

Abstract

An eight step synthesis of (±)-grandisol features a key sequence involving a high-yielding, microwave-assisted enyne metathesis to yield a 1-alkenylcyclobutene that is semihydrogenated to yield a silyl-protected grandisol. Metathesis catalyst screens revealed an intriguing trend whereby substrate conversion correlated strongly with the identity of the ligands on the catalyst. In addition, new reactivity of 1-alkenylcyclobutenes toward hydrogenation is described.
Grandisol is the primary constituent of the grandlure, a mixture of four pheromones that comprise the sex attractant of the cotton boll weevil. Cotton boll weevils cause significant damage to cotton crops, and grandlure-filled traps are one means used to protect against boll weevil infestation and its associated economic consequences. The extraction of grandlure components from large collections of dead weevils is tedious and unpleasant, making efficient synthetic routes an attractive alternative to harvesting. Furthermore, racemic grandisol has proven equally effective at attracting boll weevils as the natural enantiomer,1 rendering moot the need for enantioselective syntheses for agricultural purposes.
Historically, the alkenylcyclobutane core of grandisol has presented a challenge to synthetic chemists and has served as a proving ground for new methodologies.2 Herein we report the use of a 1,5-enyne metathesis reaction as the key step in a straightforward and efficient assembly of the carbon framework of grandisol. Our retrosynthetic analysis is described in Scheme 1. We envisioned the vinylcyclobutane core of grandisol arising from a semihydrogenation (monohydrogenation) of vinylcyclobutene 1. Substituted vinylcyclobutene 1 can be prepared from enyne 2 in a metathesis cyclization. 2 may be prepared from commercially available α-methyl-γ-butyrolactone using standard synthetic manipulations.
Scheme 1.
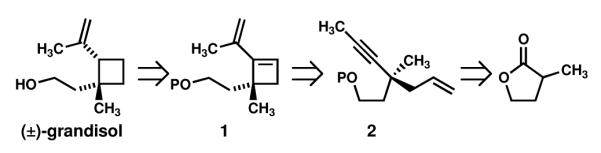
Retrosynthesis of (±)-grandisol
The hydrogenation of 1 to 3 is a formidable synthesis challenge (Scheme 2); it must proceed with regioselectivity (preferential hydrogenation of the cyclic alkene over the acyclic alkene to instead form 4), diastereoselectivity (preferential hydrogenation from the face of the cyclobutene bearing the methyl group over the ethyloxy group to instead form 5), and chemoselectivity (preferential semihydrogenation of the diene over further hydrogenation of the intermediate monoene to instead form 6). The reactivity of 1-alkenylcyclobutenes have not been extensively studied,3 and no comprehensive strategy exists for the regioselective semihydrogenation of dienes in general and of conjugated dienes in particular.4 However, for the conversion of 1 to 3 we anticipated being able to obtain the desired regioselectivity by taking advantage of the enhanced reactivity of the cyclobutene due to its inherent ring strain.5 Furthermore, we expected that increasing the steric bulk of the alcohol through its conversion to a bulky silyl ether (for instance, P = TBDPS, 1a) might promote diastereoselective hydrogenation from the opposite, less hindered face. Finally, though we hoped the inherent difference in reactivity of the strained cyclobutene would allow us to easily achieve the desired chemoselectivity, given the known sensitivity of hydrogenation reactions to substrate structure, catalyst, and solvent,6 and how little is known about the inherent reactivity of vinylcyclobutenes, we anticipated an empirical screen of reaction conditions might be necessary.
Scheme 2.

Challenges associated with selective semihydrogenation of 1
Our synthesis (Scheme 3) began with allylation of α-methyl-γ-butyrolactone followed by reduction of the lactone to yield lactol 7. Silylaytion of the open-chain form of 7 gave aldehyde 8. Alkynylation of the aldehyde with the Bestmann-Ohira reagent followed by methylation of the terminal alkyne generated enyne 9, the substrate for our key methathesis cyclization.
Scheme 3.
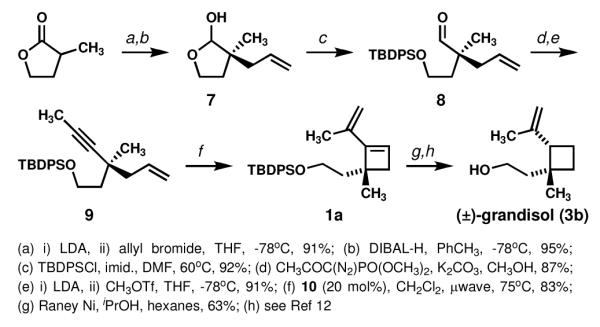
Synthesis of (±)-grandisol
When 1,5-enyne 9 was exposed to conditions previously reported by Campagne et al. to induce a metathesis cyclization for a variety of substrates7 (catalyst 10, microwave irradiation, CH2Cl2, 75 °C, 30 min), vinylcyclobutene 1a was isolated in 83% yield. Notably, this yield is higher than any reported in the literature,7 possibly due to the Thorpe-Ingold effect.8 Encouraged by this result, we performed a comprehensive catalyst screen to determine which catalyst structural feature(s) facilitated the transformation. All but one catalyst bearing a mesityl-disubstituted N-heterocyclic carbene ligand (10→13) led to complete consumption of 9 within 30 min at 75 °C as determined by crude 1H NMR (Figure 1). Of the remaining catalysts, those bearing a tricyclohexylphosphine ligand (15→17) showed moderate conversions, and those bearing an o-tolyl ligand showed poor conversions (18, 19). NMR analyses of crude reaction mixtures revealed evidence of catalyst decomposition especially in reactions catalyzed by 15→17, indicating the success of catalysts 10→13 is likely due to their enhanced stability at the elevated temperatures that are rapidly achieved with microwave irradiation. Such trends have been observed previously in microwave-assisted olefin metatheses.9
Figure 1.
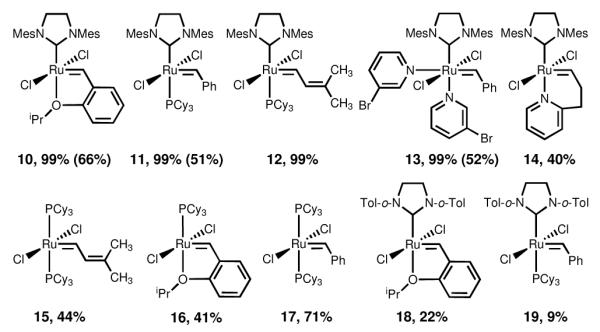
Result of catalyst screen. Percentages shown are conversion yields of the microwave reaction estimated from analysis of the crude NMR. Values in parentheses are isolated yields obtained using our sealed tube conditions (vide infra).
In an effort to expand the utility of this reaction to those without access to a microwave reactor, we developed a thermal, sealed tube protocol (sealed tube, CH2Cl2, 75 °C, 30 min) that gives lower yields than the microwave conditions described above yet significantly higher yields than the open flask yields reported in the literature.7 Three of the four catalysts that led to complete consumption of 9 were evaluated under our sealed tube conditions (Figure 1). Though the sealed tube yields are lower than what can be achieved under microwave conditions, they are synthetically useful and complement the seminal results previously reported.7
The hydrogenation of 1a proved challenging, with standard hydrogenation procedures generally leading within minutes to complete consumption of 1a and formation of significant amounts of fully hydrogenated 6a along with lesser amounts of desired 3a (see Scheme 2) and other isomers (Table 1). Simple variations on solvent, temperature, and catalyst did not lead to increased production of 3a. Based on previous observations that the adsorbed hydrogen on the surface of Raney Ni alone is sufficient to prevent full hydrogenation of alkynes,10 we hoped that complete reduction to 6a might be prevented through use of Raney Ni without a hydrogen atmosphere. We were pleased to discover that this was indeed the case; the desired isomer 3a was isolated in 63% yield.11 Conditions for cleaving the silyl protective group have been previously reported and occurs in quantitative yield.12
Table 1.
Results of Representative Regioselective Semihydrogenations of 1a After 10 minutes of Reaction Time
| Catalyst3 | Solvent | Temp | 1a1 | 3a | 4a2 | 6a |
|---|---|---|---|---|---|---|
| Pd/C | THF | 25°C | 15% | 31% | 10% | 44% |
| Pd/C | THF | 0°C | 14% | 35% | 11% | 40% |
| Pd/C | PhCH3 | 25°C | 42% | 19% | 10% | 29% |
| Pd/C | EtOH | 25°C | 0% | 35% | 12% | 53% |
| Pd/C | Hexane | 25°C | 0% | 43% | 12% | 45% |
| Pd/CaCO3 | EtOH | 25°C | 0% | 43% | 5% | 52% |
| Raney Ni4 | iPrOH | 25°C | 0% | 70% | 8% | 22% |
Percentages based on relative NMR integrations of distinctive peaks and scaled to 100%.
Product 5a was formed in <5% yield in each run.
5 mol%.
Performed in the absence of a hydrogen atmosphere.
Notably, the undesired diastereomer 5a was not observed in any significant quantity during the Raney Ni hydrogenation of 1a, indicating the stereoselectivity of the transformation is indeed controlled by the substrate. To provide further support for this hypothesis, we deprotected 1a (TBAF, THF, 81%) to form alcohol 1b and attempted the Raney Ni hydrogenation on this substrate. 1H NMR analyses of crude reaction samples throughout the reaction indicated no diastereoselectivity (essentially 1:1 production of 3b:5b) which stands in stark contrast to the diastereoselective reaction observed with 1a, which bears a large protective group.13
We have, therefore, described a synthetic sequence wherein (±)-grandisol may be prepared in 8 steps and 33% overall yield from commercially available α-methyl-γ-butyrolactone. Key advances in this synthesis include development of a non-microwave assisted method for a 1,5-enyne metathesis that proceeds in high yield and discovery of new insights into the reactivity of 1-alkenylcyclobutenes, a compound class whose reactivity has not yet been extensively studied.
Experimental Section
Procedure for Thermal, Sealed Tube Preparation of 10.
Note: sealed tube experiments should always be conducted behind a safety shield.
To a solution of 9 (467 mg, 1.19 mmol) in anhydrous CH2Cl2 (31 mL) was added 11 (150 mg, 20 mol%). The flask was then briefly purged with argon and sealed. The flask was then submerged in a preheated 75 °C oil bath and allowed to stir for 35 min. The flask was removed, cooled in an ice bath, and concentrated to give a green residue. Biotage column chromatography (2% Et2O:pentane) afforded 10 as a colorless oil (309 mg, 66%).
Supplementary Material
ACKNOWLEDGMENT
Financial support from ACS-PRF (47106-GB1), Research Corporation (CC7044/7165), and NIH-NCRR (P20 RR-016461) to B.G. is gratefully acknowledged. We thank The Milliken Foundation for providing critical instrumentation and Biotage, LLC for use of a microwave reactor. T.G. acknowledges Furman University for a research fellowship.
Footnotes
SUPPORTING INFORMATION AVAILABLE: Detailed experimental procedures for the preparation of all new compounds reported in Schemes 2 and 3 along with corresponding characterization data and the complete citation for Reference 2 (PDF). This material is available free of charge at http://pubs.acs.org.
References
- 1).Hibbard BE, Webster FX. J. Chem. Ecol. 1993;19:2129. doi: 10.1007/BF00979652. [DOI] [PubMed] [Google Scholar]
- 2).Citations for thirty-four previously reported syntheses of grandisol may be found in reference 4 in the Supporting Information. Some of these syntheses are competitive with the present synthesis in terms of efficiency, and most describe a methodological advance. In particular, we direct interested readers to five manuscripts that are representative of the variety of excellent chemistries inspired by grandisol: Refs. f, q, y, ff, and dd.
- 3).For cycloadditions, see: Park JD, Frank WC. J. Org. Chem. 1964;29:1445.Thummel RP. J. Am. Chem. Soc. 1976;98:628.Thummel RP, Nutakul W. J. Org. Chem. 1977;42:300.Thummel RP, Cravey WE, Nutakul W. J. Org. Chem. 1978;43:2473.Markgraf JH, Greeno EW, Miller MD, Zaks WJ. Tet. Lett. 1983;24:241.Doecke CW, Garratt PJ, Shahriari-Zavareh H, Zahler R. J. Org. Chem. 1984;49:1412.For an oxidation, see: Takeda A, Tsuboi S, Sakai F, Tanabe M. J. Org. Chem. 1974;21:3098.
- 4).Tungler A, Hegedüs L, Fodor K, Farkas G, Fürcht Á, Karancsi Zs. P. In: The Chemistry of Dienes and Polyenes. Rapaport Z, editor. Vol 2. John Wiley and Sons; New York: 2000. p. 992. [Google Scholar]
- 5).Wiberg KB. Angew. Chem. Int. Ed. Eng. 1986;25:312. [Google Scholar]
- 6).For a comprehensive treatment, see: Kluwer AM, Elsevier CJ. In: The Handbook of Homogeneous Hydrogenation. de Vreis JG, Elsevier CJ, editors. Wiley-VCH; Weinheim: 2007. Nishimura S. Handbook of Heterogeneous Catalytic Hydrogenation for Organic Synthesis. John Wiley and Sons; New York: 2001. Freifelder M. Catalytic Hydrogenation in Organic Synthesis: Procedures and Commentary. John Wiley and Sons; New York: 1978.
- 7).Debleds O, Campagne JJ. Am. Chem. Soc. 2008;130:1562. doi: 10.1021/ja0780986. [DOI] [PubMed] [Google Scholar]
- 8)a).Beesley RM, Ingold CK, Thorpe JF. J. Chem. Soc., Trans. 1915;107:1080.For an example in the context of a metathesis reaction see: Fürstner A, Langemann K. J. Org. Chem. 1996;61:8746. doi: 10.1021/jo961600c.
- 9).For a review of microwave-assisted olefin metatheis, see: Coquerel Y, Rodriguez J. Eur. J. Org. Chem. 2008:1125.Also see the following references therein: Efskind J, Undheim K. Tetrahedron Lett. 2003;44:2837.Michaut M, Boddaert T, Coquerel Y, Rodriguez J. Synthesis. 2007:2867.
- 10)a).Baran PS, Shenvi RA. J. Am. Chem. Soc. 2006;128:14028. doi: 10.1021/ja0659673. [DOI] [PubMed] [Google Scholar]; b) Soukup M, Widmer E. Tetrahedron Lett. 1991;32:4117. [Google Scholar]
- 11).This compound has been previously reported (see Ref. 12). 1H and 13C NMR spectra, included in the Supporting Information, match the data reported in the literature. HRMS data was not previously reported for this compound and has been included in the Supporting Information.
- 12).(a) Narasaka K, Kusama H, Hayashi Y. Bull. Chem. Soc. Jpn. 1991;64:1471. [Google Scholar]; (b) Kim D, Kwak YS, Shin KJ. Tetrahedron Lett. 1994;35:9211. [Google Scholar]
- 13).The 1H NMR analyses of these crude reaction mixtures were simplified by the fact that 5b is fragranol,14 a known natural product, whose chemical shifts are distinct from those of 3b (grandisol).
- 14).Bernard AM, Frongia A, Secci F, Delogu G, Ollivier J, Pieras PP, Salaün J. Tetrahedron. 2003;59:9433. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.