depletion with anti-CD20 antibody prior to i.r. infection with SHIVSF162P3N blunted the development of antiviral antibody response but did not select for X4 emergence (unpublished data). Collectively, the data support R5 SHIVSF162P3N infection of RM as a bona fide model to study the mechanistic basis and selective forces for HIV-1 coreceptor switching in vivo.
We performed frequent and extensive samplings of lymphoid and nonlymphoid organs in the infected macaques to more closely monitor the kinetics and site of X4 virus evolution and amplification. Surgery performed at 13 wpi, a time concomitant with emergence of the phylogenetically distinct X4 HR and dual-R Δ22-25 variants in plasma of DG08 (Fig. 5), provided a unique opportunity to gain insights into the dynamics of the two parallel R5-to-X4 evolutionary processes. Clonal analysis of envelope sequences revealed high frequencies of the dual-R Δ22-25 variant in multiple tissues sampled at this time point in infection but undetectable levels of the X4 variant in any of the tissues sites examined. The wide tissue distribution and ease of detection of the dual-R variant compared to the X4 virus by clonal analysis indicated generalized dissemination and dominance of the former virus and suggested that the dual-R switch event took place earlier than the X4 switch. With time, however, the frequency of the dual-R Δ22-25 variant in tissue cells decreased while that of the X4 HR virus increased. In this regard, we along with others have shown that viruses capable of using CXCR4 in addition to CCR5 display a loss in replicative fitness compared to the early R5 and late X4 viruses in vitro and use the CCR5 and CXCR4 coreceptors less efficiently as well (35, 50, 51, 63). Truncations of the V3 loop have also been reported to result in reduced HIV-1 envelope function (37, 73, 74). Thus, it is conceivable that while the dual-R Δ22-25 variant may have had a distinct advantage over early or coexisting R5 viruses when it first appeared because of target cell expansion through acquisition of CXCR4 usage, this advantage was lost with the subsequent emergence of the X4 variant, accounting for its diminished dominance with time.
In addition to differences in the tempo of dissemination, compartmentalization of the X4 and dual-R viruses in macaque DG08 differed as well. Similar to findings in DR16, the dual-R virus in DG08 replicated in multiple tissues (Fig. 6). These included the lymph nodes, gut, bone marrow, and spleen and, in the case of DR16, also the thymus. Furthermore, the dual-R virus in DG08 dominated in the LPLs at the switch point (13 wpi), indicating that the gut is not an exclusive site for R5 virus replication. In contrast, and consistent with findings in i.v. infected switch macaques (Table 2), X4 virus amplification in DG08 was restricted to the lymph nodes and was not found in the gut, bone marrow, or thymus, even with the use of the highly sensitive sequence-specific PCR detection method (Fig. 7B). Further supporting compartmentalization of X4 virus replication in lymph nodes is the finding that X4 virus was first detected in the inguinal LN (Fig. 7A). The most likely reason for the different anatomical location of dual-R and X4 viruses in DG08 is the tissue distribution of their target cells. X4 viruses predominately target naïve and central memory CD4 cells that express high CXCR4 levels and are enriched in peripheral lymphoid tissues. In contrast, CCR5 is expressed primarily on effector memory T cells that are abundant at mucosal effector sites such as the gut-associated lymphoid tissues, with little or no expression in the naïve population (5, 27, 36, 52, 69). This then explains the wide systemic dissemination of dual-R virus that can use both coreceptors, with the lymph nodes as the preferred site(s) of X4 virus expansion. While we showed that the gut is not an exclusive site for R5 virus replication, prior seeding and depletion of target cells by the dual-R and R5 viruses at this site may have limited the establishment of infection by the emerging X4 virus in DG08. The finding of lymph nodes as niches where the chances for X4 selection are the greatest has implications for the monitoring of X4 virus emergence in patients treated with CCR5 inhibitors, suggesting that analyzing peripheral blood may not be sufficiently sensitive.
Studies of postmortem tissues in HIV-1-infected children point to the thymus as being a major site for X4 evolution and/or amplification (56). But since antemortem thymic or lymphoid tissues were not available from these human subjects, emergence of similar X4 variants in other tissues such as lymph nodes prior to or concomitant with their appearance in the thymus cannot be excluded. Furthermore, given the hyperproliferative state of the neonatal thymus and its later involution, this organ likely plays a more important role in pediatric than adult HIV-1 infection. Studies of coreceptor switching in R5 SHIVSF162P3N-infected newborn RM should help to address this issue. Longitudinal samplings of tissue sites, especially peripheral lymph nodes, are difficult if not impractical in HIV-1-infected patients, highlighting the importance and utility of a relevant animal model in understanding the role of lymphoid and extralymphoid tissues in the evolution and amplification of X4 viruses.
In summary, we have established an animal model to study coreceptor switch after mucosal infection. Using this model, we have analyzed a large set of samples taken at frequent time intervals, including a time point that happened to capture a localized switch event in a particular macaque (DG08), providing important insights into the dynamics and anatomic sites of viral phenotype change. Because X4 emergence is known to be associated with poorer clinical prognosis and because treatment with CCR5 inhibitors may select for viruses with the ability to use CXCR4, a deeper understanding of the determinants and tissue compartments involved in HIV-1 phenotypic switch is warranted. The finding that the phenotypic characteristics as well as envelope V3 sequences required for coreceptor switch in SHIVSF162P3N-infected macaques overlap with those reported in HIV-1-infected individuals supports the use of this animal model to study the underlying selective pressures for the change in coreceptor preference in vivo. Ultradeep pyrosequencing of V3 amplicons from serial plasma, PBMCs, and tissue biopsies of infected macaques to detect rare variants could provide a more comprehensive picture of the diversity, frequency, and kinetics of the R5-to-X4 evolutionary process. Additionally, as mucosal infection of Indian RM with R5 SHIVSF162P3N recapitulates key pathogenic effects of HIV-1 infection in humans, including acute CD4+ T-cell depletion in the gut, high viral replication, and progression to AIDS with a switch in coreceptor preference, the model will also be extremely useful for advancing the discovery of T- as well as B-cell based candidate HIV vaccines.
Acknowledgments
We thank Lisa Chakrabarti for helpful discussion and critique of this work. The following were obtained through the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: reagents TAK779 (catalog no. 4983 from Takeda Chemical Industries, Ltd.) and AMD3100 (catalog no. 8128 from AnorMed, Inc.) and the TZM-bl (catalog no. 8129 from John C. Kappes, Xiaoyun Wu, and Tranzyme, Inc.) and U87.CD4 indicator cell lines (catalog no. 4035 and 4036 from HongKui Deng and Dan R. Littman). CD4-IgG2 (PRO 542) was a gift from Progenics Pharmaceuticals (Tarrytown, NY).
This work was supported by National Institutes of Health grants RO1AI46980 and R37AI41945. Additional support was provided by the Tulane National Primate Research Center Base, grant RR00164.
Footnotes
Published ahead of print on 21 October 2009.
REFERENCES
- 1.Alkhatib, G., C. Combadiere, C. C. Broder, Y. Feng, P. E. Kennedy, P. M. Murphy, and E. A. Berger. 1996. CC CKR5: a RANTES, MIP-1alpha, MIP-1beta receptor as a fusion cofactor for macrophage-tropic HIV-1. Science 272:1955-1958. [DOI] [PubMed] [Google Scholar]
- 2.Back, N. K., L. Smit, J. J. De Jong, W. Keulen, M. Schutten, J. Goudsmit, and M. Tersmette. 1994. An N-glycan within the human immunodeficiency virus type 1 gp120 V3 loop affects virus neutralization. Virology 199:431-438. [DOI] [PubMed] [Google Scholar]
- 3.Berger, E. A., R. W. Doms, E. M. Fenyo, B. T. Korber, D. R. Littman, J. P. Moore, Q. J. Sattentau, H. Schuitemaker, J. Sodroski, and R. A. Weiss. 1998. A new classification for HIV-1. Nature 391:240. [DOI] [PubMed] [Google Scholar]
- 4.Berger, E. A., P. M. Murphy, and J. M. Farber. 1999. Chemokine receptors as HIV-1 coreceptors: roles in viral entry, tropism, and disease. Annu. Rev. Immunol. 17:657-700. [DOI] [PubMed] [Google Scholar]
- 5.Bleul, C. C., L. Wu, J. A. Hoxie, T. A. Springer, and C. R. Mackay. 1997. The HIV coreceptors CXCR4 and CCR5 are differentially expressed and regulated on human T lymphocytes. Proc. Natl. Acad. Sci. U. S. A. 94:1925-1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Casper, C., L. Naver, P. Clevestig, E. Belfrage, T. Leitner, J. Albert, S. Lindgren, C. Ottenblad, A. B. Bohlin, E. M. Fenyo, and A. Ehrnst. 2002. Coreceptor change appears after immune deficiency is established in children infected with different HIV-1 subtypes. AIDS Res. Hum. Retroviruses 18:343-352. [DOI] [PubMed] [Google Scholar]
- 7.Cheng-Mayer, C., D. Seto, M. Tateno, and J. A. Levy. 1988. Biologic features of HIV-1 that correlate with virulence in the host. Science 240:80-82. [DOI] [PubMed] [Google Scholar]
- 8.Choe, H., M. Farzan, Y. Sun, N. Sullivan, B. Rollins, P. D. Ponath, L. Wu, C. R. Mackay, G. LaRosa, W. Newman, N. Gerard, C. Gerard, and J. Sodroski. 1996. The beta-chemokine receptors CCR3 and CCR5 facilitate infection by primary HIV-1 isolates. Cell 85:1135-1148. [DOI] [PubMed] [Google Scholar]
- 9.Cocchi, F., A. L. DeVico, A. Garzino-Demo, A. Cara, R. C. Gallo, and P. Lusso. 1996. The V3 domain of the HIV-1 gp120 envelope glycoprotein is critical for chemokine-mediated blockade of infection. Nat. Med. 2:1244-1247. [DOI] [PubMed] [Google Scholar]
- 10.Connor, R. I., B. K. Chen, S. Choe, and N. R. Landau. 1995. Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology 206:935-944. [DOI] [PubMed] [Google Scholar]
- 11.Connor, R. I., K. E. Sheridan, D. Ceradini, S. Choe, and N. R. Landau. 1997. Change in coreceptor use coreceptor use correlates with disease progression in HIV-1-infected individuals. J. Exp. Med. 185:621-628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De Jong, J. J., A. De Ronde, W. Keulen, M. Tersmette, and J. Goudsmit. 1992. Minimal requirements for the human immunodeficiency virus type 1 V3 domain to support the syncytium-inducing phenotype: analysis by single amino acid substitution. J. Virol. 66:6777-6780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dejucq, N., G. Simmons, and P. R. Clapham. 2000. T-cell line adaptation of human immunodeficiency virus type 1 strain SF162: effects on envelope, vpu and macrophage-tropism. J. Gen. Virol. 81:2899-2904. [DOI] [PubMed] [Google Scholar]
- 14.Delwart, E. L., and C. J. Gordon. 1997. Tracking changes in HIV-1 envelope quasispecies using DNA heteroduplex analysis. Methods 12:348-354. [DOI] [PubMed] [Google Scholar]
- 15.Deng, H., R. Liu, W. Ellmeier, S. Choe, D. Unutmaz, M. Burkhart, P. Di Marzio, S. Marmon, R. E. Sutton, C. M. Hill, C. B. Davis, S. C. Peiper, T. J. Schall, D. R. Littman, and N. R. Landau. 1996. Identification of a major co-receptor for primary isolates of HIV-1. Nature 381:661-666. [DOI] [PubMed] [Google Scholar]
- 16.Doranz, B. J., J. Rucker, Y. Yi, R. J. Smyth, M. Samson, S. C. Peiper, M. Parmentier, R. G. Collman, and R. W. Doms. 1996. A dual-tropic primary HIV-1 isolate that uses fusin and the beta-chemokine receptors CKR-5, CKR-3, and CKR-2b as fusion cofactors. Cell 85:1149-1158. [DOI] [PubMed] [Google Scholar]
- 17.Douek, D. C., R. D. McFarland, P. H. Keiser, E. A. Gage, J. M. Massey, B. F. Haynes, M. A. Polis, A. T. Haase, M. B. Feinberg, J. L. Sullivan, B. D. Jamieson, J. A. Zack, L. J. Picker, and R. A. Koup. 1998. Changes in thymic function with age and during the treatment of HIV infection. Nature 396:690-695. [DOI] [PubMed] [Google Scholar]
- 18.Dragic, T., V. Litwin, G. P. Allaway, S. R. Martin, Y. Huang, K. A. Nagashima, C. Cayanan, P. J. Maddon, R. A. Koup, J. P. Moore, and W. A. Paxton. 1996. HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5. Nature 381:667-673. [DOI] [PubMed] [Google Scholar]
- 19.Fatkenheuer, G., M. Nelson, A. Lazzarin, I. Konourina, A. I. Hoepelman, H. Lampiris, B. Hirschel, P. Tebas, F. Raffi, B. Trottier, N. Bellos, M. Saag, D. A. Cooper, M. Westby, M. Tawadrous, J. F. Sullivan, C. Ridgway, M. W. Dunne, S. Felstead, H. Mayer, and E. van der Ryst. 2008. Subgroup analyses of maraviroc in previously treated R5 HIV-1 infection. N. Engl. J. Med. 359:1442-1455. [DOI] [PubMed] [Google Scholar]
- 20.Fouchier, R. A., M. Groenink, N. A. Kootstra, M. Tersmette, H. G. Huisman, F. Miedema, and H. Schuitemaker. 1992. Phenotype-associated sequence variation in the third variable domain of the human immunodeficiency virus type 1 gp120 molecule. J. Virol. 66:3183-3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Glushakova, S., Y. Yi, J. C. Grivel, A. Singh, D. Schols, E. De Clercq, R. G. Collman, and L. Margolis. 1999. Preferential coreceptor utilization and cytopathicity by dual-tropic HIV-1 in human lymphoid tissue ex vivo. J. Clin. Invest. 104:R7-R11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guindon, S., and O. Gascuel. 2003. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52:696-704. [DOI] [PubMed] [Google Scholar]
- 23.Gulick, R. M., J. Lalezari, J. Goodrich, N. Clumeck, E. DeJesus, A. Horban, J. Nadler, B. Clotet, A. Karlsson, M. Wohlfeiler, J. B. Montana, M. McHale, J. Sullivan, C. Ridgway, S. Felstead, M. W. Dunne, E. van der Ryst, and H. Mayer. 2008. Maraviroc for previously treated patients with R5 HIV-1 infection. N. Engl. J. Med. 359:1429-1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gulick, R. M., Z. Su, C. Flexner, M. D. Hughes, P. R. Skolnik, T. J. Wilkin, R. Gross, A. Krambrink, E. Coakley, W. L. Greaves, A. Zolopa, R. Reichman, C. Godfrey, M. Hirsch, and D. R. Kuritzkes. 2007. Phase 2 study of the safety and efficacy of vicriviroc, a CCR5 inhibitor, in HIV-1-infected, treatment-experienced patients: AIDS clinical trials group 5211. J. Infect. Dis. 196:304-312. [DOI] [PubMed] [Google Scholar]
- 25.Harouse, J. M., A. Gettie, T. Eshetu, R. C. Tan, R. Bohm, J. Blanchard, G. Baskin, and C. Cheng-Mayer. 2001. Mucosal transmission and induction of simian AIDS by CCR5-specific simian/human immunodeficiency virus SHIVSF162P3. J. Virol. 75:1990-1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harrowe, G., and C. Cheng-Mayer. 1995. Amino acid substitutions in the V3 loop are responsible for adaptation to growth in transformed T-cell lines of a primary human immunodeficiency virus type 1. Virology 210:490-494. [DOI] [PubMed] [Google Scholar]
- 27.Ho, S. H., L. Shek, A. Gettie, J. Blanchard, and C. Cheng-Mayer. 2005. V3 loop-determined coreceptor preference dictates the dynamics of CD4+-T-cell loss in simian-human immunodeficiency virus-infected macaques. J. Virol. 79:12296-12303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ho, S. H., S. Tasca, L. Shek, A. Li, A. Gettie, J. Blanchard, D. Boden, and C. Cheng-Mayer. 2007. Coreceptor switch in R5-tropic simian/human immunodeficiency virus-infected macaques. J. Virol. 81:8621-8633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ho, S. H., N. Trunova, A. Gettie, J. Blanchard, and C. Cheng-Mayer. 2008. Different mutational pathways to CXCR4 coreceptor switch of CCR5-using simian-human immunodeficiency virus. J. Virol. 82:5653-5656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang, W., S. H. Eshleman, J. Toma, S. Fransen, E. Stawiski, E. E. Paxinos, J. M. Whitcomb, A. M. Young, D. Donnell, F. Mmiro, P. Musoke, L. A. Guay, J. B. Jackson, N. T. Parkin, and C. J. Petropoulos. 2007. Coreceptor tropism in human immunodeficiency virus type 1 subtype D: high prevalence of CXCR4 tropism and heterogeneous composition of viral populations. J. Virol. 81:7885-7893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jensen, M. A., F. S. Li, A. B. van 't Wout, D. C. Nickle, D. Shriner, H. X. He, S. McLaughlin, R. Shankarappa, J. B. Margolick, and J. I. Mullins. 2003. Improved coreceptor usage prediction and genotypic monitoring of R5-to-X4 transition by motif analysis of human immunodeficiency virus type 1 env V3 loop sequences. J. Virol. 77:13376-13388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kiselyeva, Y., R. Nedellec, A. Ramos, C. Pastore, L. B. Margolis, and D. E. Mosier. 2007. Evolution of CXCR4-using human immunodeficiency virus type 1 SF162 is associated with two unique envelope mutations. J. Virol. 81:3657-3661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kitchen, S. G., and J. A. Zack. 1997. CXCR4 expression during lymphopoiesis: implications for human immunodeficiency virus type 1 infection of the thymus. J. Virol. 71:6928-6934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Koot, M., I. P. Keet, A. H. Vos, R. E. de Goede, M. T. Roos, R. A. Coutinho, F. Miedema, P. T. Schellekens, and M. Tersmette. 1993. Prognostic value of HIV-1 syncytium-inducing phenotype for rate of CD4+ cell depletion and progression to AIDS. Ann. Intern. Med. 118:681-688. [DOI] [PubMed] [Google Scholar]
- 35.Kuiken, C. L., J. J. de Jong, E. Baan, W. Keulen, M. Tersmette, and J. Goudsmit. 1992. Evolution of the V3 envelope domain in proviral sequences and isolates of human immunodeficiency virus type 1 during transition of the viral biological phenotype. J. Virol. 66:4622-4627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kunkel, E. J., J. Boisvert, K. Murphy, M. A. Vierra, M. C. Genovese, A. J. Wardlaw, H. B. Greenberg, M. R. Hodge, L. Wu, E. C. Butcher, and J. J. Campbell. 2002. Expression of the chemokine receptors CCR4, CCR5, and CXCR3 by human tissue-infiltrating lymphocytes. Am. J. Pathol. 160:347-355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Laakso, M. M., F. H. Lee, B. Haggarty, C. Agrawal, K. M. Nolan, M. Biscone, J. Romano, A. P. Jordan, G. J. Leslie, E. G. Meissner, L. Su, J. A. Hoxie, and R. W. Doms. 2007. V3 loop truncations in HIV-1 envelope impart resistance to coreceptor inhibitors and enhanced sensitivity to neutralizing antibodies. PLoS Pathog. 3:e117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Landovitz, R. J., J. B. Angel, C. Hoffmann, H. Horst, M. Opravil, J. Long, W. Greaves, and G. Fatkenheuer. 2008. Phase II study of vicriviroc versus efavirenz (both with zidovudine/lamivudine) in treatment-naive subjects with HIV-1 infection. J. Infect. Dis. 198:1113-1122. [DOI] [PubMed] [Google Scholar]
- 39.Larkin, M. A., G. Blackshields, N. P. Brown, R. Chenna, P. A. McGettigan, H. McWilliam, F. Valentin, I. M. Wallace, A. Wilm, R. Lopez, J. D. Thompson, T. J. Gibson, and D. G. Higgins. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23:2947-2948. [DOI] [PubMed] [Google Scholar]
- 40.Li, Y., M. A. Rey-Cuille, and S. L. Hu. 2001. N-linked glycosylation in the V3 region of HIV type 1 surface antigen modulates coreceptor usage in viral infection. AIDS Res. Hum. Retroviruses 17:1473-1479. [DOI] [PubMed] [Google Scholar]
- 41.Lin, G., A. Bertolotti-Ciarlet, B. Haggarty, J. Romano, K. M. Nolan, G. J. Leslie, A. P. Jordan, C. C. Huang, P. D. Kwong, R. W. Doms, and J. A. Hoxie. 2007. Replication-competent variants of human immunodeficiency virus type 2 lacking the V3 loop exhibit resistance to chemokine receptor antagonists. J. Virol. 81:9956-9966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mackall, C. L., T. A. Fleisher, M. R. Brown, M. P. Andrich, C. C. Chen, I. M. Feuerstein, M. E. Horowitz, I. T. Magrath, A. T. Shad, S. M. Steinberg, et al. 1995. Age, thymopoiesis, and CD4+ T-lymphocyte regeneration after intensive chemotherapy. N. Engl. J. Med. 332:143-149. [DOI] [PubMed] [Google Scholar]
- 43.Malenbaum, S. E., D. Yang, L. Cavacini, M. Posner, J. Robinson, and C. Cheng-Mayer. 2000. The N-terminal V3 loop glycan modulates the interaction of clade A and B human immunodeficiency virus type 1 envelopes with CD4 and chemokine receptors. J. Virol. 74:11008-11016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McCaffrey, R. A., C. Saunders, M. Hensel, and L. Stamatatos. 2004. N-linked glycosylation of the V3 loop and the immunologically silent face of gp120 protects human immunodeficiency virus type 1 SF162 from neutralization by anti-gp120 and anti-gp41 antibodies. J. Virol. 78:3279-3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McCune, J. M. 1997. Thymic function in HIV-1 disease. Semin. Immunol. 9:397-404. [DOI] [PubMed] [Google Scholar]
- 46.Miedema, F., M. Tersmette, and R. A. van Lier. 1990. AIDS pathogenesis: a dynamic interaction between HIV and the immune system. Immunol. Today 11:293-297. [DOI] [PubMed] [Google Scholar]
- 47.Milich, L., B. Margolin, and R. Swanstrom. 1993. V3 loop of the human immunodeficiency virus type 1 Env protein: interpreting sequence variability. J. Virol. 67:5623-5634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moore, J. P., S. G. Kitchen, P. Pugach, and J. A. Zack. 2004. The CCR5 and CXCR4 coreceptors—central to understanding the transmission and pathogenesis of human immunodeficiency virus type 1 infection. AIDS Res. Hum. Retroviruses 20:111-126. [DOI] [PubMed] [Google Scholar]
- 49.Nolan, K. M., A. P. Jordan, and J. A. Hoxie. 2008. Effects of partial deletions within the human immunodeficiency virus type 1 V3 loop on coreceptor tropism and sensitivity to entry inhibitors. J. Virol. 82:664-673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pastore, C., R. Nedellec, A. Ramos, O. Hartley, J. L. Miamidian, J. D. Reeves, and D. E. Mosier. 2007. Conserved changes in envelope function during human immunodeficiency virus type 1 coreceptor switching. J. Virol. 81:8165-8179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pastore, C., R. Nedellec, A. Ramos, S. Pontow, L. Ratner, and D. E. Mosier. 2006. Human immunodeficiency virus type 1 coreceptor switching: V1/V2 gain-of-fitness mutations compensate for V3 loss-of-fitness mutations. J. Virol. 80:750-758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Perez-Patrigeon, S., B. Vingert, O. Lambotte, J. P. Viard, J. F. Delfraissy, J. Theze, and L. A. Chakrabarti. 2009. HIV infection impairs CCR7-dependent T-cell chemotaxis independent of CCR7 expression. AIDS 23:1197-1207. [DOI] [PubMed] [Google Scholar]
- 53.Polzer, S., M. T. Dittmar, H. Schmitz, and M. Schreiber. 2002. The N-linked glycan g15 within the V3 loop of the HIV-1 external glycoprotein gp120 affects coreceptor usage, cellular tropism, and neutralization. Virology 304:70-80. [DOI] [PubMed] [Google Scholar]
- 54.Regoes, R. R., and S. Bonhoeffer. 2005. The HIV coreceptor switch: a population dynamical perspective. Trends Microbiol. 13:269-277. [DOI] [PubMed] [Google Scholar]
- 55.Reyes, R. A., D. R. Canfield, U. Esser, L. A. Adamson, C. R. Brown, C. Cheng-Mayer, M. B. Gardner, J. M. Harouse, and P. A. Luciw. 2004. Induction of simian AIDS in infant rhesus macaques infected with CCR5- or CXCR4-utilizing simian-human immunodeficiency viruses is associated with distinct lesions of the thymus. J. Virol. 78:2121-2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Salemi, M., B. R. Burkhardt, R. R. Gray, G. Ghaffari, J. W. Sleasman, and M. M. Goodenow. 2007. Phylodynamics of HIV-1 in lymphoid and non-lymphoid tissues reveals a central role for the thymus in emergence of CXCR4-using quasispecies. PLoS ONE 2:e950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schuitemaker, H., M. Koot, N. A. Kootstra, M. W. Dercksen, R. E. de Goede, R. P. van Steenwijk, J. M. Lange, J. K. Schattenkerk, F. Miedema, and M. Tersmette. 1992. Biological phenotype of human immunodeficiency virus type 1 clones at different stages of infection: progression of disease is associated with a shift from monocytotropic to T-cell-tropic virus population. J. Virol. 66:1354-1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schuitemaker, H., N. A. Kootstra, R. E. de Goede, F. de Wolf, F. Miedema, and M. Tersmette. 1991. Monocytotropic human immunodeficiency virus type 1 (HIV-1) variants detectable in all stages of HIV-1 infection lack T-cell line tropism and syncytium-inducing ability in primary T-cell culture. J. Virol. 65:356-363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shioda, T., S. Oka, S. Ida, K. Nokihara, H. Toriyoshi, S. Mori, Y. Takebe, S. Kimura, K. Shimada, Y. Nagai, et al. 1994. A naturally occurring single basic amino acid substitution in the V3 region of the human immunodeficiency virus type 1 env protein alters the cellular host range and antigenic structure of the virus. J. Virol. 68:7689-7696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Simpson, J. G., E. S. Gray, and J. S. Beck. 1975. Age involution in the normal human adult thymus. Clin. Exp. Immunol. 19:261-265. [PMC free article] [PubMed] [Google Scholar]
- 61.Speck, R. F., K. Wehrly, E. J. Platt, R. E. Atchison, I. F. Charo, D. Kabat, B. Chesebro, and M. A. Goldsmith. 1997. Selective employment of chemokine receptors as human immunodeficiency virus type 1 coreceptors determined by individual amino acids within the envelope V3 loop. J. Virol. 71:7136-7139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tamura, K., J. Dudley, M. Nei, and S. Kumar. 2007. MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol. Biol. Evol. 24:1596-1599. [DOI] [PubMed] [Google Scholar]
- 63.Tasca, S., S. H. Ho, and C. Cheng-Mayer. 2008. R5X4 viruses are evolutionary, functional, and antigenic intermediates in the pathway of a simian-human immunodeficiency virus coreceptor switch. J. Virol. 82:7089-7099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Taylor, J. R., Jr., K. C. Kimbrell, R. Scoggins, M. Delaney, L. Wu, and D. Camerini. 2001. Expression and function of chemokine receptors on human thymocytes: implications for infection by human immunodeficiency virus type 1. J. Virol. 75:8752-8760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tersmette, M., R. A. Gruters, F. de Wolf, R. E. de Goede, J. M. Lange, P. T. Schellekens, J. Goudsmit, H. G. Huisman, and F. Miedema. 1989. Evidence for a role of virulent human immunodeficiency virus (HIV) variants in the pathogenesis of acquired immunodeficiency syndrome: studies on sequential HIV isolates. J. Virol. 63:2118-2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vajdy, M., R. Veazey, I. Tham, C. deBakker, S. Westmoreland, M. Neutra, and A. Lackner. 2001. Early immunologic events in mucosal and systemic lymphoid tissues after intrarectal inoculation with simian immunodeficiency virus. J. Infect. Dis. 184:1007-1014. [DOI] [PubMed] [Google Scholar]
- 67.van Rij, R. P., M. D. Hazenberg, B. H. van Benthem, S. A. Otto, M. Prins, F. Miedema, and H. Schuitemaker. 2003. Early viral load and CD4+ T cell count, but not percentage of CCR5+ or CXCR4+ CD4+ T cells, are associated with R5-to-X4 HIV type 1 virus evolution. AIDS Res. Hum. Retroviruses 19:389-398. [DOI] [PubMed] [Google Scholar]
- 68.van't Wout, A. B., N. A. Kootstra, G. A. Mulder-Kampinga, N. Albrecht-van Lent, H. J. Scherpbier, J. Veenstra, K. Boer, R. A. Coutinho, F. Miedema, and H. Schuitemaker. 1994. Macrophage-tropic variants initiate human immunodeficiency virus type 1 infection after sexual, parenteral, and vertical transmission. J. Clin. Invest. 94:2060-2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Veazey, R. S., K. G. Mansfield, I. C. Tham, A. C. Carville, D. E. Shvetz, A. E. Forand, and A. A. Lackner. 2000. Dynamics of CCR5 expression by CD4+ T cells in lymphoid tissues during simian immunodeficiency virus infection. J. Virol. 74:11001-11007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wei, X., J. M. Decker, S. Wang, H. Hui, J. C. Kappes, X. Wu, J. F. Salazar-Gonzalez, M. G. Salazar, J. M. Kilby, M. S. Saag, N. L. Komarova, M. A. Nowak, B. H. Hahn, P. D. Kwong, and G. M. Shaw. 2003. Antibody neutralization and escape by HIV-1. Nature 422:307-312. [DOI] [PubMed] [Google Scholar]
- 71.Westby, M., M. Lewis, J. Whitcomb, M. Youle, A. L. Pozniak, I. T. James, T. M. Jenkins, M. Perros, and E. van der Ryst. 2006. Emergence of CXCR4-using human immunodeficiency virus type 1 (HIV-1) variants in a minority of HIV-1-infected patients following treatment with the CCR5 antagonist maraviroc is from a pretreatment CXCR4-using virus reservoir. J. Virol. 80:4909-4920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wodarz, D., and M. A. Nowak. 1998. The effect of different immune responses on the evolution of virulent CXCR4-tropic HIV. Proc. Biol. Sci. 265:2149-2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wyatt, R., N. Sullivan, M. Thali, H. Repke, D. Ho, J. Robinson, M. Posner, and J. Sodroski. 1993. Functional and immunologic characterization of human immunodeficiency virus type 1 envelope glycoproteins containing deletions of the major variable regions. J. Virol. 67:4557-4565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yang, Z. Y., B. K. Chakrabarti, L. Xu, B. Welcher, W. P. Kong, K. Leung, A. Panet, J. R. Mascola, and G. J. Nabel. 2004. Selective modification of variable loops alters tropism and enhances immunogenicity of human immunodeficiency virus type 1 envelope. J. Virol. 78:4029-4036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yant, L. J., T. C. Friedrich, R. C. Johnson, G. E. May, N. J. Maness, A. M. Enz, J. D. Lifson, D. H. O'Connor, M. Carrington, and D. I. Watkins. 2006. The high-frequency major histocompatibility complex class I allele Mamu-B*17 is associated with control of simian immunodeficiency virus SIVmac239 replication. J. Virol. 80:5074-5077. [DOI] [PMC free article] [PubMed] [Google Scholar]