

Abstract
In view of the importance of Candida Drug Resistance Protein (Cdr1p) of pathogenic Candida albicans in azole resistance, we have characterized its ability to efflux variety of substrates by subjecting its entire transmembrane segment (TMS) 5 to site directed mutagenesis. All the mutant variants of putative 21 amino acids of TMS 5 and native CaCdr1p were over expressed as a GFP-tagged protein in a heterologous host Saccharomyces cerevisiae. Based on the drug susceptibility pattern, the mutant variants could be grouped into two categories. The variants belonging to first category were susceptible to all the tested drugs, as compared to those belonging to second category which exhibited resistance to selective drugs. The mutant variants of both the categories were analyzed for their ATP catalysis and drug efflux properties. Irrespective of the categories, most of the mutant variants of TMS 5 showed an uncoupling between ATP hydrolysis and drug efflux. The mutant variants such as M667A, F673A, I675A and P678A were an exception since they reflected a sharp reduction in both Km and Vmax values of ATPase activity when compared with WT CaCdr1p-GFP. Based on the competition experiments, we could identify TMS 5 residues which are specific to interact with select drugs. TMS 5 residues of CaCdr1p thus not only impart substrate specificity but also selectively act as a communication link between ATP hydrolysis and drug transport.
Keywords: Candida albicans, Cdr1p, ABC transporter, multidrug resistance, fungal transporters, alanine scanning
INTRODUCTION
An over expression of the drug efflux pump encoding CDR1 and CDR2 genes, belonging to the ATP Binding Cassette (ABC) [1–5] and CaMDR1, belonging to Major Facilitator (MFS) super families of transporters [6–8], represent one of the most significant mechanisms of azole resistance in pathogenic Candida albicans. Among the ABC transporters, Cdr1p is a major drug transporter of C. albicans whose elevated expression coincides with an increased efflux of drug substrates in azole resistant clinical isolates [4,9].
CaCdr1p is a 170 kDa multidrug resistance protein that belongs to PDR subfamily of ABC transporter super family [10]. CaCdr1p has 1501 amino acids which are organized as two homologous halves. Each half begins with a cytoplasmic hydrophilic nucleotide binding domain (NBD) that harnesses energy through ATP hydrolysis. Each NBD is followed by a highly hydrophobic transmembrane spanning domain (TMD) comprising of six transmembrane segments (TMS), which confer substrate specificity to CaCdr1p [2]. An enormous range of structurally unrelated substrates compounds such as drugs, lipids and steroids can be exported by CaCdr1p [11–13]. Experiments with purified CaCdr1p have conclusively shown that ATP binding to Cdr1p is not a prerequisite for drug binding and both the mechanisms of drug and ATP binding result in specific conformational changes which take place independent of each other [14]. A direct link between the ability of CaCdr1p to translocate fluorescent glycerophospholipids and efflux drugs has also been demonstrated by Shukla et al [15].
Several structure and function studies on human ABCB1/P-glycoprotein (P-gp) and its homologues have identified the domains and residues involved in recognition, binding and efflux of drugs. Recent cross linking studies have shown that the cytoplasmic ends of TMS 5 and 8 of human P-gp are in close proximity on one side of the drug binding pocket while those of TMS 2 and TMS 11 are close on the other side of the drug binding pocket, thus forming gates for the entry of drug substrates [16]. The multidrug transporter Pdr5p from Saccharomyces cerevisiae is proposed to have at least three different substrate binding sites, each of which appears to use different chemical properties to transport compounds [17]. In addition, several amino acid residues that alter Pdr5p substrate specificity and sensitivity to inhibitors have been shown to be distributed throughout the length of protein [18–20].
Considering chemically diverse substrates which are expelled by CaCdr1p, the exact number of residues involved in drug binding and transport are far from understood. To understand the mechanism of drug transport mediated by CaCdr1p, we have generated its battery of mutant variants that drastically affect various stages involved in drug extrusion [13]. Shukla et al., 2003 have shown that the deletion of F774 in TMS 6 resulted in mislocalization of the CaCdr1p. In the same study, by employing photo affinity substrate analogs such as [125I] iodoarylazidoprazosin (IAAP) and [3H] azidopine two drug binding sites on CaCdr1p were revealed. Alanine scanning of TMS 11 of CaCdr1p showed that at least seven residues which were critical for determining substrate specificity and drug transport were clustered on the hydrophilic face of the −α helical projection of TMS11 [13]. Together, these studies suggest that the drug binding sites in Cdr1p are scattered throughout the protein and probably more than one residue of different helices are involved in binding and extrusion of drugs. However, there is still insufficient information available to predict where exactly the most common anti fungals, such as azoles bind and how they are extruded.
Based on human P-gp where interactivity of different TMS is well demonstrated [16] and our own study with CaCdr1p-GFP where we have shown that selected residues of TMS 11 involved in drug binding appear to be clustered in a pepwheel projection [13], in this study, we have subjected TMS 5 of Cdr1p to site directed mutagenesis where all the predicted 21 residues were replaced with alanine (existing alanines were replaced by glycine). We show that most of the residues of TMS 5 when replaced displayed decreased resistance to tested drugs. Notwithstanding the variations in ATPase activities, the drug efflux of all the mutant variants was abrogated. Our study for the first time shows that substitution of some of the TMS 5 residues of CaCdr1p results in uncoupling of drug transport from ATP hydrolysis.
Experimental Procedures
Materials
Anti-GFP monoclonal antibody was purchased from BD Biosciences Clontech, Palo Alto, Calif. DNA− modifying enzymes were purchased from NEB, USA. Protease inhibitors (Phenylmethylsulfonylfluoride, leupeptin, aprotinin, pepstatin A) and the drugs miconazole (MIC), cycloheximide (CYC), ketoconazole (KTC), anisomycin (ANI), itraconazole (ITR,) R6G, β-estradiol (β-EST), progestrerone (PRG), oligomycin and other molecular grade chemicals were obtained from Sigma Chemicals Co (St. Louis, Mo.). FLC was generously provided by Ranbaxy Laboratories, India and tritylimidazole (tritylimz) and tricyclohexyltinchloride (hexyltin-Cl) were kind gifts from John Golin. The radio labeled [125I] iodoarylazidoprazosin (IAAP) (2,200 Ci/mmol) was purchased from Perkin–Elmer Life Sciences (Boston Mass.) while [3H] Fluconazole ([3H] FLC) was custom prepared by Amersham, UK.
Media and Strains
Plasmids were maintained in Escherichia coli DH5α E. coli was cultured in Luria-Bertani medium (Difco, BD Biosciences, NJ, USA) to which ampicillin was added (100μg/ml). The S. cerevisiae strain used was AD1-8u− (MATa pdr1-3 his1 ura3 Δyor1::hisG Δsnq2::hisG Δpdr5::hisG Δpdr10::hisG Δpdr11::hisG Δycf1::hisG Δpdr3::hisG Δpdr15::hisG), provided by Richard D. Cannon, University of Otago, Dunedin, New Zealand. The yeast strains used in this study are listed in the supplementary data, Table S1. The yeast strains were cultured in Yeast Extract Peptone Dextrose (YEPD) broth (BIO101, Vista, Calif.) or in SD Ura− dropout media (0.67% yeast nitrogen base, 0.2% dropout mix, and 2% glucose; Difco). For agar plates, 2.5% (w/v) bacto agar (Difco BD Biosciences, NJ.) was added to the medium.
Methods
Site specific mutagenesis of Cdr1p
Site specific mutagenesis was performed by using the Quick-Change Mutagenesis kit from Stratagene (La Jolla, Calif.) as described previously [21]. The mutations were introduced into the plasmid pPSCDR1-GFP according to the manufacturer’s instructions, and the desired nucleotide sequence alterations were confirmed by DNA sequencing of the ORF. The primers used for the purpose are listed in supplementary data, Table S2. The mutated plasmid, after linearising with Xba1, was used to transform AD1-8u− cells for uracil prototrophy by lithium acetate transformation protocol [21]. Integration was confirmed by Southern blot analysis.
Preparation of the plasma membranes and immunodetection of Cdr1p and its mutants
The plasma membranes (PM) were prepared from S. cerevisiae cells, as described previously [21]. The PM protein concentration was determined by bicinchonic acid assay using bovine serum albumin as the standard. The Western blot analysis was conducted using anti-GFP monoclonal antibody (1:5000), as described previously [21]. Proteins on immunoblots were visualized using the enhanced chemiluminescence assay system (ECL kit, Amersham Biosciences, Arlington Heights, IL.).
Confocal Microscopy
The cells were grown to late log phase in SD ura− medium, except for AD1-8u−, where uridine (0.02%) was added to the SD ura− medium. The cells were washed and resuspended in an appropriate volume of 50 mM HEPES (pH 7.0). The cells were imaged under an oil immersion objective at 100 × magnification on a confocal microscope (Radiance 2100, AGR, 3Q/BLD; Bio-RAD, UK) [8].
Flow Cytometry
Flow cytometric (FACS) analysis of CaCdr1p-GFP and its mutant variants expressed in S. cerevisiae cells was performed with FACsort flow cytometer (Becton-Dickson immunocytometry systems, San Jose, Calif.). Cells were grown to mid log phase and 106 cells were harvested and washed with 50mM HEPES (pH 7.0). Cells were resuspended in appropriate volume of 50mM HEPES (pH 7.0)/1× PBS. Ten thousand cells were analyzed in acquisition. Analysis was performed with cell quest software (Becton-Dickson immunocytometry systems) [8,21].
Drug Susceptibility
The susceptibilities of S. cerevisiae cells to different drugs were determined using spot assays. For spot assay, 3-μl samples of fivefold serial dilutions of each yeast culture (each with cells suspended in normal saline to an optical density at 600nm of 0.1 were spotted onto YEPD plates in the absence (control) or in the presence of the drugs [22].
ATPase assay
The CaCdr1p-GFP associated ATPase activity of the purified PM was measured as an oligomycin sensitive release of inorganic phosphate as described previously [21]. Briefly, plasma membrane suspension (10μg of PM protein) was incubated at 30°C in 0.1ml of medium containing 60mM Tris pH 7.5 and 8mM MgCl2 (ATPase assay buffer) and 20 μM oligomycin where indicated. To eliminate possible contributions from non specific vacuolar and mitochondrial ATPases, 50mM KNO3, and 10mM NaN3, respectively were included in the reaction mixture. The reaction was started by addition of 5mM ATP and was stopped by the addition of 0.1 ml of 5% SDS solution. The amount of inorganic phosphate released was determined immediately as described previously [23].
Efflux of substrates
Rhodamine 6G
Efflux of R6G was determined essentially using a previously described protocol [21,24]. Briefly, approximately 107 yeast cells from overnight grown culture were transferred into YEPD media and allowed to grow for 5h. Cells were pelleted and washed and then resuspended in phosphate-buffered saline (PBS) without glucose as a 2% cell suspension to which R6G was added at a final concentration of 10 μM and incubated for 1h at 30°C. The cells were then washed and resuspended in PBS and reaction was initiated by the addition of 2% glucose. Samples of 1 ml volume were withdrawn at indicated time (30 min) and centrifuged at 9000 g for 2 min. The supernatant was collected and absorption was measured at 527 nm. Glucose free controls were included in all experiments. For competition assays, the competing substrate (5X) was added to the de energized cells 5 minutes before the addition of R6G and allowed to equilibrate for an hour before initiating the efflux by the addition of glucose.
Fluconazole
The accumulation of [3H] FLC was determined by modification of method described elsewhere [25]. Cells from mid log phase were centrifuged at 3000g for 3 min and resuspended in PBS as 5% cell suspension (2.5×108 cells ml−1). 100 μl of cell suspension was incubated at 30°C and [3H] FLC was added at a final concentration of 100 nM [specific activity 19 Ci mmol−1]. The cells were incubated with [3H] FLC for 30 min, filtered rapidly and washed twice with PBS, pH 7.4 on millipore manifold filter assembly using 0.45 μm cellulose nitrate filter (Millipore, USA). The filter discs were dried and put in cocktail ‘O’ and the radioactivity was measured in a liquid scintillation counter (Beckman, USA). The accumulation was expressed as pmoles mg−1 dry weight.
Photoaffinity labeling with IAAP
The PM proteins (25μg) were incubated with the indicated drug for 10 min at 37°C in 50mM Tris-HCl (pH 7.5). The samples were brought to room temperature and 3 to 6 nM iodoarylazidoprazosin [125I] IAAP (2,200 Ci/mmol) was added and incubated for additional 5 min under subdued light. The samples were then illuminated with a UV lamp assembly (PGC, Scientifics, Gaitherberg, MD) fitted with two black light (self filtering) UV-A long wavelength F15T8BLB tubes (365 nm) for 10 min at room temperature (21 to 23°C). Following SDS-polyacrylamide gel electrophoresis (SDS-PAGE) on an 8% Tris-glycine gel at constant voltage, gels were dried and exposed to Bio-Max MR film (Eastman Kodak, Rochester, N.Y) at −80°C for 12 to 24h. The radioactivity incorporated into the CaCdr1p band was quantified using a STORM 860 phosphorimager system (Molecular Dynamics, Sunnyvale, Calif.) and the software ImageQuaNT as described previously [21]. Western analysis as described earlier was used to check equal loading of CaCdr1p-GFP [21].
Statistical analysis
Data are the means ± S.D from duplicate samples of at least three independent experiments. Differences between the mean values were analyzed by Student’s t-Test and results were considered significant when P< 0.001.
RESULTS
In the present study, we have systematically investigated the role of putative TMS 5 in drug binding and transport (Fig. 1A and B). Some of the TMS 5 residues were more conserved than the others across fungal and mammalian ABC transporters (Fig. 1C). However, all the 21 residues were subjected to alanine scanning. For this, we employed site directed mutagenesis using mutagenic oligonucleotides (supplementary data, Table S2). To avoid the introduction of new side chains, the two existing alanines in TMS 5 (A660, A666) were replaced with glycines. For functional analysis of the TMS 5 mutant variants, a heterologous hyper-expression system, where GFP tagged CaCdr1p is stably over expressed from a genomic PDR5 locus in a S. cerevisiae mutant AD1-8u−, was used [26]. The host AD1-8u− developed by Goffeau’s group [27], was derived from a Pdr1-3 mutant strain with a gain of function mutation in the transcription factor Pdr1p, resulting in constitutive hyper induction of the PDR5 promoter [26]. A single copy integration of each transformant at the PDR5 locus was confirmed by Southern hybridization (data not shown). Two positive clones of each GFP-tagged CaCdr1p-mutant variants were selected to rule out any clonal variations. We confirmed that GFP tagging of CaCdr1p (CaCdr1p-GFP) did not impair its expression and the functional activity of the protein [21].
Figure 1. Topology of yeast Cdr1p.
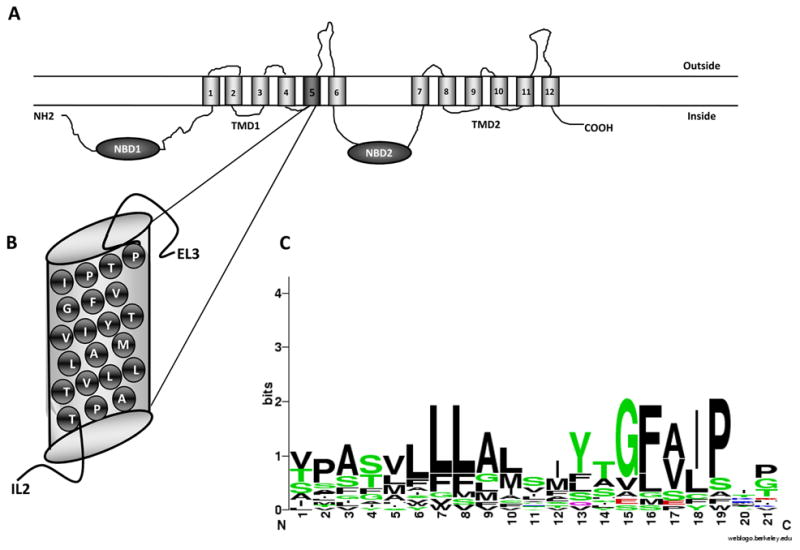
(A) Predicted topology of CaCdr1p with two NBDs and two TMDs each consisting of six TMS. Each TMS is numbered. (B) The putative TMS 5 is magnified to show the amino acid residues. (C) The sequence alignment of protein TMS 5 residues with other fungal and mammalian transporter segments. The putative TMS 5 of each transporter were predicted with the program HMMTOP [36]. The scale indicates the certainty of finding a particular amino acid at a given position and is determined by multiplying the frequency of that amino acid by the total information at that position. The residues at each position are arranged in order of predominance from top to bottom, with the highest frequency residue at the top. The height of the symbol within each stack indicates the relative frequency of each amino acid at that position. Sequence logos were generated using the “WebLogo” (http://weblogo.berkeley.edu/) Colors: such as green defines polar; blue correspond to basic; red to acidic and black to hydrophobic.
Substitutions of TMS 5 residues with alanine decrease drug resistance
Two positive clones of each mutant were selected and were screened for their sensitivity to different substrates by spot assay. Our results of drug susceptibility assay revealed that most of the cells expressing the mutant variants of TMS 5 of CaCdr1p-GFP residues when substituted with alanine became susceptible to tested drugs/substrates (Fig. 2). However, their extent of sensitivity to various drugs was variable (discussed below). Based on the drug susceptibility results, all the mutant variants could be placed into two categories: The first category included seven residues T658A, A660G, T661A, V662A, L665A, I669A and Y670A. These showed hyper susceptibility comparable to host AD1-8u− cells for all the tested drugs (Fig. 2A). The second category, comprising of fourteen mutant variants, P659A, L663A, L664A, A666G, M667A, V668A, T671A, G672A, F673A, V674A, I675A, P676A, T677A and P678A displayed a selective loss of resistance to one or more of the tested drugs (Fig. 2B). Residue A666, when replaced with glycine (A666G), displayed hyper resistant phenotype towards FLC comparable to wild type (WT) CaCdr1p-GFP expressing cells but showed considerable sensitivity to other drug substrates (Fig. 2B). P659A, T671A, V674A, and P678A mutant variants which showed significant resistance to both hexyltin-Cl and R6G were highly sensitive to most of the tested azoles, CYC, ANI and tritylimz (Fig. 2B). Interestingly, similar to L663A, L664A, G672A, F673A, I675A, P676A and T677A mutant, the above mentioned variants also showed partial resistance to FLC. The mutant variants L663A, L664A, M667A, V668A, G672A, F673A and T677A were partly resistant to R6G but were susceptible to other tested substrates and the two variants I675A and P676A exhibited partial resistance to hexyltin-Cl and FLC in comparison to other tested substrates (Fig. 2B). While most of the mutant variants of second category exhibited modest resistance to FLC but it was not the case with the two mutant variants M667A and V668A. Of note, when replaced with alanines/glycines, the abrogation in drug resistance was seen with all the variants, irrespective of the fact whether the residue of TMS 5 of CaCdr1p belonged to conserved or non-conserved category (Fig. 1C and 2). Notably, the expression of CaCdr1p as revealed by immunoblot analysis employing anti-GFP monoclonal antibody and confocal analysis was similar to WT CaCdr1p-GFP protein in all the mutant variants except in T661A which was mislocalised and thus it was not detected in the plasma membrane (PM) (Fig. 3A). As the expression of T661A in the PM was negligible, this mutant was not taken for further characterization.
Figure 2.

Drug resistance profiles of WT CaCdr1p-GFP and mutant CaCdr1p-GFP strains determined by Spot assay. Cells were freshly streaked, grown overnight and then resuspended in normal saline to an A600 of 0.1. For spot assay, 3μl of fivefold serial dilutions, namely 1 (1:5), 2 (1:25), 3 (1:125) and 4 (1:625), of each strain was spotted on to YEPD plates in the absence (control) and presence of the following drugs: FLC (0.75mg/L), CYC (.02mg/L), ITR (.08mg/L), KTC (.05mg/L), MIC (.03mg/mL), ANI (0.4mg/L), R6G (0.6mg/L), hexyltin-Cl (4mg/L), tritylimz (.015mg/L). Growth differences were recorded following incubation of the plates for 48h at 30°C. Growth was not affected by the presence of the solvents used for the drugs. (A) The mutant variants of first category which exhibit susceptible phenotype for all the tested substrates. (B) The mutant variant of second category included those which showed susceptibility to one or more of the drugs tested.
Figure 3.

Expression and drug binding assay using plasma membranes cells expressing WT CaCdr1p-GFP and the mutant variants of TMS 5 in S. cerevisiae. (A) Western analysis showing proper membrane localization of WT CaCdr1p-GFP and its mutant variants. (B) Photo affinity labeling of WT CaCdr1p-GFP and its mutant variants with [125I] IAAP in the presence and absence of NYS. The PM proteins (25μg) were photoaffinity labeled with [125I] IAAP under subdued light as described in Materials and Methods. Upper panel, lane 1, WT CaCdr1p-GFP (no drug); lane 2, WT CaCdr1p-GFP with NYS; lanes 3, 5, 7 show [125I] IAAP binding in mutant variants of CaCdr1p-GFP without drug while lanes 4, 6, 8 exhibit [125I] IAAP binding in the presence of 200μM NYS. Lower panel depicts Western analysis to show equal loading of CaCdr1p-GFP.
ATPase activity of most of the mutant variants remains unaffected
To test the possibility that the introduced mutations in TMS 5 have, in any way, altered the catalytic cycle of CaCdr1p, which might have resulted in impaired efflux of substrates, we checked the basal ATPase activity. We have used heterologous host S. cerevisiae deleted in eight ABC transporters [27] to over express CaCdr1p and thus have minimum background ATPase activity interference from other similar proteins. To eliminate possible contributions from non-specific vacuolar and mitochondrial ATPases, 50 mM KNO3, and 10mM NaN3, respectively were included in the reaction mixture [21]. Of note, our earlier studies established that there was no significant drug stimulated ATPase activity elicited by CaCdr1p in presence of drug substrates [14]. For routine ATPase assay, the purified PM from WT CaCdr1p-GFP as well as from mutant CaCdr1p-GFP variants were analyzed for their oligomycin-sensitive ATPase basal activity (without drug substrates) [21]. ATPase activities of most of the mutants at a fixed nucleotide concentration (ATP 5mM) did not show any difference between mutants and the wild type protein expressing cells (data not shown). However, a more rigorous kinetic analysis employing Lineweaver Burke plots, revealed kinetic differences in ATPase activity of various mutant variants. For example, in mutants such as T658A, A660G, V662A, A666G and V668A, there was no significant change in Km and Vmax values. However, in the other set of mutants such as P659A, L663A, L664A, L665A, I669A, Y670A, T671A, G672A, V674A, P676A and T677A, there was a modest decrease in apparent Km (not exceeding 20%), but all these mutants were able to attain Vmax in the same range as of WT CaCdr1p-GFP. Therefore, these mutants seem to uncouple ATP catalysis from drug efflux (Table 1). However, this was not the case with M667A, F673A, I675A and P678A mutants where both the Km and Vmax values showed a significant decrease (Table 1).”
Table 1.
Comparison of ATPase activity of CaCdr1p with its mutant variants #
| Strain | Km (mM) | Vmax (nmol min−1 mg protein−1) |
|---|---|---|
| CaCdr1p-GFP | 6.39 ± 0.46 | 129.87 ± 3.68 |
| T658A | 7.55 ± 0.20 | 120.49 ± 1.02 |
| P659A | 4.82 ± 0.08 | 107.45 ± 2.58 |
| A660G | 7.19 ± 0.14 | 116.84 ± 5.76 |
| V662A | 5.71 ± 0.71 | 114.13 ± 6.27 |
| L663A | 3.62 ± 0.38 | 105.19 ± 4.99 |
| L664A | 4.06 ± 0.13 | 112.41 ± 1.78 |
| L665A | 3.73 ± 0.40 | 100.70 ± 3.22 |
| A666G | 6.08 ± 0.62 | 119.69 ± 11.74 |
| M667A | 2.68 ± 0.01 | 81.64 ± 0.707 |
| V668A | 5.69 ± 0.41 | 106.81 ± 4.81 |
| I669A | 4.04 ± 0.11 | 93.07 ± 2.17 |
| Y670A | 3.62 ± 0.29 | 88.18 ± 1.91 |
| T671A | 4.36 ± 0.12 | 91.74 ± 0.59 |
| G672A | 5.21 ± 0.32 | 101.74 ± 2.89 |
| F673A | 3.92 ± 0.10 | 78.195 ± 1.72 |
| V674A | 4.75 ± 0.36 | 92.87 ± 3.64 |
| I675A | 1.58 ± 0.13 | 76.09 ± 1.43 |
| P676A | 4.83 ± 0.45 | 114.23 ± 6.34 |
| T677A | 4.90 ± 0.47 | 103.02 ± 4.85 |
| P678A | 2.70 ± 0.87 | 83.75 ± 9.42 |
ATPase activity of the PM fraction of cells expressing the wild type Cdr1p and its mutant variants were assayed as described under Materials and Methods; the values represent the average ± standard deviation of three independent experiments.
Drug binding remains unaffected in mutant variants
In order to explore whether the observed differences in drug susceptibilities accompanied by abrogated efflux activities were due to any aberration in drug binding, some of the mutants based on their selective sensitivity to drugs (Fig. 2) were analyzed for their drug binding abilities. For this we used iodoarylazidoprazosin [125I] (IAAP) which is a photo affinity analog of CaCdr1p substrate prazosin. We had earlier shown that [125I] IAAP which has been extensively used to map human P-gp drug binding sites also binds to WT CaCdr1p-GFP and can be competed out with nystatin (NYS) [21]. It was observed that the drug binding properties of the TMS 5 mutant variants of CaCdr1p-GFP remained unaffected and were similar to WT CaCdr1p-GFP (Fig. 3B).
Enhanced drug sensitivity of the TMS 5 mutants is associated with reduced efflux of substrates
Since there was no detectable difference in ATP hydrolysis, we checked if the observed drug sensitivity by CaCdr1p mutant variant expressing cells was associated with impaired drug extrusion. For this, two well known substrates of CaCdr1p such as R6G and FLC were used to monitor the drug efflux activity [28]. Both fluorescent R6G and [3H] FLC were allowed to equilibrate in the de-energized cells by passive diffusion and the energy dependent extrusion was then initiated by the addition of glucose. As depicted in Fig. 4A (inset), after glucose addition, WT CaCdr1p-GFP expressing cells demonstrated energy dependent efflux of R6G (increase in extra cellular concentration) which is not seen in AD1-8u− cells. Notably, a 40–60 % decrease in the extra cellular concentration (due to low efflux) of R6G was observed in the TMS 5 mutant variants as compared to that in the WT CaCdr1p-GFP cells, which clearly indicated impaired efflux activity by the mutant cells (Fig. 4A). The accumulation due to passive diffusion of R6G and [3H] FLC in the de energized cells in the mutant variants was comparable to that in the WT CaCdr1p-GFP (data not shown). That the efflux ability of the TMS 5 mutant cells was reduced was further confirmed by using another substrate, [3H] FLC. The inset in Fig 4B shows energy dependent decrease in accumulation of FLC in cells expressing WT CaCdr1p-GFP which was not the case with AD1-8u− cells. Fig. 4B depicts that the intracellular levels of accumulation of [3H] FLC were 43–65 % higher (due to lower levels of efflux) in the mutant cells than the WT CaCdr1p-GFP. Notably, most mutations displayed impaired efflux, irrespective of the categories based on drug resistance profile (Fig. 4A and B). All the changes observed in R6G efflux are statistically significant (P<0.001).
Figure 4.

Drug transport assays in WT CaCdr1p-GFP and mutant variants of TMS 5. (A) R6G efflux in WT CaCdr1p-GFP and mutant CaCdr1p-GFP variant expressing cells. The inset shows the extra cellular concentration of R6G in presence and absence of glucose in AD1-8u− and WT CaCdr1p-GFP. The assay was performed as described in Materials and method section. (B) FLC accumulation in WT CaCdr1p-GFP and mutant CaCdr1p-GFP variant expressing cells. The inset shows the intracellular accumulation of [3H] FLC in presence and absence of glucose in AD1-8u− and WT CaCdr1p-GFP. The results are means ± standard deviations for three independent experiments.
As shown in figure 4A and 4B, we observe that the mutant variant L665A displays an increased R6G efflux but shows no resistance to R6G in Figure 2. The opposite is true for L664A, T671A, and V674A. The mutants A660G, L665A or I669A show a decreased FLC intracellular accumulation level, but in the spot assay they do not show any resistance to FLC. Also, the resistance showed by mutant variant A666G towards FLC on spot assay was not reflected in FLC transport assay. It should be pointed out that the two parameters, drug efflux and drug sensitivity do not directly match, hence, should be considered independently. For example, susceptibility to FLC/R6G could be due to culmination of factors other than an over expression of efflux pump proteins while the efflux of the drugs is directly dependent on MDR pump proteins such as CaCdr1p in this case.
In the following experiment, R6G efflux was competed out with five fold molar excess CaCdr1p substrates such as KTC, ITR, MIC, FLC, ANI, PRG, β-EST and CYC (Fig. 5). In comparison to AD1-8u− cells where no change in the extra cellular concentration of R6G was observed (Fig. 5A), substrates such as ITR, KTC and MIC could compete with R6G efflux (up to three fold) in the cells expressing WT CaCdr1p-GFP. The other substrates like FLC, ANI, CYC, PRG and EST could not significantly compete with R6G (Fig. 5B).
Figure 5.

Competition of various drugs with R6G in WT CaCdr1p-GFP. For competition between R6G and various drugs, R6G was used at a final concentration of 10μM and a fivefold concentration of each drug (50 μM), KTC, MIC, ITR, FLC, CYC, ANI, EST and PRG was used for competition studies. KTC, MIC, ITR compete for R6G in WT CaCdr1p-GFP. (A) Efflux of R6G in AD1-8u− cells in presence of different substrates. (B) Efflux of R6G in presence of different substrates in cells expressing WT CaCdr1p-GFP. The results are means ± standard deviations for three independent experiments.
We evaluated if R6G efflux in mutant variants of TMS 5 owing to loss of residues still retained the ability to be competed with KTC, ITR and MIC. Since, the inhibition of R6G efflux varies between 40%–65% (Fig 4A); the residual activity in these mutant variants was taken as their 100% activity for fold change calculations. As shown in Fig. 6A, B and C, the ability of KTC, MIC and ITR to compete with R6G efflux was severely abrogated in most of the TMS 5 mutants resulted in an increase in extracellular concentration of R6G between 1.8 to 6.8 folds. The mutant variant T658A was an exception as its R6G efflux was comparable to that of WT CaCdr1p in presence of MIC. All the changes observed in R6G efflux in the presence of different drugs like KTC, ITR and MIC are statistically significant (P<0.001).
Figure 6.

Competition of various drugs in mutant variants of CaCdr1p-GFP with R6G. For competition between R6G and various drugs, R6G was used at a final concentration of 10μM and a fivefold concentration (50 μM) of each competing drug KTC, MIC and ITR was added. Panel A, B and C show the percentage values of R6G efflux of mutant variant expressing cells in presence of KTC, ITR and MIC, respectively. Unlike in WT CaCdr1p-GFP, R6G efflux by all the mutant variants was no longer competed out by ITR, KTC and MIC. The results are means ± standard deviations for three independent experiments.
Discussion
Cdr1p of C. albicans belongs to ABC super family of primary transporters which directly couples ATP hydrolysis to substrate/drug transport. These ABC pump proteins such as Cdr1p and Cdr2p of C. albicans not only efflux azoles and its derivatives but also extrude a variety of structurally unrelated compounds. It is the promiscuity in substrates specificity which is intriguing and challenging in designing modulators/inhibitors of CaCdr1p. To this end, it is essential to understand, how drugs bind and how their efflux is powered by ATP hydrolysis. Based on studies from human ABC multidrug transporter P-gp where TMS 11 is predicted to interact with TMS 5 and our own data where some residues of TMS 11 have been shown to be critical; in the present study, we have subjected entire TMS 5 of CaCdr1p to alanine scanning to gain an insight into its involvement in drug binding and transport. For this, all the putative 21 residues were subjected to site directed mutagenesis and replaced with alanine.
Our results of drug susceptibility by spot assays revealed that all the mutants displayed increased susceptibility to the tested drugs. Essentially, all the mutants could be clustered in two groups:1) included T658A, A660G, T661A, V662A, L665A, I669A and Y670A which displayed susceptibility to all tested drugs, 2) included P659A, T671A, V674A, P678A L663A, L664A, M667A, V668A, G672A, F673A, T677A, I675A and P676A which showed varying degree of susceptibility to different drugs. For example, the mutant variants of group 2 such as L663A, L664A, M667A, V668A, G672A, F673A and T677A were only moderately resistant to FLC and R6G while mutant variants A666G showed decreased resistance to all the tested drugs except to FLC which was comparable to WT CaCdr1p-GFP.
Golin et al. [17] have used imidazole and trialkyltin chloride derivatives to determine the minimum prerequisite of Pdr5p substrate interaction and transport. In this study, we tested the susceptibility of CaCdr1p mutant variants towards one such imidazole namely tritylimz and trialkyltin chloride derivative, i.e. hexyltin-Cl. Interestingly, most of the mutant variants of TMS 5 became hypersusceptible towards both the compounds. However, mutant variants namely, P659A, T671A, V674A, I675A, P676A and P678A exhibited varying degrees of resistance to hexyltin-Cl and in parallel showed moderate resistance to FLC and R6G. These results are interesting in the light of our earlier results where selected critical residues of TMS 11 showed opposite effect to these compounds. All the mutant variants of TMS 11 remained resistant to hexyltin-Cl while seven of them were selectively sensitive to tritylimz [13]. It is clear that unlike TMS 11, the residues of TMS 5 are involved in coordinating interactions with both hexyltin-Cl and tritylimz. Though the simple aromatic imidazole derivative, tritylimz used in this study differs dramatically in structure from the alkyltin compound, hexyltin-Cl, but they overlap in their size and hydrophobicity values and have also been shown to bind at the same site in Pdr5p [17]. This explains why both these compounds are able to evoke similar responses from the mutant variants because of their probable similar interactions with residues of TMS 5 which was not the case with TMS 11 residues. Taken together, the display of selective sensitivity by these mutant variants of TMS 5 suggests that some of the residues appear to be critical for establishing interactions with selected drugs Notably, the observed phenotypic differences displayed by the mutant variants of TMS 5 were regardless of location of residue within the helix; the degree of conservation of the residue or the nature of the side chain of the amino acid residues (Fig. 1). This is interesting in the context of TMS 11 where only selected residues are involved in drug binding which seem to cluster on one face of the helix [13].
We have earlier observed that in the WT CaCdr1p-GFP except for selected azoles such as KTC, MIC and ITR, none of the other tested substrates could compete with R6G efflux. This suggested that KTC, MIC and ITR may share binding sites with R6G on CaCdr1p-GFP. In the present study, it is evident from competition assays that efficient competitors of R6G efflux, such as KTC, MIC and ITR, became totally ineffective in most of the mutant variants of CaCdr1p-GFP. An exception to this was the residue T658 which upon mutation to alanine continued to efflux R6G in a similar manner as the WT CaCdr1p in the presence of MIC and thus ruling out its involvement towards MIC binding. Therefore, the residues of this TMS appear to be crucial to substrate recognition and extrusion. Taken together; it appears that similar to human P-gp, CaCdr1p has multiple binding sites [29]. One of the site (s) is probably responsible for translocation of R6G and the azoles like KTC, MIC and ITR while another site (s) can only interact and transport substrate such as FLC. There could still be third binding site where prazosin analog IAAP binds.
There are essentially four steps involved in drug extrusion: ATP and drug binding, ATP hydrolysis and drug release. Since our mutant variants displayed decreased resistance to selective drugs, we analyzed each step to pinpoint the defect leading to increased drug susceptibilities. For this, basal ATPase activities of all the mutant variants were measured. In most mutants, there was no significant change in Km and Vmax values. Notwithstanding the status of ATPase activity, the drug efflux in all the mutants was considerably reduced. Our drug binding results with representative CaCdr1p substrate [125I] (IAAP) also showed that the drug binding ability of TMS 5 mutant variants remained unaffected in comparison to WT CaCdr1p-GFP. In this context, it is essential to mention that since we have used a representative substrate, prazosin’s photoaffinity analog [125I] (IAAP) for drug binding, the possibility of its binding to another site(s) on CaCdr1p cannot be excluded. Taken together, it appears that most of the mutant variants of TMS 5 uncouple ATP hydrolysis from drug transport and thus may represent residues involved in cross talk between NBDs and TMDs.
The structure of bacterial ABC transporter Sav 1866, which may be considered as a functional ortholog of ScPdr5p, human P-gp, ABCG2p and CaCdr1p, has been resolved to 3Ǻ [30]. The 3D structure of Sav 1866 for the first time confirms significant contacts between the TMDs and NBDs. These contacts occur via intracellular loops that link TMS helices. This structure organization offers a possible explanation for the mechanical coupling between the NBDs and the TMDs of ABC transporters which is consistent with the genetic data that implicated ICL1 of human MDR1 to interact with NBDs [31]. The mutational studies and sequence comparisons also indicated that ICL2 of TAP1/TAP2, as well as, ICL4 of CFTR provide similar crucial contacts [32]. In addition, studies on P-gp and Yor1p using site directed mutagenesis and cross linking agents have demonstrated the close proximity of ICL2 and Q loop of NBD1 [33]. A study by Sauna et al., have shown a cross talk between NBD1 and Transmembrane helix 2 in a CaCdr1p homologue, Pdr5p of S. cerevisiae. Taken together, it is now fairly established that the TMDs of these transporters, all featured strikingly similar, short α-helices located in their cytoplasmic loops between TMS which provide the bulk of contacts to NBDs and have been labeled as coupling helices [35]. ABC transporters functionality hinges on precise coupling of ATP hydrolysis which occurs at NBDs and with drug transport which occurs at TMDs. In CaCdr1p, ICL2 links TMS 4 and 5 and ICL4 links TMS10 and 11, however, how they mechanically couple ATP hydrolysis with drug transport is not established. However, taking clues from Sav 1866 3D structure and recent study of ScPdr5p by Golin’s group, it is reasonable to speculate that ICL2 and 4 of CaCdr1p function as communicating links between TMS 4:5 and TMS 10:11, respectively. We had earlier shown that similar to TMS 5, some of the TMS 11 mutant variants also display uncoupling between ATP catalysis and drug transport. Therefore, probably some mutations in TMS 5 (present study) and 11 result in conformational changes which limit the interaction between NBDs and TMDs via ICL 2 and 4. This in turn results in futile ATP hydrolysis which is uncoupled from the drug transport. Alternatively, residues in TMS 5 have also been seen to affect the kinetics of the coupling between ATPase activities; substrate binding and efflux activity. The exact mechanism of drug transport remains speculative at this point till we have 3D structural details of CaCdr1p. These aspects are actively being pursued as second phase of work.
Supplementary Material
Acknowledgments
We thank R. D Cannon for providing us with the plasmid and the strains. We further thank Ranbaxy Laboratories Limited, India, for providing fluconazole. We thank Pankaj Pandotra for his help in FACS analyses and Shahid Jameel and Charu Tanwar for the confocal pictures.
Funding
The work presented in this paper has been supported in parts by grants to R.P. from the Department of Biotechnology, (DBT/PR4862/BRB/10/360/2004), Council of Scientific and Industrial Research (38(1122)/06/EMR-II, 22/3/2006), Department of Science and Technology (SR/SO/BB-12/2004, 5/9/2005) and Indo–French Centre for the Promotion of Advanced Research (IFC/A/3403-2/2006). S.S. and S.V.A. are supported by the intramural research program of the National Cancer Institute, NIH, and Center for Cancer Research. N.P. and M.G. acknowledge Indian Council of Medical Research, India and University Grants Commission, India, respectively for Junior and Senior Research fellowships.
References
- 1.Walmsley MB, Mckeegan KS, Walmsley AR. Structure and function in efflux pumps that confer resistance to drugs. Biochem J. 2003;376:313. doi: 10.1042/BJ20020957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prasad R, Worgifosse PD, Goffeau A, Balzi E. Molecular cloning and characterisation of a novel gene of C. albicans, CDR1, conferring multiple resistance to drugs and antifungals. Curr Genet. 1995;27:320. doi: 10.1007/BF00352101. [DOI] [PubMed] [Google Scholar]
- 3.Sanglard D, Ischer F, Monod M, Bille J. Cloning of Candida albicans genes conferring resistance to azole antifungal agents: Characterisation of CDR2, a new multidrug ABC transporter gene. Microbiology. 1997;143:405. doi: 10.1099/00221287-143-2-405. [DOI] [PubMed] [Google Scholar]
- 4.Hernaez ML, Gil C, Pla J, Nombela C. Induced expression of the Candida albicans multidrug resistance gene CDR1 in response to fluconazole and other antifungals. Yeast. 1998;14:517. doi: 10.1002/(SICI)1097-0061(19980430)14:6<517::AID-YEA250>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 5.White TC. Antifungal drug resistance in Candida albicans. ASM News. 1997;63:427. [Google Scholar]
- 6.Ben-Yaacov R, Knoller S, Caldwell GA, Becker JM, Koltin Y. Candida albicans gene encoding resistance to benomyl and methotrexate is a multidrug resistance gene. Antimicrob Agents Chemother. 1994;38:648. doi: 10.1128/aac.38.4.648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kohli AK, Gupta V, Krishnamurthy S, Hasnain SE, Prasad R. Specificity of drug transport mediated by CaMDR1: a major facilitator of Candida albicans. J Biosci. 2001;26:101. doi: 10.1007/BF02703742. [DOI] [PubMed] [Google Scholar]
- 8.Pasrija R, Banerjee D, Prasad R. Structure and function analysis of CaMdr1p, a MFS antifungal efflux transporter protein of Candida albicans: identification of amino acid residues critical for drug/H+ transport. Eukaryot Cell. 2007;6:443. doi: 10.1128/EC.00315-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Niimi M, Niimi K, Takano Y, Holmes AR, Fischer FJ, Uehara Y, Cannon RD. Regulated overexpression of CDR1 in Candida albicans confers multidrug resistance. J Antimicrob Chemother. 2004;54:999. doi: 10.1093/jac/dkh456. [DOI] [PubMed] [Google Scholar]
- 10.Gaur M, Choudhury D, Prasad R. Complete inventory of ABC proteins in human pathogenic yeast, Candida albicans. J Mol Microbiol Biotechnol. 2005;9:3. doi: 10.1159/000088141. [DOI] [PubMed] [Google Scholar]
- 11.Krishnamurthy S, Gupta V, Snehlata P, Prasad R. Characterisation of human steroid hormone transport mediated by Cdr1p, multidrug transporter of Candida albicans, belonging to the ATP binding cassette super family. FEMS Microbiol Lett. 1998;158:69. doi: 10.1111/j.1574-6968.1998.tb12802.x. [DOI] [PubMed] [Google Scholar]
- 12.Dogra S, Krishnamurthy S, Gupta V, Dixit BL, Gupta CM, Sanglard D, Prasad R. Asymmetric distribution of phosphatidylethanolamine in C. albicans: possible mediation by CDR1, A multidrug transporter belonging to ATP binding cassette (ABC) superfamily. Yeast. 1999;15:111. doi: 10.1002/(SICI)1097-0061(19990130)15:2<111::AID-YEA350>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 13.Saini P, Prasad T, Gaur NA, Shukla S, Jha S, Komath SS, Khan LA, Haq QM, Prasad R. Alanine scanning of transmembrane helix 11 of Cdr1p ABC antifungal efflux pump of Candida albicans: identification of amino acid residues critical for drug efflux. J Antimicrob Chemother. 2005;56:77. doi: 10.1093/jac/dki183. [DOI] [PubMed] [Google Scholar]
- 14.Shukla S, Rai V, Banerjee D, Prasad R. Characterization of Cdr1p, a major multidrug efflux protein of Candida albicans: purified protein is amenable to intrinsic fluorescence analysis. Biochemistry. 2006;45:2425. doi: 10.1021/bi0519147. [DOI] [PubMed] [Google Scholar]
- 15.Shukla S, Rai V, Saini P, Banerjee D, Menon AK, Prasad R. Candida Drug Resistance Protein 1, a Major Multidrug ATP Binding Cassette Transporter of Candida albicans, Translocates Fluorescent Phospholipids in a Reconstituted System. Biochemistry. 2007;46:12081. doi: 10.1021/bi700453e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Loo TW, Clarke DM. Do drug substrates enter the common drug-binding pocket of P-glycoprotein through “gates”? Biochem Biophys Res Commun. 2005;329:419. doi: 10.1016/j.bbrc.2005.01.134. [DOI] [PubMed] [Google Scholar]
- 17.Golin J, Ambudkar SV, Gottesman MM, Habib AD, Sczepanski J, Ziccardi W, May L. Studies with novel Pdr5p substrates demonstrate a strong size dependence for xenobiotic efflux. J Biol Chem. 2003;278:5963. doi: 10.1074/jbc.M210908200. [DOI] [PubMed] [Google Scholar]
- 18.Egner R, Rosenthal FE, Kralli A, Sanglard D, Kuchler K. Genetic separation of FK506 susceptibility and drug transport in the yeast Pdr5 ATP-binding cassette multidrug resistance transporter. Mol Biol Cell. 1998;9:523. doi: 10.1091/mbc.9.2.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Egner R, Bauer BE, Kuchler K. The transmembrane domain 10 of the yeast Pdr5p ABC antifungal efflux pump determines the substrate specificity and inhibitor susceptibility. Mol Microbiol. 2000;35:1255. doi: 10.1046/j.1365-2958.2000.01798.x. [DOI] [PubMed] [Google Scholar]
- 20.Tutulan-Cunita AC, Mikoshi M, Mizunuma M, Hirata D, Miyakawa T. Mutational analysis of the yeast multidrug resistance ABC transporter Pdr5p with altered drug specificity. Genes Cells. 2005;10:409. doi: 10.1111/j.1365-2443.2005.00847.x. [DOI] [PubMed] [Google Scholar]
- 21.Shukla S, Saini P, Smriti, Jha S, Ambudkar SV, Prasad R. Functional characterization of Candida albicans ABC transporter Cdr1p. Eukaryot Cell. 2003;2:1361. doi: 10.1128/EC.2.6.1361-1375.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mukhopadhyay K, Kohli A, Prasad R. Drug susceptibilities of yeast cells are affected by membrane lipid composition. Antimicrob Agents Chemother. 2002;46:3695. doi: 10.1128/AAC.46.12.3695-3705.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sarkadi B, Price EM, Boucher RC, Germann UA, Scarborough GA, Niederweis M. Expression of the human multidrug resistance cDNA in insect cells generates a high activity drug-stimulated membrane ATPase. J Biol Chem. 1992;267:4854. [PubMed] [Google Scholar]
- 24.Maesaki S, Marichal P, Bossche HV, Sanglard D, Kohno S. Rhodamine 6G efflux for the detection of CDR1-overexpressing azole-resistant Candida albicans strains. J Antimicrob Chemother. 1999;44:27. doi: 10.1093/jac/44.1.27. [DOI] [PubMed] [Google Scholar]
- 25.Prasad T, Saini P, Gaur NA, Vishwakarma RA, Khan LA, Haq QM, Prasad R. Functional analysis of CaIPT1, a sphingolipid biosynthetic gene involved in multidrug resistance and morphogenesis of Candida albicans. Antimicrob Agents Chemother. 2005;49:3442. doi: 10.1128/AAC.49.8.3442-3452.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakamura K, Niimi M, Niimi K, Holmes AR, Yates JE, Decottignies A, Monk BC, Goffeau A, Cannon RD. Functional expression of Candida albicans drug efflux pump Cdr1p in a Saccharomyces cerevisiae strain deficient in membrane transporters. Antimicrob Agents Chemother. 2001;45:3366. doi: 10.1128/AAC.45.12.3366-3374.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Decottignies A, Grant AM, Nichols JW, De Wet H, McIntosh DB, Goffeau A. ATPase and multidrug transport activities of the overexpressed yeast ABC protein Yor1p. J Biol Chem. 1998;273:12612. doi: 10.1074/jbc.273.20.12612. [DOI] [PubMed] [Google Scholar]
- 28.Sanglard D, Ischer F, Monod M, Bille J. Susceptibilities of Candida albicans multidrug transporter mutants to various antifungal agents and other metabolic inhibitors. Antimicrob Agents Chemother. 1996;40:2300. doi: 10.1128/aac.40.10.2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Safa AR. Identification and characterization of the binding sites of P-glycoprotein for multidrug resistance-related drugs and modulators. Curr Med Chem Anticancer Agents. 2004;4:1. doi: 10.2174/1568011043482142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dawson RJ, Locher KP. Structure of a bacterial multidrug ABC transporter. Nature. 2006;443:180. doi: 10.1038/nature05155. [DOI] [PubMed] [Google Scholar]
- 31.Currier SJ, Kane SE, Willingham MC, Cardarelli CO, Pastan I, Gottesman MM. Identification of residues in the first cytoplasmic loop of P-glycoprotein involved in the function of chimeric human MDR1-MDR2 transporters. J Biol Chem. 1992;267:25153. [PubMed] [Google Scholar]
- 32.Cotten JF, Ostedgaard LS, Carson MR, Welsh MJ. Effect of cystic fibrosis-associated mutations in the fourth intracellular loop of cystic fibrosis transmembrane conductance regulator. J Biol Chem. 1996;271:21279. doi: 10.1074/jbc.271.35.21279. [DOI] [PubMed] [Google Scholar]
- 33.Pagant S, Brovman EY, Halliday JJ, Miller EA. Mapping of interdomain interfaces required for the functional architecture of Yor1p, a eukaryotic ATP-binding cassette (ABC) transporter. J Biol Chem. 2008;283:26444. doi: 10.1074/jbc.M803912200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sauna ZE, Bohn SS, Rutledge R, Dougherty MP, Cronin S, May L, Xia D, Ambudkar SV, Golin J. Mutations define cross-talk between the N-terminal nucleotide-binding domain and transmembrane helix-2 of the yeast multidrug transporter Pdr5: possible conservation of a signaling interface for coupling ATP hydrolysis to drug transport. J Biol Chem. 2008;283:35010. doi: 10.1074/jbc.M806446200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dawson RJ, Hollenstein K, Locher KP. Uptake or extrusion: crystal structures of full ABC transporters suggest a common mechanism. Mol Microbiol. 2007;65:250. doi: 10.1111/j.1365-2958.2007.05792.x. [DOI] [PubMed] [Google Scholar]
- 36.Tusnady GE, Simon I. The HMMTOP transmembrane topology prediction server. Bioinformatics. 2001;17:849. doi: 10.1093/bioinformatics/17.9.849. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.