

Abstract
Human pathogenic viruses manipulate host cell translation machinery to ensure efficient expression of viral genes and to thwart host cell protein synthesis. Viral strategies include cleaving translation factors, manipulating translation factor abundance and recruitment into translation initiation complexes, or expressing viral translation factor analogs. Analyzing translation factors in herpes simplex virus type 1 (HSV-1)-infected HeLa cells, we found diminished association of the polyadenylate-binding protein (PABP) with the cap-binding complex. Although total PABP levels were unchanged, HSV-1 infection prompted accumulation of cytoplasmic PABPC1, but not its physiologic binding partner PABP-interacting protein 2 (Paip2), in the nucleus. Using glutathione S-transferase-PABP pull-down and proteomic analyses, we identified several viral proteins interacting with PABPC1 including tegument protein UL47 and infected-cell protein ICP27. Transient expression of ICP27 and UL47 in HeLa cells suggested that ICP27 and UL47 jointly displace Paip2 from PABP. ICP27 expression alone was sufficient to cause PABPC1 redistribution to the nucleus. ICP27 and UL47 did not alter translation efficiency of transfected reporter RNAs but modulated transcript abundance and expression of reporter cDNAs in transfected cells. This indicates that redistribution of PABPC1 may be involved in co- and posttranscriptional regulation of mRNA processing and/or nuclear export by HSV-1 gene regulatory proteins.
Cytoplasmic polyadenylate-binding protein (PABP), through binding the poly(A) tail on mRNAs, occupies a central role in posttranscriptional gene regulation via modulation of translation initiation (21) and mRNA metabolism (32). In addition, traces of the major form of cytoplasmic PABP, PABPC1 (32), are detected in the nucleus in some cultured cell lines (1) and may associate with maturing mRNAs (18) in that compartment. As a reflection of the central role of PABPC1 in gene regulation, many human-pathogenic viruses interfere with PABPC1 function, presumably to benefit viral translation and propagation and/or to thwart host cell gene expression. Reported RNA virus effects on PABPC1 include redistribution to the nucleus, e.g., in rotavirus-infected cells (16), functional modulation through association with viral proteins, e.g., in rubella virus infection (19), or cleavage, e.g., by enterovirus 3C/3CD proteases (20, 28).
Translation initiation occurs upon assembly of the eukaryotic initiation factor 4F (eIF4F), consisting of the m7G cap-binding protein eIF4E, the central scaffold and ribosome adaptor eIF4G, and the RNA helicase eIF4A, at the m7G cap (15). PABPC1, through bridging eIF4G and poly(A) (45), may mediate m7G cap-poly(A) synergistic stimulation of translation initiation (52). Because PABPC1 stimulates eIF4F assembly at the m7G cap and subsequent ribosomal subunit recruitment, it is a bona fide translation factor (21). PABP activity is physiologically modulated by association with PABP-interacting protein 1 (Paip1) and Paip2, which stimulate (7) or inhibit (25) PABP's function in translation initiation, respectively. Both enteroviruses and rotavirus manipulate host cell translation through eIF4G either by cleavage (11) or by antagonizing eIF4G-PABP interaction (38). Moreover, they use unconventional translation templates and initiation mechanisms with reduced requirements for intact eIF4G (enteroviruses) or eIF4G-PABP interaction (rotavirus) (37, 46). It is thus not surprising that replication strategies of these RNA viruses should include manipulation of PABPC1.
However, there also is emerging evidence for manipulation of PABPC1 in cells infected with herpesviruses, DNA viruses that employ canonical translation templates for standard, cap-dependent initiation. For example, lytic infection with Kaposi's sarcoma-associated herpesvirus (KSHV) leads to PABPC1 nuclear redistribution (2, 31), which may be causally related to binding of the KSHV protein K10/10.1 to PABP (23). Similarly, the HSV-1 immediate-early protein ICP27 (infected-cell polypeptide 27) associates with PABP (14). ICP27 is a multifunctional nucleo-cytoplasmic shuttling protein involved in transcriptional and posttranscriptional regulation of viral and cellular gene expression (42). It has been implicated in the regulation of temporal patterns of HSV-1 gene expression by repression of select immediate-early/early genes and induction of early and late genes (33, 35, 39, 40). ICP27 is an RNA-binding protein (36) involved in host cell gene regulation, e.g., through inhibition of host mRNA splicing (17) or proposed stabilization of an AU-rich element containing mRNAs (4). It also has been shown to participate in exonuclear egress of viral transcripts (41) by providing access for intronless, viral mRNAs to the host nuclear export machinery (26). Lastly, ICP27 has been implicated in translation regulation of viral transcripts (10, 13, 30). The significance of ICP27-PABP interaction in HSV-1-infected cells and the effects of HSV-1 protein(s) on PABP function are unknown.
We report here that HSV-1 infection of HeLa cells leads to redistribution of PABPC1 to the nucleus, diminished PABP association with the cap-binding initiation complex via reduced binding to eIF4G, and decreased PABP binding to its physiologic binding partner Paip2. These changes occur without effects on PABPC1, eIF4G, or Paip2 expression levels or integrity. Pull-down assays from HSV-1-infected cell lysates with recombinant glutathione S-transferase (GST)-PABP revealed that in addition to the known association with ICP27, a number of other viral proteins associate with PABP in infected cells. Most notably, the 73.8-kDa tegument protein (UL47), via binding to ICP27, exists in a complex with PABP in HSV-1-infected cells. Joint binding of ICP27 and UL47 to PABP led to displacement of Paip2 and nuclear PABPC1 accumulation in transfected cells. Transfection of either ICP27 or UL47 had opposing effects on translation of transfected reporter cDNAs but failed to alter RNA reporter translation significantly. This suggests that redistribution of PABPC1 and disruption of physiologic binding to eIF4G/Paip2 in HSV-1-infected cells may primarily affect steps in viral or host mRNA processing and/or export rather than translation initiation.
MATERIALS AND METHODS
Cells, viruses, and antibodies.
HeLa and Vero cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum. HSV-1(Kos) was grown and titrated in Vero cells. Polyclonal anti-Paip2 and anti-PABPC1 antibodies were kindly provided by Y. Svitkin and N. Sonenberg (McGill University, Montreal, Canada) and by J. Keene (Duke University, Durham, NC), respectively. Antibodies used included those against eIF4GI (catalog no. 2469), eIF4A (catalog no. 2013), eIF4E (catalog no. 2067), and GAPDH (catalog no. 2118) (Cell Signaling Technology); antibodies against Flag (catalog no. F7425/F3165), c-myc (catalog no. M4439), PABP (catalog no. P6246), Paip2 (catalog no. P0087), tubulin (catalog no. T6074), rabbit immunoglobulin G/tetramethyl rhodamine isothiocyanate [IgG/TRITC] catalog no. T6778), and mouse IgG/FITC (catalog no. F0257) (Sigma); antibody against ICP27 (catalog no. P1119) (Virusys); and antibodies against VP5 (catalog no. ab6508) (Abcam) and against HSV-1 glycoprotein (DakoCytomation).
Plasmids, recombinant proteins, and in vitro RNA synthesis.
The following annealed complementary oligonucleotides were used to insert a Flag tag into NheI-HindIII sites of pcDNA3.1 (Invitrogen): 1, 5′-CTAGCACCATGGATTACAAGGATGACGACGATAAGA; and 2, 5′-AGCTCCTTATCGTCGTCATCCTTGTAATCCATGGTG. The ICP27 open reading frame (ORF) was PCR-amplified from pM27, kindly provided by S. Rice (University of Minnesota, Minneapolis, MN) with the primer pair 3 (5′-TTGGATCCGCGACTGACATTGATATGCT) and 4 (5′-AAGCGGCCGCCTAAAACAGGGAGTTGCA) or the pair 4 and 5 (5′-GGAAGCTTACCATGGCGACTGACATTGATATGCTAATTG) for cloning Flag-tagged or untagged forms into BamHI/NotI sites of Flag-pcDNA3.1 or HindIII-NotI sites of original pcDNA3.1 vector, respectively. The UL47 ORF generated by PCR from HSV-1 genomic DNA with the primer pair 6 (5′-AAGGATCCTCGGCTCGCGAACCCGC) and 7 (5′-TTCTCGAGCTTATGGGCGTGGCG) was inserted downstream of the Flag epitope using BamHI/XhoI sites. Flag-Paip2 expression cDNA was constructed by subcloning the Paip2 ORF from a GST-Paip2 plasmid (3) into the BamHI-NotI sites of the pcDNA3.1-Flag construct. Paip2 with the mutation F118A [Paip2(F118A)] was generated by PCR from wild-type (wt) Paip2 using the primer pair 8 (5′-CGTGGATCCAAAGATCCAAGTCGCAGCAGTACTAGCCCAAGC) and 9 (5′-ACCCCAGGAACAGCCTCCTTTGCAT) and the pair 10 (5′-ATGCAAAGGAGGCTGTTCCTGGGGT) and 11 (5′-ATGCGGCCGCTCAAATATTTCCGTACTTCACCCCAGGAAC). The overlapping fragments were fused in the second PCR with the primer pair 8 and 11 and inserted into the pcDNA3.1-Flag. To generate the GST-PABP bacterial expression plasmid, the PABP ORF was PCR amplified from pTYB2-PABP (3) using the primer pair 12 (5′-GCGGATCCATGAACCCCAGTGCCCCCAG) and 13 (5′-GCGCGGCCGCTTAAACAGTTGGAACACCGGTGGC) and inserted into BamHI-NotI sites of pGEX-4T-1 (GE Healthcare). Escherichia coli BL21 cells transformed with GST- or GST-PABP expression plasmids were induced with isopropyl-β-d-thiogalactopyranoside ([IPTG] 0.5 mM) and cultured for 6 h at 30°C before harvesting and lysis by sonication. Recombinant proteins were purified using a GSTrap FF column (Amersham) and dialyzed against hypotonic buffer (10 mM HEPES at pH 7.5, 0.5 mM MgOAc2, 10 mM KOAc, and 2 mM dithiothreitol, where Ac is acetate). Construction of a TNT-RLuc reporter plasmid containing the 5′-β-globin leader, Renilla luciferase (RLuc) ORF, and poly(A) tail has been reported previously (8). For synthesis of m7G-capped, polyadenylated reporter RNA, TNT-RLuc vector DNA was linearized with BamHI and in vitro transcribed in the presence of m7G(5′)ppp(5′)G cap analog (NEB) using a MAXIscript T7 Kit (Ambion). After a 1-h incubation at 37°C, in vitro transcription reactions were treated with Turbo DNase (Ambion), and RNA was purified using an RNeasy Kit (Qiagen) and quantified on a Nanodrop instrument. Intron-containing or intronless RLuc DNA reporters were constructed by subcloning the RLuc ORF from the TNT-RLuc construct into pCI (Promega) or pcDNA3.1 mammalian expression vectors, respectively.
Transient expression of exogenous proteins, HSV-1 infection, cell extracts, and immunoblotting.
HeLa cells were transfected with pcDNA3.1 control vector or ICP27, UL47, Paip2, or Paip2(F118A) expression plasmids using Lipofectamine 2000 (Invitrogen). HSV-1 infection was carried out at a multiplicity of infection (MOI) of 3. Metabolic labeling was performed as described previously (5). After 16 to 18 h of incubation, cells were scraped, washed with cold phosphate-buffered saline (PBS), and pelleted by centrifugation at 4°C. The pellet was resuspended in an equal volume of lysis buffer (150 mM NaCl, 50 mM HEPES-KOH, pH 7.3, 10% glycerol, 1 mM EDTA, 5 mM EGTA, 0.5% NP-40, 2.5 mM dithiothreitol, and 1 mM phenylmethylsulfonyl fluoride) supplemented with protease inhibitor cocktail (Sigma) and Halt phosphatase inhibitor (Pierce). Lysates were incubated on ice for 5 to 10 min and stored in aliquots at −80°C. Thereafter, cellular debris was separated by centrifugation of the total cell lysate at 14,000 × g for 10 min, and total protein concentration was determined by Bradford assay. For Northern blot analysis, nuclear and cytoplasmic fractions were separated using NE-PER nuclear and cytoplasmic extraction reagents (ThermoScientific). Immunoblotting was carried out as described before (22).
GST-PABP, cap-Sepharose pull-downs, and IP.
Cell lysates (0.5 mg of 35S-labeled or 5 mg of mock- or HSV-1-infected cells) was used for GST-PABP pull-downs. Glutathione-Sepharose beads (GE Healthcare) were preswollen in NT2 buffer (150 mM NaCl, 50 mM Tris-HCl, pH 7.5, 1 mM MgCl2, 0.05% NP-40) supplemented with 1% bovine serum albumin and incubated with 8 to 20 μg of recombinant GST-PABP or GST overnight at 4°C. Unbound proteins were washed off three times with 1 ml of cold NT2 buffer, and beads were incubated with precleared mock- or HSV-1-infected lysates for 4 h at 4°C. Thereafter, beads were rinsed five times with NT2 buffer, and bound proteins were resolved by electrophoresis in 4 to 12% Bis-Tris NuPAGE gels (Invitrogen) for subsequent autoradiography, Coomassie staining (Denville Scientific), or Western blot analysis. The same protocol was used for 7-methyl-GTP-Sepharose (GE Healthcare) pull-downs. Immunoprecipitation (IP) reactions were carried out with protein G-Sepharose beads (GE Healthcare) coated with 10 μg of primary antibodies and 1 to 1.5 mg of mock- or HSV-1-infected cell lysates. Anti-Flag M2-agarose (Sigma) was used to precipitate Flag-tagged proteins from transfected HeLa cell lysates.
HeLa cell transfections and RLuc reporter assays.
To assess the effect of HSV-1 proteins ICP27 and/or UL47 on reporter RNA translation, 1 × 106 HeLa cells were transfected with pcDNA3.1 control, 0.1 μg of ICP27, 0.5 μg of Flag-UL47 expression vectors, or a combination of these. Total amounts of transfected DNA were kept constant by addition of pcDNA3.1 DNA if necessary. Twenty-four hours later, cells were transfected with 0.5 μg of capped RLuc reporter RNA using DMRIE-C (1,2-dimyristyloxypropyl-3-dimethyl-hydroxy ethyl ammonium bromide and cholesterol) reagent (Invitrogen); RLuc DNA reporters were cotransfected with the ICP27 and/or Flag-UL47 expression plasmids. RLuc activity was measured 8 h (RNA reporter transfections) or 24 h (cDNA reporter transfections) later in a luminometer (Turner Biosystems). The effect of HSV-1 infection on RNA/DNA reporter translation was evaluated after transfection into HeLa cells 1 h after HSV-1 or mock infection.
Northern blot analyses.
Total RNA from nuclear and cytoplasmic fractions was extracted with RNeasy columns (Qiagen) and treated with RNase-free DNase (Qiagen). The quality of the extracted total RNA samples was verified by agarose gel electrophoresis and UV spectrometry. Thereafter, RNA was subjected to gel electrophoresis on 1% agarose gels containing glyoxal and transferred to membrane (Nytran SuperCharge; Whatman) by passive diffusion. Hybridization with radioactively labeled probe was carried out with ExpressHyb solution (Clontech) according to the manufacturer's protocol. Probe corresponding to the portion of the RLuc ORF was generated by PCR with primers 14 (5′-GGGCGGTAGGCGTGTACGGTGGG) and 15 (5′-GATGGCAACATGGTTTCCACG) and labeled with [α32-P]dCTP by random priming using a High Prime system (Roche) according to the manufacturer's suggestions.
Immunofluorescence.
Immunostaining was performed essentially as described previously (24). Briefly, 105 HeLa cells were plated on 12-mm glass coverslips and incubated for 24 h, infected with HSV-1 at an MOI of 10 for 7 h, or transfected with the ICP27 and/or UL47 expression construct for 18 h. Cells were washed three times with PBS, fixed with 4% paraformaldehyde in PBS for 15 min, and permeabilized with −20°C methanol for 10 min. The cells were treated for 1 h with blocking buffer (10% goat serum in PBS) and incubated with primary antibodies diluted in blocking buffer at 4° overnight. Cells were washed and incubated for 45 min at room temperature with a mixture of FITC-conjugated anti-mouse IgG (Sigma) and TRITC-conjugated anti-rabbit IgG (Sigma) at dilutions of 1:150 and 1:500, respectively. Coverslips were washed three times and mounted on glass slides using ProLong Gold antifade reagent with 4′,6′-diamidino-2-phenylindole (DAPI; Invitrogen). Images were collected using an XI50 Olympus microscope, DP70 digital camera, and DPController/DPManager software. Images were processed and analyzed using Adobe Photoshop software.
RESULTS
Reduced association of PABP with the translation initiation complex in HSV-1 infected cells.
To evaluate the effects of HSV-1 infection on translation initiation factors in HeLa cells, we examined the relative abundance of all eIF4F components and PABP in mock- and HSV-1-infected cells. Immunoblot analysis demonstrated that the abundance and integrity of these proteins were unchanged upon HSV-1 infection (Fig. 1A). Next, to test functional interaction of translation factors, we analyzed eIF4F assembly at the m7G cap. Lysates prepared from mock- or HSV-1-infected HeLa cells were subjected to m7G cap-Sepharose pull-down, and precipitated proteins were analyzed by immunoblotting (Fig. 1B). Although equal amounts of translation initiation factors were present in input lysates (Fig. 1A), we consistently recovered slightly reduced amounts of eIF4E and eIF4G from HSV-1-infected samples. A more pronounced difference was observed for PABP's association with eIF4F, with significantly less PABP recovered by m7G cap-Sepharose pull-downs in HSV-1-infected cells (Fig. 1B). A similar finding has been reported previously (51). Since PABP associates with eIF4F through eIF4G, we performed coimmunoprecipitation (co-IP) of these two proteins. Because IP of endogenous eIF4G is inefficient with available antibodies (data not shown), lysates were prepared from mock- or HSV-1-infected HeLa cells expressing tetracycline (Tet)-inducible myc-tagged eIF4GI-b (22). IP with anti-c-myc antibody recovered equal amounts of eIF4GI from mock- and HSV-1-infected cells, but significantly less PABP was coprecipitated from infected lysates (Fig. 1C). This suggests reduced PABP association with the m7G cap-binding complex in HSV-1-infected cells (Fig. 1B). All assays were performed with antibodies specifically recognizing PABPC1.
FIG. 1.

Abundance and association of translation initiation complex components in HSV-1-infected cells. (A) Increasing amounts of mock- and HSV-1(Kos)-infected HeLa lysates were loaded to compare steady-state levels and integrity of m7G cap-binding complex components (eIF4G, eIF4A, and eIF4E) and PABP. ICP27 was included to mark HSV-1 infection. (B) Equal amounts (1 mg of total protein) of lysates prepared from mock- or HSV-1-infected HeLa cells were incubated with m7G-Sepharose, and precipitated proteins were analyzed by immunoblot. (C) Lysates prepared from mock- or HSV-1-infected HeLa cells expressing myc-tagged eIF4GI after Tet induction were subjected to IP with anti-myc antibody or isotype-matched mouse IgG1 control.
Recent reports of nuclear accumulation of cytoplasmic PABP in KHSV-infected cells undergoing lytic reactivation (2) prompted us to examine an effect of HSV-1 on the intracellular distribution of cytoplasmic PABP. HeLa cells were infected at an MOI of 10 for 7 h and subjected to immunofluorescent labeling of PABPC1 and HSV-1 glycoproteins (Fig. 2D to F). In mock-infected cells, PABPC1 was present exclusively in cytoplasm (Fig. 2A), but HSV-1 infection produced conspicuous PABPC1 redistribution to the nuclear compartment (Fig. 2D, G, and J). This effect was observed only in infected cells since nuclear PABPC1 signal overlapped with HSV-1 glycoproteins (Fig. 2F). The distribution of glyceraldehyde-3-phosphate dehydrogenase (GAPDH), a cytoplasmic component in mock-infected cells (Fig. 2C), did not change upon HSV-1 infection (Fig. 2H and I). Similarly, Paip2, an established PABP interaction partner with cytoplasmic distribution (25), also remained in this location in HSV-1-infected cells (Fig. 2B, K, and L). These findings suggest that HSV-1 infection promotes relocalization of cytoplasmic PABP, but not its native binding partner Paip2, to the nucleus.
FIG. 2.

HSV-1 infection results in PABPC1 redistribution to the nucleus. HeLa cells were mock infected (A to C) or infected at an MOI of 10 with HSV-1 for 7 h (D to L). PABPC1 (A), Paip2 (B) and GAPDH control (C) are shown in mock-infected HeLa cells. HSV-1 infection results in PABPC1 relocalization to the nucleus in infected (D; white arrow) but not uninfected (D; yellow arrows) cells in the same culture. (E) Infection was confirmed by staining with anti-HSV-1 glycoprotein antibody (gp). (F) Overlapping nuclear PABPC1/HSV-1 glycoprotein is visible in the merged image. PABPC1 (G and J), Paip2 (H), and GAPDH (K) are shown in presumably uninfected cells (yellow arrow) and infected cells (white arrow). Merged images show overlapping nuclear PABPC1/DAPI signal and markedly reduced cytoplasmic PABPC1 signal only in infected cells (I and L). pi, postinfection.
Binding of viral proteins to PABP in HSV-1-infected cells.
Reduced PABPC1 association with eIF4GI and the m7G cap-binding complex and relocation to the nucleus upon HSV-1 infection suggest an involvement of viral proteins. To address this question, we performed GST-PABPC1 pull-down experiments from metabolically labeled HeLa cells and analyzed the differences between PABP binding partners in mock- and HSV-1-infected cells (Fig. 3A). In RNase-untreated lysates this assay was encumbered by relatively high background signal that complicated interpretation of the results. For this reason, all cellular extracts in subsequent experiments were subjected to RNase treatment.
FIG. 3.

Identification of proteins interacting with PABP in HSV-1-infected cells. (A) GST-PABPC1 pull-down from metabolically labeled mock- or HSV-1-infected HeLa cell lysates. Band 1 in mock lysates and band 2 in infected lysates correspond to Paip2 and ICP27, respectively. (B) Immunoblotting of proteins bound to GST alone (negative control; lanes 1 and 3) or GST-PABP (lanes 2 and 4). (C) Co-IP of Paip2 with PABP in mock- and HSV-1-infected cells. Antibodies used for IP (isotype-matched mouse IgG1 [mIgG] or anti-PABP) are indicated in a gray-shaded box. (D) Proteins bound to GST-PABP were resolved by SDS-PAGE and stained with Coomassie blue, and bands 1 to 4 present in HSV-1-infected but not in mock-infected lysates were submitted for MS. (E) List of PABP-associated proteins identified in HSV-1-infected cell lysates. MM, molecular mass.
We detected a changing pattern of PABP-associated proteins in HSV-1-infected compared to mock-infected lysates (Fig. 3A). Several protein bands associated with GST-PABPC1 in mock lysates were weakened or absent in infected lysates while unique protein bands were present in HSV-1-infected lysates (Fig. 3A). Disappearing cellular proteins from infected GST-PABPC1 pull-downs could be due to either reduced synthesis in infected cells or virus-mediated events that discourage PABP binding. The latter scenario is exemplified by band 1 in Fig. 3A. Parallel immunoblotting showed that this band is Paip2 (data not shown). Equal amounts of Paip2 were detected by immunoblotting from total lysates prepared from mock- and HSV-1-infected cells (Fig. 3B and C, lanes 5 and 6), but the amount of protein present in GST-PABPC1 pull-downs of infected lysates was much less than the amount in mock lysates (Fig. 3B, compare lanes 2 and 4). We were concerned by the fact that we reproducibly observed less PABP bound to recombinant GST-PABPC1 in HSV-1-infected lysates (Fig. 3B, compare lanes 2 and 4), which might be the reason for reduced Paip2 co-IP. To exclude this possibility, we performed IP of mock- and HSV-1-infected lysates with anti-PABPC1 (Fig. 3C) antibody. In this experiment, similar amounts of PABPC1 were precipitated from both extracts, but no detectable Paip2 was coprecipitated in infected cells (Fig. 3C, compare lanes 2 and 4), suggesting that PABP-Paip2 interaction is indeed disrupted by HSV-1 infection. One prominent band in GST-PABPC1 pull-downs of infected lysates corresponded by size with ICP27 (Fig. 3A, band 2), which has been shown before to interact with PABP in HSV-1-infected cells (14). Immunoblot analysis confirmed that ICP27, indeed, is precipitated with GST-PABPC1 in infected extracts (Fig. 3B, lane 4).
The GST-PABPC1 pull-down assay suggested that multiple HSV-1 proteins associate with PABPC1 in infected cells. Therefore, lysates from mock- and HSV-1-infected HeLa cells were incubated with GST-PABPC1-coupled Sepharose, and bound proteins were resolved by SDS-PAGE and visualized by Coomassie blue (Fig. 3D). Bands present in infected but absent in mock lysates were analyzed by tandem mass spectrometry (MS/MS). We identified five viral proteins associating with PABP in HSV-1-infected cells (Fig. 3E). These were the HSV-1 major capsid protein VP5, ICP6, the 73.8 tegument protein (UL47), ICP27, and VP16 (Fig. 3E). The only Homo sapiens protein identified was PABP2, a known binding partner of PABPC1 (Fig. 3E) (6). We again confirmed ICP27 binding to PABP in our screen, but VP5 and UL47 were novel PABP-interacting proteins. ICP6 and VP16 did not have very high MS MS/MS scores; they may have precipitated in our assay via known interactions with eIF4G (49) and UL47 (48), respectively. Since ICP27 and UL47 associated with PABPC1 in RNase-treated extracts, we assume that binding occurs due to direct protein-protein interactions.
Validation of HSV-1 proteins binding to PABP.
To confirm association of HSV-1 proteins with PABPC1 in HSV-1-infected cells, we performed IPs with ICP27- or VP5-specific antibodies, followed by immunoblot analysis to identify coprecipitating proteins (Fig. 4). Mock-infected cell lysates and control mouse IgG1 were used to ensure binding specificity. PABPC1 was coprecipitated with anti-ICP27 from HSV-1-infected but not mock-infected lysates (Fig. 4A, lane 4); no nonspecific IP was observed with mouse IgG1 control (Fig. 4A, lane 3). Small amounts of VP5 and Paip2 observed in ICP27 IP from infected cell lysates (Fig. 4A, lane 4) indicated either direct interaction of these proteins with ICP27 or binding mediated via PABPC1. Exceedingly low levels of ICP27 and no PABPC1 was detected in VP5 IP of HSV-1-infected lysates (Fig. 4B, lane 4), suggesting a lack of direct VP5-PABPC1 binding. Thus, the presence of VP5 in GST-PABPC1 pull-downs might reflect inherent, nonspecific binding to recombinant protein or indirect association with PABPC1 mediated by ICP27. Because UL47 antibodies are unavailable commercially, we used ectopic, Flag-tagged UL47 for the evaluation of PABPC1-UL47 interactions. Flag-tagged UL47 was expressed in HeLa cells, and proteins coprecipitated with anti-Flag antibody from untransfected or Flag-UL47-transfected mock- and HSV-1-infected cells were analyzed by immunoblotting (Fig. 4C). PABPC1 was detected only in anti-Flag IP of HSV-1-infected lysates (Fig. 4C, lane 6), suggesting that additional viral proteins mediate UL47-PABP binding. ICP27 and VP5, also identified in GST-PABPC1 pull-downs, were detected in anti-Flag-UL47 IP of infected lysates. We therefore speculate that ICP27 might bridge PABP-UL47 association since it interacts with PABPC1 directly (Fig. 4A).
FIG. 4.

Confirmation of PABPC1 interactions with HSV-1 proteins ICP27, VP5, and UL47. Proteins from mock- or HSV-1-infected cells were immunoprecipitated with mouse IgG1 (mIgG) or anti-ICP27 (A), anti-VP5 (B), or anti-Flag (C) antibodies (the antibodies used for IP are indicated in shaded boxes), resolved by 4 to 12% PAGE, and probed with the indicated antibodies. For anti-Flag IP (C), cells were transfected with a Flag-tagged UL47 expression construct followed by mock or HSV-1 infection. Protein content in cell lysates is shown in input lanes. ab, antibody.
ICP27 binds UL47 directly.
To test our hypothesis, we transiently expressed ICP27, UL47, or both together in HeLa cells and analyzed their association with PABPC1 (Fig. 5). Two distinct ICP27 expression constructs (Flag tagged and untagged) were used to ensure authenticity of protein binding to Flag-Sepharose. None of the analyzed proteins was observed in anti-Flag IP of vector- or ICP27-transfected lysates, indicating the absence of nonspecific binding (Fig. 5A, lanes 1 and 2). PABPC1 was coprecipitated in the presence of only Flag-ICP27, either expressed alone or in combination with UL47 (Fig. 5A, lanes 3 and 5). UL47 was not able to bind PABP on its own (Fig. 5A, lane 4) but precipitated untagged ICP27 (Fig. 5A, lane 6), suggesting that other viral proteins are not required for ICP27-UL47 interaction. ICP27 IP performed with the same cell lysates proved this hypothesis (Fig. 5B, lane 6). Similar to PABP, its binding partner Paip2 was coprecipitated with ICP27 (Fig. 5 A, lanes 3 and 5, and B, lanes 3 and 6) although it is not clear if ICP27 interacts with Paip2 directly or whether their coprecipitation is mediated by PABP binding. Interestingly, in both anti-Flag and anti-ICP27 IPs, less Paip2 coprecipitated from lysates containing both ICP27 and UL47 (Fig. 5A, lane 5, and B, lane 6), suggesting that they jointly displace Paip2 from PABP, as observed with GST-PABPC1 pull-downs of HSV-1-infected lysates (Fig. 3B).
FIG. 5.

ICP27 binds UL47 directly. Lysates were prepared from HeLa cells transfected with various expression vectors, indicated on top, and subjected to anti-Flag (A) or anti-ICP27 IP (B) (antibodies used for IP are shown in the shaded box). Coprecipitated proteins were resolved by 4 to 12% PAGE and probed with the indicated antibody. Protein content in lysates prior to IP is shown in input lanes. mIgG, mouse IgG; ab, antibody.
To address the possibility of ICP27-Paip2 interaction, we used Paip2 mutant F118A, which is unable to bind PABP (27). If ICP27-Paip2 binding occurs via PABP, disruption of the PABP-Paip2 heterodimer should abolish co-IP of ICP27 with Paip2. cDNA constructs expressing Flag-tagged Paip2 (wt or F118A) alone or in combination with ICP27 were transfected in HeLa cells, and proteins coprecipitating on Flag-Sepharose were analyzed by immunoblotting (Fig. 6). ICP27 coprecipitates only with wt, PABP-binding-competent Paip2 (Fig. 6, lane 5). Paip2(F118A) binds neither PABP nor ICP27 (Fig. 6, lane 6), suggesting that ICP27 does not interact with Paip2 directly.
FIG. 6.

PABPC1 mediates Paip2-ICP27 interaction. HeLa cells were transfected with the indicated expression plasmids and subjected to anti-Flag IP. Protein input for corresponding lysates is shown in lanes 9 to 12. Paip2(F118A) (Paip2mut) has consistently slower electrophoretic mobility than the wt form.
ICP27 expression results in PABPC1 relocalization to the nucleus.
ICP27 and UL47 are nucleo-cytoplasmic shuttling proteins that, through their interaction with PABPC1, might be involved in its redistribution (9, 36). To test this hypothesis, we performed immunostaining of HeLa cells after transfection of ICP27 and Flag-UL47 expression constructs (Fig. 7). In accordance with published data, both UL47 (9) and ICP27 (36) assumed a predominantly nuclear distribution in transfected cells (Fig. 7A to C). ICP27 expression mediated PABPC1 redistribution to the nucleus, similar to the effect observed in HSV-1-infected cells compare Fig. 7D to F with Fig. 2G and J). UL47 expression alone did not alter PABPC1 distribution (Fig. 7G to I), reflecting its inability to associate with the protein on its own. Combined expression of ICP27 and UL47 did not significantly change PABPC1 subcellular distribution compared to ICP27 alone (Fig. 7J to L).
FIG. 7.

Effect of ectopic HSV-1 ICP27 and UL47 on PABPC1 subcellular distribution. HeLa cells were transfected with ICP27 and/or UL47 expression constructs (as indicated on the left of each panel) 18 h prior to immunostaining. UL47 (A) and ICP27 (B) colocalize in the nucleus of transfected cells (C). ICP27 expression leads to nuclear accumulation of PABPC1 in transfected (D; white arrow) cells but not their nontransfected neighbors (D; yellow arrow). Nuclear PABPC1 and ICP27 (E) colocalize in nuclei, as shown in merged images (F). In UL47-transfected cells, PABPC1 (G) and UL47 (H) do not colocalize (I). In cells cotransfected with ICP27 and UL47, PABPC1 redistribution (J; white arrow) occurs in cells expressing UL47 (K) but not in untransfected neighbors (J; yellow arrow). The merged image shows nuclear colocalization of PABPC1 and UL47 (L).
Effect of ICP27 and UL47 on translation.
A number of observations in our study suggest concerted action of HSV-1 proteins on PABPC1. All documented events, reduced PABPC1-eIF4G association, redistribution of PABPC1 to the nucleus, ICP27 and UL47 binding to PABPC1, and displacement of Paip2, have the potential to modulate PABPC1's role in translation control. To selectively assess the effect of HSV-1 PABP-binding proteins on translation efficiency, we examined translation of RLuc RNA reporters transfected into ICP27- and/or UL47-expressing HeLa cells. HSV-1 proteins were efficiently expressed 24 h after expression plasmid transfection (Fig. 8D).
FIG. 8.
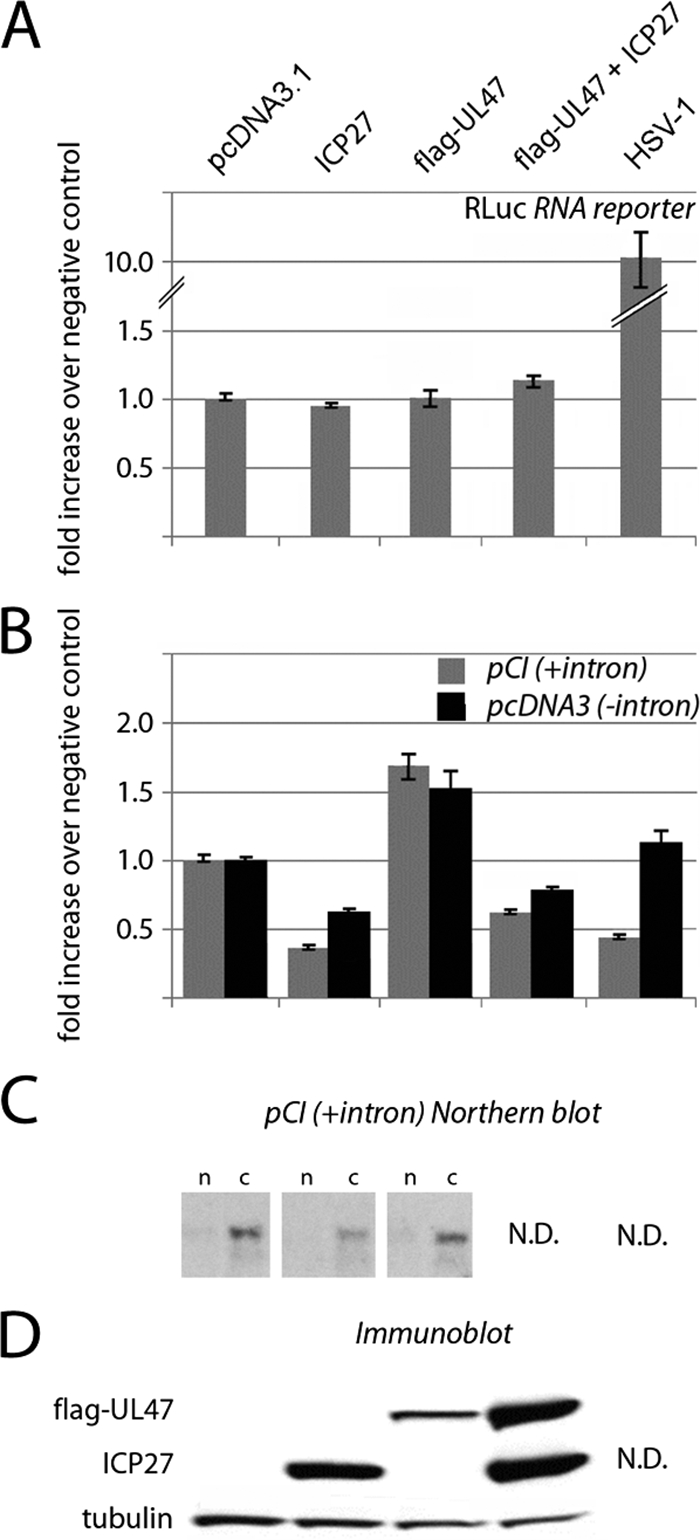
Effect of ectopic HSV-1 ICP27 and UL47 on gene expression. (A) RNA reporter cotransfection studies. HeLa cells were transfected with RLuc expression reporter RNA and pcDNA3.1 control, ICP27, or Flag-UL47 expression vectors alone or combined (see Materials and Methods for details). (B) DNA reporter cotransfection studies. HeLa cells were transfected with pCI-RLuc (with intron) or pcDNA3-RLuc (intronless) expression plasmids and pcDNA3.1 control, ICP27, or Flag-UL47 expression vectors alone or combined. (C) Northern blot analysis of RLuc mRNA in nuclear (n) and cytoplasmic (c) fractions of HeLa cells transfected with pcDNA3.1 control or ICP27 or UL47 expression constructs. (D) Immunoblotting of ectopic viral proteins. Where indicated, HSV-1 infection was performed 1 h prior to reporter RNA/DNA transfection, and RLuc levels are shown as the relative change over mock-infected or control-transfected cells.
ICP27, UL47, or their combination failed to significantly affect reporter RNA translation (Fig. 8A). Since our assay involved two tandem transfections, it is possible that not all cells contain both the exogenous viral factors and reporter RNA. However, tests with green fluorescent protein-expressing control reporters consistently showed cDNA transfection efficiencies of ≥80% (data not shown), indicating that the vast majority of reporter RNA-transfected cells express ICP27 and/or UL47. In HSV-1-infected cells, reporter RNA expression is actually stimulated, most likely due to degradation of competing host mRNAs. Thus, our assays suggest that PABPC1 redistribution does not affect conventional, m7G cap-mediated translation.
To test the effect of ICP27 and UL47 on gene-regulatory events prior to translation, we cotransfected HeLa cells with pcDNA3 control, ICP27, or UL47 expression vectors or their combination along with one of two RLuc expression cDNAs, either intronless (pcDNA-RLuc) or containing a chimeric intron upstream of the RLuc ORF (pCI-RLuc). RLuc activity measured 24 h after cDNA transfection demonstrated that ICP27 and UL47 had opposing effects on reporter expression (Fig. 8B). While ICP27 considerably reduced RLuc expression, UL47 had a stimulatory effect that was able to partially compensate for ICP27-mediated inhibition of RLuc expression in coexpressing cells (Fig. 8B). Cotransfection with ICP27 and UL47 expression plasmids consistently yielded higher UL47 levels than those achieved with the UL47 cDNA alone (Fig. 8D), albeit identical amounts of DNA were transfected. The reasons for this are unknown. Northern blot analyses of RLuc mRNA in ICP27- or UL47-expressing cells revealed that translation modulation covaried with template levels in cytoplasm (Fig. 8C). This suggests that ICP27 reduces reporter expression primarily by interfering with template synthesis or maturation. Although ICP27's effect on translation of the intron-containing reporter was more pronounced (Fig. 8B), possibly due to interference with splicing, RLuc expression was also reduced with the intronless reporter, suggesting involvement of ICP27 in processes other than splicing.
Northern blot data suggested that UL47, unlike ICP27, does not affect RLuc mRNA synthesis or maturation (Fig. 8C). We speculated that UL47-mediated stimulation of reporter expression might result from enhanced nuclear mRNA export because UL47 binds polyadenylated mRNAs, is a nucleo-cytoplasmic shuttling protein (9), and has been reported to associate with the mRNA export factor CRM1 (53). Testing the effect of the CRM1 inhibitor leptomycin B on reporter cDNA expression in UL47-expressing HeLa cells was inconclusive due to overlapping toxicities of transfection and the drug (data not shown). In contrast to ICP27 transfection, HSV-1 did not alter the expression of intronless reporter (Fig. 8B). This may reflect ICP27-independent viral effects on mRNA maturation and export, specifically favoring export of unspliced RNAs and reducing the competition for their translation.
DISCUSSION
Our data suggest that the major form of cytoplasmic PABP, PABPC1, associates with a number of HSV-1 proteins in infected cells, leading to disruption of binding to its physiologic partners eIF4G and Paip2 and to redistribution to the nucleus. Co-IP studies demonstrated that ICP27 provides the anchor for its binding partner UL47 associating with PABPC1. Other HSV-1 proteins proposed to associate with UL47, e.g., VP16 (48), or with eIF4G, e.g., ICP6 (49), were identified in our proteomic screen and may exist in a larger complex with PABPC1. Apart from ICP27 association with PABP (14), a number of modulations of translation factor function have been proposed to occur in HSV-1-infected cells. It has been suggested that formation of eIF4F at the m7G cap may be aided by ICP0-induced eIF4E phosphorylation via the mitogen-activated protein kinase-integrating kinase Mnk1 (50) and/or binding of ICP6 to eIF4G (49), particularly in quiescent cells.
We did not detect outright modification of translation factors in HSV-1-infected HeLa cells, and association of initiation complexes at the m7G cap appears to occur unchanged, with the exception of reduced PABPC1 recruitment. Also, transfection of m7G cap-dependent reporter RNAs into cells expressing PABPC1 binding proteins ICP27 and/or UL47 revealed no influence on translation. It is plausible that in HSV-1-infected cells, with the bulk of host mRNA degraded by the viral host shutoff protein (vhs) (12, 29), residual cytoplasmic PABPC1 is sufficient for translation of viral transcripts or that PABP requirements for efficient initiation are lowered. Indeed, HSV-1 infection actually stimulates translation of transfected reporter RNAs, most likely due to removal of competing host mRNAs. Our findings correspond with PABP small interfering RNA depletion studies that show only ∼10% inhibition of translation in cells with ∼85% reduction of PABP levels (54). Thus, HSV-1 interference with PABPC1 does not seem to primarily alter its function in translation initiation. Rather, our Luc reporter assays and Northern blot data with transfected cDNA reporters point to a role of PABPC1 binding proteins in modulating gene expression in HSV-1-infected cells at a step prior to or concurrent with mRNA export.
The vast majority of PABPC1 is located in the cytoplasm in uninfected HeLa cells. Thus, although nuclear shuttling of PABPC1 may occur under physiologic conditions (1), the proportion of nuclear PABPC1 appears to be minuscule. It has been proposed that PABPC1 associates with nascent poly(A) tails on maturing mRNAs, but a role for nuclear PABPC1 in mRNA metabolism remains unclear (18). Thus, there appears to be no physiologic correlate for the significant nuclear influx of PABPC1 in HSV-1-infected cells. Furthermore, since there are no defined functional roles of nuclear PABPC1 for eukaryotic mRNA processing, it may uniquely benefit viral gene expression and replication strategies.
Both UL47 (47) and ICP27 (41) are nucleocytoplasmic shuttling proteins and, thus, capable in principle of recruiting PABPC1 to the nucleus. Due to their biochemically detectable association with PABPC1 in HSV-1-infected cells, our studies focused on ICP27 and UL47. However, there is no doubt that the effects of these proteins on gene expression control depend on the larger context in HSV-1-infected cells. ICP27 and UL47, when expressed alone, had opposing effects on translation in HeLa cells transfected with reporter cDNA. ICP27 was a potent inhibitor of translation, in accordance with known roles in repression of host cell gene expression. Due to its inhibitory effect on host cell splicing (43), spliced reporters were more affected than intronless transcripts. Since reporter expression was assessed at least 24 h after ICP27 transfection, it is likely that adverse effects on splicing exert nonspecific secondary effects on gene expression not directly related to PABPC1 in our assay. Despite its effects on host cell translation, ICP27 has been shown to stimulate translation of select late viral RNAs, an effect that has been linked to its association with PABP (13). Interestingly, UL47 expression alone consistently caused a significant surge in translation from reporter cDNAs. UL47, a major structural component of the HSV-1 tegument (34) and nucleo-cytoplasmic shuttling protein (47), was reported to bind the nuclear export factor CRM1 (53). Since UL47 is also an RNA-binding protein (44), its translation-stimulatory function may be due to a role in mRNA export.
In addition to redistribution to the nucleus, our results indicate that ICP27 and its binding partner UL47 are responsible for disrupting PABPC1-Paip2 interactions in HSV-1-infected cells. Since Paip2 remains in its physiological cytoplasmic locale in HSV-1-infected cells, it is unclear whether Paip2 dissociation is a primary event following the association of HSV-1 proteins with PABPC1 or secondary to PABPC1 redistribution to the nucleus. Although co-IP data suggest that disrupting PABP-Paip2 binding requires joint binding of ICP27 and UL47 to PABPC1, ICP27 alone caused PABPC1 redistribution. This may indicate simultaneous action of multiple contributing factors associating with PABPC1 and modifying its function in HSV-1-infected cells. Paip2 primarily has been implicated in regulating PABPC1's function in translation initiation (25). With its natural binding partner, PABPC1, largely absent in the cytoplasm of HSV-1-infected cells, this function may be abolished.
In summary, our data suggest that association of ICP27 and its binding partner UL47 with PABPC1 results in its redistribution to the nucleus. Nuclear PABPC1 may support their known roles in regulation of viral and host gene expression, e.g., via an involvement in viral mRNA processing and export. For example, although both ICP27 and UL47 are RNA-binding proteins themselves, abundant nuclear PABPC1 might provide a convenient adaptor for binding to polyadenylated viral transcripts in the nucleus, facilitating viral mRNA export. Scarce nuclear PABPC1 appears to associate with nascent eukaryotic mRNA under physiologic circumstances (18), but a role in eukaryotic mRNA biogenesis and processing was difficult to decipher. Abundant nuclear PABPC1 in HSV-1-infected cells may help unravel its possible physiological roles in mRNA metabolism.
Acknowledgments
We are grateful for gifts of anti-PABPC1 antibodies (J. Keene, Duke University, Durham, NC), anti-Paip2 antibodies (Y. Svitkin and N. Sonenberg, McGill University, Montreal, Canada), and ICP27 expression plasmids (S. Rice, University of Minnesota, Minneapolis, MN). We thank I. Mohr (New York University, New York, NY) for critical discussion of our data.
This work was supported by PHS grant CA124756 (M.G.).
Footnotes
Published ahead of print on 28 October 2009.
REFERENCES
- 1.Afonina, E., R. Stauber, and G. N. Pavlakis. 1998. The human poly(A)-binding protein 1 shuttles between the nucleus and the cytoplasm. J. Biol. Chem. 273:13015-13021. [DOI] [PubMed] [Google Scholar]
- 2.Arias, C., D. Walsh, J. Harbell, A. C. Wilson, and I. Mohr. 2009. Activation of host translational control pathways by a viral developmental switch. PLoS Pathog. 5:e1000334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bradrick, S. S., E. Y. Dobrikova, C. Kaiser, M. Shveygert, and M. Gromeier. 2007. Poly(A)-binding protein is differentially required for translation mediated by viral internal ribosome entry sites. RNA 13:1582-1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown, C. R., M. S. Nakamura, J. D. Mosca, G. S. Hayward, S. E. Straus, and L. P. Perera. 1995. Herpes simplex virus trans-regulatory protein ICP27 stabilizes and binds to 3′ ends of labile mRNA. J. Virol. 69:7187-7195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Campbell, S. A., M. Mulvey, I. Mohr, and M. Gromeier. 2007. Attenuation of herpes simplex virus neurovirulence with picornavirus cis-acting genetic elements. J. Virol. 81:791-799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chiu, S. Y., F. Lejeune, A. C. Ranganathan, and L. E. Maquat. 2004. The pioneer translation initiation complex is functionally distinct from but structurally overlaps with the steady-state translation initiation complex. Genes Dev. 18:745-754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Craig, A. W., A. Haghighat, A. T. Yu, and N. Sonenberg. 1998. Interaction of polyadenylate-binding protein with the eIF4G homologue PAIP enhances translation. Nature 392:520-523. [DOI] [PubMed] [Google Scholar]
- 8.Dobrikova, E. Y., R. N. Grisham, C. Kaiser, J. Lin, and M. Gromeier. 2006. Competitive translation efficiency at the picornavirus type 1 internal ribosome entry site facilitated by viral cis and trans factors. J. Virol. 80:3310-3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Donnelly, M., and G. Elliott. 2001. Nuclear localization and shuttling of herpes simplex virus tegument protein VP13/14. J. Virol. 75:2566-2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ellison, K. S., R. A. Maranchuk, K. L. Mottet, and J. R. Smiley. 2005. Control of VP16 translation by the herpes simplex virus type 1 immediate-early protein ICP27. J. Virol. 79:4120-4131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Etchison, D., S. C. Milburn, I. Edery, N. Sonenberg, and J. W. Hershey. 1982. Inhibition of HeLa cell protein synthesis following poliovirus infection correlates with the proteolysis of a 220,000-dalton polypeptide associated with eucaryotic initiation factor 3 and a cap binding protein complex. J. Biol. Chem. 257:14806-14810. [PubMed] [Google Scholar]
- 12.Fenwick, M. L., and M. M. McMenamin. 1984. Early virion-associated suppression of cellular protein synthesis by herpes simplex virus is accompanied by inactivation of mRNA. J. Gen. Virol. 65:1225-1228. [DOI] [PubMed] [Google Scholar]
- 13.Fontaine-Rodriguez, E. C., and D. M. Knipe. 2008. Herpes simplex virus ICP27 increases translation of a subset of viral late mRNAs. J. Virol. 82:3538-3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fontaine-Rodriguez, E. C., T. J. Taylor, M. Olesky, and D. M. Knipe. 2004. Proteomics of herpes simplex virus infected cell protein 27: association with translation initiation factors. Virology 330:487-492. [DOI] [PubMed] [Google Scholar]
- 15.Gingras, A. C., B. Raught, and N. Sonenberg. 1999. eIF4 initiation factors: effectors of mRNA recruitment to ribosomes and regulators of translation. Annu. Rev. Biochem. 68:913-963. [DOI] [PubMed] [Google Scholar]
- 16.Harb, M., M. M. Becker, D. Vitour, C. H. Baron, P. Vende, S. C. Brown, S. Bolte, S. T. Arold, and D. Poncet. 2008. Nuclear localization of cytoplasmic poly(A)-binding protein upon rotavirus infection involves the interaction of NSP3 with eIF4G and RoXaN. J. Virol. 82:11283-11293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hardy, W. R., and R. M. Sandri-Goldin. 1994. Herpes simplex virus inhibits host cell splicing, and regulatory protein ICP27 is required for this effect. J. Virol. 68:7790-7799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hosoda, N., F. Lejeune, and L. E. Maquat. 2006. Evidence that poly(A) binding protein C1 binds nuclear pre-mRNA poly(A) tails. Mol. Cell. Biol. 26:3085-3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ilkow, C. S., V. Mancinelli, M. D. Beatch, and T. C. Hobman. 2008. Rubella virus capsid protein interacts with poly(A)-binding protein and inhibits translation. J. Virol. 82:4284-4294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Joachims, M., P. C. Van Breugel, and R. E. Lloyd. 1999. Cleavage of poly(A)-binding protein by enterovirus proteases concurrent with inhibition of translation in vitro. J. Virol. 73:718-727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kahvejian, A., Y. V. Svitkin, R. Sukarieh, M. N. M'Boutchou, and N. Sonenberg. 2005. Mammalian poly(A)-binding protein is a eukaryotic translation initiation factor, which acts via multiple mechanisms. Genes Dev. 19:104-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kaiser, C., E. Y. Dobrikova, S. S. Bradrick, M. Shveygert, J. T. Herbert, and M. Gromeier. 2008. Activation of cap-independent translation by variant eukaryotic initiation factor 4G in vivo. RNA 14:2170-2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kanno, T., Y. Sato, T. Sata, and H. Katano. 2006. Expression of Kaposi's sarcoma-associated herpesvirus-encoded K10/10.1 protein in tissues and its interaction with poly(A)-binding protein. Virology 352:100-109. [DOI] [PubMed] [Google Scholar]
- 24.Kedersha, N., and P. Anderson. 2007. Mammalian stress granules and processing bodies. Methods Enzymol. 431:61-81. [DOI] [PubMed] [Google Scholar]
- 25.Khaleghpour, K., Y. V. Svitkin, A. W. Craig, C. T. DeMaria, R. C. Deo, S. K. Burley, and N. Sonenberg. 2001. Translational repression by a novel partner of human poly(A) binding protein, Paip2. Mol. Cell 7:205-216. [DOI] [PubMed] [Google Scholar]
- 26.Koffa, M. D., J. B. Clements, E. Izaurralde, S. Wadd, S. A. Wilson, I. W. Mattaj, and S. Kuersten. 2001. Herpes simplex virus ICP27 protein provides viral mRNAs with access to the cellular mRNA export pathway. EMBO J. 20:5769-5778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kozlov, G., G. De Crescenzo, N. S. Lim, N. Siddiqui, D. Fantus, A. Kahvejian, J. F. Trempe, D. Elias, I. Ekiel, N. Sonenberg, M. O'Connor-McCourt, and K. Gehring. 2004. Structural basis of ligand recognition by PABC, a highly specific peptide-binding domain found in poly(A)-binding protein and a HECT ubiquitin ligase. EMBO J. 23:272-281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuyumcu-Martinez, N. M., M. E. Van Eden, P. Younan, and R. E. Lloyd. 2004. Cleavage of poly(A)-binding protein by poliovirus 3C protease inhibits host cell translation: a novel mechanism for host translation shutoff. Mol. Cell. Biol. 24:1779-1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kwong, A. D., and N. Frenkel. 1987. Herpes simplex virus-infected cells contain a function(s) that destabilizes both host and viral mRNAs. Proc. Natl. Acad. Sci. U. S. A. 84:1926-1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Larralde, O., R. W. Smith, G. S. Wilkie, P. Malik, N. K. Gray, and J. B. Clements. 2006. Direct stimulation of translation by the multifunctional herpesvirus ICP27 protein. J. Virol. 80:1588-1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee, Y. J., and B. A. Glaunsinger. 2009. Aberrant herpesvirus-induced polyadenylation correlates with cellular messenger RNA destruction. PLoS Biol. 7:e1000107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mangus, D. A., M. C. Evans, and A. Jacobson. 2003. Poly(A)-binding proteins: multifunctional scaffolds for the post-transcriptional control of gene expression. Genome Biol. 4:223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McCarthy, A. M., L. McMahan, and P. A. Schaffer. 1989. Herpes simplex virus type 1 ICP27 deletion mutants exhibit altered patterns of transcription and are DNA deficient. J. Virol. 63:18-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McLean, G., F. Rixon, N. Langeland, L. Haarr, and H. Marsden. 1990. Identification and characterization of the virion protein products of herpes simplex virus type 1 gene UL47. J. Gen. Virol. 71:2953-2960. [DOI] [PubMed] [Google Scholar]
- 35.McMahan, L., and P. A. Schaffer. 1990. The repressing and enhancing functions of the herpes simplex virus regulatory protein ICP27 map to C-terminal regions and are required to modulate viral gene expression very early in infection. J. Virol. 64:3471-3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mears, W. E., and S. A. Rice. 1996. The RGG box motif of the herpes simplex virus ICP27 protein mediates an RNA-binding activity and determines in vivo methylation. J. Virol. 70:7445-7453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pestova, T. V., C. U. Hellen, and I. N. Shatsky. 1996. Canonical eukaryotic initiation factors determine initiation of translation by internal ribosomal entry. Mol. Cell. Biol. 16:6859-6869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Piron, M., P. Vende, J. Cohen, and D. Poncet. 1998. Rotavirus RNA-binding protein NSP3 interacts with eIF4GI and evicts the poly(A) binding protein from eIF4F. EMBO J. 17:5811-5821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rice, S. A., and D. M. Knipe. 1990. Genetic evidence for two distinct transactivation functions of the herpes simplex virus alpha protein ICP27. J. Virol. 64:1704-1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sacks, W. R., C. C. Greene, D. P. Aschman, and P. A. Schaffer. 1985. Herpes simplex virus type 1 ICP27 is an essential regulatory protein. J. Virol. 55:796-805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sandri-Goldin, R. M. 1998. ICP27 mediates HSV RNA export by shuttling through a leucine-rich nuclear export signal and binding viral intronless RNAs through an RGG motif. Genes Dev. 12:868-879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sandri-Goldin, R. M. 2008. The many roles of the regulatory protein ICP27 during herpes simplex virus infection. Front. Biosci. 13:5241-5256. [DOI] [PubMed] [Google Scholar]
- 43.Sandri-Goldin, R. M., and G. E. Mendoza. 1992. A herpesvirus regulatory protein appears to act post-transcriptionally by affecting mRNA processing. Genes Dev. 6:848-863. [DOI] [PubMed] [Google Scholar]
- 44.Sciortino, M. T., B. Taddeo, A. P. Poon, A. Mastino, and B. Roizman. 2002. Of the three tegument proteins that package mRNA in herpes simplex virions, one (VP22) transports the mRNA to uninfected cells for expression prior to viral infection. Proc. Natl. Acad. Sci. U. S. A. 99:8318-8323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tarun, S. Z., Jr., and A. B. Sachs. 1996. Association of the yeast poly(A) tail binding protein with translation initiation factor eIF-4G. EMBO J. 15:7168-7177. [PMC free article] [PubMed] [Google Scholar]
- 46.Vende, P., M. Piron, N. Castagne, and D. Poncet. 2000. Efficient translation of rotavirus mRNA requires simultaneous interaction of NSP3 with the eukaryotic translation initiation factor eIF4G and the mRNA 3′ end. J. Virol. 74:7064-7071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Verhagen, J., I. Hutchinson, and G. Elliott. 2006. Nucleocytoplasmic shuttling of bovine herpesvirus 1 UL47 protein in infected cells. J. Virol. 80:1059-1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vittone, V., E. Diefenbach, D. Triffett, M. W. Douglas, A. L. Cunningham, and R. J. Diefenbach. 2005. Determination of interactions between tegument proteins of herpes simplex virus type 1. J. Virol. 79:9566-9571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Walsh, D., and I. Mohr. 2006. Assembly of an active translation initiation factor complex by a viral protein. Genes Dev. 20:461-472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Walsh, D., and I. Mohr. 2004. Phosphorylation of eIF4E by Mnk-1 enhances HSV-1 translation and replication in quiescent cells. Genes Dev. 18:660-672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Walsh, D., C. Perez, J. Notary, and I. Mohr. 2005. Regulation of the translation initiation factor eIF4F by multiple mechanisms in human cytomegalovirus-infected cells. J. Virol. 79:8057-8064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wells, S. E., P. E. Hillner, R. D. Vale, and A. B. Sachs. 1998. Circularization of mRNA by eukaryotic translation initiation factors. Mol. Cell 2:135-140. [DOI] [PubMed] [Google Scholar]
- 53.Williams, P., J. Verhagen, and G. Elliott. 2008. Characterization of a CRM1-dependent nuclear export signal in the C terminus of herpes simplex virus type 1 tegument protein UL47. J. Virol. 82:10946-10952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yoshida, M., K. Yoshida, G. Kozlov, N. S. Lim, G. De Crescenzo, Z. Pang, J. J. Berlanga, A. Kahvejian, K. Gehring, S. S. Wing, and N. Sonenberg. 2006. Poly(A) binding protein (PABP) homeostasis is mediated by the stability of its inhibitor, Paip2. EMBO J. 25:1934-1944. [DOI] [PMC free article] [PubMed] [Google Scholar]