

Abstract
Inferring the gene content of ancestral genomes is a fundamental challenge in molecular evolution. Due to the statistical nature of this problem, ancestral genomes inferred by the maximum likelihood (ML) or the maximum-parsimony (MP) methods are prone to considerable error rates. In general, these errors are difficult to abolish by using longer genomic sequences or by analyzing more taxa. This study describes a new approach for improving ancestral genome reconstruction, the ancestral coevolver (ACE), which utilizes coevolutionary information to improve the accuracy of such reconstructions over previous approaches. The principal idea is to reduce the potentially large solution space by choosing a single optimal (or near optimal) solution that is in accord with the coevolutionary relationships between protein families. Simulation experiments, both on artificial and real biological data, show that ACE yields a marked decrease in error rate compared with ML or MP. Applied to a large data set (95 organisms, 4873 protein families, and 10,000 coevolutionary relationships), some of the ancestral genomes reconstructed by ACE were remarkably different in their gene content from those reconstructed by ML or MP alone (more than 10% in some nodes). These reconstructions, while having almost similar likelihood/parsimony scores as those obtained with ML/MP, had markedly higher concordance with the coevolutionary information. Specifically, when ACE was implemented to improve the results of ML, it added a large number of proteins to those encoded by LUCA (last universal common ancestor), most of them ribosomal proteins and components of the F0F1-type ATP synthase/ATPases, complexes that are vital in most living organisms. Our analysis suggests that LUCA appears to have been bacterial-like and had a genome size similar to the genome sizes of many extant organisms.
The problem of reconstructing ancestral states is as old as the field of molecular evolution, pioneered by Fitch around 40 yr ago (Fitch 1971). This first algorithm assumed a binary alphabet and was based on the maximum parsimony (MP) criterion, i.e., find the labels to the internal nodes of a tree that minimize the number of changes or mutations along the tree edges. Over the years this basic algorithm was generalized in many ways. Sankoff (1975) showed how to efficiently solve versions of the MP problem with a nonbinary alphabet and with multiple edge weights. Algorithms for inferring ancestral sequences based on the maximum likelihood (ML) principle (instead of MP) were suggested more than 15 yr later (Barry and Hartigan 1987; Felsenstein 1993; Pagel 1999; Pupko et al. 2000; Krishnan et al. 2004; Elias and Tuller 2007), aiming to identify the ancestral sequences that are most likely, given the current data in probabilistic terms. In a manner analogous to inferring an ancestral sequence such as a protein or gene (Pupko et al. 2000; Blanchette et al. 2004; Ma et al. 2006), many studies have aimed to reconstruct ancestral genomes (genomic content or gene content; e.g., see Boussau et al. 2004; Ouzounis et al. 2006; Putnam et al. 2007). Most existing phylogenetic reconstruction algorithms can be adapted to reconstructing ancestral gene content (e.g., see Felsenstein 1993; Yang 1997; Swofford 2002).
The main problem related to reconstructing ancestral sequences and gene inventories is that, in practice, the reconstructed sequences often contain a large number of errors. A major source of this phenomenon is the existence of multiple local and/or global maxima (i.e., solutions that are different but have the same scores of “goodness”) in the solution space searched by both the ML and the MP approaches (e.g., see Fig. 1; Chor et al. 2000). Furthermore, due to the statistical nature of the problem, and as both ML/MP assume that different sites and different genes/proteins evolve independently, increasing the amount of information used (the lengths of the sequences and the number of organisms) (Li et al. 2008) does not guarantee a decrease in the error rate. Thus, in many cases, the confidence that we may assign to the most likely or most parsimonious reconstructed ancestral state is not very high.
Figure 1.

A simple example demonstrating how coevolution can be used to improve ancestral sequence reconstruction. This example includes a coevolving forest with two trees, the tree edges are solid lines, while the coevolutionary edges are dashed wavy arrows; the ancestral states (e.g., the labels at the internal nodes x1 and y1) are smaller, while the known nonancestral states are larger and in italics (for formal definitions, see Supplementary Note 1). (A) The reconstructed ancestral states of two proteins (protein x and protein y); for protein x there are two parsimonious solutions: In one solution, all of the labels of the three internal nodes, t1, t2, t3, are “0”; in the second solution, all of the labels of the three internal nodes are “1.” (B) By taking into consideration the coevolution of protein x with protein y, the “all ‘1’ ” solution is chosen for x, thus resolving the ambiguity in the labels of x by using information about protein y with which it interacts, and which has less ambiguous labels.
In this work we describe a novel approach for improving the accuracy of reconstructed ancestral genomes. Our approach is based on utilizing information embedded in the coevolution of interacting proteins. The potential utility of this strategy is intuitively simple: If the ancestral reconstruction of protein A is ambiguous (e.g., different solutions achieve similar scores), but the ancestral reconstruction of protein B is unambiguous, then, knowing that A and B have coevolved (usually due to some functional interaction) can help us disambiguate the ancestral reconstruction of A. A simple illustration of this idea is described in Figure 1. As demonstrated further on, our approach can successfully distinguish between solutions with similar parsimony/likelihood scores.
Importantly, coevolutionary relations between proteins are quite ubiquitous and have been traced and reported in numerous studies (Pazos et al. 1997; Chen and Dokholyan 2006; Marino-Ramirez et al. 2006; Barker et al. 2007; Wapinski et al. 2007; Felder and Tuller 2008; Tuller et al. 2009). One such source is protein–protein interactions (PPIs): Several studies have already demonstrated a link between coevolution and physical interactions. These investigations used the fact that interacting proteins tend to coevolve in order to successfully predict physical interactions (e.g., see Wu et al. 2003; Sato et al. 2005; Juan et al. 2008). Here, we take the opposite approach and use data on physical interactions between proteins as a proxy for their coevolution. Large-scale PPI datasets currently exist only for relatively few organisms, but as physical interactions tend to be conserved across species, at least to some extent, (e.g., see Wu et al. 2003; Liang et al. 2006; Hirsh and Sharan 2007), it is plausible to assume that existing PPIs testify to coevolutionary relations across the ancestral tree. Another source of coevolution information that we utilize is metabolic adjacency: Following the studies of Spirin et al. (2006) and Zhao et al. (2007) we assume that metabolic enzymes that catalyze consecutive reactions in the same pathway tend to coevolve. We also incorporate additional sources of coevolution information, including genomic proximity, coexpression, and gene fusion, which have all been shown to be good predictors of coevolution (Lee et al. 2004; Chen and Dokholyan 2006; Jensen et al. 2009; Tuller et al. 2009). The coevolution data from all of these sources is combined to reconstruct the evolutionary history of 95 unicellular bacteria, archaea, and eukaryotes.
Results
The ancestral coevolver algorithm (ACE)
The ancestral coevolver method incorporates information about the coevolutionary relationships between pairs of proteins and the evolutionary distance between organisms (an evolutionary tree[s]) to improve the accuracy of ancestral protein content reconstruction. In this section we briefly describe our approach, model, and definitions. More formal definitions appear in Supplementary Note 1. For simplicity, we will relate similarly to proteins and the genes that encode them.
A phylogenetic tree is a rooted binary tree (i.e., the input degree of each node is one and the output degree is two) together with leaf labels. In this work, we assume that each node in a phylogenetic tree corresponds to a different organism (i.e., this is a species tree) and one can use this phylogenetic tree to describe the evolution of each protein family. In the simplest binary case, each label is either “0” (the gene does not appear in the genome of the organism) or “1” (the protein is encoded in the genome of an organism). In the nonbinary case each label is a natural number that denotes the number of paralogous copies of a gene in the corresponding genome. The leaves in a phylogenetic tree correspond to extant species, while the internal nodes correspond to ancestral organisms.
A coevolving forest is a set of phylogenetic trees with identical topology that correspond to the same organisms, each tracking the evolution of a different gene/protein. The forest includes an additional set of coevolutionary edges that connect pairs of nodes, each node in an edge belonging to a different phylogenetic tree. Each such coevolutionary edge connects a pair of nodes that correspond to the same organism (Fig. 1). The edges that are part of the evolutionary tree (the standard evolutionary transition edges) are termed “tree edges” here. For example, Figure 1 illustrates a coevolving forest spanning two gene/protein trees (the coevolutionary edges are dashed wavy arrows, while the tree edges are continuous). In this work we assume that pairs of coevolutionary edges that connect the roots of two evolutionary trees are “inherited” by the other pairs of nodes of the evolutionary trees (as described in Fig. 1), i.e., our mathematical model is capable of dealing with cases where coevolutionary edges appear/disappear during evolution. However, overall, we assume that the events where new coevolutionary edges appear or disappear are relatively rare.
A coevolutionary forest additionally includes a weight table for each coevolutionary edge and each tree edge. These weight tables include the cost of each pair of labels at the two ends of the edge. In the case of tree edges, these weights reflect the probability of gaining/losing a copy number along the edge (Methods). For coevolutionary edges, these weights reflect the distribution of mutual occurrences of the labels of the nodes connected by the edge. As we discuss later, these tables can be further weighted to reflect our confidence in these two types of information (the evolutionary transitions vs. coevolution information).
Another basic term needed for the description of our method is the notion of a coevolutionary graph, which is an undirected graph that describes the coevolutionary relationships in the coevolutionary forest. In such a graph, each node corresponds to a tree in the coevolutionary forest, and two nodes are connected by an edge if there is at least one coevolutionary edge between their corresponding trees. For example, the coevolutionary graph corresponding to the coevolutionary forest in Figure 1A includes two nodes (as there are two evolutionary trees in the forest), and one edge that connects these nodes (as there are coevolutionary edges between the two evolutionary trees). A connected component in the coevolutionary forest is a subset of trees whose corresponding nodes in the coevolutionary graph induce a connected component (i.e., a set of nodes such that there is a path between each pair of nodes in the set. For example, in the coevolutionary forest that appears in Figure 1A the two trees induce a connected component). The coevolutionary edges used in this work are drawn from various sources of information: protein interactions, coexpression, proximity in the metabolic networks, and other measures of cofunctionality (see Methods). As it is not clear how to model the evolution of a network with many types of interactions, we decided to use the generalized MP as an objective function.
This leads us to the definition of the computational problem we are concerned with, the ancestral coevolution problem: Given a coevolutionary forest (a set of trees, a set of coevolutionary edges that connect pairs of nodes in these trees, and weight tables for each tree edge and each coevolutionary edge) we want to find labels for the internal nodes of all of the trees in the coevolutionary forest, such that the sum of the corresponding weights along all of the tree edges and the coevolutionary edges (the total cost) is minimal.
We designed an algorithm for solving this problem—the ancestral coevolver (ACE). The goal of ACE is to find labels for all of the nodes of all of the trees in a coevolutionary forest by optimizing the ancestral coevolution problem.
The sum of coevolutionary weights that are induced by choosing labels (a solution) for all of the internal nodes in a coevolutionary forest composes the coevolutionary score. In this work we normalize this score by dividing it by the minimal possible coevolutionary score. Similarly, the sum of tree weights that are induced by choosing labels (a solution) for all of the internal nodes in a coevolutionary forest is named the maximum likelihood or the maximum parsimony score (for a ML/MP problem, correspondingly). Also, in this case we normalized this score by dividing it by the minimal possible ML/MP score. By changing the relative weights assigned to the coevolutionary edges relative to those of the tree edges we can control the comparative influence of these two sources of information on the inferred labels. For sufficiently small weights of the coevolutionary edges, ACE will choose one of the solutions obtained by the common ML/MP approaches, solving the problem of multiple optima of ML/MP (the extreme case of ancestral coevolution problem without coevolutionary edges describes a conventional MP/ML problem). On the other hand, using very large coevolutionary weights will result in a solution that is mostly influenced by the coevolutionary relations (see Supplementary Notes 1 and 2; Methods). We name such weighting procedure as either ML/CE weighting or MP/CE weighting, (depending on the algorithm used).
In the next section we demonstrate three different ways for choosing the ML/CE weighting (see more details in Supplementary Notes 1, 2; Methods): The first is called the “conservative way”: choose small enough weights for the coevolutionary edges such that the solution will be one of the ML/MP solutions. This approach should be used if one strongly believes in the conventional ML/MP approach and the corresponding tree edge weight tables (this approach is demonstrated in section The Simple Parsimony Case: A Classical Example of Multiple Maxima). The second is called the “weighted average”: Choose the weights of the coevolutionary edges such that the weighted average of the coevolutionary score and the ML/MP score are optimized. If one has similar beliefs in the two sources of information, a simple average should be used; otherwise, a weighted average, reflecting the relative confidence in each of the information sources should be used (this approach is demonstrated in section The ML Case: A Detailed Biological Case Stud). The third approach is called the learning approach: In this case we “flip” a small fraction of the sites at the leaves and try to correct them by using the entire model. We chose the weights with the best performances (more details in the section The ACE Reconstructs Missing Values at the Leaves of the Evolutionary Tree Better Than ML/MP).
The general steps of the algorithm appear in Figure 2. The preprocessing steps required for generating the input to the ACE algorithm include (Fig. 2A): reconstructing a phylogenetic tree, inferring groups of orthologs, gathering functional/physical interactions between orthologs, and computing edge weights. Next, ACE is implemented (Fig. 2B). The algorithm has four main steps: (1) the coevolutionary forest is partitioned into smaller subforests; (2) an optimal labeling is inferred for each of these subforests; (3) the solutions are merged; and (4) a final step of greedy optimization is performed. A detailed description of the algorithm is presented in the Methods section, in Supplemental Figure 1, and in Supplementary Note 1. Note that an ancestral genome may include more genes than each of the genomes at the leaves (Supplementary Note 3).
Figure 2.

(A) The preprocessing step: generating an input for ACE. The input includes an evolutionary tree with tree edge weights as well as coevolutionary edges with corresponding weights (see more details in Methods). (B) The ACE algorithm has four main steps: (1) partitioning of the coevolutionary graph to smaller subgraphs, (2) finding the optimal ancestral states in these subgraph, (3) merging these subgraphs, (4) improving the solution greedily (see a detailed description in the Methods section and in Supplementary Note 1).
Here, we show an application of ACE within the MP framework. However, the optimization problem of inferring the ancestral states of a phylogenetic tree when the optimization criterion used is ML (e.g., see Pupko et al. 2000) under independently and identically distributed (i.i.d.) probabilistic models such as Jukes and Cantor (1969) (JC), Neyman (1971), or the model of Yang et al. (1995) can be formalized as a MP problem for a nonbinary alphabet with multiple edge weights (Sankoff 1975) (Supplementary Note 4). Thus, the ACE algorithm can be used for improving both ML and MP algorithms.
A comparative simulation study of ACE vs. MP/ML
To demonstrate the performance of ACE we designed a simulation incorporating coevolution, where the genes/proteins evolve along the corresponding evolutionary trees, but also coevolve with each other (Supplementary Note 5; Supplemental Fig. 2). We examined the performance of the ACE algorithm in comparison to the standard MP/ML approaches while varying the following parameters: (1) the amount of coevolutionary edges: the number of edges per node in the coevolutionary graph; (2) the introduction of errors in the weights of the tree edges and the coevolutionary graph; and (3) varying the probability of mutations along the tree edges (corresponding to the weights of the tree edges; Supplementary Note 5; Supplemental Fig. 2). The aims of the simulation are: (1) to demonstrate the robustness of the ACE algorithm to various parameters; (2) to compare the ACE approach to the conventional approaches (MP/ML) that do not use coevolutionary information (currently, there are no previous approaches that use coevolutionary information to infer genome content that can be compared to the ACE).
A summary of the simulation results is provided in Figure 3. First we investigated the effect of modifying coevolution levels, quantified by the mean number of edges per node in the coevolutionary graph. Figure 3 clearly demonstrates that ACE outperforms the MP/ML approaches, even at low levels of coevolution. Both the performance of ACE (Fig. 3A), as measured by the error rate (the frequency of ancestral states inferred incorrectly), and its superiority over the other approaches (Fig. 3C), increases with the level of coevolution. The error rate of ACE undergoes a decrease of more than 50% (to an error rate of just a few percent) when the level of coevolution increases from 0 to 5 (Fig. 3A). Note that this level of coevolution is reasonable in real biological terms: For example, there are more than five PPI interactions for each protein, on average, in the yeast PPI network (which includes 39,396 interactions and around 6400 open reading frames [ORFs]; see Methods). If we incorporate a number of sources of coevolutionary information (see above), the average degree in the resulting coevolutionary graph turns out to be much higher (e.g., the mean node degree [connections per protein] in the coevolutionary graph underlying the biological case analyzed in the next section was larger than 14).
Figure 3.

Summary of the simulation results, reporting the mean over 10 runs using coevolutionary forests with 100 trees, each with 20 leaves and branch lengths in the range of from 0.1 to 0.4. (A) The error rate (frequency of sites inferred with error, y-axis) vs. the level of coevolution (normalized by the number of trees, x-axis). The inset shows the typical behavior at very low levels of coevolution. (B) The error rate vs. errors in the weights of the tree edges. (C) Difference between the error rate of ACE and the error rate of MP or ML (in %) vs. level of coevolution. (D) The error rate vs. error in the coevolutionary graph, the latter denoting the mean number of coevolutionary edges changed between a node (representing an organism) and its descendant in the evolutionary trees; the ACE was based on the coevolutionary graph in one of the nodes. The inset shows the difference (in %) between the error rates vs. the error in coevolutionary edges. Note that the gap between ML and ACE decreases when the error in coevolutionary edges increases. (E) The error rate vs. mean weight of the tree edges. The inset shows the difference (in %). Note that the gap between ML/MP and ACE increases when the mean weight of the tree edges (the probability to gain/loss a protein) increases.
As can be seen in Figure 3B, ACE is more robust than the standard MP/ML approaches to errors in the weights of tree edges. ACE performs better at all error rates, but predominantly when the error level is particularly high. Moreover, ACE also performs better than MP/ML when coevolutionary edges disappear/appear in evolution (this process can also reflect errors in the topology of the assumed coevolutionary graph, another possible source of error) (Fig. 3D; D'haeseleer and Church 2004). Finally, Figure 3E shows that a higher probability of gene gain/loss events along the tree edges (Supplementary Note 5) improves the performance of ACE compared with the other methods. Thus, we conclude that ACE outperforms the other methods, especially in the presence of some unreliable/noisy data, as typically found in large biological datasets.
Applying ACE to biological data
Using ACE we reconstructed ancestral genomes from a large set of organisms from the three domains of life, Bacteria, Archaea, and Eukarya. Figure 2 summarizes the different steps in generating the biological inputs (Methods). We used the 95 genomes of unicellular organisms represented in the COG database (Tatusov et al. 2003; Jensen et al. 2009) (see Supplemental Fig. 3). We studied a binary case where “1” represents the presence of a gene/protein and “0” represents its absence. Using the 4873 groups of orthologs that appear in COG, we represented each of the 95 genomes by a binary string having a length of 4873 characters. We inferred the ancestral genomes by using two main versions of edge weights (Methods): (1) Binary MP; (2) binary ML (details about the results on additional versions of MP appear in Supplementary Notes 6 and 7 and Supplemental Fig. 4). In each case, we examined a range of ML/CE or MP/CE weightings and chose one of the MP/CE weightings according to the approaches mentioned before. As mentioned earlier, the two extreme cases are when the weight of the coevolutionary edges is very small relative to the tree edges and vice versa, when the weight of the tree edges is very small relative to the weight of the coevolutionary edges.
ACE reconstructs missing values at the leaves of the evolutionary tree better than ML/MP
At the first stage, to further demonstrate the advantages of ACE over conventional ML/MP we performed the following procedure:
We randomly flipped 3%–5% of the values with coevolutionary relations in 3%–5% of the genomes, from absence to presence, or vice versa.
We reconstructed the ancestral states of the coevolutionary forest based on the altered genomic contents.
We then “fixed” the values at the leaves that were flipped by choosing labels that optimize the score of the coevolutionary network given the states inferred in step 2.
Steps 1–3 were repeated seven times.
The mean error-rates of the inferred values at the leaves for the above procedure and for different MP/CE or ML/CE weightings, MP and ML, as well as for different percents of flipping are provided in Table 1.
Table 1.
Error rates for different ML/CE or MP/CE weightings, when used for correcting flipped labels at the leaves of the coevolutionary forest

aThe maximal decrease in the error rate.
bThe minimal error rate.
As can be seen, using the coevolutionary information reduces the error rate by more than 50%, where usually most of the improvement is achieved in the case of the second weighting (whose ML/MP score is also relatively high). This analysis further supports the use of coevolutionary information in addition to conventional ML/MP approaches. Additionally, as evident, the approach described here can be used for selecting an optimal ML/CE weighting.
The simple parsimony case: A classical example of multiple global maxima
To study the effect of integrating coevolution data on MP gene content reconstruction, we began with the simple parsimony case, in which the penalty for losing/gaining a protein is identical along all tree edges. In this case, there are many possible solutions, each having the same parsimony score. This case illustrates how ACE can be utilized for choosing one optimum by adding the coevolutionary biological constraints (see Fig. 4A). Out of all possible solutions, ACE chose one whose score (33,525) is identical to the best score obtained using the MP algorithm. However, the inclusion of coevolutionary constraints resulted in a biologically different solution: The number of inferred proteins was either similar or higher in most of the internal nodes, and ACE added 34 proteins to the LUCA (see Fig. 4B). As MP does not consider the fact that in some of the tree edges the probability to lose/gain a protein is low, it tends to remove ancestral proteins; ACE partially fixes this problem by adding proteins to most of the internal nodes. The function of the protein families inferred by ACE and not by MP in LUCA appear in Figure 4C. Gene families whose function is related to translation are overrepresented in this set (hypergeometric P-value = 1.97 × 10−5; Methods); gene families whose function are related to amino acid transport and metabolism are underrepresented in this set (hypergeometric P-value = 0.0065; Methods).
Figure 4.

(A) The coevolutionary score (dashed), the simple maximum parsimony score (continuous) for different MP/CE weightings. The scores are normalized to the minimal solution (i.e., “1” denotes a minimal/optimal solution): By definition, at the first MP/CE weighting, the simple parsimony score is optimal/minimal, while at the fifth MP/CE weighting the coevolutionary score is optimal/minimal. (B) The difference between the number of proteins that ACE inferred (using the first MP/CE weighting) and the number of proteins inferred by simple MP for each internal node. (C) The function of the protein families inferred by ACE and not by MP in LUCA. Gene families whose function is related to translation are overrepresented in this set (hypergeometric P-value = 1.97 × 10−5; Methods); gene families whose function is related to amino acid transport and methabolism are underrepresented in this set (hypergeometric P-value = 0.0065; Methods).
Similar results were obtained for the other version of the inputs (e.g., the nonbinary and nonsymmetric versions; see Methods; Supplementary Notes 6, 7; Supplemental Fig. 4), demonstrating that ACE can be successfully applied to different versions of the input. As is evident, in all the cases ACE added dozens of new protein families to LUCA, suggesting that common ML/MP approaches tend to underestimate the number of proteins in LUCA.
The ML case: A detailed case study
Figure 5 includes a summary of the results for the binary ML case. As can be seen in Figure 5A, the optimal labels inferred for each ML/CE weighting correspond to solutions that are very close (not more than 20% higher) both to the optimal coevolutionary solution and to the ML solution (for comparison, with 100 random labelings, the mean ML score is 1547% higher than the optimal score, and the mean coevolutionary score is 518% higher than the optimal coevolutionary score; P-value < 0.01 in both cases).
Figure 5.

(A) The coevolutionary score (solid line), the maximum likelihood score (long-dashed line), and their mean (short-dashed line) for different ML/CE weightings. In all of the ML/CE weightings the two scores are <120% higher than their optimum/minimum. The second ML/CE weighting optimizes the mean of these two scores (see short-dashed line). (B) The difference between the number of proteins inferred by ACE and the number of proteins inferred by maximum likelihood for each internal node, using the optimal weighting. (C) The function of the protein families inferred by ACE and not by ML in LUCA. Gene families whose function is related to translation or intracellular trafficking are overrepresented in this set (hypergeometric P-values = 8 × 10−5 and 0.02, respectively; Methods); gene families whose function is related to amino acid transport and metabolism and transcription are underrepresented in this set (hypergeometric P-value = 0.0001 and 0.038, respectively; Methods).
In the ML case, since there were many optima points with similar but not identical scores (as in the case of MP), the ACE solution for the first ML/CE weighting was identical to the ML solution. Thus, in this case, we examined the ML/CE weighting that optimized the sum of the two scores (short-dashed line in Fig. 5A). At a weighting level of 2, the two scores (CE and ML) are within 4% from their respective optima. This ML/CE weighting also seems the most appropriate for additional reasons: This ML/CE weighting reconstructed the gene content of LUCA to be an intermediate between the solution that only relies on coevolution and the ML solution (it adds 57 protein families to the ML reconstruction of LUCA, while the solution with extreme ML/CE weighting adds 98 to this reconstruction); thus, it represents a midway point between the two sources of information. Second, remarkably, this weighting gave the largest reduction of the error rate at the leaves of the biological data tree (see previous section).
Figure 5B depicts the difference between the number of proteins inferred by ACE and the number of proteins inferred by ML in each internal node of the evolutionary tree (see Supplemental Fig. 3). As evident, ACE added proteins to some of the nodes (e.g., LUCA and the ancestor of the Fungi), removed proteins from others (e.g., the subtree of the Archeae and the Firmicutes), and kept some nodes unchanged (e.g., the ancestor of the Cyanobacteria and the ancestor of the alpha-protobacteria).
We turned to analyze the reconstructed gene content of LUCA, which has been the subject of much controversy in recent years (see Glansdorff et al. 2008 for a recent survey of about 200 papers studying LUCA, e.g., Penny and Poole 1999; Koonin 2003). One major open question concerning LUCA is the size of its genome. ACE finds a total of 743 inferred proteins in LUCA, which is still less than the mean number of protein families in the extant organisms analyzed (1265), but is considerably higher than the number of proteins inferred by the conventional MP method (only 587) or the ML method (686). More specifically, the inferred genome size of LUCA was larger than the genomes of 16% (15/95) of the organisms in the analyzed data set. Koonin (2003) suggested that LUCA had 500–600 genes, less than the great majority (96%) of the organisms in the analyzed data set, while Ouzounis et al. (2006) suggested that LUCA had a relatively rich genome. Our results are more in line with the suggestions of Ouzounis et al. (2006), namely, that LUCA had a relatively rich genome that resembles the gene contents of extant organisms.
Fifty-seven gene families were added to LUCA by ACE compared with ML (Supplemental Table 1). Figure 5C depicts the functionality of the new proteins found by ACE. Functions that are underrepresented in the set of new proteins added by ACE are related to “amino acid transport and metabolism” and “transcription.” On the other hand, most of the new protein families are related to “energy production” and to “translation and ribosome structure”; gene families from this last category are overrepresented in this set (hypergeometric P-value = 8 × 10−5; Methods). The proteins from the first group are mainly composed of components of the F0F1-type ATP synthase, and the proteins from the second group are mainly ribosomal proteins. Interestingly, while F0F1-type ATP synthases are presently found primarily in aerobic bacteria and mitochondria, they are believed to be descended from ancient anaerobic enzymes, and thus their antiquity is highly plausible (Cross and Muller 2004). Strikingly, the additional ribosomal proteins that are conserved in extant bacteria, but absent in archaea and eukaryotes, were inferred to be encoded by LUCA. This may imply a bacterial-like LUCA from which archaea later diverged (Gogarten et al. 1989; Iwabe et al. 1989), rather than a parallel emergence of the two prokaryotic domains (Woese 1998). Furthermore, the presence of the methionyl-tRNA formyltransferase in LUCA implies that the bacterial-like ancestor already used formyl-methionine to initiate translation, and that this formylation was subsequently lost in Archaea and Eukarya as their translation system diverged from the bacterial one. Another interesting observation is that the preprotein translocase subunits SecF and SecD, which are absent in some bacteria and nonessential in others, (Wooldridge 2009) are predicted to be ancestral, as is the YidC translocase subunit, which is involved in integration of membrane proteins (Serek et al. 2004).
Analysis under extreme ML/CE weightings
In this subsection we repeated the analysis reported in the previous section for the case where the ML/CE weighting is maximal (i.e., the weights of the coevolutionary tables are dominant). In this case, ACE finds a total of 784 inferred proteins in LUCA, more than have been reported in the previous cases; 98 (14%) of the proteins are added to the result of the conventional ML; see details in Supplemental Figure 5.
In this case, the prominent functions overlap those that have been reported in the previous subsection: Proteins related to translation are overrepresented in both cases, while protein families related to metabolism are underrepresented in both cases, showing that our previous conclusions are robust to the ML/CO weighting. A few additional proteins unique to the extreme weighting are nevertheless notable. These include, for example, several proteins involved in the synthesis of the bacterial peptidoglycan cell wall and many central proteins of the DNA replication and DNA repair machineries, including DNA polymerases, ligases, helicases, and a single-strand binding protein. Thus, if one were inclined to trust the extreme ML/CE weighting, one would conclude that LUCA already possessed advanced bacterial-like surface components and robust DNA repair and replication machineries. These conclusions further reinforce the view of LUCA as an organism not unlike extant bacterial species, both in the number of genes and in its physiological capabilities.
Discussion
In this study, we describe a new approach for reconstructing ancestral genomes. Our approach captures coevolutionary dependencies between different proteins and uses this information to disambiguate the gene contents of the reconstructed ancestral genomic contents. ACE is geared to tackle an inherent problem that plagues the MP and ML approaches, whose solution space tends to be populated with multiple maxima (e.g., see Fig. 1; Chor et al. 2000). We demonstrate the superiority of the new approach over existing methods in a simulated scenario where coevolutionary information is available: Using ACE with biologically plausible levels of coevolutionary information reduces the error rate of the ancestral genome reconstruction by more than 50%.
When ACE was applied to the study of ancestral genomes, our analysis showed that it can find solutions whose likelihood/parsimony score is very similar or identical to the ML/MP scores. These solutions, however, are often significantly different from the ML/MP solutions and, since they incorporate coevolution data, are more plausible from a biological standpoint. Accordingly, ACE finds dozens of additional proteins in LUCA, many of which are ribosomal proteins or part of the ATP synthase, complexes that are essential for life.
Our coevolution-based approach is presented here in the framework of ancestral genome reconstruction due to its importance for evolutionary biology and because coevolutionary information can be readily obtained at the gene/protein level. However, its potential scope goes far beyond inferring ancestral gene content: ACE can be used to tackle any general problem of ancestral sequence reconstruction, including the reconstruction of different sites or domains in proteins, or the reconstruction of individual sites in DNA or RNA sequences (see Noller and Woese 1981; Gutell et al. 1986; Knudsen and Hein 1999; Lockless and Ranganathan 1999; Pedersen et al. 2006; Yeang and Haussler 2007; Yeang et al. 2007). The success of such future applications depends on the existence of reliable coevolutionary information at the position/site level. Coevolution information may be obtained from proximity in the three-dimensional structure of the protein itself, from information on coevolution of amino acids sharing specific binding site(s) or information about binding sites shared between interacting proteins. Finally, as already partially demonstrated here, the ACE approach can be generalized in the future to more complex reconstruction models, e.g., using nonbinary alphabets, dependency between adjacent sites, and various probabilistic models (Akerborg et al. 2009).
Additionally, we intend to design algorithms (e.g., algorithms that are based on the belief propagation approach) that may improve the accuracy and running time of the basic ACE algorithm described in this work.
Methods
Coevolutionary information
The coevolutionary edges used here were gathered from three sources of information: (1) Proximity in the protein interaction network of E. coli and S. cerevisiae (for example, Wu et al. [2003], Sato et al. [2005], and Juan et al. [2008] reported a relation between coevolution and protein interactions). (2) Proximity in the metabolic networks of the analyzed organisms (for example, Spirin et al. [2006] and Zhao et al. [2007] reported the relation between coevolution and proximity in metabolic networks). (3) Various physical and functional interactions that were downloaded from String (Jensen et al. 2009) (http://string.embl.de/; for example, Chen and Dokholyan [2006] and Tuller et al. [2009] reported the relation between coevolution and similar functionality).
A coevolutionary edge was added to the model if the following two conditions were satisfied: (1) The corresponding pair of orthologs physically or functionally interact according to the three sources of information described above (see more details below). (2) The two orthologs exhibit a pattern of co-occurrence that is significantly different from random in the genome of the organisms studied (in our case, the ratio between the highest and lowest probability in the co-occurrence distribution table is at least 4.25). The initial set of coevolutionary edges included more than 70,000 edges, and after the filtering it included 10,000 edges.
The weights in the tables of the coevolutionary edges were computed according to the co-occurrence probabilities of the corresponding pairs of proteins.
The following subsections include more details about the three sources of coevolutionary information used in this work.
The protein interaction networks of S. cerevisiae and E. coli
The protein interaction network of S. cerevisiae was obtained from recently published manuscripts (Gavin et al. 2006; Krogan et al. 2006; Reguly et al. 2006) and from public databases (Xenarios et al. 2002; Christie et al. 2004). High-throughput mass spectrometry data (Gavin et al. 2006; Krogan et al. 2006) was translated into binary protein–protein interactions using the spoke model (Bader and Hogue 2002). The final yeast protein–protein network included 39,396 interactions. The E. coli network was generated based on Mori et al. (2000), Xenarios et al. (2002), and Arifuzzaman et al. (2006). The final E. coli protein–protein network included 16,756 interactions. As coevolutionary edges, we only considered edges that appeared in the two protein interaction networks. E. coli and S. cerevisiae are the only organisms in our data set whose PPI networks have been reconstructed on a large scale. These interactions were mapped to the corresponding COGs by mapping to E. coli genes. In the final set of coevolutionary edges, 256 were based on this source of information.
The metabolic networks of the organisms analyzed
We used the representation of Ma and Zeng (2003) for constructing the metabolic networks, i.e., two enzymes are connected with an edge if they catalyze successive (or the same) steps in a metabolic pathway (see the Introduction for the motivation for using adjacency as evidence for coevolution). Each metabolic network was reconstructed by the following stages: First we parsed all data pertaining reactions, compounds, and enzymes from KEGG release 46 (Kanehisa 2002) and created a list of the existing enzymes in each species in our collection, the reactions they catalyze, the reaction products and substrates, and their directionality.
The metabolic network of each organism was generated from its list of enzymes as follows: Each enzyme is represented as a node in the network. Let E1 = [e11, e11,.., e1n] denote the set of enzymes that catalyze reaction R1, and E2 = [e21, e21,.., e2n] denote the set of enzymes that catalyze reaction R2. If a product of R1 is a substrate of R2, then undirected edges are assigned between all nodes of E1 and all nodes of E2. Edges are also assigned within E1 nodes and within E2 nodes. As coevolutionary edges, we considered enzymes that are connected in at least 10% of the metabolic networks in KEGG that are larger than 50% of the E. coli's metabolic network. The set of enzymes were mapped to COG groups by mapping to E. coli and S. cerevisiae genes (these data were downloaded from KEGG) (Kanehisa 2002). In the final set of coevolutionary edges, 250 were based on this source of information.
Other types of cofunctionality (the String database)
As an additional source of coevolutionary edges we used information downloaded from the String database (Jensen et al. 2009). This database includes various sources of coevolutionary/functional relations that are different from the two types of information mentioned above. Specifically, it includes information on genomic neighborhood, coexpression, gene fusion, coexpression, and more. Each coevolutionary relation in this data set was based on a composite score that is a weighted average of these sources of information (more details about the different component of a score were not available). Most of the edges (98%) in the final file set of coevolutionary edges were based on this source of information.
The phylogenetic tree
The phylogenetic tree is based on the following sources of information. The subtree related to alpha-proteobacteria was downloaded from Boussau et al. (2004). The subtree related to Cyanobacteria was downloaded from Shi and Falkowski (2008). The subtree related to fungi was downloaded from Wapinski et al. (2007). The reconstruction of other parts of the tree and the merging of all these trees were based on maximum likelihood (Dagan and Martin 2007) and iTOL (Letunic and Bork 2007). The final phylogenetic tree included 95 organisms from the three domains of life: eukaryotes (18 organisms), prokaryotes (65 organisms), and archaea (12 organisms). The list of organism names and their taxa i.d. appear in Supplemental Table 2.
We used Neyman's two-state model (Neyman 1971), a version of Jukes Cantor (JC) model (Jukes and Cantor 1969) for inferring the edge lengths of the tree by maximum likelihood. This was done by PAML (Yang 1997). These edge lengths correspond to the probabilities that a protein family will appear/vanish along the corresponding lineage.
Clusters of orthologs
Clusters of orthologs were gathered from various sources. COG mapping to most of the organisms (83 out of 95) were downloaded from the String database (Jensen et al. 2009). Some of the fungi and the cyanobacteria (see Supplemental Table 2) do not appear in the String database; the clusters in the missing fungi were generated in the following way: (1) We downloaded the data set of fungal clusters of orthologs from Wapinski et al. (2007) and the cluster of cyanobacterial orthologs from Shi and Falkowski (2008). (2) We considered the fungi/cyanobacteria that appear in the COG data set and mapped each fungal/cyanobacterial cluster to the COG that has at least 90% overlap with it. (3) After the mapping, we reconstructed ortholog clusters in the organism that are missing in COG from their corresponding cluster in the fungal/cyanobacterial data set. The smallest genome (M. genitalium) in the analyzed data set included 393 genes (sum of copy numbers from each COG family), while the largest genome (B. japonicum) included 6162 genes (Supplemental Table 3 includes the COGs distributions in all the analyzed genomes).
Annotation of ancestral proteins and enrichment P-values
The annotation of ancestral proteins was based on COG annotations (Tatusov et al. 2003) and the Gene Ontology (GO) annotations of S. cerevisiae and E. coli (http://www.geneontology.org/index.shtml).
Enrichment P-values for the proteins that were added by the ACE were computed according to the COG annotations. For each function, we computed an hypergeometric P-value based on the total number of COG families that have this function in LUCA, the total number of COG families added by the ACE, and the number of COG families with this function that were added by the ACE.
The ancestral coevolver (ACE) algorithm
The input to ACE is a set of phylogenetic trees (a tree for each protein) with the same topology and with coevolutionary edges between pairs of internal nodes of the trees (corresponding to pairs of protein that coevolve). ACE has three main steps (Fig. 2; Supplemental Fig. 1): (1) By removing some of the coevolutionary edges, the input (set of trees and edges between them) is partitioned into smaller groups of trees such that there are edges only between pairs of trees that are in the same group. (2) Optimal labels (not considering the removed edges) are assigned to the internal nodes of each of these smaller coevolutionary forests by an algorithm that is a generalization of algorithms for finding ancestral states by maximum likelihood or maximum parsimony (Fitch 1971; Sankoff 1975; Pupko et al. 2000); in total, these labels are an approximate solution for the input coevolutionary forest. (3) Finally, the solution is further improved in a greedy manner.
We begin by briefly describing stage 2 of the algorithm and then explain why stage 1 is necessary. Similarly to many algorithms for computing the optimal labels of internal tree nodes (by MP or ML criterion) (Fitch 1971; Sankoff 1975; Pupko et al. 2000), our algorithm has two phases: In the first phase, it traverses the coevolutionary forest from the leaves to the root; in the second phase, it traverses the coevolutionary forest from the root to the leaves.
However, our algorithm is performed jointly for all the trees in each connected component.
Let e = (i, j) denote a coevolutionary edge or a tree edge. Let  denote the cost corresponding to assigning α at node i, and β at node j where (i, j) is a coevolutionary edge. Similarly, if e = (i, j) is a tree edge, we use
denote the cost corresponding to assigning α at node i, and β at node j where (i, j) is a coevolutionary edge. Similarly, if e = (i, j) is a tree edge, we use  to denote the cost of assigning α and β at the two nodes of e.
to denote the cost of assigning α and β at the two nodes of e.
Let 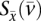 denote the optimal cost (the minimal sum of weights) of the sub-coevolutionary forest that corresponds to the coevolutionary subforest whose roots are
denote the optimal cost (the minimal sum of weights) of the sub-coevolutionary forest that corresponds to the coevolutionary subforest whose roots are  , such that all the nodes in such a vector of roots correspond to the same ancestral organism in the phylogenetic trees, and when assigning
, such that all the nodes in such a vector of roots correspond to the same ancestral organism in the phylogenetic trees, and when assigning  in these roots. For example, consider Figure 1, the optimal cost when considering the coevolutionary forest whose roots are
in these roots. For example, consider Figure 1, the optimal cost when considering the coevolutionary forest whose roots are 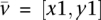 , and when assigning
, and when assigning 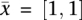 in these roots is 0.51 (due to the coevolutionary edge).
in these roots is 0.51 (due to the coevolutionary edge).
In the first phase, the algorithm traverses the coevolutionary forest from the leaves to the root and computes the cost for each set of internal nodes, , such that all of these nodes correspond to the same ancestral organism in all of the phylogenetic trees, after the costs of the two sets of internal nodes that are descendant of were computed (see Supplemental Fig. 1 B; Supplementary Note 1). In the second phase, the algorithm traverses the subforest from the roots to the leaves, and chooses optimal labels for each set of internal nodes, given the optimal labels of its corresponding set of parents (see Supplemental Fig. 1B; Supplementary Note 1).
As the running time of the algorithm is exponential with the size of the coevolutionary forest, the algorithm has an initial stage (stage 1), where the input graph is partitioned into small enough connected components. The algorithm used at this stage, Partite, recursively clusters the trees to groups according to the edge weights between them (thus minimizing the number of edges between different clusters), such that the number of trees in each cluster is small enough (less than a predetermined parameter K; Supplemental Fig. 1C; Supplementary Note 8). We used hierarchical k-means (MacQueen 1967) for clustering (but obviously other clustering algorithms can be used too).
The input to the Partite algorithm is a weighted graph whose edges correspond to the edges in the coevolutionary graph. The weights of the graph edges can be any measure that represents that strength of the coevolution between the corresponding proteins (trees/subtrees) in a certain part of the evolution. We used the ratio between the maximal and the minimal values of the edges in the weight tables. The clustering parameter K induces a trade-off between accuracy and speed: larger maximal cluster/subgraph (K) increases the running time, but also increases the accuracy of the solution.
The final stage of the ancestral coevolver algorithm is a greedy stage (Supplemental Fig. 1F; Supplementary Note 1). In each step of the greedy algorithm, it considers all of the edges; then, it chooses an edge (tree edge or coevolutionary edge) and new labels to its ends in a way that gives improvement of the cost of the coevolutionary forest. In each step, the new labels are updated; the algorithm stops when it does not find new labels that improve the cost of the coevolutionary forest.
The total running time of the algorithm is exponential with the size of the largest connected component, but behaves quite linearly with the other parameters (including the number of trees). For detailed explanations see Supplementary Note 1. The typical running time for an input with several hundred trees and largest connected component of size eight trees is up to a few minutes. For inputs with several thousand trees and largest connected component of size 9, the running time is a few hours. So, as long as one takes care to partition the coevolutionary graph to connected components of sufficiently small size in the preprocessing phase (which can always be done by a corresponding choice of a clustering algorithm), the algorithm can be run within a reasonable time frame.
The models of evolution used in the analysis of the biological inputs
We checked several models for the weights of the tree edges. In the binary parsimony case, there are two possible states for each character (i.e., for each COG): “0” denotes that the COG is not encoded in a genome, and “1” denotes that the COG is encoded in the genome. The penalty to switch from “1” to “0” and vice versa is identical (e.g., 1) in all the branches.
In the binary likelihood case, as in the binary parsimony case, there are two similar possible characters for each state (COG). However, in this case, each tree edge, (α, β), has a different length (gain/loss probability, pα,β) that induces a different weight table (see the previous section about how pα,β can be computed). The penalty (the corresponding entry in the weight matrix) for a change from “0” to “1” or vice versa, in this case, is −log(pα,β); if there is no change, the cost is −log(1−pα,β). These models appear in the main text. We decided to use models where the penalty for gain or loss of a gene is identical following the work of Mirkin et al. (2003), who showed that this is the most reasonable/suitable penalty in our context.
We also checked additional models whose results are reported in Supplemental Figure 4 and Supplementary Notes 6 and 7: In the nonbinary parsimony case, there are a few possible states for each character (COG). Each state is a positive integer that denotes the number of genes from a COG in a certain genome (note that only 6% of the states in the analyzed biological input are larger than “1”). Let Cx denote the number of genes from a COG C in node x. In the symmetric nonbinary case, the penalty for a change in the number of genes corresponding to a COG C along the tree edge (α,β) is −log(pα,β)*|Cα−Cβ|. In the nonbinary nonsymmetric case, the penalty for losing a gene from a COG C along the tree edge (α,β) where a is the ancestor of b is −2log(pα,β)*|Cα−Cβ|, while the penalty for gaining a COG is −log(pα,β)*|Cα−Cβ|.
In the nonbinary cases, we use binary coevolutionary edge weight tables (Cx > 0 or Cx = 0). This was done for two main reasons: (1) The coevolution between a pair of COGs usually corresponds to the relation(s) between any protein from one COG to any protein from the other COG. (2) Considering the limited number of analyzed organisms, a too-large coevolutionary table would include too many parameters (weights) that need to be estimated.
Weighting of the edge weights in the analysis of the biological inputs
In the biological analysis, we checked five specific values of the ML/CE weighting and MP/CE weighting. These values are described in Supplementary Note 2.
Acknowledgments
T.T. was supported by the Edmond J. Safra Bioinformatics program at Tel Aviv University and by the Yeshaya Horowitz Association through the Center for Complexity Science, and is currently a Koshland Scholar at the Weizmann Institute. U.G., M.K., and E.R. are supported by the James S. McDonnell Foundation. U.G. is also supported by the Bi-national Science Foundation. M.K. and E.R. were also supported by grants from the Israel Science Foundation and the Israeli Ministry of Science and Technology (M.K.).
Footnotes
[Supplemental material is available online at http://www.genome.org.]
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.096115.109.
References
- Akerborg O, Sennblad B, Arvestad L, Lagergren J. Simultaneous Bayesian gene tree reconstruction and reconciliation analysis. Proc Natl Acad Sci. 2009;106:5714–5719. doi: 10.1073/pnas.0806251106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arifuzzaman M, Maeda M, Itoh A, Nishikata K, Takita C, Saito R, Ara T, Nakahigashi K, Huang HC, Hirai A, et al. Large-scale identification of protein–protein interaction of Escherichia coli K-12. Genome Res. 2006;16:686–691. doi: 10.1101/gr.4527806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bader GD, Hogue CW. Analyzing yeast protein-protein interaction data obtained from different sources. Nat Biotechnol. 2002;20:991–997. doi: 10.1038/nbt1002-991. [DOI] [PubMed] [Google Scholar]
- Barker D, Meade A, Pagel M. Constrained models of evolution lead to improved prediction of functional linkage from correlated gain and loss of genes. Bioinformatics. 2007;23:14–20. doi: 10.1093/bioinformatics/btl558. [DOI] [PubMed] [Google Scholar]
- Barry D, Hartigan J. Statistical analysis of humanoid molecular evolution. Stat Sci. 1987;2:191–210. [Google Scholar]
- Blanchette M, Green ED, Miller W, Haussler D. Reconstructing large regions of an ancestral mammalian genome in silico. Genome Res. 2004;14:2412–2423. doi: 10.1101/gr.2800104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boussau B, Karlberg EO, Frank AC, Legault BA, Andersson SG. Computational inference of scenarios for alpha-proteobacterial genome evolution. Proc Natl Acad Sci. 2004;101:9722–9727. doi: 10.1073/pnas.0400975101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Dokholyan NV. The coordinated evolution of yeast proteins is constrained by functional modularity. Trends Genet. 2006;22:416–419. doi: 10.1016/j.tig.2006.06.008. [DOI] [PubMed] [Google Scholar]
- Chor B, Hendy MD, Holland BR, Penny D. Multiple maxima of likelihood in phylogenetic trees: An analytic approach. Mol Biol Evol. 2000;17:1529–1541. doi: 10.1093/oxfordjournals.molbev.a026252. [DOI] [PubMed] [Google Scholar]
- Christie KR, Weng S, Balakrishnan R, Costanzo MC, Dolinski K, Dwight SS, Engel SR, Feierbach B, Fisk DG, Hirschman JE, et al. Saccharomyces Genome Database (SGD) provides tools to identify and analyze sequences from Saccharomyces cerevisiae and related sequences from other organisms. Nucleic Acids Res. 2004;32:D311–D314. doi: 10.1093/nar/gkh033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross RL, Muller V. The evolution of A-, F-, and V-type ATP synthases and ATPases: Reversals in function and changes in the H+/ATP coupling ratio. FEBS Lett. 2004;576:1–4. doi: 10.1016/j.febslet.2004.08.065. [DOI] [PubMed] [Google Scholar]
- D'haeseleer P, Church GM. Estimating and improving protein interaction error rates. Proc IEEE Comput Syst Bioinform Conf. 2004;2004:216–223. doi: 10.1109/csb.2004.1332435. [DOI] [PubMed] [Google Scholar]
- Dagan T, Martin W. Ancestral genome sizes specify the minimum rate of lateral gene transfer during prokaryote evolution. Proc Natl Acad Sci. 2007;104:870–875. doi: 10.1073/pnas.0606318104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias I, Tuller T. Reconstruction of ancestral genomic sequences using likelihood. J Comput Biol. 2007;14:216–237. doi: 10.1089/cmb.2006.0101. [DOI] [PubMed] [Google Scholar]
- Felder Y, Tuller T. Discovering local patterns of co-evolution. In: Nelson CE, Vialette S, editors. RECOMB-CG. Springer-Verlag; Heidelberg, Germany: 2008. pp. 55–71. [Google Scholar]
- Felsenstein J. PHYLIP (phylogeny inference package) version 3.5c. Distributed by the author. Department of Genetics, University of Washington; Seattle, WA: 1993. [Google Scholar]
- Fitch WM. Toward defining the course of evolution: Minimum change for a specified tree topology. Syst Zool. 1971;20:406–416. [Google Scholar]
- Gavin AC, Aloy P, Grandi P, Krause R, Boesche M, Marzioch M, Rau C, Jensen LJ, Bastuck S, Dumpelfeld B, et al. Proteome survey reveals modularity of the yeast cell machinery. Nature. 2006;440:631–636. doi: 10.1038/nature04532. [DOI] [PubMed] [Google Scholar]
- Glansdorff N, Xu Y, Labedan B. The last universal common ancestor: Emergence, constitution and genetic legacy of an elusive forerunner. Biol Direct. 2008;3:29. doi: 10.1186/1745-6150-3-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gogarten JP, Kibak H, Dittrich P, Taiz L, Bowman EJ, Bowman BJ, Manolson MF, Poole RJ, Date T, Oshima T, et al. Evolution of the vacuolar H+-ATPase: Implications for the origin of eukaryotes. Proc Natl Acad Sci. 1989;86:6661–6665. doi: 10.1073/pnas.86.17.6661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutell RR, Noller HF, Woese CR. Higher order structure in ribosomal RNA. EMBO J. 1986;5:1111–1113. doi: 10.1002/j.1460-2075.1986.tb04330.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsh E, Sharan R. Identification of conserved protein complexes based on a model of protein network evolution. Bioinformatics. 2007;23:e170–e176. doi: 10.1093/bioinformatics/btl295. [DOI] [PubMed] [Google Scholar]
- Iwabe N, Kuma K, Hasegawa M, Osawa S, Miyata T. Evolutionary relationship of archaebacteria, eubacteria, and eukaryotes inferred from phylogenetic trees of duplicated genes. Proc Natl Acad Sci. 1989;86:9355–9359. doi: 10.1073/pnas.86.23.9355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen LJ, Kuhn M, Stark M, Chaffron S, Creevey C, Muller J, Doerks T, Julien P, Roth A, Simonovic M, et al. STRING 8—a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 2009;37:D412–D416. doi: 10.1093/nar/gkn760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juan D, Pazos F, Valencia A. High-confidence prediction of global interactomes based on genome-wide coevolutionary networks. Proc Natl Acad Sci. 2008;105:934–939. doi: 10.1073/pnas.0709671105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jukes TH, Cantor CR. Evolution of protein molecules. In: Munro HN, editor. Mammalian protein metabolism. Academic Press; New York: 1969. pp. 21–123. [Google Scholar]
- Kanehisa M. The KEGG database. Novartis Found Symp. 2002;247:91–101. [PubMed] [Google Scholar]
- Knudsen B, Hein J. RNA secondary structure prediction using stochastic context-free grammars and evolutionary history. Bioinformatics. 1999;15:446–454. doi: 10.1093/bioinformatics/15.6.446. [DOI] [PubMed] [Google Scholar]
- Koonin EV. Comparative genomics, minimal gene-sets and the last universal common ancestor. Nat Rev Microbiol. 2003;1:127–136. doi: 10.1038/nrmicro751. [DOI] [PubMed] [Google Scholar]
- Krishnan NM, Seligmann H, Stewart C, Koning APJ, Pollock DD. Ancestral sequence reconstruction in primate mitochondrial DNA: Compositional bias and effect on functional inference. Mol Biol Evol. 2004;21:1871–1883. doi: 10.1093/molbev/msh198. [DOI] [PubMed] [Google Scholar]
- Krogan NJ, Cagney G, Yu H, Zhong G, Guo X, Ignatchenko A, Li J, Pu S, Datta N, Tikuisis AP, et al. Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature. 2006;440:637–643. doi: 10.1038/nature04670. [DOI] [PubMed] [Google Scholar]
- Lee I, Date SV, Adai AT, Marcotte EM. A probabilistic functional network of yeast genes. Science. 2004;306:1555–1558. doi: 10.1126/science.1099511. [DOI] [PubMed] [Google Scholar]
- Letunic I, Bork P. Interactive Tree Of Life (iTOL): An online tool for phylogenetic tree display and annotation. Bioinformatics. 2007;23:127–128. doi: 10.1093/bioinformatics/btl529. [DOI] [PubMed] [Google Scholar]
- Li G, Steel M, Zhang L. More taxa are not necessarily better for the reconstruction of ancestral character states. Syst Biol. 2008;57:647–653. doi: 10.1080/10635150802203898. [DOI] [PubMed] [Google Scholar]
- Liang Z, Xu M, Teng M, Niu L. Comparison of protein interaction networks reveals species conservation and divergence. BMC Bioinformatics. 2006;7:457. doi: 10.1186/1471-2105-7-457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockless SW, Ranganathan R. Evolutionarily conserved pathways of energetic connectivity in protein families. Science. 1999;286:295–299. doi: 10.1126/science.286.5438.295. [DOI] [PubMed] [Google Scholar]
- Ma H, Zeng AP. Reconstruction of metabolic networks from genome data and analysis of their global structure for various organisms. Bioinformatics. 2003;19:270–277. doi: 10.1093/bioinformatics/19.2.270. [DOI] [PubMed] [Google Scholar]
- Ma J, Zhang L, Suh BB, Raney BJ, Burhans RC, Kent WJ, Blanchette M, Haussler D, Miller W. Reconstructing contiguous regions of an ancestral genome. Genome Res. 2006;16:1557–1565. doi: 10.1101/gr.5383506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacQueen JB. Proceedings of the 5th Berkeley Symposium on Mathematical Statistics and Probability. University of California Press; Berkeley, CA: 1967. Some methods for classification and analysis of multivariate observations; pp. 281–297. [Google Scholar]
- Marino-Ramirez L, Bodenreider O, Kantz N, Jordan IK. Co-evolutionary rates of functionally related yeast genes. Evol Bioinform Online. 2006;2:295–300. [PMC free article] [PubMed] [Google Scholar]
- Mirkin BG, Fenner TI, Galperin MY, Koonin EV. Algorithms for computing parsimonious evolutionary scenarios for genome evolution, the last universal common ancestor and dominance of horizontal gene transfer in the evolution of prokaryotes. BMC Evol Biol. 2003;3:2. doi: 10.1186/1471-2148-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori H, Isono K, Horiuchi T, Miki T. Functional genomics of Escherichia coli in Japan. Res Microbiol. 2000;151:121–128. doi: 10.1016/s0923-2508(00)00119-4. [DOI] [PubMed] [Google Scholar]
- Neyman J. Molecular studies of evolution: A source of novel statistical problems. In: Gupta S, Jackel Y, editors. Statistical decision theory and related topics. Academic Press; New York: 1971. pp. 1–27. [Google Scholar]
- Noller HF, Woese CR. Secondary structure of 16S ribosomal RNA. Science. 1981;212:403–411. doi: 10.1126/science.6163215. [DOI] [PubMed] [Google Scholar]
- Ouzounis CA, Kunin V, Darzentas N, Goldovsky L. A minimal estimate for the gene content of the last universal common ancestor—exobiology from a terrestrial perspective. Res Microbiol. 2006;157:57–68. doi: 10.1016/j.resmic.2005.06.015. [DOI] [PubMed] [Google Scholar]
- Pagel M. The maximum likelihood approach to reconstructing ancestral character states of discerete characters on phylogenies. Syst Biol. 1999;48:612–622. [Google Scholar]
- Pazos F, Helmer-Citterich M, Ausiello G, Valencia A. Correlated mutations contain information about protein–protein interaction. J Mol Biol. 1997;271:511–523. doi: 10.1006/jmbi.1997.1198. [DOI] [PubMed] [Google Scholar]
- Pedersen JS, Bejerano G, Siepel A, Rosenbloom K, Lindblad-Toh K, Lander ES, Kent J, Miller W, Haussler D. Identification and classification of conserved RNA secondary structures in the human genome. PLoS Comput Biol. 2006;2:e33. doi: 10.1371/journal.pcbi.0020033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penny D, Poole A. The nature of the last universal common ancestor. Curr Opin Genet Dev. 1999;9:672–677. doi: 10.1016/s0959-437x(99)00020-9. [DOI] [PubMed] [Google Scholar]
- Pupko T, Pe'er I, Shamir R, Graur D. A fast algorithm for joint reconstruction of ancestral amino acid sequences. Mol Biol Evol. 2000;17:890–896. doi: 10.1093/oxfordjournals.molbev.a026369. [DOI] [PubMed] [Google Scholar]
- Putnam NH, Srivastava M, Hellsten U, Dirks B, Chapman J, Salamov A, Terry A, Shapiro H, Lindquist E, Kapitonov VV, et al. Sea anemone genome reveals ancestral eumetazoan gene repertoire and genomic organization. Science. 2007;317:86–94. doi: 10.1126/science.1139158. [DOI] [PubMed] [Google Scholar]
- Reguly T, Breitkreutz A, Boucher L, Breitkreutz BJ, Hon GC, Myers CL, Parsons A, Friesen H, Oughtred R, Tong A, et al. Comprehensive curation and analysis of global interaction networks in Saccharomyces cerevisiae. J Biol. 2006;5:11. doi: 10.1186/jbiol36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankoff D. Minimal mutation trees of sequences. SIAM J Appl Math. 1975;28:35–42. [Google Scholar]
- Sato T, Yamanishi Y, Kanehisa M, Toh H. The inference of protein–protein interactions by co-evolutionary analysis is improved by excluding the information about the phylogenetic relationships. Bioinformatics. 2005;21:3482–3489. doi: 10.1093/bioinformatics/bti564. [DOI] [PubMed] [Google Scholar]
- Serek J, Bauer-Manz G, Struhalla G, van den Berg L, Kiefer D, Dalbey R, Kuhn A. Escherichia coli YidC is a membrane insertase for Sec-independent proteins. EMBO J. 2004;23:294–301. doi: 10.1038/sj.emboj.7600063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi T, Falkowski PG. Genome evolution in cyanobacteria: The stable core and the variable shell. Proc Natl Acad Sci. 2008;105:2510–2515. doi: 10.1073/pnas.0711165105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spirin V, Gelfand MS, Mironov AA, Mirny LA. A metabolic network in the evolutionary context: Multiscale structure and modularity. Proc Natl Acad Sci. 2006;103:8774–8779. doi: 10.1073/pnas.0510258103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swofford DL. PAUP*. Phylogenetic analysis using parsimony (*and other methods) Sinauer Associates; Sunderland, MA: 2002. [Google Scholar]
- Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV, Krylov DM, Mazumder R, Mekhedov SL, Nikolskaya AN, et al. The COG database: An updated version includes eukaryotes. BMC Bioinformatics. 2003;4:41. doi: 10.1186/1471-2105-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuller T, Kupiec M, Ruppin E. Co-evolutionary networks of genes and cellular processes across fungal species. Genome Biol. 2009;10:R48. doi: 10.1186/gb-2009-10-5-r48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wapinski I, Pfeffer A, Friedman N, Regev A. Natural history and evolutionary principles of gene duplication in fungi. Nature. 2007;449:54–61. doi: 10.1038/nature06107. [DOI] [PubMed] [Google Scholar]
- Woese C. The universal ancestor. Proc Natl Acad Sci. 1998;95:6854–6859. doi: 10.1073/pnas.95.12.6854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wooldridge K. Bacterial secreted proteins: Secretory mechanisms and role in pathogenesis. Caister Academic Press; Norfolk, UK: 2009. [Google Scholar]
- Wu J, Kasif S, DeLisi C. Identification of functional links between genes using phylogenetic profiles. Bioinformatics. 2003;19:1524–1530. doi: 10.1093/bioinformatics/btg187. [DOI] [PubMed] [Google Scholar]
- Xenarios I, Salwinski L, Duan XJ, Higney P, Kim SM, Eisenberg D. DIP, the Database of Interacting Proteins: A research tool for studying cellular networks of protein interactions. Nucleic Acids Res. 2002;30:303–305. doi: 10.1093/nar/30.1.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. PAML: A program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci. 1997;13:555–556. doi: 10.1093/bioinformatics/13.5.555. [DOI] [PubMed] [Google Scholar]
- Yang Z, Kumar S, Nei M. A new method of inference of ancestral nucleotide and amino acid sequences. Genetics. 1995;141:1641–1650. doi: 10.1093/genetics/141.4.1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeang CH, Haussler D. Detecting coevolution in and among protein domains. PLoS Comput Biol. 2007;3:e211. doi: 10.1371/journal.pcbi.0030211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeang CH, Darot JF, Noller HF, Haussler D. Detecting the coevolution of biosequences–an example of RNA interaction prediction. Mol Biol Evol. 2007;24:2119–2131. doi: 10.1093/molbev/msm142. [DOI] [PubMed] [Google Scholar]
- Zhao J, Ding GH, Tao L, Yu H, Yu ZH, Luo JH, Cao ZW, Li YX. Modular co-evolution of metabolic networks. BMC Bioinformatics. 2007;8:311. doi: 10.1186/1471-2105-8-311. [DOI] [PMC free article] [PubMed] [Google Scholar]