

Abstract
Three photaffinity labeled derivatives of epothilone D were prepared by total synthesis, using efficient novel asymmetric synthesis methods for the preparation of two important synthetic building blocks. The key step for the asymmetric synthesis of (S,E)-3-(tert-butyldimethylsilyloxy)-4-methyl-5-(2-methylthiazol-4-yl)pent-4-enal involved a ketone reduction with (R)-Me-CBS-oxazaborolidine. For the synthesis of (5S)-5,7-di-[(tert-butyldimethylsilyl)oxy]-4,4-dimethylheptan-3-one an asymmetric Noyori reduction of a β-ketoester was employed. The C26 hydroxyepothilone D derivative was constructed following a well-established total synthesis strategy and the photoaffinity labels were attached to the C26 hydroxyl group. The photoaffinity analogues were tested in a tubulin assembly assay and for cytotoxicity against MCF-7 and HCT-116 cancer cell lines. The 3- and 4-azidobenzoic acid analogues were found to be as active as epothilone B in a tubulin assembly assay, but demonstrated significantly reduced cellular cytotoxicity compared to epothilone B. The benzophenone analogue was inactive in both assays. Docking and scoring studies were conducted that suggested that the azide analogues can bind to the epothilone binding site, but that the benzophenone analogue undergoes a sterically driven ligand rearrangement that interrupts all hydrogen bonding and therefore protein binding. Photoaffinity labeling studies with the 3-azidobenzoic acid derivative did not identify any covalently labeled peptide fragments, suggesting that the phenylazido side chain was predominantly solvent-exposed in the bound conformation.
Introduction
Since the isolation of the epothilones from Sorangium cellulosum by Höfle and collaborators at the GBF,1 and the determination of their biological mode of action by researchers at Merck,2 the epothilones have been explored for their potential as anti-tumor agents. The epothilones have been the subject of numerous elegant total syntheses and hundreds of analogues have been synthesized, some with improved biological profiles.3–10 The epothilones have the same mechanism of action, microtubule hyperstabilization, as paclitaxel, which is a highly successful drug in the treatment of a variety of cancers.2 The epothilones are cytotoxic to cancer cell lines and exhibit excellent activity against multi-drug resistant cell lines, in which the activity of paclitaxel is diminished.2,11 Patupilone (epothilone B),12 sagopilone (a synthetic analog of epothilone B),13 21-amino epothilone B,14 and 9,10-didehydroepothilone D15 are presently undergoing clinical trials and the aza analogue of epothilone B, ixabepilone,16 has received FDA approval for the treatment of metastatic breast cancer.16
Information on the nature of the binding interactions between the epothilones and β-tubulin comes from the 2.9 Å electron crystal structure of protofilament sheets of bovine brain tubulin stabilized by epothilone A.17 Further binding studies were carried out using NMR to analyze epothilone bound to unpolymerized α,β-tubulin dimer in solution.18 Photoaffinity labeling represents another approach to identify sites of interaction between a small molecule and a protein and has been used successfully in the study of paclitaxel-tubulin interactions.19–23 This work focuses on the design of photoaffinity labels of epothilone D that could be used as tools to determine those amino acid residues that are in close proximity to the epothilone molecule. With a better understanding of the epothilone binding site, the structure of the epothilones can be modified to obtain the maximum selectivity for a potential cancer chemotherapeutic agent. Herein, the synthetic approach to photoaffinity labels attached to C26 hydroxyepothilone D is described (Scheme 1). This position was selected because Nicolaou and collaborators had shown that this site tolerates structural modifications without loss of activity.24 The biological results obtained with C26 hydroxyepothilone D photoaffinity labels in tubulin assembly and cytotoxicity assays are also presented.
Scheme 1.

The disconnections for the total synthesis of the C26 hydroxyepothilone D are detailed in Scheme 1 and follow a well-precedented strategy,24,25 using three key precursors: the C1–C6 ketone A, the C7–C12 Wittig ester B, and the C13–C20 thiazole aldehyde C. Novel efficient asymmetric synthesis methods for the preparation of two of the three building blocks, required for the total synthesis, were developed. The key step for the asymmetric synthesis of (S,E)-3-(tert-butyldimethylsilyloxy)-4-methyl-5-(2-methylthiazol-4-yl)pent-4-enal (C) involved a ketone reduction with (R)-Me-CBS-oxazaborolidine.26 For the synthesis of (5S)-5,7-di-[(tert-butyldimethylsilyl)oxy]-4,4-dimethylheptan-3-one (A) an asymmetric Noyori reduction27 of a β-ketoester was employed.
Results and Discussion
The synthesis of thiazole aldehyde C28 began with the reaction of aldehyde 229 with β-ketophosphonate 3 (Scheme 2).30 These precursors underwent the Paterson modification31 of the Horner-Emmons-Wadsworth reaction using barium hydroxide and provided the desired enone 4 with >20:1 E:Z selectivity. The stereochemistry at the C15 position was then established using Corey’s (R)-Me-CBS-oxazaborolidine26 to reduce the enone 4. On small scale the reduction was carried out in good yield and with high enantiomeric excess (95% ee) using (R)-Me-CBS-oxazaborolidine and borane dimethylsulfide. Upon scale-up, however, a decrease in ee to 89% was observed. The reaction conditions were studied and the catalyst loading and the addition rate were determined to be the key parameters for obtaining high enantiomeric excess. (R)-Me-CBS-oxazaborolidine (0.5 equiv) and borane dimethylsulfide (1.5 equiv) were combined and enone 4 was transferred dropwise via syringe pump over 15 hours. Following work-up and chromatography the required S-alcohol 5 was obtained from the optimized conditions in 98% yield and 92–95% enantiomeric excess. The enantiomeric excess was determined on a chiral column using racemic 5 as the standard. The resulting secondary alcohol 5 was protected as its TBS ether, followed by selective deprotection of the primary TBS group using HF in the presence of glass splinters.32,33 The primary alcohol was then oxidized with the Dess-Martin periodinane and provided known aldehyde 6.34 We then followed the Nicolaou group protocol for the synthesis of intermediate 7.35,36
Scheme 2.

a) Ba(OH)2.8H2O, THF, then 2 in THF: H2O (40:1) 0 °C; b) (R)-2-Me-CBS-oxazaborolidine (0.5 eq), BH3.Me2S (1.5 eq), CH2Cl2, 0 °C, then 4, 0 °C, 2 h, then ethanolamine; c) TBSOTf, 2,6-lutidine, CH2Cl2, −78 °C; d) HF (40% aq.), glass splinters, MeCN:Et2O (1:1), 0 °C; e) Dess-Martin periodinane, CH2Cl2.
The key step for the synthesis of fragment A was the asymmetric Noyori reduction27 of β-ketoester 9 as shown in Scheme 3. The anion of methyl isobutyrate was acylated with 3-(benzyloxy)propanoyl chloride to furnish the β-keto ester 9. The best results for the asymmetric Noyori reduction37,38 were obtained when the catalyst was generated in situ from bis-(2-methylallyl)cycloocta-1,5-diene ruthenium and (S)-BINAP in deoxygenated acetone in the presence of HBr. High enantiomeric excess (>95% ee) was consistently obtained through hydrogenation with low catalyst loading (1.4 mol%) in deoxygenated methanol, however, due to the hindered nature of the β-ketoester the conversion was incomplete. The optimized hydrogenation conditions for the hindered β-ketoester required heating at 60 °C at 60 psi for 110 hours to maximize the yield. Following chromatography, β-hydroxyester 10 was obtained in 92% yield and 95% enantiomeric excess as determined by chiral HPLC. β-Hydroxyester 10 was subsequently debenzylated via catalytic hydrogenation with 10% Pd/C and the resulting diol was protected as the bis-TBS ether. Conversion of the ester to the corresponding ethylphenyl sulfone39 followed by reductive cleavage of the sulfone provided the C1–C6 ketone 11.34,40 This synthesis constitutes one of the shortest ways to access the C1–C6 fragment. Another advantage of this method is low catalyst loading and the fact that the catalyst can be recycled.
Scheme 3.

a) Lithium isopropylcyclohexylamine, THF, −78 °C, 3-(benzyloxy)propanoyl chloride; b) RuBr2(S)-binap, H2, MeOH, 60 psi, 60 °C, 96 hours; c) H2, 10% Pd/C, THF; d) TBSOTf, 2,6-lutidine, CH2Cl2; e) PhSO2Et, n-BuLi, −78 °C; f) Na(Hg), Na2HPO4, MeOH; g) LDA, −78 °C, ketone 11, −78 °C to −40 °C, aldehyde 7, 2 min.
The subsequent chemistry shown in Scheme 3 for the synthesis of 26-hydroxyepothilone D follows the work by Nicolaou and collaborators.24,25 The primary alcohol in 13 was acylated with 3-azidobenzoic acid using the peptide coupling reagent O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HBTU) in the presence of diisopropylethylamine (DIPEA) (Scheme 4). The TBS groups were then removed using TFA to provide the desired 3-azido photoaffinity analogue 14 of C26 hydroxyepothilone D. This process was repeated to generate the 4-azido photoaffinity analogue 15, and the benzophenone analogue 16 (Scheme 4).
Scheme 4.

The 3- and 4-azido analogues and the benzophenone analogues of 14–16 were tested for tubulin assembly-promoting activity and cytotoxicity against the MCF-7 breast cancer and the HCT-116 colon cancer cell lines (Table 1). The azido compounds were as active as epothilone B in the tubulin assembly assay, and demonstrated cytotoxicity against both cell lines but were significantly less active than epothilone B. This may be due to differences in transport across the cell membrane. The benzophenone analogue did not promote tubulin assembly, nor did it show cytotoxicity in either cancer cell line. Photolabeling studies were carried out with the 3-azido analogue with bovine brain tubulin polymerized in the presence of the analogue. After UV irradiation and subsequent examination of tryptic digests of the protein for adducts by LC-MS and LC-MS/MS studies no covalently labeled peptide fragments could be identified, suggesting that the phenylazido side chain was predominantly solvent-exposed in the bound conformation.
Table 1.
Cytotoxicity and Tubulin Assembly ED50 Valuesa
| Compound | ED50, nM MCF-7 | ED50, nM HCT-116 | ED50, μM Tubulin Assembly |
|---|---|---|---|
| 14 | >200 | 400 | 2.0 |
| 15 | >200 | 500 | 2.5 |
| 16 | >1000 | >1000 | >20 |
| Epothilone B | 1.0 | 1.8 | 2.0 |
Assays were carried out as described in reference 41.
Docking and scoring studies were conducted to probe tubulin binding modes and ligand-receptor interactions for epothilone B as well as for compounds 14, 15 and 16. Docking validation was carried out by comparing the docked configuration of epothilone A to the experimental configuration (RMSD = 1.575 Å). The initial conformations of epothilone B and compounds 14, 15 and 16 were prepared by modifying the experimental (X-ray) bound configuration of epothilone A,17 and optimizing the resulting geometries using the MMFF94s force field in MOE (Chemical Computing Group, Inc.). Figure 2 (a) shows the docked configuration of epothilone B in the tubulin binding site; Figure 2 (b) and (c) show the docked configurations of compounds 14 and 15 (respectively). More details on the docking and scoring protocols are provided in the Experimental Section. Comparing all three predicted bound configurations illustrates that epothilone B, 14 and 15 all exhibit similar key hydrogen-bonding interactions with the tubulin active site. The relatively minor rearrangements of azido compounds 14 and 15 in the active site with respect to epothilone B do not disrupt these interactions to any significant extent; hence their similar activities in the tubulin assembly assay. Although hydrogen bonding between the thiazole ring and His227 is not predicted to occur in compound 15, the docking suggests that the loss of this hydrogen bond is not detrimental to activity as long as the other key interactions are preserved. However, in the case of compound 16 (Figure 2 (d)), van der Waals clashes between the receptor and the benzophenone moiety lead to sterically driven ligand rearrangement that disrupts the entire hydrogen-bonding network and consequently destroys tubulin assembly-promoting activity for this compound.
Figure 2.
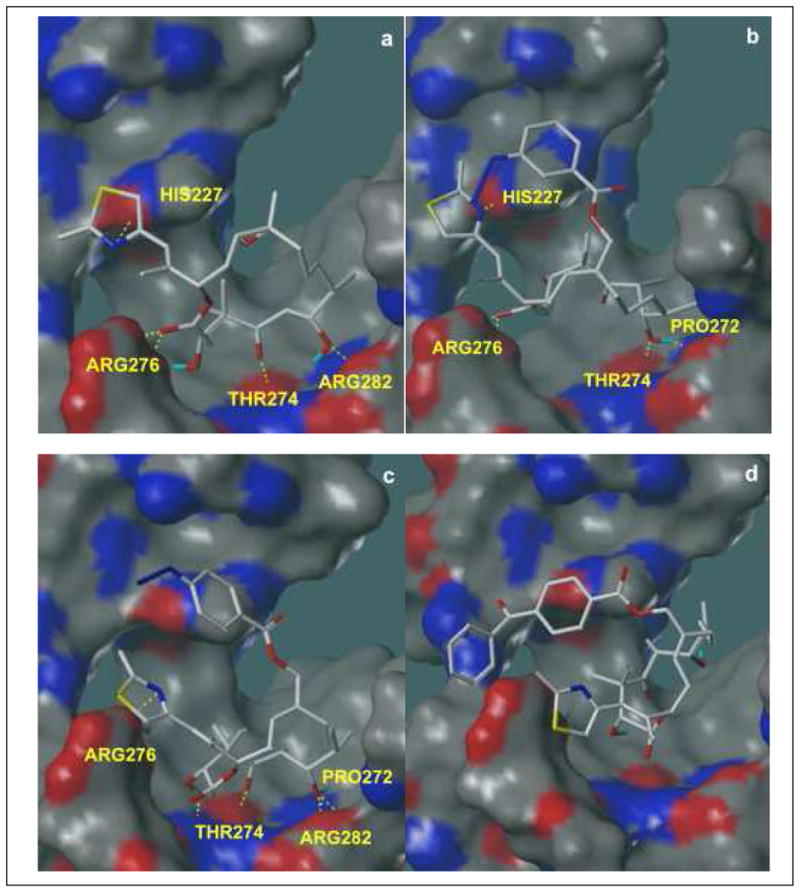
Docked configurations (Surflex-Dock, Tripos, Inc.) of epothilone B (a), compound 14 (b), compound 15 (c) and compound 16 (d) shown with MOLCAD (Tripos, Inc.) electron density surfaces of the tubulin active site (1TVK1), onto which hydrogen bond donor and acceptor regions have been mapped. Red areas represent hydrogen bond donors; blue areas represent hydrogen bond acceptors; and gray indicates regions in which no hydrogen bonding takes place. Key ligand-receptor interactions are shown in these pictures. In the docked conformation of epothilone B (a), hydrogen bonding occurs between the thiazole nitrogen and the imidazole NH of His227; between the C1 carbonyl and two amino groups in Arg276; between the C3 carbonyl and backbone NH of Thr274; and between C5 OH and one amino group in Arg282. In the docked conformation of 14 (b), hydrogen bonding occurs between the thiazole nitrogen and the imidazole NH of His227; between the C1 carbonyl and two amino groups in Arg276; between the C7 OH and backbone carbonyl of Pro272; and between C7 OH and backbone NH of Thr274. In the docked configuration of 15 (c), hydrogen bonding occurs between the thiazole nitrogen and one of the amino groups of Arg276; between the C3 OH and the backbone carbonyl of Thr274; between the C5 carbonyl and backbone NH of Thr274; between the C7 OH and the two amino groups in Arg282; and between the C7 OH and backbone carbonyl of Pro272. Overall, the hydrogen bonding networks between receptor and ligand are preserved in (b) and (c). However, all of these interactions disappear in the docked conformation of 16 (d) due to sterically driven ligand rearrangement. As shown, the molecule rearranges to avoid steric clashes between the receptor and the bulky group on C12.
Experimental Section
Diethyl 5-(tert-Butyldimethylsilyloxy)-3-oxopentan-2-ylphosphonate (3)
Diethyl ethanephosphonate (38.6 g, 232 mmol, 3.20 equiv) was placed in THF (150 mL) along with 4Å molecular sieves. The reaction temperature was lowered to −78 °C and n-BuLi (160 mL, 225 mmol, 3.10 equiv) was added dropwise over two hours via a dropping funnel. The reaction mixture was stirred at −78 °C for an additional hour and then methyl 3-(tert-butyldimethylsilyloxy)propanoate (prepared by silylation42 of commercially available methyl 3-hydroxypropanoate) was added dropwise via cannula. The reaction was stirred at −78 °C for two hours and then quenched with methanol (10 mL). Saturated aqueous NH4Cl (80 mL) was added and the mixture was allowed to warm to room temperature. The organic layer was decanted and concentrated. The aqueous layer was filtered to remove the molecular sieves. Brine was added and the aqueous layer was washed four times with EtOAc. The organic layer was dried over Na2SO4 and concentrated. Three columns on silica gel using a gradient of 10–25% ethyl acetate in hexanes provided 17.9 g of the β-ketophosphonate (70% yield). This was necessary as the product ran together with the starting phosphonate and the mixed fractions were subjected to further column chromatography. Distillation was attempted on a small scale, but resulted in decomposition: 1H NMR (400 MHz, CDCl3) δ 4.17-4.12 (m, 4 H), 3.93-3.90 (t, J = 6.3 Hz, 2 H), 3.40-3.25 (dq, J = 7.1, 7.1 Hz, 1 H), 2.94-2.84 (m, 2 H), 1.38-1.33 (m, 6 H), 0.94 (s, 3 H), 0.89 (s, 9 H), 0.06 (s, 6 H); 13C NMR (100 MHz, CDCl3) δ 205.2, 62.8, 59.2, 48.1, 46.9, 46.3, 26.1, 18.4, 16.6, −5.2; IR (neat) 2960, 2925, 2850, 1711, 1480, 1396 cm−1; MS (CI) = 353 (M+H+); HRMS m/e calc’d for C15H34O5PSi (M+H+): 353.1905, found 353.1911.
(E)-5-(tert-Butyldimethylsilyloxy)-2-methyl-1-(2-methylthiazol-4-yl)pent-1-en-3-one (4)
Barium hydroxide octahydrate (12.7 g, 40.2 mmol, 0.800 equiv) was added to β-ketophosphonate 3 (17.7 g, 50.2 mmol, 1.00 equiv) in THF (143 mL) and stirred at room temperature until it dissolved. The mixture was then cooled to 0 °C and aldehyde 229 (6.40 g, 50.2 mmol, 1.00 equiv) dissolved in THF (143 mL) and water (3.27 mL) was added dropwise. The reaction mixture was stirred at 0 °C for two hours. The mixture was quenched with saturated aqueous sodium bicarbonate and passed through Celite. After the addition of brine, the aqueous layer was extracted five times with ethyl acetate and the combined organics were dried over sodium sulfate and concentrated. Flash column chromatography of the crude material on silica gel using a gradient of 20%–30% ether in hexanes gave 11.15 g of the enone 4 (68%): 1H NMR (400 MHz, CDCl3) δ 7.56 (s, 1 H), 7.36 (s, 1 H), 4.00 (t, J = 6.7 Hz, 2 H), 3.05 (t, J = 6.7 Hz, 2 H), 2.78 (s, 3 H), 2.25 (s, 3 H), 0.88 (s, 9 H), 0.07 (s, 6 H); 13C NMR (100 MHz, CDCl3) δ 201.5, 165.8, 152.3, 138.0, 131.9, 121.7, 60.1, 41.1, 26.3, 19.7, 18.7, 13.7; IR (neat) 3100, 2960, 2940, 2860, 1665, 1625, 1468 cm−1; MS (+FAB) = 326 (M+H+); HRMS m/e calc’d for C16H28NO2SSi (M+H+): 326.1610, found 326.1625.
(S,E)-5-(tert-Butyldimethylsilyloxy)-2-methyl-1-(2-methylthiazol-4-yl)pent-1-en-3-ol (5)40
(R)-Me-CBS-oxazaborolidine (16.6 mL, 16.6 mmol, 1.0 M solution in toluene, 0.500 equiv) was placed in a 500 mL flask and the toluene was removed in vacuo. The residual solid was dissolved in CH2Cl2 (76 mL) and was cooled to 0 °C. Borane dimethylsulfide complex (50.0 mL, 49.8 mmol, 1.0 M in CH2Cl2, 1.50 equiv) was added dropwise and this mixture was stirred for one hour. Enone 4 (11.0 g, 33.2 mmol, 1.00 equiv) dissolved in CH2Cl2 (65 mL) was added dropwise via syringe pump over a period of fifteen hours, followed by a CH2Cl2 rinse (20 mL) over 4 hours. The reaction was allowed to warm to room temperature over 4 hours, and methanol (20 mL) was carefully added followed by ethanolamine (20 mL). Twelve hours later additional ethanolamine (20 mL) was added. The reaction mixture was stirred for another 24 hours at room temperature and then quenched with saturated aqueous ammonium chloride. The organic and aqueous layers were separated and aqueous layer was extracted three times with CH2Cl2. The organic layer was subsequently washed with brine. The combined aqueous layer was washed once with CH2Cl2. The combined organic layer was then dried over sodium sulfate and concentrated. The crude material was then purified by column chromatography on silica gel using a 20–30% ethyl acetate in hexanes gradient as eluent. The desired alcohol was obtained in 98% yield (10.8 g) and in 93% enantiomeric excess. The enantiomeric excess was determined by chiral HPLC on a Chiracel OD-H column at 254 nm using 90:10 hexanes/isopropanol at 0.8 mL/min as the mobile phase. The retention times major: 5.5 minutes, minor: 7.5 minutes were compared to the racemic alcohol for verification: 1H NMR (400 MHz, CDCl3) δ 6.95 (s, 1 H), 6.63 (s, 1 H), 4.42-4.38 (m, 1 H), 3.95-3.86 (m, 2 H), 3.55 (d, J = 2.7 Hz, 1 H), 2.73 (s, 3 H), 2.07 (s, 3 H), 1.85 (q, J = 5.7 Hz, 2 H), 0.93 (s, 9 H), 0.11 (s, 3 H), 0.10 (s, 3 H); 13C NMR (100 MHz, CDCl3) δ 164.8, 153.6, 142.1, 118.6, 115.8, 62.7, 37.4, 26.2, 19.7, 18.6, 15.3, −5.1; IR (neat) 3400-3350 b, 2950, 2940, 2850, 1510, 1470, 1256 cm−1; MS (+FAB) = 328 (M+H+); HRMS m/e calc’d for C16H30NO2SSi (M+H+): 328.1767, found 328.1789; [α]D20 −7.2° (c = 0.76, CHCl3).
(±)-(E)-5-(tert-Butyldimethylsilyloxy)-2-methyl-1-(2-methylthiazol-4-yl)pent-1-en-3-ol ((±)-5)
The enone 4 (10 mg, 0.031 mmol, 1.0 equiv) was dissolved in CH2Cl2 (0.5 mL) and the temperature was lowered to −78 °C. DIBAL-H (0.062 mL, 0.062 mmol, 1.0 M, 2.0 equiv) was added dropwise and the reaction was stirred for 1.25 hours. The reaction was quenched with methanol (0.5 mL) and additional CH2Cl2 (5.0 mL) and saturated potassium sodium tartrate solution (5.0 mL) were added. The mixture was stirred for two hours and then extracted four times with CH2Cl2. The combined organic layer was dried over Na2SO4 and concentrated. Purification by preparative chromatography on silica gel using 70:30 ethyl acetate/hexanes gave 7 mg (70% yield) of the racemic material which was used as an HPLC standard to monitor the enantiomeric excess of the CBS reduction: 1H NMR (500 MHz, CDCl3) δ 6.96 (s, 1 H), 6.64 (s, 1 H), 4.42-4.39 (m, 1 H), 3.93-3.86 (m, 2 H), 3.77 (t, J = 6.6 Hz, 1 H), 2.74 (s, 3 H), 2.08 (s, 3 H), 1.89-1.87 (m, 2 H), 0.93 (s, 9 H), 0.11 (s, 6 H); 13C NMR (125 MHz, CDCl3) δ 164.8, 153.6, 142.1, 118.6, 115.8, 62.7, 37.4, 26.2, 19.7, 18.6, 15.3, −5.1; IR (neat) 3400-3350 b, 2924, 2854, 1463, 1259, 1094 cm−1; MS (+FAB) = 328 (M+H+); HRMS m/e calc’d for C16H30NO2SSi (M+H+): 328.1767, found 328.1776.
(S,E)-4-(3,5-bis(tert-Butyldimethylsilyloxy)-2-methylpent-1-enyl)-2-methylthiazole (5a)33
Alcohol 5 (10.8 g, 33.0 mmol, 1.00 equiv) dissolved in CH2Cl2 (200 mL) was cooled to 0 °C and 2,6-lutidine (8.20 mL, 70.3 mmol, 2.13 equiv) was added followed by TBSOTf (11.8 mL, 51.2 mmol, 1.55 equiv). The temperature was gradually warmed to room temperature and after stirring for 1.5 hours the reaction mixture was quenched with saturated aqueous ammonium chloride, followed by washing with saturated NaHCO3 and brine. The aqueous layer was extracted once with dichloromethane. The combined organics were dried over sodium sulfate and concentrated. Flash column chromatography of the crude using silica gel and 95:5 hexanes/EtOAc as eluent provided 13.70 g (94% yield) of the bis-silyl ether of 5: 1H NMR (400 MHz, CDCl3) δ 6.92 (s, 1 H), 6.49 (s, 1 H), 4.35-4.32 (dd, J = 8.0, 4.6 Hz, 1 H), 3.69-3.64 (m, 2 H), 2.71 (s, 3 H), 2.00 (s, 3 H), 1.78-1.73 (m, 2 H), 0.91 (s, 18 H), 0.08 (s, 3 H), 0.07 (s, 3 H), 0.05 (s, 3 H), 0.03 (s, 3 H); 13C NMR (100 MHz, CDCl3) δ 164.8, 153.6, 143.0, 119.0, 115.4, 75.5, 60.1, 40.3, 26.3, 26.3, 26.1, 19.6, 18.6, 14.2, −4.2, −4.7, −4.9, −4.9; IR (neat) 2952, 2940, 2855, 1470, 1380, 1360, 1256 cm−1; MS (+FAB) = 441 (M+H+); HRMS m/e calc’d for C22H43NO2SSi2 (M+H+): 441.2553, found 441.2525; [α]D20 −4.9° (c = 0.88, CHCl3).
(S,E)-3-(tert-Butyldimethylsilyloxy)-4-methyl-5-(2-methylthiazol-4-yl)pent-4-en-1-ol (5b)33
(S,E)-4-(3,5-bis(tert-butyldimethylsilyloxy)-2-methylpent-1-enyl)-2-methylthiazole (13.16 g, 29.79 mmol) was placed in a 500 mL plastic bottle with acetonitrile (120 mL) and ether (120 mL) and the temperature was lowered to 0 °C. Glass splinters (133 mg, catalytic) were added followed by 40% aqueous HF (20 mL). The reaction was stirred at 0 °C for 2 hours and 40% aqueous HF (20 mL) was again added. After stirring for one hour, solid sodium bicarbonate (84.0 g) was added over 15 minutes. The mixture was stirred for 30 minutes and water was then added to dissolve the solids (800 mL). Brine was also added (100 mL). The mixture was extracted four times with CH2Cl2, and the organic layer was again washed with brine. The organic layer was dried over sodium sulfate and concentrated. Column chromatography on silica gel using a gradient of 20–50% ether in hexanes provided 8.0 grams (82% yield) of the primary alcohol (S,E)-3-(tert-butyldimethylsilyloxy)-4-methyl-5-(2-methylthiazol-4-yl)pent-4-en-1-ol: 1H NMR (400 MHz, CDCl3) δ 6.93 (s, 1 H), 6.52 (s, 1 H), 4.42-4.39 (dd, J = 7.3, 4.6 Hz, 1 H), 3.80-3.72 (m, 2H), 2.72 (s, 3 H), 2.43 (m, 1 H), 2.04 (s, 3 H), 1.91-1.80 (m, 2 H), 0.93 (s, 9 H), 0.12 (s, 3 H), 0.05 (s, 3 H); 13C NMR (100 MHz, CDCl3) δ 170.9, 165.0, 153.4, 142.0, 119.2, 115.8, 109.5, 60.8, 39.0, 26.2 (3 C), 19.6, 18.5, 14.8, −4.2, −4.8; IR (neat) 3400-3350 b, 2960, 2940, 2860, 1500, 1475, 1256, 1082 cm−1; MS (+FAB) = 328 (M+H+); HRMS m/e calc’d for C16H30NO2SSi (M+H+): 328.1767, found 328.1738; [α]D20 −33 (c = 2.5, CHCl3).
(S,E)-3-(tert-Butyldimethylsilyloxy)-4-methyl-5-(2-methylthiazol-4-yl)pent-4-enal (6)34
(S,E)-3-(tert-Butyldimethylsilyloxy)-4-methyl-5-(2-methylthiazol-4-yl)pent-4-en-1-ol (6.70 g, 20.5 mmol, 1.00 equiv) was dissolved in CH2Cl2 and the Dess-Martin periodinane (11.3 g, 26.6 mmol, 1.30 equiv) was added. After stirring at room temperature for 6 hours the reaction mixture was concentrated and subjected to column chromatography using 30% ether in hexanes. This provided 6.5 g (98% yield) of the desired aldehyde 6: 1H NMR (400 MHz, CDCl3) δ 9.81 (s, 1 H), 6.97 (s, 1 H), 6.59 (s, 1 H), 4.71 (dd, J = 8.3, 4.0 Hz, 1 H), 2.75 (dd, J = 7.1, 3.0 Hz, 1 H), 2.74 (s, 3 H), 2.54 (dd, J = 4.0, 2.0 Hz, 1 H), 2.07 (s, 3 H), 0.90 (s, 9 H), 0.11 (s, 3H), 0.06 (s, 3H); IR (neat) 2960, 2920, 2855, 2715, 1728, 1500, 1468, 1466 cm−1; MS (+FAB) = 326 (M+H+) HRMS m/e calc’d for C16H28NO2SSi (M+H+): 326.1610, found 326.1606; [α]D20 −17 (c = 3.0, CHCl3).
Methyl 5-(Benzyloxy)-2,2-dimethyl-3-oxopentanoate (9)
To a solution of isopropylcyclohexylamine (7.3 mL, 45 mmol 1.5 equiv) in THF (40 mL) at −30 °C was added dropwise n-butyllithium (16.1 mL, 38.6 mmol, 1.3 equiv) and stirred for 30 min. The temperature was raised to 0 °C for 15 min and then cooled to −78 °C for 15 min. Methyl isobutyrate (3.75 mL, 32.7 mmol, 1.10 equiv) dissolved in THF (5 mL) was added dropwise to the reaction mixture. After stirring for 30 min. 3-(benzyloxy)propanoyl chloride (5.0 g, 29.7 mmol, 1 equiv.) in THF (5 mL) was added dropwise. The reaction mixture was stirred for an additional hour until the disappearance of the starting material (TLC: 80:20 hexanes/EtOAc). The reaction was quenched with HCl (20 mL, 20%) and warmed to room temperature. The reaction mixture was extracted with ether (3 times) and the combined organic phase was washed twice with sodium bicarbonate and once with brine. The combined aqueous layer was cross-extracted twice with ether. The two combined organic layers were dried (Na2SO4) and concentrated under reduced pressure. Flash column chromatography on silica gel (gradient hexanes/EtOAc) provided 5.1 g (65%) of the target compound. 1H NMR (400 MHz, CDCl3) δ 7.38-7.32 (m, 5 H), 4.52 (s, 2 H), 3.74 (t, J = 6 Hz, 2 H), 3.69 (s, 3 H), 2.76 (t, J = 6 Hz, 2 H), 1.40 (s, 6 H). 13C NMR (100 MHz, CDCl3) δ 206.5, 174.4, 138.6, 128.8, 128.0, 127.2, 73.7, 65.8, 52.9, 38.8, 22.2. IR (neat) 2940, 2850, 1740, 1710, 1450, 1360, 1145, 1100 cm−1; MS (CI) = 282 (M+NH4+); HRMS m/e calc’d for C15H21O4 (M+H+): 265.1423, found 265.1155.
Methyl (3S)-5-(Benzyloxy)-2,2-dimethyl-3-hydroxypentanoate (10)
Acetone and MeOH were degassed five times using the freeze-thaw method and placed under argon. Bis-(2-methylallyl)cycloocta-1,5-diene ruthenium (II) complex (91 mg, 0.28 mmol, 1.0 equiv) and ((S)-BINAP, 177 mg, 0.284 mmol, 1.00 equiv) were combined in a Schlenk flask with acetone (24 mL) and HBr solution [2.0 mL, (0.25 mL 48% HBr, 5.1 mL acetone)]. The mixture was stirred for 1.5 hours and the acetone was removed under reduced pressure. A solution of β-ketoester 9 (5.36 g, 22.7 mmol, 71.0 equiv) in MeOH (22.8 mL) was degassed four times and transferred to a Parr hydrogenation flask. The catalyst was then added to the Parr flask using MeOH. The reaction mixture was hydrogenated for 110 hours at 60 psi and 60 °C. The resulting suspension was concentrated under reduced pressure and then taken up in ether. The reaction mixture was filtered twice to remove the catalyst and concentrated. Final purification by column chromatography (silica gel, 5–15% EtOAc in hexane, 1% MeOH, 1% TEA) provided 4.57 g (85% yield) of β-hydroxy ester 10 in 95% enantiomeric excess. The enantiomeric excess was determined by chiral HPLC on a Chiracel OD-H column at 254 nm, using 99:1 hexanes/isopropanol at 0.8 mL/min, retention times major: 19.5 min and minor: 22.3 min, compared to the racemic alcohol for verification. 1H NMR (400 MHz, CDCl3) δ 7.29-7.21 (m, 5 H), 4.56 (d, J = 11.9 Hz, 1 H), 4.53 (d, J = 11.8 Hz, 1 H), 3.85 (m, 1 H), 3.68-3.58 (m, 2 H), 3.62 (s, 3 H), 3.11 (d, J = 4.0 Hz, 1 H), 1.65-1.61 (m, 2 H), 1.13 (s, 3 H), 1.11 (s, 3 H); 13C NMR (100 MHz, CDCl3) δ 178.2, 138.4, 128.8, 128.1, 76.0, 73.8, 69.7, 52.3, 47.4, 31.8, 22.1, 20.8; IR (neat) 3500, 2950, 2860, 1721, 1468, 1450, 1431, 1385, 1365, 1265, 1190, 1135, 1090 cm−1; MS (CI) = 267 (M+H+); HRMS m/e calc’d for C15H23O4 (M+H+): 267.1596, found 267.1617; [α]D20 −2.8 (c = 2.0, CHCl3).
Methyl (3S)-3,5-Di[(tert-butyldimethylsilyl)oxy]-2,2-dimethylpentanoate (10a)40
β-Hydroxy ester 10 (6.90 g, 1.00 equiv) was dissolved in THF (75 mL) and transferred to a Parr hydrogenation vessel under argon. 10% Pd/C (1.73 g, 0.250 equiv) was added and the flask was purged for an additional 10 minutes with argon. The hydrogenation reaction was carried out at 58-52 psi for 22 hours. The resulting mixture was filtered and the residue washed with EtOAc (300 mL). The filtrate was concentrated under reduced pressure to provide 4.50 g (99% yield) of the crude diol, which was carried on to the next step without further purification. The diol (4.50 g, 25.6 mmol, 1.00 equiv) was dissolved in CH2Cl2 (85 mL) and the temperature was lowered to 0 °C. 2,6-Lutidine (14.9 mL, 128 mmol, 5.00 equiv) was added followed by TBSOTf (17.6 mL, 76.7 mmol, 3.00 equiv). The reaction mixture was gradually warmed to room temperature and stirred overnight. The reaction was quenched with ammonium chloride (25 mL). The reaction mixture was extracted four times with CH2Cl2 and the combined organic layer was washed with brine, dried over Na2SO4, and concentrated. Purification via column chromatography on silica gel using a gradient of 0–5% EtOAc in hexanes, provided 9.27 g (90% yield) of 10a: 1H NMR (400 MHz, CDCl3) δ 4.07 (dd, J = 2.3, 2.2 Hz, 1 H), 3.72-3.59 (m, 2 H), 3.67 (s, 3 H), 1.66-1.55 (m, 2 H), 1.18 (s, 3 H), 1.11 (s, 3 H), 0.90 (s, 9 H), 0.89 (s, 9 H), 0.10 (s, 3 H), 0.06 (s, 6 H), 0.05 (s, 3 H); 13C NMR (100 MHz, CDCl3) δ 178.0, 73.8, 60.7, 52.1, 48.7, 37.3, 26.4, 26.3, 22.1, 20.7, 18.7, −3.5, −3.9, −4.9; IR (neat) 2940, 2914, 2870, 2840, 1721, 1458, 1422, 1379, 1351, 1242, 1180, 1122, 1087 cm−1; MS (CI) = 405 (M+H+); HRMS m/e calc’d for C20H45O4Si2 (M+H+): 405.2856, found 405.2850; [α]D20 −5.6 (c = 3.0, CHCl3).
(5S)-5,7-Di-[(tert-butyldimethylsilyl)oxy]-4,4-dimethylheptan-3-one (11)40
Ethylphenyl sulfone (3.4 g, 20 mmol, 5.5 equiv) was dissolved in THF (50 mL) and the temperature was lowered to −78 °C. n-BuLi (13 mL, 18 mmol, 5.0 equiv) was added dropwise and the pale yellow solution that formed was stirred at −78 °C for 1.5 hours. The ester 10a in THF (15 mL) was added dropwise at −78 °C. The bath was removed and the reaction was stirred at room temperature for 20 hours. The reaction was quenched with 1:1 saturated NH4Cl and water (30 mL) and the aqueous layer was extracted five times with Et2O. The organic layer was dried over Na2SO4 and concentrated. The crude was then dissolved in MeOH (50 mL) and then temperature was lowered to 0 °C. NaH2PO4 (2.00 g, 14.4 mmol, 4.00 equiv) was added followed by Na(Hg) (4.39 g). The pink colored suspension, which developed, was stirred at 0 °C for 1 hour and 15 min, and 1:1 saturated NH4Cl and water was added followed by dilution with Et2O. The mercury metal was filtered using a funnel and a plug of glass wool. The filtrate was extracted five times with Et2O. The combined organic layer was washed with H2O and brine, dried over Na2SO4 and concentrated. Column chromatography on silica gel using 5% Et2O in hexanes provided 1.3 g (90%) of the ketone: 1H NMR (400 MHz, CDCl3) δ 4.08 (dd, J = 3.0, 3.0 Hz, 1 H), 3.66-3.63 (m, 2 H), 2.60-2.50 (m, 2 H), 1.56-1.49 (m, 2 H), 1.13 (s, 3 H), 1.07 (s, 3 H), 1.01 (t, J = 7.1 Hz, 3 H), 0.91 (s, 9 H), 0.90 (s, 9 H), 0.11 (s, 3 H), 0.07 (s, 3 H), 0.06 (s, 3 H), 0.05 (s, 3 H); 13C NMR (100 MHz, CDCl3) δ 216.0, 73.8, 60.5, 53.4, 37.7, 32.0, 26.5, 26.3, 22.6, 20.4, 18.7, 18.6, 8.1, −3.6, −3.7, −4.9; IR (neat) 2939, 2910, 2840, 1698, 1459, 1380, 1355, 1248, 1087 cm−1; MS (CI) = 403 (M+H+); HRMS m/e calc’d for C21H47O3Si2 (M+H+): 403.3064, found 403.3051; [α]D20 −7.0 (c = 1.8, CHCl3).
(4S,7R,8S,9S,16S,E)-4,8-bis(tert-Butyldimethylsilyloxy)-13-(hydroxymethyl)-5,5,7,9-tetramethyl-16-((E)-1-(2-methylthiazol-4-yl)prop-1-en-2-yl)oxacyclohexadec-13-ene-2,6-dione (13)25
(4S,7R,8S,9S,16S,E)-4,8-bis(tert-Butyldimethylsilyloxy)-5,5,7,9-tetramethyl-16-((E)-1-(2-methylthiazol-4-yl)prop-1-en-2-yl)-13-(trityloxymethyl)oxacyclohexadec-13-ene-2,6-dione,25 (50.0 mg, 0.0512 mmol, 1.00 equiv) was placed in ether (1.0 mL) and the temperature was lowered to −20 °C. Formic acid (1.0 mL) was added dropwise over 15 minutes. The reaction was stirred at −10 °C for 10 min and then at −5 to 0 °C for 6 hours. The reaction was quenched with water (2 mL) and solid sodium bicarbonate was added until the pH was neutral. The mixture was extracted four times with ether. The organic layer was dried over MgSO4 and concentrated. Column chromatography on silica gel using a gradient of 20–50% ether in hexanes gave 22 mg (60% yield) of the mono deprotected product 13 that was fully characterized and found to match the results obtained by the Nicolaou group25 for this derivative: 1H NMR (500 MHz, CDCl3) δ 6.99 (s, 1 H), 6.60 (s, 1 H), 5.51 (dd, J = 9.4, 6.8 Hz, 1 H), 5.05 (d, J = 8.4 Hz, 1 H), 4.18-4.13 (m, 1 H), 4.06 (d, J = 9.0 Hz, 1 H), 4.02 (d, J = 13.0 Hz, 1 H), 3.92 (d, J = 13.6 Hz, 1 H), 3.08-3.01 (m, 1 H), 2.80-2.78 (m, 1 H), 2.78-2.76 (m, 1 H), 2.74 (s, 3 H), 2.71 (dd, J = 16.3, 10.1 Hz, 1 H), 2.47-2.41 (m 1 H), 2.23-2.18 (m, 1 H), 2.13 (s, 3 H), 2.03-1.97 (m, 1 H), 1.85-1.63 (m, 2 H), 1.62-1.50 (m, 3 H), 1.21 (s, 3 H), 1.16 (s, 3 H), 1.12 (d, J = 6.8 Hz, 3 H), 0.99 (d, J = 6.9 Hz, 3 H), 0.96 (s, 9 H), 0.86 (s, 9 H), 0.13 (s, 3 H), 0.12 (s, 3 H), 0.09 (s, 3 H), −0.01 (s, 3 H); 13C NMR (125 MHz, CDCl3) δ 215.1, 171.0, 164.6, 152.3, 143.8, 138.3, 128.5, 127.7, 120.3, 119.6, 116.0, 79.4, 76.0, 66.4, 64.3, 53.3, 48.1, 39.3, 37.3, 32.0, 31.3, 28.1, 27.3, 26.3 (3 C), 26.0 (3 C), 24.3, 19.1, 18.5 (2 C), 17.7, 15.1, −3.4, −3.7, −3.8, −5.7; IR (neat) 2929, 2856, 1742, 1695, 1463, 1381, 1255 cm−1; MS (+FAB) = 736.4 (M+H+); HRMS m/e calc’d for C39H70NO6SSi2 (M+H+): 736.4462, found 736.4463; [α]D20 −22 (c = 0.40, CHCl3).
((2S,9S,10S,11R,14S,E)-10,14-Dihydroxy-9,11,13,13-tetramethyl-2-((E)-1-(2-methylthiazol-4-yl)prop-1-en-2-yl)-12,16-dioxooxacyclohexadec-4-en-5-yl)methyl 3-Azidobenzoate (14)
Alcohol 13 (11.0 mg, 0.0150 mmol, 1.00 equiv) was placed in a flask with m-azidobenzoic acid (10.0 mg, 0.0600 mmol, 4.00 equiv), HBTU (18.0 mg, 0.0450 mmol, 3.00 equiv) and CH3CN (0.40 mL). The temperature was lowered to 0 °C and DIEA (0.020 mL, 0.090 mmol, 6.0 equiv) was added. The reaction was gradually warmed to room temperature and was stirred overnight. The reaction was quenched with saturated NaHCO3, extracted three times with ether, dried over MgSO4, and concentrated. Column chromatography on silica gel using a gradient of 5–20% ether in hexanes gave 9 mg (69% yield) of the acylated product: 1H NMR (500 MHz, CDCl3) δ 7.84 (d, J = 7.2 Hz, 1 H), 7.73 (s, 1 H), 7.49 (t, J = 7.8 Hz, 1 H), 7.23 (d, J = 8.0 Hz, 1 H), 7.00 (s, 1 H), 6.61 (s, 1 H), 5.64 (dd, J =12.1, 9.0 Hz, 1 H), 5.12 (d, J = 9.5 Hz, 1 H), 4.87 (d, J = 12.5 Hz, 1 H), 4.70 (d, J = 12.5 Hz, 1 H), 4.10 (d, J = 8.9 Hz, 1 H), 3.92 (d, J = 8.9 Hz, 1 H), 3.06 (m, 1 H), 2.84-2.79 (m, 2 H), 2.72-2.69 (m, 1 H), 2.74 (s, 3 H), 2.49-2.42 (m, 1 H), 2.31-2.22 (m, 1 H), 2.16 (s, 3 H), 2.10-2.08 (m, 1 H), 1.80-1.73 (m, 1 H), 1.70-1.54 (m, 1 H), 1.35-1.28 (m, 3 H), 1.22 (s, 3 H), 1.16 (s, 3 H), 1.13 (d, J = 6.7 Hz, 3 H), 0.99 (d, J = 6.8 Hz, 3 H), 0.97 (s, 9 H), 0.87 (s, 9 H), 0.13 (s, 3 H), 0.11 (s, 3 H), 0.07 (s, 3 H), −0.09 (s, 3 H); 13C NMR (125 MHz, CDCl3) δ 215.1, 170.9, 165.0, 151.7, 140.5, 138.1, 132.0, 129.7 (2 C), 125.9 (2 C), 123.3, 120.0 (2 C), 119.8, 116.2, 78.1, 75.9, 68.2, 67.9, 53.3, 40.5, 38.6, 37.9, 32.0, 31.8, 30.2, 28.6, 26.3 (3 C), 26.0 (3 C), 24.4, 22.6, 19.1, 18.5 (2 C), 17.7, 15.1, 14.0, −3.6 (2 C), −3.8 (2 C); IR (neat) 2958, 2925, 2854, 2111, 1728, 1462, 1259, 1096, 1020, 801 cm−1; MS (+FAB) = 881.5 (M+H+); HRMS m/e calc’d for C46H73N4O7SSi2 (M+H+): 881.4739, found 881.4759; [α]D20 −76 (c = 0.075, CHCl3). The acylated product (4.0 mg, 0.0045 mmol, 1.0 equiv) was placed in a flask and the temperature was lowered to −20 °C. A TFA solution (0.100 mL, 20% v/v TFA in CH2Cl2) was added and the reaction was stirred at −20 °C for 6 hours. The reaction was then quenched into saturated sodium bicarbonate. The mixture was extracted three times with ethyl acetate, dried over Na2SO4, and concentrated. Column chromatography on silica gel using a gradient of 0–1% CH3OH in CH2Cl2 gave 2.0 mg (68% yield) of the final product 14: 1H NMR (500 MHz, CDCl3) δ 7.83-7.81 (m, 1 H), 7.70 (t, J = 1.9 Hz, 1 H), 7.45 (t, J = 7.9 Hz, 1 H), 7.22 (dd, J = 2.3, 2.3 Hz, 1 H), 6.94 (s, 1 H), 6.61 (s, 1 H), 5.60 (dd, J = 9.4, 5.6 Hz, 1 H), 5.31 (d, J = 9.1 Hz, 1 H), 4.84 (d, J = 12.6 Hz, 1 H), 4.71 (d, J = 12.7 Hz, 1 H), 4.34 (d, J = 9.8 Hz, 1 H), 3.72-3.71 (m, 1 H), 3.63 (d, J = 5.5 Hz, 1 H), 3.19 (dq, J = 6.9, 2.2 Hz, 1 H), 3.00 (bs, 1 H), 2.74-2.68 (m, 1 H), 2.71 (s, 3 H), 2.52-2.47 (m, 1 H), 2.46-2.40 (m, 1 H), 2.38-2.31 (m, 1 H), 2.29 (dd, J = 14.5, 2.6 Hz, 1 H), 2.20-2.16 (m, 1 H), 2.09 (s, 3 H), 1.78-1.76 (m, 2 H), 1.42-1.38 (m, 3 H), 1.38 (s, 3 H), 1.20 (d, J = 6.8 Hz, 3 H), 1.09 (s, 3 H), 1.03 (d, J = 7.0 Hz, 1 H); 13C NMR (125 MHz, CDCl3) δ 220.5, 170.2, 165.3, 165.0, 151.7, 140.5, 138.5, 136.7, 131.9, 129.8, 125.9, 125.2, 119.9, 119.3, 115.6, 77.9, 73.9, 72.0, 68.3, 53.6, 41.5, 39.6, 37.9, 32.0, 31.5, 30.0, 28.4, 25.2, 22.8, 19.0, 17.6, 15.9, 15.7, 13.2; IR (neat) 2160, 1705, 1695, 1325 cm−1; MS (+FAB) = 653.4 (M+H+); HRMS m/e calc’d for C34H45N4O7S (M+H+): 653.3009, found 653.2984; [α]D20 −42 (c = 0.15, CHCl3).
((2S,9S,10S,11R,14S,E)-10,14-Dihydroxy-9,11,13,13-tetramethyl-2-((E)-1-(2-methylthiazol-4-yl)prop-1-en-2-yl)-12,16-dioxooxacyclohexadec-4-en-5-yl)methyl 4-Azidobenzoate (15)
Alcohol 13 (12.0 mg, 0.0160 mmol, 1.00 equiv) was placed in a flask with p-azidobenzoic acid (11.0 mg, 0.0650 mmol, 4.00 equiv), HBTU (18.0 mg, 0.0490 mmol, 3.00 equiv) and CH3CN (0.30 mL). The temperature was lowered to 0 °C and DIEA (0.019 mL, 0.098 mmol, 6.0 equiv) was added. The reaction was gradually warmed to room temperature and was stirred overnight. The reaction was quenched with saturated NaHCO3, extracted three times with ethyl acetate, dried over Na2SO4, and concentrated. Column chromatography on silica gel using a gradient of 10–50% ether in hexanes gave 8.5 mg (65% yield) of the acylated product: 1H NMR (500 MHz, CDCl3) δ 7.84 (d, J = 7.5 Hz, 1 H), 7.72 (s, 1 H), 7.45 (t, J = 7.8 Hz, 1 H), 7.23 (d, J = 8.6 Hz, 1 H), 7.00 (s, 1 H), 6.60 (s, 1 H), 5.63 (dd, J = 12.1, 9.0 Hz, 1 H), 5.10 (d, J = 9.3 Hz, 1 H), 4.86 (d, J = 12.8 Hz, 1 H), 4.69 (d, J = 12.3 Hz, 1 H), 4.08 (d, J = 9.1 Hz, 1 H), 3.91 (d, J = 9.0 Hz, 1 H), 3.04 (m, 1 H), 2.85-2.76 (m, 2 H), 2.74 (s, 3 H), 2.72-2.69 (m, 1 H), 2.50-2.45 (m, 1 H), 2.28-2.25 (m, 1 H), 2.16 (s, 3 H), 2.10-2.03 (m, 1 H), 1.80-1.73 (m, 1 H), 1.70-1.51 (m, 1 H), 1.34-1.18 (m, 3 H), 1.22 (s, 3 H), 1.16 (s, 3 H), 1.13 (d, J = 6.8 Hz, 3 H), 0.99 (d, J = 7.0 Hz, 3 H), 0.96 (s, 9 H), 0.86 (s, 9 H), 0.13 (s, 3 H), 0.12 (s, 3 H), 0.10 (s, 3 H), −0.09 (s, 3 H); 13C NMR (125 MHz, CDCl3) δ 215.5, 170.2, 165.3, 151.2, 140.5, 132.0, 129.8 (2 C), 125.9 (2 C), 123.7, 123.3, 120.0 (2 C), 119.8, 116.2, 79.1, 75.9, 68.2, 67.9, 53.3, 39.4, 38.6, 37.6, 32.0, 31.8, 30.1, 28.6, 26.3 (3 C), 26.0 (3 C), 24.7, 22.6, 19.1, 18.6, 18.5, 17.1, 15.1, 14.0, −3.4, −3.6, −3.8, −5.6; IR (neat) 2954, 2925, 2112, 1732, 1463, 1259, 1094, 798 cm−1; MS (+FAB) = 881.4 (M+H+); HRMS m/e calc’d for C46H73N4O7SSi2 (M+H+): 881.4739, found 881.4746; (M+H)+; [α]D20 −11 (c = 0.15, CHCl3). The acylated compound (8.5 mg, 0.0097 mmol, 1.0 equiv) was placed in a flask and the temperature was lowered to −20 °C. TFA solution (0.200 mL, 20% v/v TFA in CH2Cl2) was added and the reaction was stirred at −20 °C for 5 hours. The reaction was then placed in the freezer overnight at −4 °C. The reaction was quenched into saturated sodium bicarbonate. The mixture was extracted three times with ethyl acetate, dried over Na2SO4, and concentrated. Column chromatography on silica gel using a gradient of 0–1% CH3OH in CH2Cl2 gave 4.5 mg (71% yield) of the final product 15: 1H NMR (500 MHz, CDCl3) δ 7.82 (d, J = 6.7 Hz, 1 H), 7.70 (t, J = 1.7 Hz, 1 H), 7.44 (t, J = 7.9 Hz, 1 H), 7.23 (dd, J = 2.2, 2.3 Hz, 1 H), 6.95 (s, 1 H), 6.60 (s, 1 H), 5.60 (dd, J = 11.7, 5.6 Hz, 1 H), 5.29 (d, J = 7.2 Hz, 1 H), 4.84 (d, J = 12.6 Hz, 1 H), 4.71 (d, J = 12.7 Hz, 1 H), 4.34 (d, J = 10.9 Hz, 1 H), 3.71 (bs, 1 H), 3.69-3.67 (m, 1 H), 3.18 (dq, J = 13.6, 2.1 Hz, 1 H), 3.02 (bs, 1 H), 2.72-2.69 (m, 1 H), 2.71 (s, 3 H), 2.52-2.46 (m, 1 H), 2.45-2.38 (m, 1 H), 2.37-2.32 (m, 1 H), 2.28 (dd, J = 14.5, 2.6 Hz, 1 H), 2.21-2.17 (m, 1 H), 2.09 (s, 3 H), 1.78-1.70 (m, 2 H), 1.42-1.38 (m, 3 H), 1.38 (s, 3 H), 1.20 (d, J = 6.8 Hz, 3 H), 1.09 (s, 3 H), 0.94 (d, J = 7.0 Hz, 3 H); 13C NMR (125 MHz, CDCl3) δ 221.1, 170.7, 165.8, 165.5, 152.2, 141.0, 139.0, 137.1, 132.3, 130.3, 126.4, 125.7, 120.4, 119.7, 116.1, 78.4, 74.4, 72.5, 68.8, 54.1, 42.0, 40.0, 38.4, 32.4, 32.0, 30.1, 28.8, 25.7, 23.3, 19.4, 18.0, 16.5, 16.1, 13.6; IR (neat) 2950, 2140, 1730, 1305, 1265 cm−1; MS (+FAB) = 653.4 (M+H+); HRMS m/e calc’d for C34H45N4O7S (M+H+): 653.3009, found 653.3017; [α]D20 −60 (c = 0.10, CHCl3).
((2S,9S,10S,11R,14S,E)-10,14-Dihydroxy-9,11,13,13-tetramethyl-2-((E)-1-(2-methylthiazol-4-yl)prop-1-en-2-yl)-12,16-dioxooxacyclohexadec-4-en-5-yl)methyl 4-Benzoylbenzoate (16)
Alcohol 13 (12.0 mg, 0.0163 mmol, 1.00 equiv) was placed in a flask with benzoylbenzoic acid (15.0 mg, 0.0653 mmol, 4.00 equiv), HBTU (18 mg, 0.049 mmol, 3.0 equiv) and CH3CN (0.20 mL). The temperature was lowered to 0 °C and DIEA (0.020 mL, 0.098 mmol, 6.0 equiv) was added. The reaction was gradually warmed to room temperature and was stirred overnight. The reaction was quenched with saturated NaHCO3, extracted three times with ethyl acetate, dried over Na2SO4, and concentrated. Column chromatography on silica gel using a gradient of 0–50% ether in hexanes gave 8 mg (53% yield) of the acylated product: 1H NMR (500 MHz, CDCl3) δ 8.18 (d, J = 8.3 Hz, 2 H), 7.84 (dd, J = 16.1, 8.3 Hz, 4 H), 7.64 (t, J = 7.4 Hz, 1 H), 7.53 (t, J = 7.4 Hz, 2 H), 7.00 (s, 1 H), 6.61 (s, 1 H), 5.67 (dd, J = 9.4, 6.6 Hz, 1 H), 5.12 (d, J = 9.3 Hz, 1 H), 4.90 (d, J = 12.5 Hz, 1 H), 4.74 (d, J = 12.6 Hz, 1 H), 4.09 (d, J = 8.5 Hz, 1 H), 3.93 (d, J = 8.9 Hz, 1 H), 3.07 (m, 1 H), 2.80-2.74 (2 H), 2.74 (s, 3 H), 2.70-2.68 (m, 1 H), 2.56-2.49 (m, 1 H), 2.20-2.12 (m, 1 H), 2.20 (s, 3 H), 2.10-2.03 (m, 1 H), 1.82-1.76 (m, 2 H), 1.38-1.18 (m, 3 H), 1.22 (s, 3 H), 1.17 (s, 3 H), 1.13 (d, J = 6.8 Hz, 3 H), 1.10 (d, J = 6.9 Hz, 3 H), 0.97 (s, 9 H), 0.87 (s, 9 H), 0.14 (s, 3 H), 0.13 (s, 3 H), 0.10 (s, 3 H), −0.08 (s, 3 H); 13C NMR (125 MHz, CDCl3) δ 215.1, 195.9, 170.9, 165.5, 164.6, 152.3, 141.2, 138.5, 136.9, 136.7, 133.3, 132.9, 130.0 (2 C), 129.7 (2 C), 129.5 (2 C), 128.4 (2 C), 119.8 (2 C), 116.2, 79.1, 75.8, 68.3, 67.9, 53.3, 39.5, 37.9, 32.0, 31.8, 30.9, 30.5, 27.5, 26.9, 26.3 (3 C), 26.0 (3 C), 24.4, 22.6, 19.1, 18.6, 18.5, 15.1, 14.0, −3.3, −3.6, −3.8, −5.6; IR (neat) 3019, 2926, 2854, 1720, 1662, 1463, 1259, 1215, 1102, 769 cm−1; MS (+FAB) = 944.4 (M+H+); HRMS m/e calc’d for C53H78NO8SSi2 (M+H+): 944.4987, found 944.5009; [α]D20 −5.3 (c = 0.15, CHCl3). The acylated product (8.0 mg, 0.0085 mmol, 1.0 equiv) was placed in a flask and the temperature was lowered to −20 °C. TFA solution (0.100 mL, 20% v/v TFA in CH2Cl2) was added and the reaction was stirred at −20 °C for 5 hours and stored in the freezer at −4 °C overnight. The reaction was then quenched into saturated sodium bicarbonate. The mixture was extracted three times with ethyl acetate, dried over Na2SO4, and concentrated. Column chromatography on silica gel using a gradient of 0–1% CH3OH in CH2Cl2 gave 4.0 mg (67% yield) of the final product 16: 1H NMR (500 MHz, CDCl3) δ 8.16 (d, J = 6.7 Hz, 2 H), 7.85 (t, J = 7.6 Hz, 4 H), 7.66 (t, J = 7.4 Hz, 1 H), 7.53 (t, J = 7.1 Hz, 2 H), 6.95 (s, 1 H), 6.61 (s, 1 H), 5.63 (dd, J = 9.5, 5.3 Hz, 1 H), 5.31 (d, J = 9.0 Hz, 1 H), 4.88 (d, J = 12.7 Hz, 1 H), 4.74 (d, J = 12.8 Hz, 1 H), 4.34 (d, J = 9.8 Hz, 1 H), 3.74 (bs, 1 H), 3.62 (bs, 1 H), 3.19 (dq, J = 14.0, 2.2 Hz, 1 H), 3.01 (bs, 1 H), 2.72-2.69 (m, 1 H), 2.70 (s, 3 H), 2.52-2.47 (m, 1 H), 2.46-2.40 (m, 1 H), 2.38-2.33 (m, 1 H), 2.30 (dd, J = 14.5, 2.6 Hz, 1 H), 2.23-2.19 (m, 1 H), 2.10 (s, 3 H), 1.81-1.78 (m, 2 H), 1.44-1.39 (m, 3 H), 1.38 (s, 3 H), 1.21 (d, J = 6.8 Hz, 3 H), 1.10 (s, 3 H), 1.04 (d, J = 7.0 Hz, 1 H); 13C NMR (125 MHz, CDCl3) δ 220.5, 195.9, 170.2, 165.4, 165.1, 151.7, 141.3, 138.5, 136.9, 136.7, 133.2, 132.9, 130.0 (2 C), 129.7 (2 C), 129.4 (2 C), 128.4 (2 C), 125.0, 119.4, 115.7, 78.0, 73.9, 72.1, 68.2, 53.5, 41.6, 39.5, 37.9, 32.0, 31.6, 28.3, 25.2, 22.8, 19.0, 17.8, 15.9, 15.7, 13.2; IR (neat) 2995, 2955, 1730, 1665, 1280 cm−1; MS (+FAB) = 716.4 (M+H+); HRMS m/e calc’d for C41H50NO8S (M+H+): 716.3257, found 716.3254; [α]D20 −60 (c = 0.20, CHCl3).
Photoaffinity labeling of tubulin
Tubulin was purified from bovine brains by a procedure described previously.43 Photoaffinity labeling of tubulin was performed as described.44 To a solution of 0.1 mmol of tubulin (10 mg) in 100 mM Pipes, pH 7 containing 1 mM MgSO4, 1 mM EGTA and 1 mM dithiothreitol at 0 °C was added 0.5 mmol of a concentrated stock solution in DMSO of 14, 15, or 16 such that the DMSO content did not exceed 0.1%. The mixture was then spread on plastic weighing boats (to maximize surface area) and irradiated by exposure to UV light of 253.7 nm (Rayonet photochemical reactor 253.7-nm lamp) for 30 min at a distance of 6 cm, achieving 600–700 mW/cm2.
Preparation of tryptic peptides
The pH of the tubulin-epothilone derivative mixture was raised to pH 8.5 with the addition of small aliquots of a 1 M NH4HCO3 solution and tryptic digestion was performed directly using TPCK-treated proteomics grade trypsin at 37°C for 24 h at an enzyme/substrate ratio of 1:100. The tryptic digest was taken to dryness, dissolved in a small volume of 50 mM glycine-HCL buffer, pH 10, containing 1.0 mM MgC12, and analyzed by reverse-phase HPLC (4.6 × 150 mm C18 column; 5–95% H2O/0.1%CF3COOH-CH3CN/CF3COOH gradient; 220 nm detection). Fractions corresponding to chromatographic peaks were pooled, lyophilized, and examined by mass spectrometry using a Q-Tof-2 instrument. The masses of all MH+ and MH2+ ions were parsed and compared against a library of tryptic fragments of all isoforms of tubulin present in bovine tubulin compiled previously by us (data obtained from two dimensional LC-ion trap MS/MS experiments performed on a LTQ Linear ion-trap MS instrument). All masses that were flagged as a mismatch (deviation of greater than 50 ppm from expected mass) were subjected to MS/MS experiments on the Q-Tof2 instrument, and neutral loss, b and y ion assignments were attempted. No MS or MS/MS ions indicative of true adduction of the epothilone derivatives were found.
Docking and Scoring
Docking and scoring calculations were carried out using Surflex-Dock45–48 and CScore49 in the SYBYL 8.0 discovery software suite (Tripos, Inc.). In Surflex-Dock, the 1TVK.pdb co-crystallized ligand (epothilone A)17 was used to guide the protomol generation process. Default parameters of 0.5 and 0 were used for docking threshold and bloat, respectively. The maximum number of conformations per compound fragment and the maximum number of poses per ligand were both set to their default values of 20, and the maximum number of rotatable bonds per molecule was set to 100. Molecule fragmentation was disabled. Post-dock minimizations were done on each molecule to enhance the quality of results, and all four CScore consensus scoring functions were implemented.
Supplementary Material
Figure 1.

The structures of epothilones A–D.
Acknowledgments
This work was supported by the National Institute of Health grants CA79641 and CA105305. E.A.R. and J.F.G. were supported by NIH Training Grant GM 07775. E.A.R. was also supported by the Army Breast Cancer Initiative Predoctoral Fellowship Program DAMD 17-001-0303 and the American Association for Pharmaceutical Education Predoctoral Fellowship Program.
Footnotes
Supporting Information Available: 1H and 13C NMR spectra are provided for compounds 3, 4, 9, 10, 14, 15, and 16. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Gerth K, Bedorf N, Hoefle G, Irschik H, Reichenbach H. J Antibiot. 1996;49:560–563. doi: 10.7164/antibiotics.49.560. [DOI] [PubMed] [Google Scholar]
- 2.Bollag DM, McQueney PA, Zhu J, Hensens O, Koupal L, Liesch J, Goetz M, Lazarides E, Woods CM. Cancer Res. 1995;55:2325–2333. [PubMed] [Google Scholar]
- 3.Nicolaou KC, Roschangar F, Vourloumis D. Angew Chem, Int Ed. 1998;37:2014–2045. doi: 10.1002/(SICI)1521-3773(19980817)37:15<2014::AID-ANIE2014>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 4.Harris CR, Danishefsky SJ. J Org Chem. 1999;64:8434–8456. [Google Scholar]
- 5.Altmann KH, Wartmann M, O’Reilly T. Biochim Biophys Acta, Rev Cancer. 2000;1470:M79–M91. doi: 10.1016/s0304-419x(00)00009-3. [DOI] [PubMed] [Google Scholar]
- 6.Nicolaou KC, Ritzen A, Namoto K. Chem Commun. 2001:1523–1535. doi: 10.1039/b104949f. [DOI] [PubMed] [Google Scholar]
- 7.Wartmann M, Altmann KH. Curr Med Chem: Anti-Cancer Agents. 2002;2:123–148. doi: 10.2174/1568011023354489. [DOI] [PubMed] [Google Scholar]
- 8.Watkins EB, Chittiboyina AG, Avery MA. Eur J Org Chem. 2006:4071–4084. [Google Scholar]
- 9.Watkins EB, Chittiboyina AG, Jung JC, Avery MA. Curr Pharm Des. 2005;11:1615–1653. doi: 10.2174/1381612053764742. [DOI] [PubMed] [Google Scholar]
- 10.Feyen F, Cachoux F, Gertsch J, Wartmann M, Altmann KH. Acc Chem Res. 2008;41:21–31. doi: 10.1021/ar700157x. [DOI] [PubMed] [Google Scholar]
- 11.Kowalski RJ, Giannakakou P, Hamel E. J Biol Chem. 1997;272:2534–2541. doi: 10.1074/jbc.272.4.2534. [DOI] [PubMed] [Google Scholar]
- 12.Smit WM, Sufliarsky J, Spanik S, Wagnerov M, Kaye S, Oza AM, Gore M, Williams KJ, Johri A, Ten Bokkel Huinink WW. J Clin Oncol. 2005;23:16S. doi: 10.1200/JCO.2008.20.4826. [DOI] [PubMed] [Google Scholar]
- 13.McMeekin S, Patel R, Verschraegan C, Celano P, Burke J, Plaxe S, Ghatage P, Giurescu M, Stredder C, Wang Y. 33rd Congress of the European Society for Medical Oncology; 2008. [Google Scholar]
- 14.Sessa C, Perotti A, Llado A, Cresta S, Capri G, Voi M, Marsoni S, Corradino I, Gianni L. Ann Oncol. 2007;18:1548–1553. doi: 10.1093/annonc/mdm198. [DOI] [PubMed] [Google Scholar]
- 15.Stopeck A, Moulder S, Jones S, Cohen J, McDowell M, Cropp G, Zhong Z, Wells S, Hannah A, Burris H. J Clin Oncol. 2007;25:2571. [Google Scholar]
- 16.Bhushan S, Walko CM. Ann Pharmacother. 2008;42:1252–1261. doi: 10.1345/aph.1L058. [DOI] [PubMed] [Google Scholar]
- 17.Nettles JH, Li H, Cornett B, Krahn JM, Snyder JP, Downing KH. Science. 2004;305:866–869. doi: 10.1126/science.1099190. [DOI] [PubMed] [Google Scholar]
- 18.Reese M, Sanchez-Pedregal VM, Kubicek K, Meiler J, Blommers MJJ, Griesinger C, Carlomagno T. Angew Chem, Int Ed. 2007;46:1864–1868. doi: 10.1002/anie.200604505. [DOI] [PubMed] [Google Scholar]
- 19.Dasgupta D, Park H, Harriman GCB, Georg GI, Himes RH. J Med Chem. 1994;37:2976–2980. doi: 10.1021/jm00044a019. [DOI] [PubMed] [Google Scholar]
- 20.Combeau C, Commercon A, Mioskowski C, Rousseau B, Aubert F, Goeldner M. Biochemistry. 1994;33:6676–6683. doi: 10.1021/bi00187a038. [DOI] [PubMed] [Google Scholar]
- 21.Rao S, Krauss NE, Heerding JM, Swindell CS, Ringel I, Orr GA, Horwitz SB. J Biol Chem. 1994;269:3132–3134. [PubMed] [Google Scholar]
- 22.Rao S, Orr GA, Chaudhary AG, Kingston DGI, Horwitz SB. J Biol Chem. 1995;270:20235–20238. doi: 10.1074/jbc.270.35.20235. [DOI] [PubMed] [Google Scholar]
- 23.Rao S, He L, Chakravarty S, Ojima I, Orr GA, Horwitz SB. J Biol Chem. 1999;274:37990–37994. doi: 10.1074/jbc.274.53.37990. [DOI] [PubMed] [Google Scholar]
- 24.Nicolaou KC, Ninkovic S, Finlay MRV, Sarabia F, Li T. Chem Commun. 1997:2343–2344. [Google Scholar]
- 25.Nicolaou KC, Finlay MRV, Ninkovic S, Sarabia F. Tetrahedron. 1998;54:7127–7166. [Google Scholar]
- 26.Corey EJ, Helal CJ. Angew Chem, Int Ed. 1998;37:1986–2012. doi: 10.1002/(SICI)1521-3773(19980817)37:15<1986::AID-ANIE1986>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 27.Noyori R, Ohkuma T, Kitamura M, Takaya H, Sayo N, Kumobayashi H, Akutagawa S. J Am Chem Soc. 1987;109:5856–5858. [Google Scholar]
- 28.Reiff EA, Nair SK, Reddy BSN, Inagaki J, Henri JT, Greiner JF, Georg GI. Tetrahedron Lett. 2004;45:5845–5847. [Google Scholar]
- 29.Shafiee A, Mazloumi A, Cohen VI. J Heterocycl Chem. 1979;16:1563–1566. [Google Scholar]
- 30.Arimoto H, Asano S, Uemura D. Tetrahedron Letters. 1999;40:3583–3586. [Google Scholar]
- 31.Paterson I, Yeung KS, Smaill JB. Synlett. 1993:774–776. [Google Scholar]
- 32.Pilcher AS, DeShong P. J Org Chem. 1993;58:5130–5134. [Google Scholar]
- 33.Schinzer D, Bauer A, Bohm OM, Limberg A, Cordes M. Chem--Eur J. 1999;5:2483–2491. [Google Scholar]
- 34.Nicolaou KC, Ninkovic S, Sarabia F, Vourloumis D, He Y, Vallberg H, Finlay MRV, Yang Z. J Am Chem Soc. 1997;119:7974–7991. [Google Scholar]
- 35.Nicolaou KC, Hepworth D, King NP, Finlay MRV, Scarpelli R, Pereira MMA, Bollbuck B, Bigot A, Werschkun B, Winssinger N. Chem-Eur J. 2000;6:2783–2800. doi: 10.1002/1521-3765(20000804)6:15<2783::aid-chem2783>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 36.Nicolaou KC, Hepworth D, Finlay MRV, Paul King N, Werschkun B, Bigot A. Chem Commun. 1999:519–520. [Google Scholar]
- 37.Genet JP, Ratovelomanana-Vidal V, Cano de Andrade MC, Pfister X, Guerreiro P, Lenoir JY. Tetrahedron Lett. 1995;36:4801–4804. [Google Scholar]
- 38.Taber DF, Silverberg LJ. Tetrahedron Lett. 1991;32:4227–4230. [Google Scholar]
- 39.Paquette LA, Galemmo RA, Jr, Caille JC, Valpey RS. J Org Chem. 1986;51:686–695. [Google Scholar]
- 40.Mulzer J, Mantoulidis A, Oehler E. J Org Chem. 2000;65:7456–7467. doi: 10.1021/jo0007480. [DOI] [PubMed] [Google Scholar]
- 41.Liu Y, Ali SM, Boge TC, Georg GI, Victory S, Zygmunt J, Marquez RT, Himes RH. Comb Chem High Throughput Screen. 2002;5:39–48. doi: 10.2174/1386207023330615. [DOI] [PubMed] [Google Scholar]
- 42.Li LS, Wu YL. Tetrahedron Lett. 2002;43:2427–2430. [Google Scholar]
- 43.Algaier J, Himes RH. Biochem Biophys Res Commun. 1988;954:235–243. doi: 10.1016/0167-4838(88)90078-7. [DOI] [PubMed] [Google Scholar]
- 44.Nath JP, Eagle GR, Himes RH. Biochemistry. 1985;24:1555–1560. doi: 10.1021/bi00327a040. [DOI] [PubMed] [Google Scholar]
- 45.Jain AN. J Comput-Aided Mol Des. 2007;21:281–306. doi: 10.1007/s10822-007-9114-2. [DOI] [PubMed] [Google Scholar]
- 46.Pham TA, Jain AN. J Med Chem. 2006;49:5856–5868. doi: 10.1021/jm050040j. [DOI] [PubMed] [Google Scholar]
- 47.Jain AN. Curr Opin Drug Discovery Dev. 2004;7:396–403. [PubMed] [Google Scholar]
- 48.Jain AN. J Med Chem. 2003;46:499–511. doi: 10.1021/jm020406h. [DOI] [PubMed] [Google Scholar]
- 49.Meng EC, Shoichet BK, Kuntz ID. J Comput Chem. 1992;13:505–524. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.