

Abstract
Staphylococcus aureus pneumonia is a common, potentially life-threatening infection caused by this human pathogen. The only therapies available to treat S. aureus pneumonia are antibiotics, a modality that is jeopardized by the organism's remarkable ability to acquire antimicrobial resistance. S. aureus alpha-hemolysin is a pore-forming cytotoxin that is essential for the pathogenesis of pneumonia. Strains lacking this cytotoxin are avirulent in a murine model of pneumonia; likewise, vaccine-based strategies that antagonize the toxin afford protection against lethal disease. Disruption of the function of this toxin therefore provides a potent mechanism to prevent and/or treat S. aureus pneumonia. β-Cyclodextrin derivatives are small molecules with a sevenfold symmetry that mirrors the heptameric alpha-hemolysin. These compounds block the assembled alpha-hemolysin pore, compromising toxin function. We report that a modified β-cyclodextrin compound, IB201, prevents alpha-hemolysin-induced lysis of human alveolar epithelial cells. This protective effect does not result from the ability of the β-cyclodextrin to impair formation of the oligomeric alpha-hemolysin on the cell surface, supporting a role for this molecule in blockade of the lytic pore. An examination of IB201 in murine S. aureus pneumonia demonstrated that administration of this compound prevents and treats disease, protecting against mortality. Consistent with the vital importance of alpha-hemolysin in pneumonia caused by methicillin-sensitive and highly virulent methicillin-resistant S. aureus strains, IB201 protects against lethal challenge with both types of isolates. These observations, coupled with a favorable safety profile of β-cyclodextrin compounds, provide a novel strategy that may be developed to combat S. aureus pneumonia.
Staphylococcus aureus is a gram-positive human pathogen that is able to cause a multitude of diseases ranging in severity (21). Of the many infections and toxinoses mediated by S. aureus, pneumonia is among the most prominent, accounting for an estimated 50,000 staphylococcal infections per year in the United States alone (18, 20). As one of the leading etiologic agents of ventilator-associated pneumonia, S. aureus has long plagued the intensive care environment (14, 19). Further, this pathogen is now increasingly recognized as an important cause of community-acquired pneumonia, displaying the capacity to infect a population of otherwise healthy adults and children (9, 10, 19). A number of these reported cases of community-acquired disease have occurred on the backdrop of an intercurrent respiratory viral infection, often caused by influenza (8, 9, 15). The fulminant nature of coinfection with influenza and S. aureus is evident in mortality rates that approach or exceed 50%, highlighting an apparent synergy of these pathogens in the lung environment. Both the reliance of an aging population on intensive care therapies and the current threat of epidemic or pandemic influenza underscore the large populations of diverse individuals that are at significant risk for the development of S. aureus pneumonia.
Complicating the clinical management of staphylococcal pneumonia is the fact that over half of S. aureus isolates are currently classified as methicillin-resistant S. aureus (MRSA), harboring genes that render these isolates insensitive to a once potent class of antimicrobial agents (14). Recent clinical observations have documented that mortality from MRSA pneumonia can exceed 50%, defining the severity of disease caused by this organism (1). The estimated direct medical cost to treat a patient suffering with S. aureus pneumonia is in excess of $35,000, imparting a significant burden on the economy (30). The combined risks of a changing disease epidemiology and widespread drug resistance among S. aureus strains mandate the development of novel strategies to both prevent and treat disease. Recent investigations highlighting the pore-forming cytotoxin alpha-hemolysin (Hla) as essential for the pathogenesis of S. aureus pneumonia have provided opportunities to design and investigate new strategies to combat this disease (5, 6, 28).
S. aureus strains lacking Hla display a profound defect in virulence in a murine model of pneumonia (4, 5). Consistent with this observation, antagonism of the toxin through a number of distinct immunization strategies has been demonstrated to provide protection against disease (6, 28). Hla displays a detrimental effect on the lung epithelium, resulting in cellular injury and death as well as the generation of proinflammatory mediators (23, 29). The toxin also has the capacity to target the pulmonary vascular endothelium, as treatment of isolated pulmonary arteries ex vivo results in increased vascular resistance and vascular leakage (31, 32). In addition to its role in the lung, this toxin plays a critical role in S. aureus pathogenesis in intraperitoneal, intramammary, and corneal models of infection (3, 7, 27). Hla is secreted by the majority of S. aureus strains as a water-soluble monomer (2, 26). This monomeric form binds to susceptible host cell membranes and through a well-detailed series of intra- and intermolecular interactions, subsequently assembles into a stable homoheptameric transmembrane pore with a 2-nm internal diameter (11, 17, 33).
With the essential nature of alpha-toxin in mind, Karginov et al. used structure-inspired drug design to demonstrate that a hepta-6-substituted β-cyclodextrin derivative, termed IB201, is able to prevent alpha-toxin-mediated hemolysis of rabbit red blood cells (rRBCs), a cell type that is highly sensitive to the lytic action of the toxin (16). Previous investigations had demonstrated the utility of unsubstituted β-cyclodextrin as an adapter molecule, capable of lodging in the central pore of alpha-hemolysin and facilitating the use of the toxin as a biosensor (12, 13). An investigation of IB201 revealed that this molecule blocks ion conductance through the assembled hemolysin pore, consistent with the ability of the cyclodextrins to insert into the pore itself. The inhibitory effect of IB201 on ion conductance and red blood cell hemolysis are both observed in the low micromolar concentration range, demonstrating the potency of this molecule as an inhibitor of the S. aureus alpha-hemolysin. Interestingly, McCormick et al. have recently utilized methyl-β-cyclodextrin plus cholesterol to inhibit the activity of S. aureus alpha-hemolysin, revealing that this treatment affords protection against toxin-induced corneal erosions in a rabbit model of S. aureus keratitis (22). We report herein that the β-cyclodextrin derivative IB201 is able to prevent alpha-toxin-mediated alveolar epithelial cell lysis in vitro and is also able to prevent mortality associated with S. aureus pneumonia in a murine model of infection.
MATERIALS AND METHODS
Bacterial strains, culture, and plasmid constructs.
For mouse lung infections, S. aureus strains Newman and LAC/USA300 were grown at 37°C in tryptic soy broth to an optical density at 660 nm (OD660) of 0.5. Fifty-milliliter culture aliquots were centrifuged and washed in phosphate-buffered saline (PBS) prior to resuspension. For mortality studies, S. aureus Newman was resuspended in 750 μl (3 × 108 to 4 × 108 CFU per 30-μl volume), while S. aureus LAC/USA300 was resuspended in 1,250 μl (2 × 108 CFU per 30-μl volume). For bacterial load and histopathology experiments, S. aureus Newman was resuspended in 1,100 μl (2 × 108 CFU per 30-μl volume). For cytotoxicity studies, 5 ml of culture prepared as described above was resuspended in 10 ml of F12K medium (Invitrogen). A 100-μl suspension was used per assay well. To generate a polyhistidine-tagged version of full-length, active Hla, a PCR product encoding the mature polypeptide was amplified from S. aureus Newman chromosomal DNA, cloned into pET24b (Novagen), and then transformed into Escherichia coli BL21(DE3) (28).
Chemicals.
Per-6-[Nα-Boc-l-ornithinyl)amino]-β-cyclodextrin trifluoroacetic acid salt (IB201) was custom synthesized by CycloLab (Budapest, Hungary). For in vitro assays, IB201 was resuspended in sterile PBS containing 0.1% dimethyl sulfoxide (DMSO) (PBSD). For in vivo assays, IB201 was resuspended in sterile PBS.
Animals and procedures.
Animal experiments were reviewed, approved, and supervised by the Institutional Animal Care and Use Committee at the University of Chicago. For lung infection, 7-week-old C57BL/6J mice (Jackson Laboratory) were anesthetized before inoculation of 30 μl of S. aureus suspension prepared as described above into the left nare. Animals were placed into the cage in a supine position for recovery and were observed for the time periods indicated in each figure. A small percentage of animals routinely succumbed within the first 6 h after inoculation, likely from the combined effects of aspiration and anesthesia. These animals were not included in subsequent statistical analyses. All animal experiments were performed on groups of 15 mice per condition.
For studies of the effects of IB201 treatment, animals infected with S. aureus received scheduled doses of the compound in a 100-μl volume via the intravenous (i.v.) route at the concentrations indicated 2 h following infection and then at 12-h intervals thereafter for a total of six doses. Control mice received 100 μl PBS according to the same schedule.
For studies of the effects of prophylactic administration of IB201 or the effects of a single dose of IB201 postinfection as depicted in Fig. 4, infected mice received 100 μl IB201 (to deliver 10 mg/kg of body weight) in a single dose either 2 h prior to infection with S. aureus or at 2, 6, or 12 h postinfection. Control mice received a single treatment with 100 μl PBS 2 h prior to the time of infection.
FIG. 4.
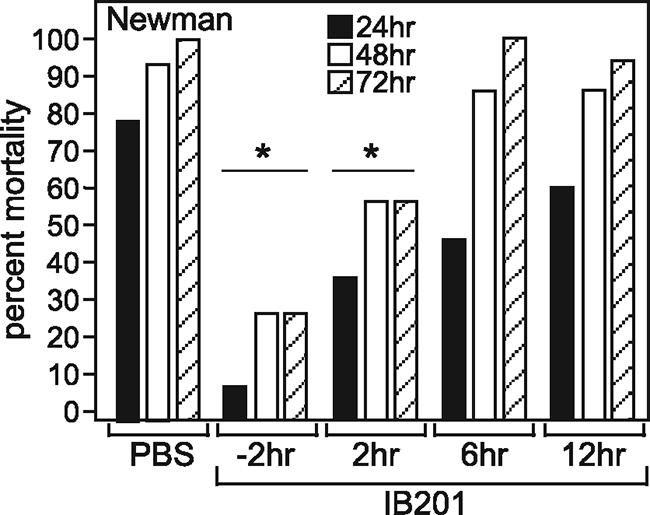
Efficacy of a single dose of β-cyclodextrin derivative IB201 in S. aureus pneumonia. (A) IB201 (10 mg/kg) was administered as a single dose at the indicated time points before or after infection with S. aureus Newman. Mortality was then recorded at 24, 48, and 72 hours postinfection. Values were statistically significantly different (P < 0.035) for animals treated either 2 h before or 2 h after infection (15 animals per group) compared to the control (PBS-treated) animals (statistical significance is denoted by an asterisk). The experimental results shown are representative of the results of two independent experiments.
To evaluate the pathological correlates of pneumonia, infected animals were killed via forced CO2 inhalation before removal of both lungs. The right lung was homogenized for enumeration of lung bacterial load via serial dilution and plating techniques. The left lung was placed in 10% formalin, paraffin embedded, thin sectioned, stained with hematoxylin-eosin, and analyzed by microscopy.
Live/dead and cytotoxicity assays.
A549 cells were washed and plated in F12K medium supplemented with 10% fetal bovine serum at a density of 1.5 × 104 cells per well of a 96-well plate. For both assays, washed A549 cells were cultured with 100 μl of staphylococcal suspension per well in F12K medium with either control PBS or the indicated concentrations of IB201 in triplicate wells. After 3.5 h of incubation at 37°C, either cells were treated with live/dead (green/red) reagent (Invitrogen), or lactate dehydrogenase (LDH) activity was determined (Roche) according to the manufacturer's recommendations. Prior to live/dead staining, cells were gently rinsed two times with PBS to remove staphylococci. Microscopic images of stained cells were obtained using a microscope (Eclipse TE2000U; Nikon). LDH activity was measured on a spectrophotometer and is depicted as the percent maximal lysis achieved with detergent treatment of the A549 cells. Results are representative of a minimum of two independent experiments. Fifty percent inhibitory concentration (IC50) calculations were performed using SigmaPlot software.
Oligomerization and rabbit red blood cell hemolysis assay.
Radiolabeled Hla was synthesized by in vitro transcription and translation performed using a construct encoding full-length Hla in the pET24b vector (Novagen) in an E. coli S30 extract (Promega) supplemented with T7 RNA polymerase, rifampin, and [35S]methionine according to the manufacturer's instructions. One hundred twenty microliters of 12.5% rabbit red blood cells in K-PBSA/βME (20 mM K-phosphate [monobasic], 150 mM NaCl [pH 7.4], 1 mg/ml bovine serum albumin [BSA], 1 mM β-mercaptoethanol [βME]) was incubated with 30 μl of the radiolabeled Hla mix in the presence of either PBS control or IB201 (50 μM) for 1 hour at 20°C. Following incubation, samples were centrifuged at 13,000 rpm for 5 min. Supernatants were removed, and the absorbance of the supernatants was measured at 475 nm. Percent hemolysis was calculated using the supernatant reading from an equivalent number of cells that had been lysed in 1% Triton X-100. The corresponding pellets were washed with 500 μl K-PBSA/βME and centrifuged as described above. The samples were then resuspended in 90 μl of 1× Laemmli buffer, divided into three separate aliquots, and incubated at either 37°C, 50°C, or 90°C as indicated for 10 min prior to loading 12 μl of the sample on 10% sodium dodecyl sulfate (SDS)-polyacrylamide gels for electrophoresis. Gels were dried, and then the results were visualized using a phosphorimager.
Statistical analysis.
Statistical significance of mortality studies was assessed using Fisher's exact test; significance of LDH release assay results, bacterial recovery from lungs, and red blood cell hemolysis was calculated using the two-tailed Student t test.
RESULTS AND DISCUSSION
β-Cyclodextrin inhibitor IB201 prevents Hla-mediated hemolysis without impairing toxin oligomerization.
The formation of an oligomeric alpha-hemolysin (Hla) pore within the membrane of susceptible cells leads to the efflux of cytoplasmic potassium and other small molecules. Initial investigations of the β-cyclodextrin derivative IB201, depicted in Fig. 1A, revealed that this molecule is able to functionally block the central Hla pore in artificial membranes, terminating an established potassium ion current (16). This blockade of the pore correlates with a significant reduction in the lytic activity of the toxin. A series of elegant studies have demonstrated that Hla forms a heat- and SDS-stable oligomer, a structure that is required for lytic capacity (25, 33, 34). To investigate whether the disruption of pore conductance and lytic function of Hla by IB201 results from an alteration in the heptameric state of the assembled toxin, we examined the effects of IB201 on Hla oligomerization in vitro. [35S]methionine-labeled Hla was generated in an E. coli S30 extract in vitro transcription/translation assay utilizing a pET-based construct encoding the mature form of Hla with an appended C-terminal histidine tag (28). Labeled, active Hla was incubated with rabbit red blood cells (rRBCs) either in control PBS or in the presence of 50 μM IB201. Following a 1-h incubation with Hla, insoluble cellular components were pelleted, and the remaining supernatants were analyzed to quantitate toxin-induced hemolysis. In the presence of PBS, labeled toxin leads to the lysis of greater than 95% of the rRBCs when measured as the OD475 of assay supernatants. Conversely, the addition of IB201 provides marked protection against Hla-mediated rRBC lysis (Fig. 1B). To examine the effects of this compound on oligomer formation, cell pellets from the above assays were divided into three equal parts and subjected to treatment at 37°C, 50°C, or 90°C. The oligomer formed by the radiolabeled wild-type toxin is SDS and heat stable to 66°C (24, 35), while mutant forms of the toxin that are defective in stable oligomer formation have a reduced capacity to remain in the assembled form at 50°C (34). Membrane-associated, labeled toxin from the hemolysis assays depicted in Fig. 1B was visualized by SDS-polyacrylamide gel electrophoresis (PAGE) after treatment at the indicated temperatures (37°C, 50°C, and 90°C), and the presence or absence of the oligomer was assessed. The addition of Hla to rRBCs in the presence of control PBS led to the formation of an oligomer at 37°C and 50°C, consistent with previously published observations (Fig. 1C, Hla7). Incubation of the rRBCs at 90°C led to disruption of the oligomer. When Hla is incubated with rRBCs in the presence of IB201, a stable oligomer forms in a fashion identical to that seen without treatment, suggesting that the modified β-cyclodextrin does not prevent toxin-induced hemolysis by disrupting the formation of the heptameric pore. In this assay, the monomeric form of membrane-bound Hla produced in vitro migrates as two bands, seen in previous studies (17, 28). Together, these data provide additional support to confirm that the principal mechanism by which IB201 protects against Hla-induced lysis is to block the transmembrane pore that is formed by the toxin. As a consequence, cell lysis is impaired, despite the fact that the toxin is membrane-bound in the fully functional oligomeric form.
FIG. 1.

β-Cyclodextrin IB201 prevents alpha-toxin-induced hemolysis without disrupting formation of the fully assembled heptameric toxin. (A) Structure of β-cyclodextrin derivative IB201. (B) Hemolysis assays were performed with active [35S]methionine-labeled toxin synthesized in vitro added to 12.5% rabbit red blood cells (rRBC) in K-PBSA/βME. The addition of 50 μM IB201 significantly reduced hemolysis as measured by the absorbance at 475 nm (Abs475) of assay supernatants (P ≤ 1.7 × 10−7). Each condition was analyzed in triplicate, and the results shown are representative of two independent experiments. (C) Active [35S]methionine-labeled toxin was synthesized in vitro and added to rRBC in the presence of PBS or 50 μM IB201 at 37°C, 50°C, or 90°C. Toxin oligomerization (designated Hla7) was evident with both PBS and IB201. IB201 did not disrupt binding of the labeled toxin to rRBCs, indicated by the presence of the Hla monomer in all lanes. The results shown are representative of the results of two independent experiments.
β-Cyclodextrin IB201 prevents Hla-mediated alveolar epithelial cell injury.
The β-cyclodextrin derivative IB201 was next examined for its ability to prevent injury to cultured A549 human alveolar epithelial cells. We have previously demonstrated that A549 cell injury and death in this model critically depends on Hla, as S. aureus strains lacking Hla do not result in cellular injury (6). Further, antagonism of the toxin via anti-Hla antibodies in this culture model is fully protective (6, 28). Uninfected A549 cells retain a green fluorophore when examined with a live (green)/dead (red) staining reagent (Fig. 2A). Upon coculture of A549 cells with live S. aureus strain Newman in the presence of PBS control for 3.5 h, cell death was apparent, as indicated by an increase in the number of red fluorescent dead cells (Fig. 2B). As the β-cyclodextrin IB201 was solubilized in a PBS-0.1% DMSO (PBSD) vehicle for these studies, we examined the effects of PBSD on cell death in the coculture model. As shown in Fig. 2C, the addition of the PBSD control led to a degree of cell death similar to that evident with PBS alone, indicating that there are no untoward effects of DMSO on cell viability in this in vitro system. When cells were treated with 5 μM IB201 (in the PBSD vehicle), they were completely protected from Hla-induced cell death (Fig. 2D). A value for cell death (a percentage) is recorded in the lower right-hand corner of each panel.
FIG. 2.

β-Cyclodextrin derivative IB201 protects human alveolar epithelial cells from S. aureus injury. Live (green)/dead (red) staining of A549 alveolar epithelial cells was imaged by fluorescence microscopy 3.5 h after infection. (A to D) Cells were uninfected (A) or cocultured with S. aureus Newman in medium treated with PBS (B), PBS plus 0.1% DMSO (PBSD) (C), or 5 μM IB201 in PBSD (D). The percentage of dead cells for each experimental condition was calculated by scoring live versus dead cells in four independent fields and expressing the number of dead cells as a fraction of the total; these values are noted in the bottom right-hand corner of each panel. Images for each condition are representative of cells visualized in two independent experiments. Bars, 20 μm. (E) LDH release by A549 cells was observed by cells cocultured with S. aureus Newman in medium treated with the indicated concentrations of IB201; an asterisk indicates a significant reduction in LDH release for specific concentration (P < 0.05). (F) Optical density readings were taken at 600 nm (OD600) to measure the growth of S. aureus strain Newman in the presence of either PBS plus 0.1% DMSO or 5 μM IB201. The values in panels E and F represent means ± standard deviations (SD) (error bars). The data shown are representative of the data in three independent experiments.
To appreciate the concentration range in which IB201 affords protection of A549 cells in vitro, we treated S. aureus-infected cells with 0.05 to 5 μM IB201 and quantified cell injury in a lactate dehydrogenase (LDH) release assay. IB201 conferred significant protection across a concentration range from 0.25 to 5.0 μM when added to cocultures of A549 cells with S. aureus Newman (Fig. 2E) (P < 0.0008). The IC50 was calculated to be 0.54 μM. To ensure the protection IB201 provides is in fact due to its ability to antagonize the toxin and not merely the result of an inhibitory effect on bacterial growth, we analyzed the growth of S. aureus Newman in tryptic soy broth supplemented with either control PBSD or 5 μM IB201. As shown in Fig. 2F, the presence of IB201 did not alter bacterial growth kinetics. Together, these results demonstrate the in vitro efficacy of IB201 in protecting human alveolar epithelial cells against cell death caused by Hla. The low micromolar concentration of IB201 required to elicit this protection is very favorable, highlighting the potential for further investigation of this and related β-cyclodextrin derivatives as clinically useful small-molecule inhibitors of Hla.
IB201 prevents mortality resulting from methicillin-sensitive S. aureus pneumonia.
To determine the ability of IB201 to protect mice from staphylococcal pneumonia, we infected 7-week-old C57BL/6J mice via the intranasal route (i.n.) with 3 × 108 to 4 × 108 CFU of S. aureus Newman. Two hours postinfection, we administered PBS or 10, 5, 1, or 0.1 mg of IB201 per kg via i.v. injection to groups of 15 mice. Mice were then given additional doses of PBS or IB201 at the corresponding concentrations 12 h after infection and every 12 h thereafter for a total of five doses. We monitored mortality, a result of acute lethal disease secondary to S. aureus pneumonia over a 72-h time course. Mice that received either 5 or 10 mg/kg of IB201 were significantly protected from S. aureus pneumonia at 48 h and 72 h (Fig. 3A) (P < 0.008). When mice were given 1 mg/kg of IB201, they were significantly protected from mortality only until 48 h postinfection (P < 0.038). This reduction in mortality indicates that multiple doses of IB201 ranging from 5 to 10 mg/kg are able to effectively treat S. aureus pneumonia in a murine model of infection.
FIG. 3.

IB201 protects against S. aureus pneumonia. (A) IB201 was administered to mice via retro-orbital injection 2 h after infection, 12 h after infection, and then every 12 h thereafter. They were challenged with S. aureus Newman via the i.n. route, and mortality was recorded at 24, 48, and 72 hours postinfection (P < 0.039; 15 animals per group) (statistical significance is denoted by an asterisk). (B) CFU recovery from the right lung was determined and demonstrated a significant decrease in bacterial burden in mice that received IB201 (P = 0.008; 15 animals per group). The symbols show the values for individual mice, and the short horizontal lines indicate the mean bacterial load for that group. (C) Histopathology of S. aureus Newman-infected lung tissue from mice that were treated with either PBS or IB201. Scale bars on low-magnification images (left) represent 0.1 cm, while scale bars on high-magnification images (right) represent 20 μm. Experimental results shown in panels A, B, and C are representative of the results in two independent experiments.
To examine the effects of IB201 on S. aureus survival and proliferation within the lung, we examined CFU recovery from the right lung of infected mice that had received a 10-mg/kg dose of IB201 2 h and 12 h after infection with S. aureus Newman. Lung tissues were harvested for processing 24 h postinfection, and IB201-treated animals were compared to animals receiving PBS alone. IB201 facilitated a nearly 1.5-log-unit reduction in S. aureus recovery from the lung relative to that observed in controls (Fig. 3B; P < 0.008; groups of 15 mice). Interestingly, this result parallels the diminished recovery of S. aureus seen in mice infected with Hla-deficient strains or in mice that received either vaccines or immunotherapy targeting Hla (5, 6, 28). In aggregate, these observations suggest that Hla may potentiate lethal staphylococcal lung injury through its ability to lead to a favorable environment that facilitates bacterial survival and/or proliferation within the lung.
IB201 does not impact significantly on pathological features of lung injury in S. aureus pneumonia.
Our previous investigations on the role of Hla in S. aureus pneumonia revealed that antibody-mediated antagonism of the toxin markedly improved the histopathologic manifestations of disease, as the signs of tissue inflammation and destruction were focal in nature in contrast to the widespread insults evident with control treatments (6, 28). This result was observed following active immunization of mice with a nontoxic Hla variant as well as in passive vaccination studies employing rabbit polyclonal anti-Hla sera or two newly described anti-Hla mouse monoclonal antibodies. To examine the impact of IB201 treatment on pathological manifestations of lung injury, we examined hematoxylin-eosin-stained thin sections of lungs from S. aureus-infected mice that had either received treatment with PBS control or 10 mg/kg IB201. Interestingly, while the bacterial burden of S. aureus in the lungs of IB201-treated mice was significantly reduced (Fig. 3B), widespread inflammation persisted in the lungs of treated animals as manifest by the accumulation of cellular infiltrates in the alveolar space and consolidation of the lung tissue (Fig. 3C). The histopathologic appearance of lung injury was essentially indistinguishable from that seen in control mice, with the exception that aggregates of S. aureus were readily visualized in the lungs of control animals but not IB201-treated animals (Fig. 3C, magnified images; S. aureus aggregates indicated by the green arrow).
This result was surprising, as antibody-mediated antagonism of Hla in our previous studies always led to a reduction in the appearance of lung injury, commensurate with the robust protection that these strategies provided against mortality from S. aureus pneumonia. In the case of treatment with IB201, however, we observe that the compound affords a similar degree of protection from mortality and a significant reduction in S. aureus load in the tissues yet does not alter the features of lung injury apparent histologically. The effects of anti-Hla antibodies in the prevention of disease are perhaps threefold. First, antibodies may block toxin binding to susceptible host cells. Second, these antibodies may have the ability to prevent oligomerization of the toxin on the cell membrane. Third, antibodies may have the ability to structurally block the pore itself. It is probable that an array of polyclonal antibodies may possess several or all of these activities, while monoclonal antibodies, such as those we have recently described, may only harbor one such activity. In contrast, IB201 permits the full assembly of the heptameric toxin on the cell membrane, exerting its antagonistic capacity entirely through a functional blockade of the pore. These disparate observations may suggest that if Hla is able to assemble on the surface of susceptible cells, the oligomer itself may induce cellular inflammatory responses, irrespective of the integrity of the pore. Thus, antibodies that primarily preclude Hla binding to host cells or prevent oligomerization may have a more potent ability to minimize lung injury than β-cyclodextrin derivatives that cannot prevent toxin assembly. This observation highlights the interesting possibility that the assembled oligomer may stimulate cellular signal transduction cascades that contribute to inflammation, independent of the injury that may result from compromise of the membrane via the 2-nm Hla pore. Together, these studies imply that IB201 and related compounds may serve as a useful tool to more carefully dissect the molecular mechanisms by which the Hla pore and the oligomeric structure exact cellular injury and drive host tissue responses.
Early treatment with IB201 is required to prevent mortality from S. aureus pneumonia.
As the development of both preventative and therapeutic strategies for S. aureus pneumonia is needed, we were curious as to whether a single dose of IB201 would be able to prevent or treat S. aureus pneumonia when given at various times prior to and following infection. A single 10-mg/kg dose of IB201 was given to mice at either 2 h prior to infection or 2, 6, or 12 h after infection with 3 × 108 to 4 × 108 CFU of S. aureus. Mortality was then observed over a 72-h time course. Mortality was significantly reduced when the β-cyclodextrin was given 2 h before and 2 h after infection (Fig. 4; P < 0.035), highlighting the role of this compound in mitigating early stages of disease. Animals that did not receive IB201 until 6 and 12 h postinfection showed a trend toward a delay in the time to death; however, this observation failed to reach statistical significance. To rule out the possibility that additional doses of IB201 would be of utility when the first dose is administered later during the course of infection (i.e., 6 or 12 h postinfection), we repeated this experiment and gave 10 mg/kg of IB201 at 6 h or 12 h after infection and then every 12 h thereafter. While mortality was again delayed in this experiment, 100% mortality was reached by 72 h in both treatment groups similar to that observed in animals that received PBS (data not shown). These findings support our previous hypothesis that Hla is essential in the early stages of infection and that after initial tissue damage, antagonizing the action of the toxin is no longer effective in halting the progression of disease.
IB201 prevents mortality resulting from methicillin-resistant S. aureus pneumonia.
The recent emergence of community-acquired methicillin-resistant S. aureus (CA-MRSA) has led to a changing epidemiology of disease, expanding the cohort of people that are normally susceptible to S. aureus pneumonia and simultaneously increasing the mortality associated with this disease. With this in mind, we decided to test the efficacy of IB201 against a clinical CA-MRSA isolate, LAC/USA300. This isolate remains one of the most prominent circulating CA-MRSA isolates in the United States, and is known to be associated with lethal S. aureus pneumonia. We have previously demonstrated the high-virulence phenotype of LAC/USA300 in the murine model of S. aureus pneumonia. This isolate secretes approximately twice as much Hla as S. aureus Newman does, resulting in rapid and complete mortality in groups of experimental animals (6). C57BL/6J mice were infected with 2 × 108 CFU of LAC/USA300 via the i.n. route and then treated 2 h after infection and at 12-h intervals thereafter with either PBS or 10, 5, 1, or 0.1 mg/kg IB201. Mice that received either 10 or 5 mg/kg IB201 were significantly protected from mortality associated with CA-MRSA pneumonia throughout the 3-day experimental time course (P = 0.0002 and 0.02, respectively), while mice that received 1 mg/kg were afforded protection through 48 h postinfection (P = 0.01) (Fig. 5).
FIG. 5.
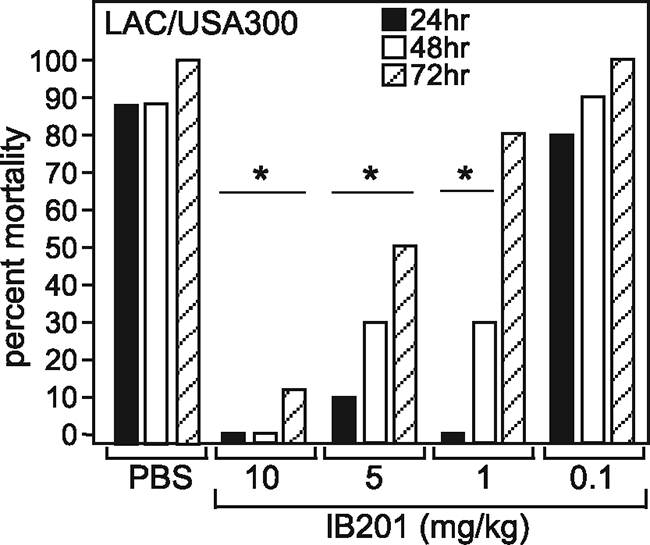
β-Cyclodextrin derivative IB201 protects against S. aureus pneumonia caused by highly virulent CA-MRSA isolate LAC/USA300. (A) IB201 (10, 5, 1, or 0.1 mg/kg) administered 2 h after infection and every 12 h thereafter conferred protection against mortality in mice infected via the i.n. route with S. aureus LAC/USA300 (10 animals per group). Values were statistically significantly different (P ≤ 0.02) from the values for the control (PBS-treated) animals (statistical significance is denoted by an asterisk). The experimental results shown are representative of the results of two independent experiments.
Current clinical S. aureus disease is rather frightening when one considers the widespread distribution of methicillin-resistant isolates that are capable of causing a wide array of infections, even in individuals that are otherwise healthy. Preventative and therapeutic strategies that specifically disrupt a facet of the host-pathogen interaction during S. aureus infection have the potential to provide a powerful means by which to decrease the morbidity and mortality associated with disease. In the case of staphylococcal pneumonia, the strict requirement for Hla in pathogenesis has allowed for the design of a number of new vaccine-based modalities to target disease, each of which has proven efficacious in an animal model and can now be considered for further development and translation for human clinical investigation. The studies reported herein describe the utility of the β-cyclodextrin derivative, IB201, in the prevention and treatment of S. aureus pneumonia. While distinct from the potent immunologic approaches we have described previously, this small-molecule inhibitor of the toxin affords a similar high level of protection against lethal disease caused by both methicillin-sensitive and methicillin-resistant clinical isolates. Functioning to block the pore formed by the assembled, heptameric toxin, IB201 is thus able to exploit the essential nature of Hla in lung infections and provide a novel mechanism by which to combat deadly staphylococcal pneumonia.
Acknowledgments
This work was supported by Innovative Biologics, Inc.; an NIH award (1R43AI082749-01) to V.A.K.; and the Departments of Pediatrics and Microbiology at the University of Chicago. J.B.W. acknowledges membership within and support from the Region V “Great Lakes” Regional Center of Excellence for Bio-defense and Emerging Infectious Diseases Research (NIH award 1-U54-AI-057153). The authors have no conflicting financial interests.
We thank O. Schneewind for critical discussions and comments on the manuscript, K. Alexander for assistance with pharmacologic calculations, the Department of Pathology at the University of Chicago for histology support, and S. Bond for microscopy support.
Footnotes
Published ahead of print on 5 October 2009.
REFERENCES
- 1.Athanassa, Z., I. I. Siempos, and M. E. Falagas. 2008. Impact of methicillin resistance on mortality in Staphylococcus aureus VAP: a systematic review. Eur. Respir. J. 31:625-632. [DOI] [PubMed] [Google Scholar]
- 2.Bhakdi, S., and J. Tranum-Jensen. 1991. Alpha-toxin of Staphylococcus aureus. Microbiol. Rev. 55:733-751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bramley, A. J., A. H. Patel, M. O'Reilly, R. Foster, and T. J. Foster. 1989. Roles of alpha-toxin and beta-toxin in virulence of Staphylococcus aureus for the mouse mammary gland. Infect. Immun. 57:2489-2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bubeck Wardenburg, J., T. Bae, M. Otto, F. R. DeLeo, and O. Schneewind. 2007. Poring over pores: alpha-hemolysin and Panton-Valentine leukocidin in Staphylococcus aureus pneumonia. Nat. Med. 13:1405-1406. [DOI] [PubMed] [Google Scholar]
- 5.Bubeck Wardenburg, J., R. J. Patel, and O. Schneewind. 2007. Surface proteins and exotoxins are required for the pathogenesis of Staphylococcus aureus pneumonia. Infect. Immun. 75:1040-1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bubeck Wardenburg, J., and O. Schneewind. 2008. Vaccine protection against Staphylococcus aureus pneumonia. J. Exp. Med. 205:287-294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Callegan, M. C., L. S. Engel, J. M. Hill, and R. J. O'Callaghan. 1994. Corneal virulence of Staphylococcus aureus: roles of alpha-toxin and protein A in pathogenesis. Infect. Immun. 62:2478-2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Finelli, L., A. Fiore, R. Dhara, L. Brammer, D. K. Shay, L. Kamimoto, A. Fry, J. Hageman, R. Gorwitz, J. Bresee, and T. Uyeki. 2008. Influenza-associated pediatric mortality in the United States: increase of Staphylococcus aureus coinfection. Pediatrics 122:805-811. [DOI] [PubMed] [Google Scholar]
- 9.Frazee, B. W. 2007. Severe methicillin-resistant Staphylococcus aureus community-acquired pneumonia associated with influenza-Louisiana and Georgia, December 2006-January 2007. Ann. Emerg. Med. 50:612-616. [DOI] [PubMed] [Google Scholar]
- 10.Fridkin, S. K., J. C. Hageman, M. Morrison, L. T. Sanza, K. Como-Sabetti, J. A. Jernigan, K. Harriman, L. H. Harrison, R. Lynfield, and M. M. Farley. 2005. Methicillin-resistant Staphylococcus aureus disease in three communities. N. Engl. J. Med. 352:1436-1444. [DOI] [PubMed] [Google Scholar]
- 11.Gouaux, E., M. Hobaugh, and L. Song. 1997. Alpha-hemolysin, gamma-hemolysin, and leukocidin from Staphylococcus aureus: distant in sequence but similar in structure. Protein Sci. 6:2631-2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gu, L. Q., and H. Bayley. 2000. Interaction of the noncovalent molecular adapter, beta-cyclodextrin, with the staphylococcal alpha-hemolysin pore. Biophys. J. 79:1967-1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gu, L. Q., O. Braha, S. Conlan, S. Cheley, and H. Bayley. 1999. Stochastic sensing of organic analytes by a pore-forming protein containing a molecular adapter. Nature 398:686-690. [DOI] [PubMed] [Google Scholar]
- 14.Hidron, A. I., J. R. Edwards, J. Patel, T. C. Horan, D. M. Sievert, D. A. Pollock, and S. K. Fridkin for the National Healthcare Safety Network Team and Participating National Healthcare Safety Network Facilities. 2008. Antimicrobial-resistant pathogens associated with healthcare-associated infections: annual summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2006-2007. Infect. Control Hosp. Epidemiol. 29:996-1011. [DOI] [PubMed] [Google Scholar]
- 15.Kallen, A. J., J. Brunkard, Z. Moore, P. Budge, K. E. Arnold, G. Fosheim, L. Finelli, S. E. Beekmann, P. M. Polgreen, R. Gorwitz, and J. Hageman. 2009. Staphylococcus aureus community-acquired pneumonia during the 2006 to 2007 influenza season. Ann. Emerg. Med. 53:358-365. [DOI] [PubMed] [Google Scholar]
- 16.Karginov, V. A., E. M. Nestorovich, F. Schmidtmann, T. M. Robinson, A. Yohannes, N. E. Fahmi, S. M. Bezrukov, and S. M. Hecht. 2007. Inhibition of S. aureus alpha-hemolysin and B. anthracis lethal toxin by beta-cyclodextrin derivatives. Bioorg. Med. Chem. 15:5424-5431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kawate, T., and E. Gouaux. 2003. Arresting and releasing staphylococcal alpha-hemolysin at intermediate stages of pore formation by engineered disulfide bonds. Protein Sci. 12:997-1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klevens, R. M., M. A. Morrison, J. Nadle, S. Petit, K. Gershman, S. Ray, L. H. Harrison, R. Lynfield, G. Dumyati, J. M. Townes, A. S. Craig, E. R. Zell, G. E. Fosheim, L. K. McDougal, R. B. Carey, and S. K. Fridkin. 2007. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA 298:1763-1771. [DOI] [PubMed] [Google Scholar]
- 19.Kollef, M. H., A. Shorr, Y. P. Tabak, V. Gupta, L. Z. Liu, and R. S. Johannes. 2005. Epidemiology and outcomes of health-care-associated pneumonia: results from a large US database of culture-positive pneumonia. Chest 128:3854-3862. [DOI] [PubMed] [Google Scholar]
- 20.Kuehnert, M. J., H. A. Hill, B. A. Kupronis, J. I. Tokars, S. L. Solomon, and D. B. Jernigan. 2005. Methicillin-resistant Staphylococcus aureus hospitalizations, United States. Emerg. Infect. Dis. 11:868-872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lowy, F. D. 1998. Staphylococcus aureus infections. N. Engl. J. Med. 339:520-532. [DOI] [PubMed] [Google Scholar]
- 22.McCormick, C. C., A. R. Caballero, C. L. Balzli, A. Tang, and R. J. O'Callaghan. 2009. Chemical inhibition of alpha-toxin, a key corneal virulence factor of Staphylococcus aureus. Invest. Ophthalmol. Vis. Sci. 50:2848-2854. [DOI] [PubMed] [Google Scholar]
- 23.McElroy, M. C., H. R. Harty, G. E. Hosford, G. M. Boylan, J. F. Pittet, and T. J. Foster. 1999. Alpha-toxin damages the air-blood barrier of the lung in a rat model of Staphylococcus aureus-induced pneumonia. Infect. Immun. 67:5541-5544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McNiven, A. C., P. Owen, and J. P. Arbuthnott. 1972. Multiple forms of staphylococcal alpha-toxin. J. Med. Microbiol. 5:113-122. [DOI] [PubMed] [Google Scholar]
- 25.Menzies, B. E., and D. S. Kernodle. 1994. Site-directed mutagenesis of the alpha-toxin gene of Staphylococcus aureus: role of histidines in toxin activity in vitro and in a murine model. Infect. Immun. 62:1843-1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.O'Reilly, M., J. C. de Azavedo, S. Kennedy, and T. J. Foster. 1986. Inactivation of the alpha-haemolysin gene of Staphylococcus aureus 8325-4 by site-directed mutagenesis and studies on the expression of its haemolysins. Microb. Pathog. 1:125-138. [DOI] [PubMed] [Google Scholar]
- 27.Patel, A. H., P. Nowlan, E. D. Weavers, and T. Foster. 1987. Virulence of protein A-deficient and alpha-toxin-deficient mutants of Staphylococcus aureus isolated by allele replacement. Infect. Immun. 55:3103-3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ragle, B. E., and J. Bubeck Wardenburg. 2009. Anti-alpha-hemolysin monoclonal antibodies mediate protection against Staphylococcus aureus pneumonia. Infect. Immun. 77:2712-2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rose, F., G. Dahlem, B. Guthmann, F. Grimminger, U. Maus, J. Hanze, N. Duemmer, U. Grandel, W. Seeger, and H. A. Ghofrani. 2002. Mediator generation and signaling events in alveolar epithelial cells attacked by S. aureus alpha-toxin. Am. J. Physiol. Lung Cell. Mol. Physiol. 282:L207-L214. [DOI] [PubMed] [Google Scholar]
- 30.Rubin, R. J., C. A. Harrington, A. Poon, K. Dietrich, J. A. Greene, and A. Moiduddin. 1999. The economic impact of Staphylococcus aureus infection in New York City hospitals. Emerg. Infect. Dis. 5:9-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seeger, W., M. Bauer, and S. Bhakdi. 1984. Staphylococcal alpha-toxin elicits hypertension in isolated rabbit lungs. Evidence for thromboxane formation and the role of extracellular calcium. J. Clin. Invest. 74:849-858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Seeger, W., R. G. Birkemeyer, L. Ermert, N. Suttorp, S. Bhakdi, and H. R. Duncker. 1990. Staphylococcal alpha-toxin-induced vascular leakage in isolated perfused rabbit lungs. Lab. Invest. 63:341-349. [PubMed] [Google Scholar]
- 33.Song, L., M. R. Hobaugh, C. Shustak, S. Cheley, H. Bayley, and J. E. Gouaux. 1996. Structure of staphylococcal alpha-hemolysin, a heptameric transmembrane pore. Science 274:1859-1866. [DOI] [PubMed] [Google Scholar]
- 34.Walker, B., and H. Bayley. 1995. Key residues for membrane binding, oligomerization, and pore forming activity of staphylococcal alpha-hemolysin identified by cysteine scanning mutagenesis and targeted chemical modification. J. Biol. Chem. 270:23065-23071. [DOI] [PubMed] [Google Scholar]
- 35.Walker, B., and H. Bayley. 1995. Restoration of pore-forming activity in staphylococcal alpha-hemolysin by targeted covalent modification. Protein Eng. 8:491-495. [DOI] [PubMed] [Google Scholar]