

Abstract
Objective
The post-translational regulation of GTP cyclohydrolase I (GCH-1), the rate-limiting enzyme for tetrahydrobiopterin (BH4) synthesis, remains elusive. Here, we identified specific phosphorylation sites on GCH-1 and characterized the function of these sites.
Methods and Results
Mass spectrometry studies showed overexpressed rat GCH-1 was phosphorylated at serine (S) 51, S167 and threonine (T) 231 in HEK293 cells whereas a computational analysis of GCH-1 revealed 8 potential phosphorylation sites [S51, S72, T85, T91, T103, S130, S167 and T231]. GCH-1 activity and BH4 were significantly decreased in cells transfected with the phospho-defective mutants (S72A, T85A, T91A, T103A or S130A) and increased in cells transfected with the T231A mutant. BH4 and BH2 were increased in cells transfected with S51E, S72E, T85E, T91E, T103D or T130D mutants, but decreased in cells transfected with the T231D mutant, while cells transfected with the S167A or the S167E mutant had increased BH2. Additionally, cells transfected with the T231A mutant had reduced GCH-1 nuclear localization and nuclear GCH-1 activity.
Conclusion
Our data suggest GCH-1 activity is regulated either positively by phosphorylation S51, S72, T85, T91, T103 and S130, or negatively at T231. Such information might be useful in designing new therapies aiming at improving BH4 bioavailability.
Keywords: GTP Cyclohydrolase I, tetrahydrobiopterin, phosphorylation
GTP cyclohydrolase I (GCH-1) is the first and rate-limiting enzyme for tetrahydrobiopterin (BH4) biosynthesis.1 BH4 is an essential cofactor for aromatic amino acid hydroxylases, nitric oxide synthase (NOS) isoforms, and glyceryl-ether monooxygenase.2 Mutations in GCH-1, resulting in BH4 deficiency, have been shown to cause phenylketonuria and Dopa-responsive dystonia (DRD). BH4 critically regulates NOS activity. BH4 shifts the NOS heme iron into a high spin state and increases the affinity of the enzyme for arginine. BH4 also facilitates the electron transfer from NOS reductase and structurally stabilizes NOS dimmers (review1). When BH4 is limited, reduction of O2 by NOS is not coupled to L-arginine oxidation resulting in superoxide anion (O2•−) generation rather than nitric oxide (•NO) production. BH4 is easily oxidized to 6, 7, 8-trihydrobiopterin or 7, 8-dihydrobiopterin (BH2), making it useless for NOS. Increased BH4 depletion or oxidation has been linked to hypertension, atherosclerosis, diabetes, cardiac hypertrophy, and myocardial ischemia.3 The role of GCH-1 in cardiovascular physiology in regulating BH4 and NOS activity has been investigated in vitro and in vivo. In endothelial cells, GCH-1 gene transfer increases BH4 more than 10-fold over baseline levels, accompanied by a marked increase in eNOS-dependent •NO production.4 Transgenic mice made to express human GCH-1 in vascular endothelial cells have a 3-fold increase in vascular BH4 and a marked decrease in endothelial O2•− production, which preserves •NO bioavailability compared with wild-type littermates.5 Overexpression of eNOS in transgenic mice increases eNOS-dependent O2•− production.6 Yet, when the eNOS transgenic mice are crossed with GCH-1 transgenic mice, O2•− generation is normalized.6
GCH-1 appears to be finely regulated by phosphorylation. GCH-1 phosphorylation was increased in cells stimulated by Angiotensin II, platelet-derived growth factor and TPA and the increase in phosphorylation was correlated with increased enzyme activity and BH4 production.7 Over-expressed GCH-1 has been shown to exist in a phosphorylated form in mast cells and phorbol ester stimulates GCH-1 phosphorylation and BH4 production which could be inhibited by PKC inhibitors.8 A more recent study showed that shear stress increases GCH-1 phosphorylation at Serine-81 and enzyme activity in human endothelial cells by a casein kinase II-dependent mechanism.9 Although phosphorylation of GCH-1 appears to regulate activity, systematic studies examining how these potential phosphorylation sites regulate enzyme activity have not been performed.
The purpose of the present study is to identify potential phosphorylation sites in GCH-1 and to determine the functional role of specific phosphorylation sites.
Materials and Methods
An expanded Materials and Methods section can be found in the supplemental materials (available online at http://atvb.ahajournals.org).
Plasmids Construction
GCH-1 cDNA from Sprague Dawley rats was cloned into pcDNA5/FRT/TO/Topo/TA (Invitrogen) with a FLAG epitope. Flag-GCH-1 phospho-defective mutants [Serine/Theonine (S/T) to alanine (A)] or phospho-mimic mutants [S/T to glutamic acid (E) or aspartic acid (D)] were generated using QuikChange® II Site-Directed Mutagenesis Kits (Stratagene). GCH-1 mutants were named as S51A, S51E or S51D and so forth. GCH-1-GFP and T231A-GCH-1-GFP were also constructed.
Cells
Flag-GCH-1 and its mutants were co-transfected with POG44 (Invitrogen) into Flp-In™ T-REx™-293 cells (Invitrogen) to establish stable cell lines or transiently into bovine aortic endothelial cells (BAEC). The cells expressing wild type (WT) or its mutants were named as WT-GCH-1, S51A-GCH-1, S51D-GCH-1 cells and so forth. Cells were treated with/without tetracycline (1 μg/ml) for 24 hours in all experiments to regulate the plasmid expression.
Determination of Subcellular Localization of GCH-1
Nuclear and cytoplasmic extracts were prepared from the cells using NE-PER™ Nuclear and Cytoplasmic Extraction Reagents (Pierce, Rockford, IL) according to the manufacturer’s instructions.
Mass Spectrometry Analysis
Top down mass spectrometry was used to analyze intact FLAG-GCH-1 from HEK 293 cells. FLAG-GCH-1 was desalted using an offline reverse phase protein microtrap (Michrom Bioresources, Inc, CA) and introduced to the massspectrometer using an automated chip-based nanoESI source (Triversa NanoMate, Advion BioSciences, Ithaca, NY)(ESI/FTMS). Bottom up mass spectrometry was used with in solution trypsin digestion of FLAG-GCH-1. After digestion, the resulting peptide mixture was separated by a nano-2DLC chromatographic system with a C18 column (Eksigent, Dublin, CA) and subsequently analyzed on-line using an LTQ mass spectrometer (Thermo Scientific Inc., Bremen, Germany). In addition, bottom up mass spectrometry was used with in-gel digestion. FLAG-GCH-1 was immunoprecipitated and isolated by SDS PAGE. The GCH-1 band was excised, destained and digested. The resulting peptides were enriched for phosphopeptides by immobilized metal ion affinity chromatography and analyzed on a Voyager DE-Pro matrix-assisted laser desorption ionization time-of-flight mass spectrometer (MALDI-TOF MS) (Applied Biosystem, Foster, CA).
Statistical Analysis
Data are expressed as mean ± SEM. Significance of differences between the means was determined by unpaired two-tailed t-tests or ANOVA with the appropriate post-hoc test. A value of P < 0.05 was considered to be significant. A non-parametric test, the Mann-Whitney test was also used to assess levels of significance.
Results
GCH-1 is Phosphorylated at S167, S51 and T231
Immunoprecipitation (IP) of FLAG-GCH-1 from WT-GCH-1 cells and western blot analysis for phospho-threonine (T) or phospho-serine (S) showed both threonine phosphorylation (strong signal) and serine phosphorylation (relatively weak signal) in GCH-1 (Figure 1A). Reverse IP for phospho-threonine and western blot for anti-FLAG showed strong GCH-1 bands (Figure 1A and supplement Figure I). Since the serine phosphorylation signal was relatively weak (possibly due to the sensibility of phospho-antibodies), reverse IP was not performed. It is well known that Phosphorylation alters protein surface charges and protein isoelectric points prompting a shift in horizontal migration in 2D gels. We observed at least 4 distinct horizontally migrating spots (Figure 1B) that were identified to be GCH-1 by MS. After GCH-1 was run on a 1D gel, coomassie staining revealed a robust band at ~30kD (Figure 1C). Western blot analysis, performed in parallel, confirmed this band to be FLAG-GCH-1 (data not shown). MALDI-TOF MS analysis of this band identified 21 peptides covering 72% of GCH-1 sequence. The molecular ions for peptide REDPKpT231RE (residues 226–233) and peptide LARIVEIYpS167R (residues 159–168) were detected in their phosphorylated form with [M+H]+ at m/z 825.3322 and 1299.7010 respectively. The mass of their non-phosphorylated peptides were 745 and 1219.7157 respectively (mass loss ~80 Da), indicating that T231 and S167 were phosphorylated. ESI/FTMS analysis of FLAG-GCH-1 revealed that it is phosphorylated with the relative percentage of un-and phosphorylated forms of approximately 80% and 20% (Figure 1D). The accurate molecular weights measured for the unphosphorylated GCH-1 is 28093.35, which matched to the DNA-predicted sequence with one acetylation. The tandem mass spectrometry (MS/MS) data acquired on one single charge state (M32+) of GCH-1 protein ions using CAD unambiguously confirmed the protein sequence and located the acetylation to the N-terminus (data not shown). In-solution digestion of GCH-1 using trypsin resulted in 30 peptides covering 81% of the sequence. ESI/LC/MS analysis of the tryptic peptides identified one phosphopeptide A[54–79]R with an experimental molecular weight of 2936.26 that matched well with the calculated molecular weight of the phosphorylated A[54–79]R. MS/MS of phosphorylated A[54–79]R localized the phosphorylation site to Ser59 corresponding to Ser51 in endogenous GCH-1 (Figure 2).
Figure 1.

GCH-1 was phosphorylated in Flp-In 293 cells. (A) FLAG-GCH-1 cells were immunoprecipitated and immunoblotted with phospho-threonine, phospho-serine or FLAG antibodies. (B) The immunoprecipitated FLAG-GCH-1 was separated by IPG gel followed by SDS/PAGE analysis. The gel was then silver-stained and the spots were validated as GCH-1 by MS. (C) The immunoprecipitated FLAG-GCH-1 was separated by SDS/PAGE and visualized by coomassie staining. (D) High resolution ESI/FTMS analysis of GCH-1 overexpressed in HEK293 cells suggesting it is monophosphorylated. Inset, isotopically resolved GCH-1 intact protein ions of +32 charge state (M+32). Circles represent the theoretical isotopic distribution of the GCH-1 protein ions with an acetylation. pGCH-1, monophosphorylated GCH-1. +Na/+K, sodium/potassium adducts of GCH-1 protein ions. Expt’l MW, experimental most abundant molecular weight; Calc’d MW: calculated most abundant molecular weight.
Figure 2.
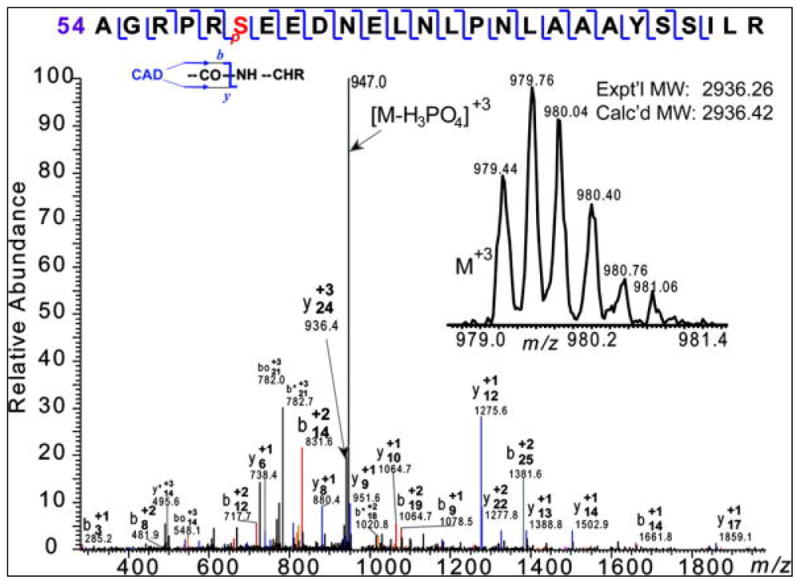
Confirmation of a phosphorylation site at S59 of FLAG-GCH-1 (corresponding to site S51 in endogenous GCH-1). CAD spectrum of an isolated +3 charge state of a tryptic peptide S[54–59]R at m/z 979 (inset), corresponding to a monophosphorylated A[54–79]R with a phosphorylated site at Ser59 (pS). Expt’l MW, experimental most abundant molecular weight; Calc’d MW: calculated most abundant molecular weight.
Prediction of GCH-1 Phosphorylation Sites
To identify all potential phosphorylation sites, we used bioinformatics tools combined with literature searches. PredPhospho, NetPhosK 2.0 and Scansites together with Pubmed were used to predict where rat GCH-1 might be phosphorylated (Supplemental Table 2). Seven sites (S51, S72, T85, T91, S103, T130 and T231) appeared in the output of all three analyses. Exceptions to this were S68 and S167 that were predicted to be phosphorylated by PredPhospho but not by others; and T97, which was predicted to be phosphorylated only by NetPhosK. Based on this information, we chose to examine the roles of 7 potential sites (S51, S72, T85, T91, S103, T130 and T231) and the S167 site (based on our MS data) in regulating GCH-1 activity. Each site was further examined for solvent accessibility as well as its position in the 3D crystal structure of GCH-1 using PHD software (http://cubic.bioc.columbia.edu). We learned that all of the selected sites, except S130 and S167, had high solvent accessibility (data not shown). Homology studies revealed that all of the 8 sites were highly conserved in rat, mouse and human, except for T91 where serine replaces threonine in the human GCH-1 sequence (Supplemental Figure II).
GCH-1 Activity of Phospho-Deficient GCH-1 Mutants
To examine the functional consequence of these 8 predicted phosphorylation sites on GCH-1 activity, we mutated the S or T to D/E or A (gain or loss of function) by site-directed mutagenesis. As shown in Fig 3A and Fig 4A, tetracycline (1 μg/ml, 24 hr) significantly and consistently increased GCH-1 and its mutants’ expression. As expected, overexpression of GCH-1 significantly increased total cellular GCH-1 activity, which was ~28 fold greater than in unstimulated cells (Figure 3B). Interestingly, cells expressing S72A-, T85A-, T91A-, T103A- or S130A-GCH-1 mutants had significantly reduced GCH-1 activity compared with WT-GCH-1 cells; particularly in the T85A-GCH-1 cells, where GCH-1 activity was reduced by 67%. Surprisingly, T231A-GCH-1 cells demonstrated enhanced GCH-1 activity (~23% more than in WT-GCH-1 cells). However, no noteworthy changes in GCH-1 activity were observed in S51A-GCH-1 cells or S167A-GCH-1 cells compared with GCH-1 activity in WT-GCH-1 cells. These results suggest that GCH-1 may be regulated at multiple sites of phosphorylation, which could influence activity either positively or negatively.
Figure 3.

The effects of phospho-defective mutations on GCH-1 expression, activity, BH4 and BH2. (A) Western blot analysis with anti-FLAG or GAPDH antibody. (B) GCH-1 activity (normalized by protein concentration and expressed as pmol neopterin per mg protein per hour). (C) BH4 levels. (D) BH2 levels. The pterin measured was normalized by protein concentration and expressed as pmol per mg protein. N=3–4, #=p<0.05 vs. GCH-1 cells without tetracycline; *=p<0.05 and NS=not significant vs. GCH-1 cells with tetracycline.
Figure 4.

The effects of GCH-1 phospho-mimic mutations on pterin production. (A) Western blot analysis with anti-FLAG antibody; (B) BH4 and (C) BH2 levels. N=4, #=p<0.05 vs. GCH-1 cells without tetracycline, *=p<0.05 and NS= not significant vs. WT-GCH-1 with tetracycline.
Pteridine Production by GCH-1 Phospho-Deficient Mutants
Since BH4 is the ultimate end-product of GCH-1 activity and the other enzymes in BH4 biosynthetic pathway are not limiting, any alteration in GCH-1 activity should result in a corresponding change in BH4 production. Tetracycline stimulation of WT-GCH-1 cells significantly increased BH4 and BH2 production compared with non-stimulated cells (Figure 3C, 3D). Similar to the results of GCH-1 activity, cells expressing GCH-1 phospho-deficient mutants, except the S51A and the S167A-GCH-1 mutant, had correspondingly reduced BH4 levels, compared with WT-GCH-1 cells. The most reduced BH4 levels were observed in T85A-GCH-1 and S130A-GCH-1 cells (decreased by 50~55%). In contrast, T231A-GCH-1 cells produced nearly 2-times BH4 of WT-GCH-1 cells, although GCH-1 activity in these cells was only about 23% more than in WT-GCH-1 cells. Interestingly, there was some disparity in BH2 levels among cells expressing the different mutants. No changes in BH2 levels were observed in cells expressing the S51A-, S72A-, T85A-, T91A-, T103A- or S130A- GCH-1 mutant, while cells expressing the S167A and T231A-GCH-1 mutants had elevated BH2 levels (50% and 171%, respectively) compared to cells expressing WT-GCH-1 (Figure 3D). These results suggest that phosphorylation of GCH-1 differentially influences biosynthesis of BH4 and the accumulation of BH2. Further, in BAEC, GCH-1 overexpression resulted in a 10-fold increase of BH4 over baseline levels. The T85A mutation significantly blunted whereas the T231A mutation further enhanced BH4 production compared to WT-GCH-1 expressing BAEC (Supplement Figure III).
Pteridine Production by Phospho-Mimic GCH-1 Mutants
To determine whether mutating GCH-1 to mimic phosphorylation induces a “gain of function” effect on BH4 production, we measured BH4 and BH2 in cells stably expressing phospho-mimic GCH-1 mutants. As expected, BH4 levels were increased in cells expressing S72E-, T85D-, T91E-, T103D- or S130D-GCH-1 mutants and significantly decreased in cells expressing the T231D-GCH-1 mutant compared with WT-GCH-1 cells. Cells expressing the S167E-GCH-1 mutant had no significant change in BH4 production (Figure 4B). Interestingly, cells expressing the S51E-GCH-1 mutant produced high BH4 concentrations, indicating that phosphorylation of S51 may also positively regulate BH4 biosynthesis. Cells expressing T231D-GCH-1 mutant had appreciably less BH2 than cells expressing WT-GCH-1, while cells expressing any of the other mutants had increased BH2 levels compared with WT-GCH-1 cells (Figure 4C).
The Effect of Mutation on Nuclear GCH-1 Content and Subcellular Activity
Although most of the GCH-1 protein resides in the cytoplasm, GCH-1 has also been found in the nucleus.10 Interestingly, our proteomic studies of GCH-1 revealed that 20% of the proteins that GCH-1 interacts with are nuclear proteins.11 As shown in Figure 5, there was a fair amount of GCH-1 expression in the nucleus of WT-GCH-1 cells. We used GAPDH and HSP90 as cellular markers to control for contamination in the nuclear fractions. As the same amount of protein was loaded in each lane, it was clear that the contamination of cytosolic proteins in the nuclear fraction was very low (Figure 5D). Interestingly, the T231A-GCH-1 cells had significantly less GCH-1 in nucleus compared to WT-GCH-1 cells; however these cells didn’t appreciably increase GCH-1 cytoplasmic content. Transfection of T231D-GCH-1 mutant resulted in a substantial increase in the cytoplasmic expression of GCH-1 compared with the levels in WT-GCH-1 cells. Even though there was more GCH-1 in the nucleus of T231D-GCH-1 cells than in the T231A-GCH-1 cells, the amount of nuclear GCH-1 in T231D-GCH-1 cells was still less than in WT-GCH-1 cells (Figure 5, Supplemental Figure IV). Using constructs of GFP fusion protein with WT-GCH-1 and T231A-GCH-1, we found that GCH-1 was distributed both in the cytosol and the nucleus. However, the nuclear localization of GCH-1 was significantly reduced in the T231A-GCH-1 cells (Figure 5C). To determine if the subcellular localization affects GCH-1 activity, cytoplasmic and nuclear extracts from these cells were examined for GCH-1 activity. Cytoplasmic GCH-1 activity in WT-GCH-1 cells was about 3 times the GCH-1 activity in nuclear fractions (Figure 6A and 6B). For the most part, the pattern of subcellular GCH-1 activity was similar to the whole cell GCH-1 activity. The one exception was the T231A mutant, which had markedly decreased GCH-1 activity in the nucleus (Figure 6A, 6B and Supplemental Figure IV).
Figure 5.

The effects of GCH-1 phosphorylation mutation on GCH-1 nuclear translocation. (A) Western blot analysis of FLAG expression in cytoplasmic and nuclear fractions from GCH-1 or its mutant’s cells. (B) Coomassie staining of cytoplasmic and nuclear proteins from cells expressing WT-GCH-1, T231A- and T231D-GCH-1 (upper panel); Western blot analysis of FLAG expression in cytoplasmic and nuclear fractions from cells expressing WT-GCH-1, T231A- and T231D-GCH-1. Nuclear fractions were also probed for nuclear marker Histone H1 to assure equal loading of proteins (lower panel). (C) Nuclear localization of GCH-1 and T231A by immunofluorescence staining. HEK293 cells were overexpressed with vector-GFP, GCH-GFP and T231A-GFP and stained with DAPI. (D) Equal amount of cytosolic and nuclear proteins from WT-GCH-1 cells with tetracycline stimulation were run on a SDS/PAGE and probed for GAPDH, HSP90, Histone H1 and FLAG by western blot analysis to determine purity of nuclear fraction.
Figure 6.

The effect of GCH-1 phosphorylation mutation on subcellular GCH-1 activity. The cytoplasmic and nuclear GCH-1 activity for cells expressing WT-GCH-1 or its phospho-defective mutants were shown in (A) and (B) respectively. N=3, #=p<0.05 vs. GCH-1 cells without tetracycline;*=p<0.05 and NS= not significant vs. GCH-1 with tetracycline.
Discussion
The present study was designed to systematically determine how multiple potential phosphorylation sites might regulate GCH-1 activity. Although previous works indicate GCH-1 was phosphorylated8 and shear stress could phosphorylate human GCH-1 at serine 81 (corresponding to S72 in the rat sequence) to increase BH4 production,9 our data indicate that GCH-1 activity is likely regulated at multiple sites. We have employed top-down mass spectrometry to observe all possible modifications on GCH-1, which confirmed that HEK 293 cell expressed GCH-1 is phosphorylated. Subsequently, we have applied in-solution and in-gel digestion of GCH-1 via the bottom-up mass spectrometry and identified three phosphorylation sites (S51, S167, and T231). Then we have taken a mutagenesis approach and characterized the function of these potential phosphorylation sites with respect to GCH-1 activity, biosynthesis of BH4 and BH2 and enzyme localization. We found that GCH-1 contains eight putative phosphorylation sites; the phosphorylation mimic mutation at S51, S72, T85, T91, T103 and S130 increased enzyme activity while phosphorylation mimic mutation at T231 decreased GCH-1 activity; and mutation at T231 alters GCH-1 nuclear localization.
We found that GCH-1 was phosphorylated at both threonine and serine sites (Figure 1A). MS data confirm that GCH-1 is phosphorylated at S51, S167 and T231. S167 lies within the core domain of GCH-1, which has been predicted to be a good candidate for phosphorylation in a previous study.12 Although S167 is buried within the enzyme, it may be exposed during conformational changes when GCH-1 binds to other interacting partners, e.g. BH2, GFRP or phenylalanine. 2D gels of cell lysates from WT-GCH-1 cells revealed that GCH-1 migrates horizontally in several distinct spots (Figure 1B). As horizontal migration is a function of “electro charge,” such patterns suggest GCH-1 may possess multiple phosphorylation sites. As the level of GCH-1 phosphorylation was only partially decreased in the T231A-GCH-1 cells compared with the WT-GCH-1 cells (data not shown), phosphorylation at T231 may down-regulate GCH-1 activity and other threonine sites may also be involved. Indeed, GCH-1 possesses 13 threonines, 4 of which are potential candidates. As the commercially available GCH-1 antibodies (tried so far) do not work well for immunoprecipitation, we limited our study to overexpressed FLAG-GCH-1 rather than endogenous GCH-1 phosphorylation. Although 8 potential phosphorylation sites were predicted by software analysis only 3 of them have been confirmed by state-of-the-art proteomic approaches. We speculate that not all the potential phosphorylation sites are phosphorylated under basal conditions in HEK cells and that the regulation of phosphorylation may be cell-specific and stimuli-specific. In addition, our data using top down mass spectrometry showed that only about 20% of GCH-1 in HEK cells under basal conditions is phosphorylated. Phosphorylated amino acid residues have been shown to interfere with trypsin cleavage13 which might prevent MS detection of the appropriate phosphorylated peptide fragment. In addition, phospho moieties on threonine can be easily lost during MS analysis. In future studies detection of other phosphorylation sites may be possibly by using a combination of different digestive enzymes and/or more advanced equipment that provides for gentle dissociation.
In our studies, we took a mutagenesis approach to determine how potential phosphorylation sites in GCH-1 might influence enzyme activity with the understanding of the limitations and potential controversies. Although classical signal transduction analysis holds that one should look for peptide fragments containing phosphorylated S, T & Y residues, site-directed mutagenesis has been used to change the charge of proteins to mimic phosphorylation/dephosphorylation. Using this approach we can gain insight into how a change in the charge of a protein at a particular location alters protein function even in the absence of direct proof of protein phosphorylation. To minimize the number of mutated constructs that could be required, we analyzed GCH-1 sequences with three different kinds of phosphorylation prediction software. We identified 8 putative phosphorylation sites. Using this approach, the S51 and T231 sites were identified by all three software programs, while the S167 site was predicted by only one software program. More importantly, MS confirmed that all three predicted sites were phosphorylated. Based on this success we speculate that the remaining 5 phosphorylation sites may be involved in regulating GCH-1 activity.
In our mutagenesis study, we found that five alanine mutations (S72A, T85A, T91A, T103A and S130A) decreased cellular GCH-1 activity and BH4 (Figure 3). T85, T91 and S130 are located at the active site of the enzyme14 (Supplemental Figure II), thus, it is logical that a change in charge might affect GCH-1 activity. T103 stands between two α-helices and its phosphorylation may induce structural changes that would in turn, influence enzyme activity. Clinically, mutations in the GCH-1 gene are common causes of BH4 deficiency, leading to inherited neurological diseases like DRD. Interestingly, S81 and T94 were found either deleted or mutated into lysine in certain DRD patients15 and these two sites correspond to sites S72 and T85 in the rat sequence. When we mutated GCH-1 to substitute alanine for these amino acids GCH-1 activity decreased. A recent report shows that laminar shear stress increased human endothelial cell GCH-1 activity by a mechanism involving phosphorylation of GCH-1 at S81.9 In our study, mutations resulting in D/E substitutions for S51, S72, T85, T91, T103 or T130 obviously increased cellular BH4 production (Figure 3B). This indicates that phosphorylation at these sites may positively regulate GCH-1 activity and BH4 biosynthesis.
BH4 constitutes about 80% of total biopterin and can be oxidized to BH2 naturally by hydroxylation, autooxidation or oxidative stress.2 BH2 can also be converted back to BH4 by dihydrobiopterin reductase or dihydrofolate reductase. High BH4 concentrations may be accompanied by an increase in hydroxylation rates as we observed in cells expressing S51E, S72E, T85D, T91E, T103D, S130D or the T231A mutant. Each of these cell lines had higher levels of BH4 with a corresponding increase in BH2 levels (Figure 3 and 4). Protein crystallization studies of GCH-1 showed that T231 participated in direct hydrogen bonding to BH2.14 Accordingly, the alanine mutation may reduce BH2 binding, resulting in an increased release of BH2 that may, in turn, stimulate GCH-1 activity.
Although MALDI-TOF MS studies indicated that GCH-1 was phosphorylated at S167, mutation at this site did not affect GCH-1 activity or BH4 biosynthesis (Figure 3B, 3C and 4B). However, both S167A-GCH-1 and S167E-GCH-1 cells had increased BH2 levels, indicating that this site may influence BH2 metabolism. Our LC/MS/MS data unambiguously showed that GCH-1 was phosphorylated at S51 (Figure 2). Interestingly, mutating S51 to A has no effect on GCH-1 activity, BH4 and BH2 biosynthesis, but mutating it to E increased both cellular BH4 and BH2 production. Intriguingly, the S51 site (a. a RPRSEEDN) is also a consensus recognition sequence for PKC and CKII, two kinases previously reported to phosphorylate GCH-1.7, 9 Although site directed mutagenesis has been used for many years now to “mimic” phosphorylation/dephosphorylation, it is rare that in nature an enzyme is fully phosphorylated/dephosphorylated. In addition, subtle changes in protein structure caused by D/E or A substitutions might also affect enzyme function differently from dephosphorylation or phosphorylation in nature, which might explain the discrepancies between A mutants and D/E mutants.
Nuclear localization of GCH-1 has been observed in COS-1 cells,10 epidermal keratinocytes and melanocytes.16 Nuclear GCH-1 is also reported to be enzymatically active in these cells. iNOS and nNOS are cytosolic proteins, whereas eNOS resides mainly in the plasma membrane and the perinuclear Golgi complex, and it’s enzyme activity is controlled tightly by its subcellular localization.17 Recently, eNOS, iNOS and nNOS have also been found in the nucleus to mediate nuclear function via •NO signaling.17–20 Nuclear localization of GCH-1 may make BH4 more available to the NOS isoforms in the nucleus to help maintain NOS-dependent •NO generation. Consistent with this idea, we observed that GCH-1 was localized in the nucleus and that the enzyme was active. Further studies are required to determine why and how GCH-1 translocates to the nucleus.
In summary, we identified 8 candidate phosphorylation sites in GCH-1 and characterized the function of these potential sites with respect to GCH-1 activity, biosynthesis of BH4 and BH2 and enzyme localization. Our report is the first to show that GCH-1 is regulated by multiple phosphorylation sites, both positively and negatively. To our knowledge, this is also the first study to identify S51, T231 and S167 as important regulatory phosphorylation sites on GCH-1. These findings should provide new insight into our understanding of the cellular mechanisms regulating BH4 production (or lack thereof) in cardiovascular and neurological diseases such as hypertension, coronary heart disease, diabetes and DRD.
Supplementary Material
Acknowledgments
SOURCES OF FUNDING
This work was supported by National Institutes of Health Grant HL080468 (to Y. S.), HL71214 (to K. A. P.), HL067244 (to J.V.V.), American Heart Association Postdoctoral Fellowship Award 09POST2250335 (to J. D.) and Wisconsin Partnership Funds for a Healthy Future (to Y. G.).
We gratefully appreciate the technical help by Dr. Bassam Wakim at Protein and Nucleic Acid Facility of Medical College of Wisconsin and Lisa Xu, Matt Lawrence and Huseyin Guner in Human Proteomics Program of UW-Madison School of Medicine and Public Health.
Footnotes
DISCLOSURES
The authors declared that they have no relevant conflicts of interest.
References
- 1.Werner-Felmayer G, Golderer G, Werner ER. Tetrahydrobiopterin biosynthesis, utilization and pharmacological effects. Curr Drug Metab. 2002;3:159–173. doi: 10.2174/1389200024605073. [DOI] [PubMed] [Google Scholar]
- 2.Thony B, Auerbach G, Blau N. Tetrahydrobiopterin biosynthesis, regeneration and functions. Biochem J. 2000;347(Pt 1):1–16. [PMC free article] [PubMed] [Google Scholar]
- 3.Moens AL, Kass DA. Therapeutic potential of tetrahydrobiopterin for treating vascular and cardiac disease. J Cardiovasc Pharmacol. 2007;50:238–246. doi: 10.1097/FJC.0b013e318123f854. [DOI] [PubMed] [Google Scholar]
- 4.Cai S, Alp NJ, McDonald D, Smith I, Kay J, Canevari L, Heales S, Channon KM. GTP cyclohydrolase I gene transfer augments intracellular tetrahydrobiopterin in human endothelial cells: effects on nitric oxide synthase activity, protein levels and dimerisation. Cardiovasc Res. 2002;55:838–849. doi: 10.1016/s0008-6363(02)00460-1. [DOI] [PubMed] [Google Scholar]
- 5.Alp NJ, Mussa S, Khoo J, Cai S, Guzik T, Jefferson A, Goh N, Rockett KA, Channon KM. Tetrahydrobiopterin-dependent preservation of nitric oxide-mediated endothelial function in diabetes by targeted transgenic GTP-cyclohydrolase I overexpression. J Clin Invest. 2003;112:725–735. doi: 10.1172/JCI17786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bendall JK, Alp NJ, Warrick N, Cai S, Adlam D, Rockett K, Yokoyama M, Kawashima S, Channon KM. Stoichiometric relationships between endothelial tetrahydrobiopterin, endothelial NO synthase (eNOS) activity, and eNOS coupling in vivo: insights from transgenic mice with endothelial-targeted GTP cyclohydrolase 1 and eNOS overexpression. Circ Res. 2005;97:864–871. doi: 10.1161/01.RES.0000187447.03525.72. [DOI] [PubMed] [Google Scholar]
- 7.Lapize C, Pluss C, Werner ER, Huwiler A, Pfeilschifter J. Protein kinase C phosphorylates and activates GTP cyclohydrolase I in rat renal mesangial cells. Biochem Biophys Res Commun. 1998;251:802–805. doi: 10.1006/bbrc.1998.9552. [DOI] [PubMed] [Google Scholar]
- 8.Hesslinger C, Kremmer E, Hultner L, Ueffing M, Ziegler I. Phosphorylation of GTP cyclohydrolase I and modulation of its activity in rodent mast cells. GTP cyclohydrolase I hyperphosphorylation is coupled to high affinity IgE receptor signaling and involves protein kinase C. J Biol Chem. 1998;273:21616–21622. doi: 10.1074/jbc.273.34.21616. [DOI] [PubMed] [Google Scholar]
- 9.Widder JD, Chen W, Li L, Dikalov S, Thony B, Hatakeyama K, Harrison DG. Regulation of Tetrahydrobiopterin Biosynthesis by Shear Stress. Circ Res. 2007;101:830–838. doi: 10.1161/CIRCRESAHA.107.153809. [DOI] [PubMed] [Google Scholar]
- 10.Elzaouk L, Laufs S, Heerklotz D, Leimbacher W, Blau N, Resibois A, Thony B. Nuclear localization of tetrahydrobiopterin biosynthetic enzymes. Biochim Biophys Acta. 2004;1670:56–68. doi: 10.1016/j.bbagen.2003.10.015. [DOI] [PubMed] [Google Scholar]
- 11.Du J, Xu H, Wei N, Wakim B, Halligan B, Pritchard KA, Jr, Shi Y. Identification of proteins interacting with GTP cyclohydrolase I. Biochem Biophys Res Commun. 2009;385:143–147. doi: 10.1016/j.bbrc.2009.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Funderburk CD, Bowling KM, Xu D, Huang Z, O’Donnell JM. A typical N-terminal extensions confer novel regulatory properties on GTP cyclohydrolase isoforms in Drosophila melanogaster. J Biol Chem. 2006;281:33302–33312. doi: 10.1074/jbc.M602196200. [DOI] [PubMed] [Google Scholar]
- 13.Kim S, Choi H, Park ZY. Improved detection of multi-phosphorylated peptides by LC-MS/MS without phosphopeptide enrichment. Mol Cells. 2007;23:340–348. [PubMed] [Google Scholar]
- 14.Maita N, Hatakeyama K, Okada K, Hakoshima T. Structural basis of biopterin-induced inhibition of GTP cyclohydrolase I by GFRP, its feedback regulatory protein. J Biol Chem. 2004;279:51534–51540. doi: 10.1074/jbc.M409440200. [DOI] [PubMed] [Google Scholar]
- 15.Saunders-Pullman R, Blau N, Hyland K, Zschocke J, Nygaard T, Raymond D, Shanker V, Mohrmann K, Arnold L, Tabbal S, deLeon D, Ford B, Brin M, Chouinard S, Ozelius L, Klein C, Bressman SB. Phenylalanine loading as a diagnostic test for DRD: interpreting the utility of the test. Mol Genet Metab. 2004;83:207–212. doi: 10.1016/j.ymgme.2004.07.010. [DOI] [PubMed] [Google Scholar]
- 16.Chavan B, Gillbro JM, Rokos H, Schallreuter KU. GTP cyclohydrolase feedback regulatory protein controls cofactor 6-tetrahydrobiopterin synthesis in the cytosol and in the nucleus of epidermal keratinocytes and melanocytes. J Invest Dermatol. 2006;126:2481–2489. doi: 10.1038/sj.jid.5700425. [DOI] [PubMed] [Google Scholar]
- 17.Jagnandan D, Sessa WC, Fulton D. Intracellular location regulates calcium-calmodulin-dependent activation of organelle-restricted eNOS. Am J Physiol Cell Physiol. 2005;289:C1024–1033. doi: 10.1152/ajpcell.00162.2005. [DOI] [PubMed] [Google Scholar]
- 18.Gobeil F, Jr, Zhu T, Brault S, Geha A, Vazquez-Tello A, Fortier A, Barbaz D, Checchin D, Hou X, Nader M, Bkaily G, Gratton JP, Heveker N, Ribeiro-da-Silva A, Peri K, Bard H, Chorvatova A, D’Orleans-Juste P, Goetzl EJ, Chemtob S. Nitric oxide signaling via nuclearized endothelial nitric-oxide synthase modulates expression of the immediate early genes iNOS and mPGES-1. J Biol Chem. 2006;281:16058–16067. doi: 10.1074/jbc.M602219200. [DOI] [PubMed] [Google Scholar]
- 19.Saini R, Patel S, Saluja R, Sahasrabuddhe AA, Singh MP, Habib S, Bajpai VK, Dikshit M. Nitric oxide synthase localization in the rat neutrophils: immunocytochemical, molecular, and biochemical studies. J Leukoc Biol. 2006;79:519–528. doi: 10.1189/jlb.0605320. [DOI] [PubMed] [Google Scholar]
- 20.Klinz F-J, Herberg N, Arnhold S, Addicks K, Bloch W. Phospho-eNOS Ser-1176 is associated with the nucleoli and the Golgi complex in C6 rat glioma cells. Neuroscience Letters. 2007;421:224–228. doi: 10.1016/j.neulet.2007.05.039. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.