

Abstract
ProPhylER (Protein Phylogeny and Evolutionary Rates) is a next-generation curated proteome resource that uses comparative sequence analysis to predict constraint and mutation impact for eukaryotic proteins. Its purpose is to inform any research program for which protein function and structure are relevant, by the predictive power of evolutionary constraint analyses. ProPhylER currently has nearly 9000 clusters of related proteins, including more than 200,000 sequences. It serves data via two interfaces. The “ProPhylER Interface” displays predictive analyses in sequence space; the “CrystalPainter” maps evolutionary constraints onto solved protein structures. Here we summarize ProPhylER's data content and analysis pipeline, demonstrate the use of ProPhylER's interfaces, and evaluate ProPhylER's unique regional analysis of evolutionary constraint. The high accuracy of ProPhylER's regional analysis complements the high resolution of its single-site analysis to effectively guide and inform structure–function investigations and predict the impact of polymorphisms.
Through evolution, all proteins have been subjected to extensive structure–function experiments by the action of mutation and natural selection on their encoding genes. The results of these experiments are recorded in the sequence variation among extant homologs (Zuckerkandl and Pauling 1965; anticipated by Crick 1958). If the homologs have maintained the same function since their divergence from a common ancestor, substitutions compatible with that function are likely to be represented in extant sequences; conversely, conservation among extant homologs implies that evolutionary variation has been consistently rejected owing to functional or structural constraint (Kimura 1983). The logic and utility of evolutionary variation analysis thus parallel those of genetic analysis: Just as a deleterious phenotype of a mutated site implies the site's necessity for normal function, so does evolutionary constraint at a site quantify its structural or functional importance. Appropriate comparative sequence analysis provides the means to interpret the results of evolution's experiments, and to leverage them as predictions for the benefit of a wide diversity of biomedical research (Sidow 2002; Fay and Wu 2003; Ng and Henikoff 2006).
ProPhylER (Protein Phylogeny and Evolutionary Rates; http://www.prophyler.org) is an interactive resource that puts evolution's results at the fingertips of researchers, by comprehensively quantifying evolutionary constraint in eukaryotic proteins. ProPhylER makes specific predictions of the importance of protein regions without relying on domain annotations, and estimates the functional impact of every possible amino acid substitution in a given protein. Underlying ProPhylER's predictive power are high-quality, curated multiple sequence alignments of closely related homologs that are analyzed with statistically rigorous, comparative analytic methods to quantify constraint. Here we describe ProPhylER's data generation and provide content statistics to demonstrate the resulting quality of ProPhylER's protein alignments and analytic data.
ProPhylER analyzes constraints at two complementary resolutions: in regions across the protein that roughly correspond to small functional domains, and for each individual amino acid. The first type of regional analysis is ESF (evolution-structure-function) (Simon et al. 2002), which detects evolutionarily constrained regions in proteins. We previously used ESF to discover novel structural and functional regions in proteins (Simon et al. 2002; Ko et al. 2003; Jackson et al. 2006; Hughes et al. 2008) and to discover regions responsible for functional differences between paralogs (Simon et al. 2002; Jackson et al. 2006). A second type of regional analysis calculates phylogenetically averaged (Stone and Sidow 2007) physicochemical properties across the protein to produce de novo identification of physicochemically unusual regions. We previously used such profiles to discover novel transmembrane domains (Hughes et al. 2008).
To analyze constraint at the single-site level, ProPhylER uses MAPP (multivariate analysis of protein polymorphism) (Stone and Sidow 2005), which estimates the variance in physicochemical properties observed at a given position in a multiple sequence alignment to predict the impact of every possible amino acid substitution. MAPP predicts the impact of mutations in test sets with 80% accuracy and is effective in distinguishing strong from weak mutant alleles (Stone and Sidow 2005). We previously used MAPP to discover and verify novel disease-associated polymorphisms (Kashuk et al. 2005; Lin et al. 2006). ProPhylER presents the results of the application of these methods to a novel comprehensive, curated, database of eukaryotic proteins via powerful, interactive interfaces. Below we include a tutorial with examples that illustrate both the functionality of the interfaces and the utility of ProPhylER's constraint analyses in informing and guiding experiments.
ProPhylER aims to benefit anyone interested in the function, structure, or disease association of a protein, by identifying the features of the protein that have been important throughout its evolutionary history. Cell biologists and biochemists conducting structure–function studies on proteins can use ProPhylER to guide and inform targeted mutagenesis, even if nothing else is known about the protein. Cancer biologists who have found lesions in tumor-suppressor genes can evaluate whether they affect important sites or regions of the proteins. Structural biologists can use ProPhylER data to annotate newly solved protein structures with constraint information. Geneticists performing linkage or association studies can evaluate polymorphisms in coding regions for their likelihood of disrupting protein function. Thus, in addition to summarizing and displaying a protein's evolutionary variation, ProPhylER facilitates experimental dissection of proteins and interpretation of natural variation in coding regions.
Results
ProPhylER's dataflow pipeline
There are three phases in ProPhylER's dataflow and analysis pipeline (Fig. 1). The first phase creates clusters of homologous sequences that have likely retained the same function, while partitioning sequences away whose functions have likely diverged (Fig. 1A). We first generate seed clusters based on pairwise alignment scores of protein sequences from 13 sequenced genomes: human, mouse, opossum, chicken, frog, stickleback, zebrafish, sea squirt, fruit fly, mosquito, nematode, budding yeast, and fission yeast. Initializing the clusters from this limited set of sequences (instead of all eukaryotic proteins) simplifies the incorporation of a phylogenetic criterion during cluster building (see Methods). The seed clusters are then augmented with all eukaryotic protein sequences from UniProt, which greatly increases the total sequence information in ProPhylER and allows us to take advantage of UniProt's substantial domain and feature annotation.
Figure 1.

The ProPhylER dataflow pipeline. (Solid arrows) Automated steps; (dashed arrows) curated steps. (A) Generating clusters of functionally conserved homologs. (A, step i) Single-linkage clusters are built from all-by-all BLAST searches of (gray-filled circles) protein sequences from 13 fully sequenced genomes. Edges of the clusters (lines of varying thickness joining sequences) are similarity scores between cluster members. (A, step ii) The MinCut routine (line through clusters) separates clusters at their weakest edges (lowest scores). (A, step iii) Manual curation rejoins overcut clusters (dashed circle). (A, step iv) Each eukaryotic sequence in UniProt (black-filled circles) is added to its best-matching cluster. (B) Building alignments and trees for ProPhylER clusters. (B, step i) The initial alignment is built (bars), and sequences containing or creating excessive alignment gaps are flagged for potential removal (underlined numbers). (B, step ii) Manual curation removes any problem sequences. (B, step iii) The remaining cluster sequences are realigned, and a maximum-likelihood phylogenetic tree is built. It is compared to its corresponding species tree, and each internal node is annotated as either a (white node) speciation or (black node) gene duplication event. (C) Predictive analyses are generated using the information in cluster alignments and trees, and are displayed with ProPhylER's graphical user interfaces.
The second phase of the pipeline produces high-quality multiple sequence alignments and phylogenetic trees for each ProPhylER cluster (Fig. 1B). Building high-quality alignments is the most critical step in the pipeline, as they are essential for reliable trees and accurate analyses. We build alignments using the highly sensitive program ProbCons (Do et al. 2005), which also produces a reliability score for each alignment column that is useful for tree-building and assessing alignment quality (see below). Next we produce maximum-likelihood trees for all ProPhylER clusters with the program SEMPHY (Friedman et al. 2002), using the most reliable columns from the multiple sequence alignment. Trees are necessary along with alignments for the predictive analyses described below. After a cluster's tree is built, it is automatically compared with a species tree for the cluster, and each internal node is annotated as being from either a speciation or gene duplication event. This allows for tree-based, and therefore objective, determination of orthology (sequences or subgroups related by speciation) and paralogy (related by gene duplication). The final alignments and annotated trees are made available for viewing or downloading via the ProPhylER Interface.
The final pipeline phase automatically analyzes each cluster for evolutionary constraints, physicochemical properties, and mutation impact predictions (Fig. 1C), using our aforementioned published methodologies. The results of these analyses are presented to the user via ProPhylER's graphical user interfaces and are described in detail below.
Curated pipeline steps
In developing the ProPhylER pipeline, we found that the automated clustering and alignment steps introduced errors that degraded the performance and quality of all downstream steps and analyses. To correct this, we added major rounds of curation after each of these critical steps.
Clustering is problematic either when non-homologs are grouped, which reduces the sensitivity of subsequent constraint analyses (Stone and Sidow 2005), or when true homologs are separated, which reduces sequence diversity and affects constraint analysis specificity (Mayrose et al. 2004). ProPhylER's clustering is intentionally stringent to avoid grouping non-homologs (see Methods), so the goal of the first round of curation is to merge homologous clusters. Curators examine every cluster, compare it to clusters containing similar sequences, and identify those to be merged based on degree of similarity, phylogenetic composition, and sequence annotation.
The goal of the second round of curation is to remove sequences that reduce alignment quality. Clusters based on similarity searches of sequence databases often contain translations of incorrect gene predictions or partial cDNA sequences. These sequences disrupt otherwise well-aligned regions, and in extreme cases cause alignment failure. Other problem sequences are genuine but highly diverged homologs that have undergone such extensive evolution that their alignment to the other cluster sequences is uncertain. For such sequences the assumption of functional conservation becomes questionable, and their inclusion may reduce the sensitivity of subsequent analyses. Curators examine every alignment and remove sequences that introduce excessive alignment gaps, or that stand out as being anomalously divergent, after taking into account cluster phylogeny and sequence annotation. For some large clusters, the curator may break the alignment down into subalignments. This both makes the alignment and subsequent pipeline steps more manageable, and allows for biologically interesting comparisons between subalignments. This process is described in the Supplemental material.
ProPhylER's content
ProPhylER currently has analyses for 8967 protein clusters. On average, the clusters contain 24 sequences from 17 different species. The species with the most sequences (13,861) in analyzed ProPhylER clusters is human; many model organisms are also well represented (Supplemental Table S1). A good indication of a cluster's diversity is its phylogenetic scope (narrowest common taxonomic rank). ProPhylER's best-represented scope is Eukaryotes, with 2887 clusters (Fig. 2A). Since the proteins in these clusters existed in the last common ancestor of all eukaryotes well over a billion years ago, and yet still align well among diverse organisms, they are among the most conserved of all proteins and perform basic functions of eukaryotic cell biology. The second-best represented scope is Vertebrates, with 2349 clusters, mostly representing proteins that originated in the vertebrate lineage after its divergence from other animals. The third scope is Metazoa, which is enriched in proteins necessary for multicellular function and differentiation. Eukaryotic clusters contain an average of 43.1 sequences (Fig. 2B). Amniote clusters (containing sequences only from mammals and birds/reptiles) tend to be smallest, having an average of 6.3 sequences. In general, the older the protein, the more sequences its cluster tends to contain. This is because proteins originating earlier in evolution are carried by a larger number of species, and are thus better represented in the sequence databases.
Figure 2.

ProPhylER cluster statistics. (A) ProPhylER clusters by phylogenetic scope. (Right) A bar chart showing the number of ProPhylER clusters (horizontal axis) by major taxonomic groups (vertical axis; the groups chosen are mammal-centric, but the clusters do not necessarily include mammals). (Left) A tree relating example species from the taxonomic groups in the bar chart (branch lengths not to scale). (B) Diversity of ProPhylER clusters. A bar chart shows the average number of sequences per cluster (black bar, top horizontal axis) and the average cluster tree length in substitutions per site (gray bar, lower horizontal axis) grouped by phylogenetic scope (vertical axis). (C) ProPhylER alignment quality. Histogram showing the percentage of alignment columns with a ProbCons reliability score of 50 or higher per sequence for all ProPhylER alignments (vertical axis), by decile (horizontal axis), (gray bars) before curation and (black bars) after curation. (D) ProPhylER cluster diversity. Histogram showing the total tree length (in substitutions per site) of all ProPhylER clusters (vertical axis), by decile (horizontal axis).
Sensitivity of ProPhylER's constraint analyses depends on the quality of the multiple alignments of cluster sequences. The alignment program ProbCons produces a reliability score between 0 and 100 for each alignment column, and we use a reliability score of 50 as the threshold for including an alignment column in the subsequent tree-building step. The percentage of columns in an alignment above this threshold provides a measure of alignment quality, as well as a means of comparing alignments before and after curation (Fig. 2C). Curated alignments have a median 51% columns exceeding this threshold (compared to a median 33% for uncurated alignments), and 90% of curated alignments have at least 21% of columns exceeding the threshold (compared to 11% of columns for the top 90% uncurated alignments). Divergent sequences introduce gaps in the alignment, so another measure of alignment quality is alignment expansion per sequence. This is calculated as the ratio of the length of the alignment to the length of its longest sequence, normalized by the number of sequences, and reflects the relative proportion of gaps the average sequence introduces to the alignment (Supplemental Fig. S1). The median expansion per sequence for curated alignments is 0.7% (compared to a median of 1.3% for uncurated alignments), and the top 90% curated alignments have <1.8% expansion per sequence (compared to 2.9% for the top 90% uncurated alignments). These measurements demonstrate the quality gained from the effort invested in ProPhylER's alignment curation.
Specificity of ProPhylER's analyses depends on the evolutionary diversity represented within its clusters. Diversity is expressed in substitutions per site and is calculated as the sum of the branch lengths in the cluster's phylogenetic tree. Site-by-site analysis is accurate down to about one substitution per site (Stone and Sidow 2005), while regional analysis, because it leverages correlations in constraints among neighboring residues, maintains accuracy down to about 0.5 substitutions per site (see below). The diversity of ProPhylER clusters ranges considerably, from 57.9 to 0.5 substitutions per site (Fig. 2D), which is our lower cutoff for analyzing clusters. Almost 90% of clusters capture more than 1 substitution per site, and thus have sufficient diversity for both site-by-site analyses and regional analyses. Plotting the tree length of clusters along with their number of sequences reveals a striking linear relationship: tree length scales by a factor of 1/5 with number of sequences across all phylogenetic scopes (Fig. 2B). In other words, regardless of phylogenetic scope of the cluster, the average sequence contributes 0.2 substitutions per site to the tree relating the sequences in the alignment.
ProPhylER's graphical user interfaces
ProPhylER is searchable using either a peptide sequence (via BLAST) or with a database identifier (from UniProt, Ensembl, PDB, or ProPhylER itself). A successful search brings up a page with a summary of the matching ProPhylER cluster, as well as a link to launch a ProPhylER Interface session for that cluster, which interactively displays evolutionary constraints, mutation impact predictions, the alignment, the tree, and other useful data for the protein. If a PDB structure containing a sequence similar to the query is also identified (or if a PDB identifier is used as the search query), the results page will display a link to launch a CrystalPainter session, which interactively displays that structure color-coded with position-specific evolutionary constraints from ProPhylER. In this section, we provide a tutorial of ProPhylER's user interfaces using links to example Interface and CrystalPainter sessions.
The ProPhylER Interface
The ProPhylER Interface is a Java applet that allows interactive browsing and downloading of the data for a single cluster. The following URL opens a web browser session for the cadherin-15 (Shimoyama et al. 1998) cluster that illustrates the functionality and utility of the ProPhylER Interface: http://www.prophyler.org/cgi-bin/example1.cgi. Figure 3 shows a screenshot of the same Interface session, which can also be initiated by entering the UniProt identifier “P55291” from the search page (http://www.prophyler.org/cgi-bin/search_form.cgi). The Interface has five “Views,” windows that can be displayed by selecting one of the tabs on the left of the Interface (Fig. 3A). The session opens displaying the Main view, which includes the most important features and analyses for the protein. All features are shown in relation to a reference sequence—usually the sequence specified in the search—but are based on the coordinates of the underlying multiple sequence alignment. Features are displayed in two panels that examine the protein at different resolutions.
Figure 3.

The ProPhylER Interface. (A) Tabs to the left of the window control the Interface view. The Main view opens by default and is portrayed here. (B) Profiles of regional evolutionary constraint (black line) and physicochemical properties (hydropathy shown, blue line). Features to plot are selected from the list on the left. The vertical axis of the plot shows the level of constraint or property relative to the protein average. The horizontal axis is position in the protein in the coordinates of the reference sequence. (C) A track showing the evolutionarily constrained regions (ECRs) of the protein. The numbers in the track rank ECRs by level of constraint, and the intensity of the dark shading is proportional to the magnitude of constraint. (D) A track showing domains in the reference sequence (pink bars) annotated by SWISS-PROT, if any. (E) A shaded window over the profile in the upper panel controls the view in the lower panel. The view is changed by clicking and holding the computer mouse over the shaded window and dragging it left or right. (F) Tabs switch the view in the lower panel between the default Summary view and the Alignment view. (G) The top of the lower panel displays the reference sequence. The single-position constraint values are plotted as gray bars above the sequence. The relative constraint value is indicated on the vertical axis. (H) A summary of the alignment in logos format. Selecting the “grid” option to the left of the summary displays a grid on which each amino acid is shaded according to its prevalence at each position. (I) Grid displaying color-coded values at every protein position indicating the impact of each possible mutation predicted by the MAPP procedure. (Shades of blue) Low-impact mutations; (yellow, orange, or red) increasingly high-impact mutations. (J) Grid displaying the relative importance of six physicochemical properties at each position in the protein. The intensity of the dark shading is proportional to the importance of the property, as estimated by the MAPP procedure.
The upper panel presents the regional perspective, dominated by the Evolutionary Constraint Profile (Fig. 3B), which is produced using ESF analysis (Simon et al. 2002). This analysis is a feature unique to ProPhylER, and we explore its underpinnings in detail below. Peaks in the profile correspond to the evolutionarily constrained regions (ECRs), the most highly ranked of which are the functionally and/or structurally most important regions of the protein. The track directly above the plot ranks the ECRs by their degree of constraint (Fig. 3C). In the case of cadherin-15, the top-ranked ECR occurs near the C terminus of the protein, and corresponds to the cytoplasmic, catenin-interacting region (Shimoyama et al. 1998). Other highly ranked ECRs occur in the cadherin domains (indicated in the “SwissProt domains” track above the ECRs track) (Fig. 3D), which are typically involved in homophilic adhesion (Ivanov et al. 2001). Interestingly, despite the high structural similarity between different cadherin domains (Boggon et al. 2002), there are substantial differences in their evolutionary constraint, suggesting differences in functional importance. Physicochemical properties, averaged appropriately across all sequences of the underlying alignment (Hughes et al. 2008), may be plotted along with constraint (Fig. 3B). Selecting “Hydropathy” for caderin-15 reveals a strongly hydrophobic region (that is also a constrained region; peak in blue line on plot below ECR 12), and which corresponds to the predicted transmembrane domain (Shimoyama et al. 1998). A shaded window (Fig. 3E), initially centered on the top-ranked ECR, can be dragged left or right to control the positioning of the 60-amino-acid-wide view in the lower panel.
The lower panel has single-site resolution. Tabs on the left of the lower panel (Fig. 3F) toggle between the default Summary view and the Alignment view. Both views display the reference sequence and a plot of single-site evolutionary constraints at the top of the pane (Fig. 3G) and allow searches for specific sequence motifs. The Summary view, which contains a logo representation of the underlying alignment (Fig. 3H), is dominated by the color-coded MAPP mutation impact predictions (Fig. 3I). MAPP assigns a P-value for each possible amino acid substitution (reflecting the probability it will be tolerated), which can be viewed by moving the cursor on the grid. Several human polymorphisms in cadherin-15 illustrate the use and power of MAPP predictions. The polymorphisms R60C, R92W, and A122V are associated with mild to severe intellectual disability (ID) and have diminished adhesion function in a cell culture assay (Bhalla et al. 2008). Consistent with these phenotypes, the respective MAPP P-values are 4.9 × 10−4, 1.9 × 10−3, and 8.1 × 10−6. Two additional polymorphisms, K103R and M109T, are not associated with ID and have wild-type adhesion function (Bhalla et al. 2008). MAPP predicts that these substitutions should have little to no impact, with P-values of 0.92 and 0.73, respectively. The Summary view of the lower panel also contains a MAPP-generated estimation of the most important physicochemical property for each position (Fig. 3J).
In addition to the Main view, tabs to the left of the window control the display of five additional views: “Gene Tree” displays the annotated phylogenetic tree for the cluster; “Taxonomy” displays the species tree; the Selector view allows switching to a different reference sequence; and “Downloads” allows the user to save the raw data in tabular format.
CrystalPainter
CrystalPainter displays PDB protein structures color-coded with ProPhylER's site-specific evolutionary constraint values, using Jmol (http://www.jmol.org/). Presenting constraints in this direct structural context gives an immediate impression of the biologically important regions of a protein, particularly those on the surface such as active sites or ligand binding patches (Sander and Schneider 1991; Lichtarge et al. 1996; Armon et al. 2001; Dean et al. 2002; Simon et al. 2002). The following URL initiates a ProPhylER session for the heterodimeric transcription factor E2F–DP bound to DNA (Zheng et al. 1999), which illustrates the functionality of the CrystalPainter (http://www.prophyler.org/cgi-bin/example2.cgi). The top of the resulting page shows a summary of the two ProPhylER clusters for the peptides associated with this structure (i.e., one for the E2F cluster, the other for the DP cluster), along with buttons to launch ProPhylER Interface sessions for each. Below that is another button to launch the CrystalPainter, which opens in a new browser window. The CrystalPainter page (Fig. 4A) is divided into several sectors. The interactive Jmol window and button controls to manipulate the views dominate the page. Beneath the Jmol window are links to download the ProPhylER-modified structure in PDB format for viewing off-line, or to save a “snapshot” of the current Jmol window in JPG format.
Figure 4.

CrystalPainter. (A) A screenshot of the CrystalPainter page for PDB ID 1CF7. Major features include: (i) the Jmol window, where the molecular image is manipulated; (ii) links to download a coded structure file or an image of the Jmol window; (iii) button controls to modify the display settings; and (iv) instructions for mouse controls and navigating the Jmol window. (B) Image capture from CrystalPainter. The peptide chains have been displayed as “Spacefill” to highlight the conserved residues on the surface (shades of blue) and the structure reoriented to view the DNA-binding surface. (C) Chain A has been displayed as “Backbone” and Chain B as “Spacefill,” and the structure reoriented to better view the dimerization surface of Chain B. (D) Chain B has been displayed as “Backbone” and Chain A as “Spacefill,” and the structure reoriented to better view the dimerization surface of Chain A.
The data color-coded onto the structure by CrystalPainter are site-specific evolutionary constraints. It is immediately obvious that the most constrained peptide residues (those colored in shades of blue) occur in the core of the E2F–DP heterodimer, while less constrained residues (colored yellow, orange, or red) tend to occur on the periphery (Fig. 4A). Choosing the “Spacefill” or “Surface” display styles for each peptide chain and orienting the complex to view the DNA-binding site (Fig. 4B) reveals that the residues contacting the DNA are the most constrained. Choosing “Backbone” for the E2F (Chain A) or DP (Chain B) molecules separately while keeping the other displayed as “Spacefill” (Fig. 4C,D) shows that the dimerization surface of each protein is also highly constrained. While these evolutionary constraint patterns are expected for this complex (Zheng et al. 1999), they serve as a proof of principle to demonstrate that that CrystalPainter clearly reveals functionally important regions of proteins with solved structures, particularly if those regions occur on the protein surface.
For almost 9000 distinct protein families, whether previously studied or not, ProPhylER data are available via these two interfaces, putting massively comprehensive analytical results on protein structure, function, and evolution at the fingertips of researchers. The regional profile, which guides users to the evolutionarily most important regions of the protein, is especially important for the usefulness of ProPhylER as a resource. We therefore assessed the underlying methodology with rigorous comparisons to experimental results and mutation phenotype data.
ProPhylER's regional analysis of evolutionary constraints
Evolutionary constraint predicts biological importance
To test the accuracy of ProPhylER's regional analysis (ESF) for revealing true biological constraints, we used five comprehensive mutation data sets as “gold standards” to independently ascertain structural and functional importance. The proteins represented in three of the five sets were the subjects of comprehensive mutagenesis experiments that included functional assays for each mutant: HIV-1 protease (Loeb et al. 1989), bacteriophage T4 lysozyme (Rennell et al. 1991), and Escherichia coli lactose repressor (Markiewicz et al. 1994; Suckow et al. 1996). A fourth protein, beta-globin, has many known human germline mutations, and clinical descriptions of each mutation's effect (http://globin.cse.psu.edu/globin/hbvar/). The fifth protein, the TP53 (also known as p53) tumor suppressor, has many thousands of documented cancer-associated somatic missense mutations (Hollstein et al. 1994; Olivier et al. 2002). The phenotype information associated with the five data sets allowed us to generate numerical scores reflecting the tolerance to mutation at each position of the proteins. For protease, lysozyme, lactose repressor, and beta-globin, we used the quantitative phenotype data reported for each mutant to generate these scores. For TP53, we used the inverse of the frequency of cancer-associated mutation at each site. These scores provided the benchmarks of biological constraint that we used to quantify the predictive accuracy of ProPhylER's evolutionary constraint analyses.
Using methods analogous to the ProPhylER pipeline (above), we collected closely related homologous sequences, made multiple sequence alignments and phylogenetic trees, and calculated single-site evolutionary rate values for each of the five sets. To establish the accuracy of these single-site rate values, we treated them as predictions of constraint: Low rates (i.e., more conserved) predict biological constraint; high rates (less conserved) predict lack of constraint. We treated each corresponding mutation tolerance score from the aforementioned mutation data sets as a measurement of the “true” constraint—we considered tolerance scores below the protein median to be constrained, and values at or above the median to be unconstrained. We then tabulated frequencies of true-positive, true-negative, false-positive, and false-negative predictions, and used them to produce ROC plots (Fig. 5A–E; Hanley and McNeil 1982). We integrated these plots to calculate accuracy scores for the evolutionary rate values. Single-site evolutionary rates accurately predict constraint as defined by mutation data for all five proteins, with scores ranging from 0.75 for beta-globin to 0.83 for HIV protease (Table 1).
Figure 5.

Regional evolutionary constraints accurately reflect biological importance. (A–E) Receiver operating characteristics (ROC) plots for the prediction of mutation impact by single-site evolutionary rates (light blue dots), regional evolutionary rates (dark blue diamonds), and “regional” rates from randomly permuted alignments (black Xs), for beta-globin (A), lactose repressor (B), lysozyme (C), p53 (D), and protease (E). For the full range of rate value thresholds of constraint prediction, the true-positive rate (TP/[TP + FN]) is plotted along the vertical axis against the false-positive rate (FP/[FP + TN]) on the horizontal axis. Random predictions would fall along the light-black diagonal lines. (F–J, blue) Evolutionary constraint profiles and (red) mutation impact profiles for beta-globin (F), lactose repressor (G), lysozyme (H), p53 (I), and protease (J). The normalized constraint and mutation impact values are plotted on the vertical axes, versus protein position on the horizontal axes. (K–O) Molecular structures of the five proteins color-coded with regional evolutionary rates: (blues) low rates (constrained regions); (green) average rates; (orange and red) high rates (unconstrained regions). (K) Human hemoglobin tetramer (PDB ID 4HHB; Fermi et al. 1984). The two beta-globin subunits are color-coded by evolutionary rate: (magenta) the two heme groups bound by beta-globins; (brown) their coordinated iron atoms. (Gray) The two alpha-globin subunits and their associated heme groups and iron atoms. (L) E. coli lactose repressor dimer in repressing conformation (PDB ID 1EFA; Bell and Lewis 2000). The repressor dimer is shown bound to operator DNA (gray), as well as the anti-inducer orthonitrophenylfucoside (which binds in the same pocket as the inducer) (magenta). (M) Bacteriophage T4 lysozyme mutant covalently bound to substrate-product intermediate (PDB ID 148L; Kuroki et al. 1993). (Magenta) The ligand, a glycosyl intermediate of E. coli cell wall cleavage, is bound in the active site of the enzyme. (N) Human tumor suppressor p53 core domain bound to DNA (PDB ID 1TSR; Cho et al. 1994). (Gray) DNA; (magenta) the bound zinc atom. (O) HIV-1 protease dimer (PDB ID 4HVP; Miller et al. 1989). (Magenta) A peptide inhibitor is shown bound in the substrate-binding pocket.
Table 1.
Accuracy of regional and single-site evolutionary constraints

aRegional accuracy values averaged from 1000 independent, random permutations, ±1 standard deviation.
We then used ESF analysis to produce regional evolutionary rate profiles from the single-site rate values for each of the five proteins. In order to directly compare the rate profiles and mutation tolerance scores, we applied the same sliding-windows averaging method to the mutation tolerance scores, producing profiles of the regional tolerance of mutation (Fig. 5F–J; note that these profiles have been inverted about the mean to show evolutionary constraint and mutation impact, in order to be consistent with the profiles displayed in the ProPhylER Interface). Evolutionarily constrained regions (ECRs) closely correspond to mutation-sensitive regions. Mapping regional rate values for the five proteins to their crystal structures (Fig. 5K–O) demonstrates that the highest-ranked ECRs (blue-shaded regions of the protein molecules) also correspond to the most structurally and functionally important regions of each protein (Fig. 5K–O). Thus, ECRs qualitatively reflect biological constraints acting on proteins, ascertained in vivo with mutation data and from three-dimensional structures of functional complexes. We quantified the accuracy of regional rate profiles in predicting biologically constrained regions as above, using the mutation tolerance profiles as measurements of the “true” regional constraint. Accuracy values exceed 0.9 for all five proteins (where 1 is the maximum possible value), demonstrating that evolutionary rate profiles are highly accurate at predicting regions of biological importance (Fig. 5A–E; Table 1).
Local correlation of constraints improves the detection of biologically important regions
For each of the five proteins analyzed, the prediction of regional constraint is more accurate than the prediction of single-site constraint (Table 1). Evolutionary rates in proteins are locally correlated, reflecting local correlations in structural and functional constraints (Fitch and Markowitz 1970): Sites common to local folds, functional domains, ligand binding sites, and so on, evolve at similar rates. A simple permutation test demonstrates that ProPhylER's ESF analysis leverages these local correlations of biological constraint to gain predictive accuracy. We repeated the analyses of the five test proteins after first randomly permuting, in tandem, the columns of the multiple sequence alignments (the source of the evolutionary rate values) and their corresponding mutation tolerance scores. Because the order of the columns in the alignment has been randomized, any potential correlation of biological constraints between neighboring positions has been eliminated. We generated evolutionary rate profiles and mutation tolerance profiles from 1000 randomly permuted alignments for each of the five proteins. The accuracy of the permuted regional analyses is reduced in each case, to levels no better than those for the single-site analyses (Fig. 5A–E; Table 1). These results show that ProPhylER's ESF analyses capture a true regional signal of constraint.
The benefits of regional evolutionary analysis persist even at high levels of sequence variation
These results raise the question of whether the improvement in accuracy for regional analysis is simply a matter of compensation for limited sampling of sequence variation in the single-site analysis. In the hypothetical extreme of an unlimited number of sequences in an alignment, would there be sufficient variation in a single column such that the benefit of a regional analysis would become negligible?
To begin to address this question, we took advantage of our large alignment for beta-globin, which contains 162 sequences and has an average of 8.9 substitutions per site. From this we made 10,000 random, independent subalignments, containing between four and 161 sequences, and ranging between 0.2 and 8.9 substitutions per site. We repeated the accuracy calculations, comparing evolutionary rate profiles generated from these alignments to the beta-globin mutation tolerance profile, and observed the relationship between accuracy and alignment sequence variation. Not surprisingly, the accuracy of both regional and single-site analyses improves with increasing sequence variation in the alignment (Fig. 6A). In addition, the standard deviation of the accuracy scores for both analyses decreases with increasing sequence variation, indicating that the analyses become less sensitive to specific alignment composition with more variation. However, both analyses appear to reach near-maximum accuracy when the alignments contain more than four substitutions per site; little increase in accuracy is observed at higher levels of sequence variation. Furthermore, the gain in accuracy of regional analysis relative to single-site analysis remains proportionally the same over the entire range of sequence variation in the alignments, indicating that the advantage of a regional analysis does not diminish with increasing sequence variation in the alignment.
Figure 6.

Effect of alignment sequence variation on accuracy. (A) The relationship between accuracy of evolutionary rates analysis (vertical axis) and average substitutions per site in the alignment (horizontal axis) for 10,000 random subalignments of beta-globin. The accuracy of regional evolutionary rates analyses (gray points, black line, and error bars) is compared with accuracy of single-site evolutionary rates analyses (black points, gray line, and error bars). Accuracy scores were binned by alignment substitutions per site, and the average for each bin was plotted (lines). The error shown is plus and minus one standard deviation. (B) The relationship between accuracy and average substitutions per analysis window. Shading and symbols are as in A.
To specifically address whether the improvement in accuracy for regional analysis is due to greater sampling of sequence variation, we normalized the data by the sequence variation sampled in each analysis window (Fig. 6B). This has no effect on the single-site analysis (where the “window size” is 1), but shifts the plot of accuracy versus sequence variation for the regional analysis to the right. Above four substitutions per window, the regional analysis is significantly more accurate than the single-site analysis. At higher levels of sequence variation, the accuracy of the regional analysis continues to improve, but there is no further improvement in the accuracy of the single-site analysis. Thus, the improved accuracy of a regional analysis really depends on the inclusion of neighboring positions and is not simply the result of compensating for lack of sampled sequence variation in a single-column “window.”
Discussion
ProPhylER leverages the results of evolution's exhaustive experiments to provide accurate predictive analyses of structural and functional constraints in proteins. ProPhylER's strength is its rigorous methodology and stringent quality control in cluster building, multiple sequence alignment, and constraint inference. ProPhylER makes all of its analyses, alignments, and phylogenetic trees available via powerful and straightforward user interfaces. ProPhylER provides scientists with tools and analyses to guide and inform experiments on protein structure and function, and with predictions for the impact of specific mutations and polymorphisms.
Other phylogenetic resources are available, but because they have different goals, they have only limited overlap with ProPhylER. Many resources provide orthologous groups of protein and/or gene sequences (Tatusov et al. 1997; Remm et al. 2001; Li et al. 2003; Deluca et al. 2006; Heinicke et al. 2007; Schneider et al. 2007; Flicek et al. 2008; Matsuya et al. 2008; Wheeler et al. 2008); their relative success at grouping orthologs has been examined (Altenhoff and Dessimoz 2009). These resources focus on formally defined orthologs from fully sequenced genomes, and thus are useful for gene annotation and “phylogenomic” comparisons (Brown and Sjölander 2006). Other resources, such as Pfam (Finn et al. 2008), identify domains conserved between otherwise unrelated protein families. These are useful for assigning putative functions to similar domains identified in novel proteins. The Conserved Domains Database (CDD) (Marchler-Bauer et al. 2009) makes the added useful distinction of domains conserved across protein families (indicating shared generic process) or within families (indicating shared specific function). Several resources provide curated alignments and/or phylogenetic trees. These are intended primarily for the classification and annotation of gene families. TreeFam (Li et al. 2006), in particular, is an excellent source for rigorous, curated phylogenetic trees of metazoan proteins. Of the handful of resources that provide single-site constraint detection, ConSurf (Landau et al. 2005) and ConSeq (Berezin et al. 2004) use the most robust methodology. In fact, starting with identical alignments and trees, the accuracy of their single-site constraints would be at least as good as ProPhylER's (Supplemental Fig. S2; Mayrose et al. 2004). However, for de novo analyses, these resources select sequences on the basis of a cutoff BLAST score. While fast, this encourages the inclusion of sequences of doubtful quality and functional conservation, which limits sensitivity and specificity of constraint detection. This problem also exists for the several resources that predict the impact of mutations or polymorphisms, including SIFT (Ng and Henikoff 2003) and PolyPhen (Ramensky et al. 2002). Thus, because ProPhylER's results are produced by rigorous methodology applied to stringently curated alignments of closely related homologs, it is unique in its utility for a wide variety of biomedical questions that involve protein structure and function.
Despite the considerable curatorial effort invested in launching ProPhylER, maintaining the resource will emphasize automation. Because we used proteins derived from fully sequenced genomes as cluster seeds, we do not anticipate a major change with time to the numbers of, or relationships among, ProPhylER's clusters. As new protein sequences become available in the public databases, it is likely that most of them will be homologous to existing ProPhylER clusters, so incorporating them into ProPhylER will be a matter of automated profile alignment (Pietrokovski 1996; Edgar 2004) and reanalysis, with minimal need for curator oversight. Future effort will thus focus on the development of new features, such as the integration of ProPhylER's protein-specific constraint into genomic evolutionary constraint analyses (The ENCODE Project Consortium 2007).
To our knowledge, ProPhylER is unique in providing de novo regional constraint annotation for proteins. Here we have demonstrated that, because biological constraints are locally correlated to maintain specific structural folds or functional interfaces, constrained regions in a protein can be determined with even greater accuracy than can be achieved for determining individual constrained residues. Using evolutionary constraint profiles to identify and rank these regions from most constrained (by inference most important) to least constrained (by inference less important) can be the first step in characterizing novel proteins and provides the researcher with a natural prioritization for conducting structure–function studies. Within the most constrained regions of a protein, the most constrained residues can be targeted for experimental analysis.
The power of comparative sequence analysis to detect structural and functional constraints potentially approaches that of directed mutagenesis and comes at a far smaller cost in time and effort. However, it is generally less appreciated and relatively underutilized. One clear limitation of the power of evolutionary analysis is the often-poor quality of sequences in the public databases, coupled with the inability of fully automated clustering and aligning procedures to reliably distinguish the good from the bad: Analyses are only as good as the underlying sequences and alignments. Our solution for ProPhylER was to curate each alignment to remove spurious sequences. Another limitation is the slippery operational meaning of “orthology” (Fitch 2000; Altenhoff and Dessimoz 2009). Restricting alignments to true orthologs (sequences related only by speciation events, with no intervening gene duplications) greatly limits scope and diversity, and affects specificity (Stone et al. 2005); but indiscriminately including every alignable paralog introduces functional divergence and affects sensitivity (Stone and Sidow 2005). Valid evolutionary inference requires the comparison of sequences that are related by descent and are functionally conserved. Pragmatically, this should be the sole criterion for sequence inclusion when the goal is constraint detection. This allows paralogs if the assumption of their functional conservation is reasonable, but also disallows functionally divergent orthologs. ProPhylER combines stringent automated clustering with curation to limit most of its clusters to homologous sequences that are likely to have retained the same precise biochemical function. The only exceptions are (as of now) 356 large protein families broken into subsets to allow interesting comparisons of paralogs (see Supplemental material), and in these cases, the subsets meet the strict homology criterion. Clearly the decisions whether to include paralogs or questionable sequences, or to group paralogs into subsets, all come down to judgment calls, but in ProPhylER's case, they are informed judgments by qualified curators. These investments in quality control advance ProPhylER toward realizing the full power of comparative sequence analysis of proteins.
Methods
Cluster creation
We retrieved protein sequences as gene translations from 13 fully sequenced genomes. We downloaded from Ensembl sequences from Homo sapiens, Mus musculus, Monodelphis domestica, Gallus gallus, Xenopus tropicalis, Danio rerio, Gasterosteus aculeatus, Ciona intestinalis, Drosophila melanogaster, Anopheles gambiae, and Caenorhabditis elegans; we obtained sequences for Saccharomyces cerevisiae from the Saccharomyces Genome Database; and we obtained sequences from Schizosaccharomyces pombe from the Sanger Institute. To create initial clusters, we performed all-against-all similarity searches among these sequences with WU-BLAST (BLASTP 2.0MP-WashU [04-May-2006], W Gish, pers. comm.), using an E-value cutoff based on sequence length and employing the “postsw” option to give local alignments (Smith and Waterman 1981) for all pairs of matching sequences. We scored each pair of sequences, awarding for each site where they aligned well to each other, and penalizing for each site where they aligned well to other sequences but not to each other. We built “loose” clusters with a greedy single-linkage algorithm, grouping sequences based on transitive similarity inferred from these scores. We then applied a stringent Normalized MinCut algorithm (Shi and Malik 2000) to iteratively break the clusters at their weakest connections (lowest scores). The algorithm involves solving eigenvalue problems, for which we use the Fortran library ARPACK (http://www.caam.rice.edu/software/ARPACK/). We included a heuristic that evaluates each proposed cut by examining the phylogenetic composition of the child clusters—that is, the species representation and number of sequences per species—and halts the process when the probability of separating orthologs becomes high. To correct for the stringency of the MinCut step, we then curated the clusters, using the criteria described above. To create the final clusters, we augmented the initial clusters with all eukaryotic sequences from UniProt. We performed WU-BLAST searches of cluster sequences using each UniProt sequence as query, and assigned the query to its closest-matching cluster above an E-value threshold. We repeated the above curation step at this point because the UniProt sequences provided additional information that helped clarify the relationships between clusters.
Alignment and tree building
Prior to building multiple alignments for the final clusters, we used several automated steps to identify and remove potential problem sequences. First, global pairwise alignments were created between all sequences in a given cluster from the same species, and isoforms were defined as pairs that are identical across their full length (duplicates), or identical across most of their length but interrupted by large stretches of low similarity (probable splice isoforms). A single isoform was chosen to include for alignment: the SWISS-PROT-derived isoform if present, otherwise, the longest isoform. Next, each cluster's sequences were multiply aligned using ProbCons (Do et al. 2005), and sequences that introduced numerous gaps to the alignment were marked for potential removal in the subsequent curation step. In cases in which ProbCons failed because of sequence number, length, or alignability, the sequences were aligned with MUSCLE (Edgar 2004).
We then curated each of the preliminary alignments as described above, evaluating the marked sequences for inclusion or removal, as well as removing any other obvious problem sequences. We then made final, multiple alignments of the remaining sequences using ProbCons. We built gene trees for each cluster with SEMPHY (Friedman et al. 2002), restricting analysis to the most confident positions in the multiple sequence alignment. To annotate the nodes in the gene trees as duplications or common species ancestors, we devised an algorithm that compared each gene tree to the NCBI taxonomy tree (Wheeler et al. 2008). The taxonomy is not a bifurcating species tree, so the algorithm considered multifurcations as consistent with any sequence of bifurcations. The algorithm classified nodes in the gene tree as consistent or inconsistent with the taxonomy. Consistent nodes were labeled as common species ancestors. Inconsistencies in branching patterns were further classified into definite duplications (when a node was an ancestor of different genes from the same organism) and possible duplications (all other cases).
Evolutionary constraint profiles
To produce profiles of evolutionary constraint, we first calculate single-site evolutionary rates in the alignment using maximum parsimony (ProtPars; J Felsenstein. 2005. Phylogeny Inference Package [PHYLIP], version 3.6. Distributed by the author. Department of Genome Sciences, University of Washington, Seattle). See Supplemental material and Supplemental Figure S2 for a comparison of different rate estimation methods. The guide tree topology is held constant (to that of the gene tree, above) for the calculation at each site. We normalize these rates to an average value of 1 across the alignment. We produce regional evolutionary rates using a modified version of ESF analysis (Simon et al. 2002). We apply sliding-windows weighted averaging to the normalized single-site rate values: The relative weight (wti) for the rate value at position i in the sliding window is:
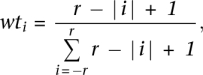 |
where r is the extent of the window to either side of the center position (so, the total window size is 2r + 1). The relative weight is thus greatest at the center position and decreases linearly on either side to the edges of the window. We assign the sum of all the weighted rate values in the window to the position in the alignment corresponding to the center of the window. (Note that this is mathematically equivalent to performing two successive rounds of arithmetic averaging using a window size of r + 1 [cf. Simon et al. 2002].) For ProPhylER we use a window size 17 positions wide (r = 8), which we determined empirically to provide the optimum balance between regional accuracy, feature resolution, and ease of interpreting profile shape (Supplemental material; Supplemental Fig. S3). To convert rates to constraints, we normalize the values to range between 0 and 1, and then invert by subtracting from 1 (because a region of low evolutionary rate is under high evolutionary constraint).
Physicochemical property profiles
To produce physicochemical property profiles, we first calculate values for each amino acid in each sequence of the multiple alignment for each of five properties: residue volume (Zamyatnin 1972), hydropathy (Kyte and Doolittle 1982), polarity (Engelman et al. 1986), and the hydrophobic moment assuming either alpha-helical or beta-strand conformations (Eisenberg et al. 1984). Next, we multiply the values for each sequence by a weighting factor that reflects the fractional contribution of that sequence to the total evolutionary diversity represented in the alignment (Stone and Sidow 2007). The property score for each alignment position is the sum of these values. We then smooth these single-position values using the same sliding-windows weighted averaging routine applied to the rate values, above, and normalize them to vary between 0 and 1.
MAPP impact predictions
We predict the impact of every possible amino acid substitution in an alignment using MAPP as described (Stone and Sidow 2005). Briefly, MAPP converts amino acid information at each alignment position into a single vector that summarizes the evolutionary importance of six physicochemical properties (hydropathy, polarity, charge, volume, and free energy in both alpha-helix and beta-strand conformations). It repeats the analysis after substituting the wild-type amino acid with the mutant under consideration and determines the significance of the difference between the two vectors using an F-test. We interpret the significance of this difference (reported as a P-value) as the probability the substitution will be tolerated.
The CrystalPainter
To color-code evolutionary constraint information onto molecular structures, we first produce a pairwise alignment between the PDB peptide sequence and its best-matching ProPhylER sequence (determined by BLAST), thereby mapping PDB residue numbers to ProPhylER cluster alignment coordinates. Next, we normalize that cluster's evolutionary constraint values to vary between 0 (most constrained) and 99 (least constrained). We then produce a modified PDB file with these normalized values replacing the values in the B-factor field for each atom of the corresponding residue. Most structure-viewing tools (including Jmol, used by ProPhylER's CrystalPainter) have the option to color the molecular image according to the B-factor values.
Mutation data sets
For details of the conversion of published mutation phenotypes to the numeric scores we used in accuracy calculations, see the Supplemental material.
We identified beta-globin homologs in a BLASTP search of SWISS-PROT, using human beta-globin as query (UniProt accession no. P68871). We identified homologs of lactose repressor in BLASTP searches of the Comprehensive Microbial Resource (http://blast.jcvi.org/cmr-blast/) and NCBI's bacterial genomes, using E. coli lactose repressor as query (UniProt accession no. P03023). For subsequent alignment, we used only “reciprocal best hits”—that is, sequences that were the top BLAST hit from their host species, and also identified lactose repressor as the top hit when used to query E. coli proteins. To identify lysozyme homologs, we used bacteriophage T4 lysozyme (UniProt accession no. P00720) as query in BLASTP or TBLASTN searches on Tulane University's T4-like Genome website (http://phage.bioc.tulane.edu/). To identify p53 homologs, we used human p53 (UniProt accession no. P04637) as query in local WU-BLAST searches of proteins from UniProt and Ensembl. We identified protease homologs in a BLASTP search of SWISS-PROT, using HIV-1 protease as query (UniProt accession no. P20892, residues 487 to 585).
We produced multiple sequence alignments and phylogenetic trees for the five sets as described above. We calculated single-site evolutionary rates using Bayesian Rate4Site (Mayrose et al. 2004), and produced regional evolutionary rate profiles from these as described above. We produced regional mutation tolerance profiles by applying to the single-position mutation tolerance scores the same sliding-windows weighted-averaging routine used to produce regional rate profiles. We determined accuracy as described in the main text.
Alignments, trees, evolutionary rates, and mutation tolerance scores for each of the five sets can be found in Supplemental materials.
Analyses of modified alignments
To create permuted alignments, we randomized the order of the amino acid columns and reassembled the alignments in the random column order. We also reordered the single-position mutation tolerance scores according to the same random order. As a result, any given column contained the same specific amino acids and was associated with the same mutation score before and after permutation. The phylogenetic tree also remains the same before and after permutation. For each data set, we created 1000 permuted alignments. We determined the accuracy of evolutionary rate profiles on permuted alignments in the same way as with nonpermuted alignments, except that we binned the sensitivity values and plotted the average on the ROC plots and calculated the average accuracy.
To create random subalignments of the 162 beta-globin sequences, we first picked random numbers between four and 161, then randomly drew that number of sequences from the full alignment (maintaining their relative alignment). We then recalculated branch lengths on the corresponding sub-trees with SEMPHY, keeping their topologies consistent with that of the full tree.
Footnotes
[Supplemental material is available online at http://www.genome.org. ProPhylER's interfaces, analyses, and data can be accessed at http://www.prophyler.org.]
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.097121.109.
References
- Altenhoff AM, Dessimoz C. Phylogenetic and functional assessment of orthologs inference projects and methods. PLoS Comput Biol. 2009;5:e1000262. doi: 10.1371/journal.pcbi.1000262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armon A, Graur D, Ben-Tal N. ConSurf: An algorithmic tool for the identification of functional regions in proteins by surface mapping of phylogenetic information. J Mol Biol. 2001;307:447–463. doi: 10.1006/jmbi.2000.4474. [DOI] [PubMed] [Google Scholar]
- Bell CE, Lewis M. A closer view of the conformation of the Lac repressor bound to operator. Nat Struct Biol. 2000;7:209–214. doi: 10.1038/73317. [DOI] [PubMed] [Google Scholar]
- Berezin C, Glaser F, Rosenberg J, Paz I, Pupko T, Fariselli P, Casadio R, Ben-Tal N. ConSeq: The identification of functionally and structurally important residues in protein sequences. Bioinformatics. 2004;20:1322–1324. doi: 10.1093/bioinformatics/bth070. [DOI] [PubMed] [Google Scholar]
- Bhalla K, Luo Y, Buchan T, Beachem MA, Guzauskas GF, Ladd S, Bratcher SJ, Schroer RJ, Balsamo J, DuPont BR, et al. Alterations in CDH15 and KIRREL3 in patients with mild to severe intellectual disability. Am J Hum Genet. 2008;83:703–713. doi: 10.1016/j.ajhg.2008.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boggon TJ, Murray J, Chappuis-Flament S, Wong E, Gumbiner BM, Shapiro L. C-cadherin ectodomain structure and implications for cell adhesion mechanisms. Science. 2002;296:1308–1313. doi: 10.1126/science.1071559. [DOI] [PubMed] [Google Scholar]
- Brown D, Sjölander K. Functional classification using phylogenomic inference. PLoS Comput Biol. 2006;2:e77. doi: 10.1371/journal.pcbi.0020077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho Y, Gorina S, Jeffrey PD, Pavletich NP. Crystal structure of a p53 tumor suppressor–DNA complex: Understanding tumorigenic mutations. Science. 1994;265:346–355. doi: 10.1126/science.8023157. [DOI] [PubMed] [Google Scholar]
- Crick FHC. On protein synthesis. Symp Soc Exp Biol. 1958;12:138–163. [PubMed] [Google Scholar]
- Dean AM, Neuhauser C, Grenier E, Golding GB. The pattern of amino acid replacements in alpha/beta-barrels. Mol Biol Evol. 2002;19:1846–1864. doi: 10.1093/oxfordjournals.molbev.a004009. [DOI] [PubMed] [Google Scholar]
- Deluca TF, Wu IH, Pu J, Monaghan T, Peshkin L, Singh S, Wall DP. Roundup: A multi-genome repository of orthologs and evolutionary distances. Bioinformatics. 2006;22:2044–2046. doi: 10.1093/bioinformatics/btl286. [DOI] [PubMed] [Google Scholar]
- Do CB, Mahabhashyam MS, Brudno M, Batzoglou S. ProbCons: Probabilistic consistency-based multiple sequence alignment. Genome Res. 2005;15:330–340. doi: 10.1101/gr.2821705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg D, Schwarz E, Komaromy M, Wall R. Analysis of membrane and surface protein sequences with the hydrophobic moment plot. J Mol Biol. 1984;179:125–142. doi: 10.1016/0022-2836(84)90309-7. [DOI] [PubMed] [Google Scholar]
- The ENCODE Project Consortium. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman DM, Steitz TA, Goldman A. Identifying nonpolar transbilayer helices in amino acid sequences of membrane proteins. Annu Rev Biophys Biophys Chem. 1986;15:321–353. doi: 10.1146/annurev.bb.15.060186.001541. [DOI] [PubMed] [Google Scholar]
- Fay JC, Wu CI. Sequence divergence, functional constraint, and selection in protein evolution. Annu Rev Genomics Hum Genet. 2003;4:213–235. doi: 10.1146/annurev.genom.4.020303.162528. [DOI] [PubMed] [Google Scholar]
- Fermi G, Perutz MF, Shaanan B, Fourme R. The crystal structure of human deoxyhaemoglobin at 1.74 Å resolution. J Mol Biol. 1984;175:159–174. doi: 10.1016/0022-2836(84)90472-8. [DOI] [PubMed] [Google Scholar]
- Finn RD, Tate J, Mistry J, Coggill PC, Sammut SJ, Hotz HR, Ceric G, Forslund K, Eddy SR, Sonnhammer EL, et al. The Pfam protein families database. Nucleic Acids Res. 2008;36:D281–D288. doi: 10.1093/nar/gkm960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitch WM. Homology: A personal view on some of the problems. Trends Genet. 2000;16:227–231. doi: 10.1016/s0168-9525(00)02005-9. [DOI] [PubMed] [Google Scholar]
- Fitch WM, Markowitz E. An improved method for determining codon variability in a gene and its application to the rate of fixation of mutations in evolution. Biochem Genet. 1970;4:579–593. doi: 10.1007/BF00486096. [DOI] [PubMed] [Google Scholar]
- Flicek P, Aken BL, Beal K, Ballester B, Caccamo M, Chen Y, Clarke L, Coates G, Cunningham F, Cutts T, et al. Ensembl 2008. Nucleic Acids Res. 2008;36:D707–D714. doi: 10.1093/nar/gkm988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman N, Ninio M, Pe'er I, Pupko T. A structural EM algorithm for phylogenetic inference. J Comput Biol. 2002;9:331–353. doi: 10.1089/10665270252935494. [DOI] [PubMed] [Google Scholar]
- Hanley JA, McNeil BJ. The meaning and use of the area under a receiver operating characteristic (ROC) curve. Radiology. 1982;143:29–36. doi: 10.1148/radiology.143.1.7063747. [DOI] [PubMed] [Google Scholar]
- Heinicke S, Livstone MS, Lu C, Oughtred R, Kang F, Angiuoli SV, White O, Botstein D, Dolinski K. The Princeton Protein Orthology Database (P-POD): A comparative genomics analysis tool for biologists. PLoS One. 2007;2:e766. doi: 10.1371/journal.pone.0000766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollstein M, Rice K, Greenblatt MS, Soussi T, Fuchs R, Sorlie T, Hovig E, Smith-Sorensen B, Montesano R, Harris CC. Database of p53 gene somatic mutations in human tumors and cell lines. Nucleic Acids Res. 1994;22:3551–3555. [PMC free article] [PubMed] [Google Scholar]
- Hughes I, Binkley J, Hurle B, Green ED NISC Comparative Sequencing Program. Sidow A, Ornitz DM. Identification of the Otopetrin Domain, a conserved domain in vertebrate otopetrins and invertebrate otopetrin-like family members. BMC Evol Biol. 2008;8:41. doi: 10.1186/1471-2148-8-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov DB, Philippova MP, Tkachuk VA. Structure and functions of classical cadherins. Biochemistry (Mosc) 2001;66:1174–1186. doi: 10.1023/a:1012445316415. [DOI] [PubMed] [Google Scholar]
- Jackson PJ, Douglas NR, Chai B, Binkley J, Sidow A, Barsh GS, Millhauser GL. Structural and molecular evolutionary analysis of Agouti and Agouti-related proteins. Chem Biol. 2006;13:1297–1305. doi: 10.1016/j.chembiol.2006.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashuk CS, Stone EA, Grice EA, Portnoy ME, Green ED, Sidow A, Chakravarti A, McCallion AS. Phenotype–genotype correlation in Hirschsprung disease is illuminated by comparative analysis of the RET protein sequence. Proc Natl Acad Sci. 2005;102:8949–8954. doi: 10.1073/pnas.0503259102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M. The neutral theory of molecular evolution. Cambridge University Press; Cambridge, UK: 1983. [Google Scholar]
- Ko DC, Binkley J, Sidow A, Scott MP. The integrity of a cholesterol-binding pocket in Niemann-Pick C2 protein is necessary to control lysosome cholesterol levels. Proc Natl Acad Sci. 2003;100:2518–2525. doi: 10.1073/pnas.0530027100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroki R, Weaver LH, Matthews BW. A covalent enzyme-substrate intermediate with saccharide distortion in a mutant T4 lysozyme. Science. 1993;262:2030–2033. doi: 10.1126/science.8266098. [DOI] [PubMed] [Google Scholar]
- Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- Landau M, Mayrose I, Rosenberg Y, Glaser F, Martz E, Pupko T, Ben-Tal N. ConSurf 2005: The projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res. 2005;33:W299–W302. doi: 10.1093/nar/gki370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Stoeckert CJ, Jr, Roos DS. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13:2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Coghlan A, Ruan J, Coin LJ, Heriche JK, Osmotherly L, Li R, Liu T, Zhang Z, Bolund L, et al. TreeFam: A curated database of phylogenetic trees of animal gene families. Nucleic Acids Res. 2006;34:D572–D580. doi: 10.1093/nar/gkj118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtarge O, Bourne HR, Cohen FE. An evolutionary trace method defines binding surfaces common to protein families. J Mol Biol. 1996;257:342–358. doi: 10.1006/jmbi.1996.0167. [DOI] [PubMed] [Google Scholar]
- Lin RJ, Blumenkranz MS, Binkley J, Wu K, Vollrath D. A novel His158Arg mutation in TIMP3 causes a late-onset form of Sorsby fundus dystrophy. Am J Ophthalmol. 2006;142:839–848. doi: 10.1016/j.ajo.2006.06.003. [DOI] [PubMed] [Google Scholar]
- Loeb DD, Swanstrom R, Everitt L, Manchester M, Stamper SE, Hutchison CA., III Complete mutagenesis of the HIV-1 protease. Nature. 1989;340:397–400. doi: 10.1038/340397a0. [DOI] [PubMed] [Google Scholar]
- Marchler-Bauer A, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, Fong JH, Geer LY, Geer RC, Gonzales NR, Gwadz M, et al. CDD: Specific functional annotation with the Conserved Domain Database. Nucleic Acids Res. 2009;37:D205–D210. doi: 10.1093/nar/gkn845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markiewicz P, Kleina LG, Cruz C, Ehret S, Miller JH. Genetic studies of the lac repressor. XIV. Analysis of 4000 altered Escherichia coli lac repressors reveals essential and non-essential residues, as well as “spacers” which do not require a specific sequence. J Mol Biol. 1994;240:421–433. doi: 10.1006/jmbi.1994.1458. [DOI] [PubMed] [Google Scholar]
- Matsuya A, Sakate R, Kawahara Y, Koyanagi KO, Sato Y, Fujii Y, Yamasaki C, Habara T, Nakaoka H, Todokoro F, et al. Evola: Ortholog database of all human genes in H-InvDB with manual curation of phylogenetic trees. Nucleic Acids Res. 2008;36:D787–D792. doi: 10.1093/nar/gkm878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayrose I, Graur D, Ben-Tal N, Pupko T. Comparison of site-specific rate-inference methods for protein sequences: Empirical Bayesian methods are superior. Mol Biol Evol. 2004;21:1781–1791. doi: 10.1093/molbev/msh194. [DOI] [PubMed] [Google Scholar]
- Miller M, Schneider J, Sathyanarayana BK, Toth MV, Marshall GR, Clawson L, Selk L, Kent SB, Wlodawer A. Structure of complex of synthetic HIV-1 protease with a substrate-based inhibitor at 2.3 Å resolution. Science. 1989;246:1149–1152. doi: 10.1126/science.2686029. [DOI] [PubMed] [Google Scholar]
- Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng PC, Henikoff S. Predicting the effects of amino acid substitutions on protein function. Annu Rev Genomics Hum Genet. 2006;7:61–80. doi: 10.1146/annurev.genom.7.080505.115630. [DOI] [PubMed] [Google Scholar]
- Olivier M, Eeles R, Hollstein M, Khan MA, Harris CC, Hainaut P. The IARC TP53 database: New online mutation analysis and recommendations to users. Hum Mutat. 2002;19:607–614. doi: 10.1002/humu.10081. [DOI] [PubMed] [Google Scholar]
- Pietrokovski S. Searching databases of conserved sequence regions by aligning protein multiple-alignments. Nucleic Acids Res. 1996;24:3836–3845. doi: 10.1093/nar/24.19.3836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramensky V, Bork P, Sunyaev S. Human non-synonymous SNPs: server and survey. Nucleic Acids Res. 2002;30:3894–3900. doi: 10.1093/nar/gkf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remm M, Storm CE, Sonnhammer EL. Automatic clustering of orthologs and in-paralogs from pairwise species comparisons. J Mol Biol. 2001;314:1041–1052. doi: 10.1006/jmbi.2000.5197. [DOI] [PubMed] [Google Scholar]
- Rennell D, Bouvier SE, Hardy LW, Poteete AR. Systematic mutation of bacteriophage T4 lysozyme. J Mol Biol. 1991;222:67–88. doi: 10.1016/0022-2836(91)90738-r. [DOI] [PubMed] [Google Scholar]
- Sander C, Schneider R. Database of homology-derived protein structures and the structural meaning of sequence alignment. Proteins. 1991;9:56–68. doi: 10.1002/prot.340090107. [DOI] [PubMed] [Google Scholar]
- Schneider A, Dessimoz C, Gonnet GH. OMA Browser—exploring orthologous relations across 352 complete genomes. Bioinformatics. 2007;23:2180–2182. doi: 10.1093/bioinformatics/btm295. [DOI] [PubMed] [Google Scholar]
- Shi J, Malik J. Normalized cuts and image segmentation. IEEE Trans Pattern Anal Mach Intell. 2000;22:888–905. [Google Scholar]
- Shimoyama Y, Shibata T, Kitajima M, Hirohashi S. Molecular cloning and characterization of a novel human classic cadherin homologous with mouse muscle cadherin. J Biol Chem. 1998;273:10011–10018. doi: 10.1074/jbc.273.16.10011. [DOI] [PubMed] [Google Scholar]
- Sidow A. Sequence first. Ask questions later. Cell. 2002;111:13–16. doi: 10.1016/s0092-8674(02)01003-6. [DOI] [PubMed] [Google Scholar]
- Simon AL, Stone EA, Sidow A. Inference of functional regions in proteins by quantification of evolutionary constraints. Proc Natl Acad Sci. 2002;99:2912–2917. doi: 10.1073/pnas.042692299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith TF, Waterman MS. Identification of common molecular subsequences. J Mol Biol. 1981;147:195–197. doi: 10.1016/0022-2836(81)90087-5. [DOI] [PubMed] [Google Scholar]
- Stone EA, Sidow A. Physicochemical constraint violation by missense substitutions mediates impairment of protein function and disease severity. Genome Res. 2005;15:978–986. doi: 10.1101/gr.3804205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone EA, Sidow A. Constructing a meaningful evolutionary average at the phylogenetic center of mass. BMC Bioinformatics. 2007;8:222. doi: 10.1186/1471-2105-8-222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone EA, Cooper GM, Sidow A. Trade-offs in detecting evolutionarily constrained sequence by comparative genomics. Annu Rev Genomics Hum Genet. 2005;6:143–164. doi: 10.1146/annurev.genom.6.080604.162146. [DOI] [PubMed] [Google Scholar]
- Suckow J, Markiewicz P, Kleina LG, Miller J, Kisters-Woike B, Muller-Hill B. Genetic studies of the Lac repressor. XV: 4000 single amino acid substitutions and analysis of the resulting phenotypes on the basis of the protein structure. J Mol Biol. 1996;261:509–523. doi: 10.1006/jmbi.1996.0479. [DOI] [PubMed] [Google Scholar]
- Tatusov RL, Koonin EV, Lipman DJ. A genomic perspective on protein families. Science. 1997;278:631–637. doi: 10.1126/science.278.5338.631. [DOI] [PubMed] [Google Scholar]
- Wheeler DL, Barrett T, Benson DA, Bryant SH, Canese K, Chetvernin V, Church DM, Dicuccio M, Edgar R, Federhen S, et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2008;36:D13–D21. doi: 10.1093/nar/gkm1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamyatnin AA. Protein volume in solution. Prog Biophys Mol Biol. 1972;24:107–123. doi: 10.1016/0079-6107(72)90005-3. [DOI] [PubMed] [Google Scholar]
- Zheng N, Fraenkel E, Pabo CO, Pavletich NP. Structural basis of DNA recognition by the heterodimeric cell cycle transcription factor E2F-DP. Genes & Dev. 1999;13:666–674. doi: 10.1101/gad.13.6.666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuckerkandl E, Pauling L. Molecules as documents of evolutionary history. J Theor Biol. 1965;8:357–366. doi: 10.1016/0022-5193(65)90083-4. [DOI] [PubMed] [Google Scholar]