

Abstract
Object A novel method of estimating metabolite T1 relaxation times using MR spectroscopic imaging (MRSI) is proposed. As opposed to conventional single-voxel metabolite T1 estimation methods, this method investigates regional and gray matter (GM)/white matter (WM) differences in metabolite T1 by taking advantage of the spatial distribution information provided by MRSI.
Material and methods The method, validated by Monte Carlo studies, involves a voxel averaging to preserve the GM/WM distribution, a non-linear least squares fit of the metabolite T1 and an estimation of its standard error by bootstrapping. It was applied in vivo to estimate the T1 of N-acetyl compounds (NAA), choline, creatine and myo-inositol in eight normal volunteers, at 1.5 T, using a short echo time 2D-MRSI slice located above the ventricles.
Results WM-T1,NAA was significantly (P < 0.05) longer in anterior regions compared to posterior regions of the brain. The anterior region showed a trend of a longer WM T1 compared to GM for NAA, creatine and myo-Inositol. Lastly, accounting for the bootstrapped standard error estimate in a group mean T1 calculation yielded a more accurate T1 estimation.
Conclusion The method successfully measured in vivo metabolite T1 using MRSI and can now be applied to diseased brain.
Keywords: MR spectroscopic imaging, Relaxation time T1 estimation, Bootstrap, Monte Carlo simulation
Introduction
Magnetic resonance imaging and spectroscopy is a powerful tool for non-invasively assessing anatomic and metabolic changes that occur in brain diseases. In clinical spectroscopic studies and especially for magnetic resonance spectroscopic imaging (MRSI) data acquisition, short repetition times (TR) are often required to meet scan time constraints, but accurate metabolite longitudinal relaxation time values (T1) are then needed to correct the metabolite concentrations for the T1-weighted effect. The metabolite T1 values are likely to be important for quantifying results that make comparisons between patients and normal controls. Moreover, the knowledge of 1H metabolite longitudinal relaxation times can by itself give insight into the properties of a given region of interest.
In many previous studies, the estimations of the metabolite T1s were performed using single voxel acquisitions. Short echo time spectra coming from either progressive saturation [1–3] or inversion recovery experiments [4–6] were collected and T1 values were usually derived from amono-exponential fit. The inversion recovery experiments typically used long repetition times (TRs are usually equal to 6 s) with varying inversion times [4–6], which is prohibitively long for MRSI experiments. These single voxel approaches assume a single T1 over the whole voxel regardless of its tissue composition. To obtain white matter or gray matter T1 values and good SNR, large (usually greater than or equal to 8 cc), and relatively heterogeneous single voxels were typically acquired. In most cases, gray matter (GM)-T1 results were obtained from voxels containing 60–70% of GM, while white matter were obtained from voxels containing 70–90% of WM. At the same time, different MRSI studies [7–10] using linear regression demonstrated how metabolite concentrations (and thus metabolite signal intensities) can be different according to their tissue origin. Thus a common concern about the single voxel studies is whether the content of GM and WM in the examined voxel has an influence on the metabolite T1 results. Moreover attempting to reduce the size of the single spectroscopic voxel to reduce the voxel tissue heterogeneity would result in increasing the number of averages and the scan time. In contrast, MRSI techniques offer the possibility to acquire simultaneously several spectra over a wide brain region and at a resolution allowing tissue analysis. The first goal of this paper was, therefore, to develop a MRSI post-processing method to estimatemetabolite T1s while accounting for the voxels’ tissue content. To date, no published studies investigated the use of MRSI data to estimate metabolite T1 relaxation times.
Then, as for any quantitative measurement based on model fitting, an assessment of the precision of the T1 estimation is desirable. A benefit of MRSI is that it provides several spectra and thus several data points for the metabolite T1 fit which can be resampled in a bootstrap manner to estimate standard error. Therefore, a second goal of this study was to develop an approach to obtain metabolite T1 standard error by bootstrapping.
The last contribution of the paper was to apply the new techniques to measure metabolite T1s in different regions and tissues of the brains of healthy subjects.
The proposed method estimates metabolite T1 relaxation times by using 2D MRSI data at different repetition times. The progressive saturation method was chosen instead of inversion recovery method for scan time concern. While conventional techniques spend time in averaging single voxel acquisitions to obtain good SNR, we use this time to acquire multi-voxel data and investigate regional and tissue specific metabolite T1 differences. The post-processing takes advantage of the combination of segmented MRI and spatially distributed spectroscopic data to investigate either WM versus GM metabolite T1 values and/or regional differences in longitudinal relaxation times. The proposed method relies on three major concepts:
Increasing the SNR by averaging voxels according to their WM/GM content and their location (for example anterior vs. posterior) since the SNR of the metabolite signals is very low using the proposed acquisition parameters (number of excitations (NEX) and number of voxels in the slice) at 1.5 T.
Estimating metabolite T1 for gray and white matter using a non-linear least squares algorithm. The underlying model function used in the fitting procedure associates WM/GM content of a voxel to the metabolite signal intensity.
Using a bootstrap technique to assess uncertainty on the metabolite T1s and taking into account this confidence when calculating a group mean value.
This paper presents the techniques developed to utilize MRSI data for metabolite T1 measurement. A validation of these techniques is then proposed through Monte Carlo data simulations, demonstrating the statistical performance of the proposed method. Finally the fitting procedure is applied to 2D conventional MRSI data acquired at 1.5 T from eight healthy subjects.
MR data acquisitions
Study subjects
A total of eight healthy volunteers (five females and three males, mean age 31.5±9.5 years) were examined to validate our method. Written and informed consentwas obtained from all participating subjects. The study was approved by the UCSF Committee on Human Research.
MR parameters
The healthy volunteers were scanned on a Signa 1.5 T clinical imager from GE Medical Systems (GE Healthcare Technologies, Waukesha, WI, USA) using a quadrature head coil. Short echo time (TE = 35 ms) 2D MRSI data sets (12×12, 1 cc resolution) were acquired using a PRESS volume selection at five different TRs (TE = 0.850, 1, 2, 4, 8s).The number of excitations (NEX) acquired were as follows: NEX=3 for TE = 0.850s, NEX=2 for TR = 1s, NEX=1 for TR = 2, 4 and 8s. Oblique Fast Spin Echo images were used to guide the positioning of the spectroscopic acquisition. Care was taken that the PRESS box avoided the ventricles and was centered in the anterior-posterior middle of the corpus callosum body. T1-weighted 3D spoiled gradient recalled (SPGR) images were also acquired for segmentation of the anatomic images. The setup and data acquisition time for the anatomic and spectroscopic imaging was approximately 55 min.
Methods
The first goal of the analysis was the formulation of an approach to estimate regional metabolite T1s in cortical gray matter and white matter.
The model function
With the assumption made by several previous MRSI studies [7–9] of a linear relationship between the voxel WM/GM content and the metabolite peak signal intensity, we applied the following simplified model for the measured signal intensity S (for example S =signal amplitude of NAA) in the nth MRSI voxel
 |
where n runs through all the voxels in a given brain region (for example anterior and posterior). pWMn and pGMn are the calculated fraction of WM and GM, respectively within the nth spectroscopic voxel, SWM0 and SGM0 are respectively the signal intensity of a fully relaxed (TR ≫ 10s) resonance pertaining to assumed pure WM and pure GM, f is a function that characterizes the T1-weighting at a certain TR. We used the usual mono-exponential function [2], 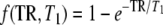 . S denotes either a resonance intensity corresponding to only a part of a metabolite or a whole metabolite signal intensity depending on whether T1 values are assumed to differ between different parts of the molecule. We then, for example, split the creatine signal into two parts, assuming that the CH3 singlet at 3.02 ppm has a different T1 than the CH2 singlet at 3.91 ppm. Note that this model does not necessarily require the WM and GM T1s to be distinct, but rather relaxes the constraint of having only one T1 independent of the tissue type.
. S denotes either a resonance intensity corresponding to only a part of a metabolite or a whole metabolite signal intensity depending on whether T1 values are assumed to differ between different parts of the molecule. We then, for example, split the creatine signal into two parts, assuming that the CH3 singlet at 3.02 ppm has a different T1 than the CH2 singlet at 3.91 ppm. Note that this model does not necessarily require the WM and GM T1s to be distinct, but rather relaxes the constraint of having only one T1 independent of the tissue type.
The T1 estimation procedure
Metabolite T1s are obtained after the following steps:
Step 1. Calculation of pWMn and pGMn
To compute the fraction of WM and GM, pWMnand pGMn of Eq. 1, for each spectroscopic voxel, the anatomic T1-weighted images (SPGR) were segmented into GM, WM and cerebrospinal fluid (CSF) compartments using the Automated Segmentation Tool, FAST [11]. Then these segmentation masks were resampled using nearest neighbor interpolation into the coordinate system of the T2-weighted image used for the prescription of the spectroscopic grid. The transformation used was calculated from the position and orientation of the T1 and T2 images, assuming there was no motion between the scans. Finally, for each voxel, the fraction of WM and GM within a voxel was computed from these segmentation masks by counting the number of WM or GM pixels within a spectroscopic voxel. Thus, the WM and GM proportion maps are at the spectroscopic resolution as shown in Fig. 1. The chemical shift displacement was minimized by exciting a larger region than desired (using a larger PRESS selected volume than usual) and then using very selective saturation (VSS) pulses to eliminate extra signals and to obtain the final, desired selected region [12].
Fig. 1.

a Segmentation of the white matter, b Percentage of white matter at the spectroscopic resolution, c Segmentation of the gray matter, d Percentage of gray matter at the spectroscopic resolution, e Example of original spectra coming from the four highlighted voxels TR = 1s, NEX = 2, SNRNAA ≈ 2
Step 2. Voxel averaging
At 1.5 T, the SNR (defined for example as the ratio of NAA time-domain singlet amplitude to the standard deviation of the time domain noise) of our acquisition was very low, typically between 1.5 and 2.8. Consequently the estimation of the metabolite intensity S is not accurate. For each TR, we proposed to generate Ngen new signals from the Ntotal original signals of the MRSI grid by averaging Navg chosen signals to increase the SNR. The Navg signals are randomly selected among the nearest neighbors of the voxel in terms of WM/GM content to track the original WM/GM distribution while reaching a greater SNR. For each averaged signal k, the new fractions pWMk and pGMk are also calculated by averaging the fractions corresponding to the Navg chosen signals, see Fig. 2. It is possible, with this technique to have more voxels than originally, Ngen ≥ Ntotal as we can draw several sets of Navg voxels for one original voxel. Figure 2b, c show the SNR gain between the original in vivo spectra and the averaged spectra.
Fig. 2.

a Graph showing the WM-GM percentage distribution of the original Ntotal voxels (black) as well as the Ngen averaged voxels. b Example of four spectra coming from the original voxels (Navg = 0, SNRNAA ≈ 2). c Example of four spectra coming from the averaged voxels (Navg = 6, SNRNAA ≈ 4.4)
Note that to perform regional analysis, this averaging procedure is applied on a specific part of the spectroscopic grid (for example in the anterior part of the slice) by working only with the voxels belonging to the region of interest.
Step 3. Quantification of S
The signal intensity S of Eq. 1 has to be estimated for each metabolite of interest, at each TR value, and for n running through all the Ngen averaged voxels. A quantification or a peak picking method can be used as is done for single voxel studies. We applied the time-domain algorithm QUEST [13] using a simulated basis set suitable to the acquisition. In a preprocessing step, the residual water was eliminated using the HLSVD method [14]. At 1.5 T, six metabolite patterns were estimated: the whole metabolite pattern for N-Acetyl compounds (NAAt = NAA + NAAG, mainly located at 2.02 ppm that we will call summarily NAA), choline compounds (3.21 ppm), glutamate (Glu, ∼2.3 ppm), and myo-Inositol (mI, ∼ 3.6 ppm), and the two patterns for creatine (Cr-CH3, 3.03 ppm, Cr-CH2, 3.91 ppm). The metabolites were simulated using the NMR-SCOPE[15] program of the jMRUI software package [16]. The background contamination coming from broad macromolecules, lipids and surrounding broad pattern of metabolites of low concentration were automatically modeled and taken into account in the semi-parametric approach utilized in QUEST [17]. The T1s of the NAA singlet (T1,NAA), Cr-CH3 (T1,Cr-CH3), Cho (T1,Cho) and mI (T1,mI) were investigated. Note that the T1 of Cr-CH2 was not estimated because of its poor signal quality due to its proximity to the water in the spectrum. To ensure the quality of the four-parameter fit, good quantification results were selected prior to step 4. Voxel results were selected following these two criteria:
estimated relative Cramér-Rao lower bound [18] of the metabolite amplitude, (rCRB) < 15%.
metabolite peaks that had smaller than 9Hz linewidth at half peak height.
After this selection, we have Nfinal,TR points at each TR for the metabolite T1 fit.
Step 4. Four-parameter fit
The four parameters SWMO, SGM0, TWM1, TGM1 of Eq. 1 were fitted using a non-linear least squares algorithm. We used the lsqnonlin method from the Optimization Toolbox of Matlab (The MathWorks, Natick, MA, USA). See Fig. 3. If there are Nfinal,TR selected results and there is a number NTR of repetition times, one has 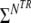 Nfinal,TR (usually 100–300) data points to fit the four parameters.
Nfinal,TR (usually 100–300) data points to fit the four parameters.
Fig. 3.

Map showing an in vivo example of the four parameter fit for NAA compounds. In this case 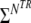 Nfinal,TR = 480 data points (5 TR, 96 points per TR) were available for the four parameters fit
Nfinal,TR = 480 data points (5 TR, 96 points per TR) were available for the four parameters fit
Estimating metabolite T1 uncertainties using bootstrap
Bootstrap is an empirical, non-parametric statistical technique based on data resampling. It is used to make statistical inference such as the variance estimate of some fitted parameters. Although well-known and used in other MR modalities such as fMRI [19] or diffusion tensor MRI [20], it has rarely been applied, to our knowledge, to MRS parameters [21]. This computer-based method relies on the drawing of some bootstrap samples [22]. In our case, a bootstrap sample consists, for each TR, in a random sample of size Nfinal,TR, say Sj/*(TR), 1 ≤ j ≤ Nfinal,TR, where Sj/*(TR) is drawn with replacement from the original data points Sj (TR). To avoid some downward bias on the estimated standard error, we applied a bootknife approach [23] which is a resampling technique combining the features of jackknife and bootstrap. For each TR, one point is first randomly omitted from the original sample of size Nfinal,TR (jacknife), then, from the remaining sample with size Nfinal−1, a bootstrap sample of size Nfinal,TR with replacement (bootstrap) is drawn. This is done at each TR and the four parameter fitting procedure is then applied to the bootknife samples to obtain a ^TWM1 and a ^TGM1 replications. This operation is done B times (typically we used B = 200), see Fig. 4, and a standard error (SE) can be estimated as follows:
 |
, where ¯T1 is the mean value of the estimates over the B bootknife samples and ^Tb1 is the bth estimation of T1 (or replication) fitted from the bth bootknife sample set.
Fig. 4.

The bootstrap algorithm adapted from [22] for estimating standard error of metabolite T1; The T1 estimate is fitted from the original sample and can be either WM or GM T1. The bootstrap replications ^Tb1, b=1 … B, are used to calculate its standard error estimate (SE). B is usually between 25 and 200
To take into account the confidence in the T1 value estimation from a subject when calculating the mean T1 value over a group of subjects, we proposed using the estimated standard errors in a weighted average calculated as follows:
Weighted sample mean:
 |
, where nbofscans corresponds to the number of subjects scanned in the study and contributing to the group mean value.
The corresponding individual standard deviation of this mean value is estimated with:
 |
, where Til is the estimated T1 for a given metabolite and subject i, ^SEi is the bootstrap estimate of the standard error of the T1 estimation for the subject i. Figure 5 summarizes the steps used in the proposed method for the case of noisy signals.
Fig. 5.

Scheme of the proposed procedure for a metabolite T1 fitting where “SE” stands for bootstrap standard error estimates
Monte carlo simulations
Simulations were conducted to validate the method. The bias and standard deviation of metabolite T1 estimates were determined using Monte Carlo studies for different Navg, with or without a macromolecular background contamination. We show the ability of the bootstrap technique to estimate the metabolite T1 standard error. We used previously described MRSI data simulation programs [24]. In this simulation technique, aMRSI k-space is generated by performing the product of the k-space distribution of a given object and its corresponding spectroscopic signal. The effect of the PRESS box selection is also taken into account. For our simulations, one object for WM and one for GM were generated and the results summed. A short echo time signal containing NAA, choline, creatine, Glu, and mI was created for each TR and each WM or GM object. A background signal reproducing the effect of macromolecular contamination in the short echo time signal and mimicking, in the metabolite region, the macromolecule baseline signal found in [3] was also added to the signal. The metabolite relative amplitudes, T1, SNR and the background components that were used in the Monte Carlo studies are summarized in Table 1. Without noise, for one voxel with a certain WM/GM content and one TR, the simulated metabolite signal amplitude was set exactly to Eq. 1. White Gaussian distributed noise signals were added to each noise-free signal of the simulated MRSI grid. These steps were repeated to obtain a total of NMC = 100 noisy MRSI sets of signals. Metabolite T1 values were obtained using the proposed method for those 100 realizations. For each metabolite, we obtained a gold standard SE (standard error) on T1 by calculating the standard deviation of all of the T1 estimations. This gold standard SE was compared to the mean value of the standard errors calculated by the proposed bootstrap approach for each realization.
Table 1.
Parameters used in the Monte Carlo simulation studies
| Metabolite | WM | GM | ||||
|---|---|---|---|---|---|---|
| Concentration (a.u.) | T1 (s) | SNR at TR = 8s | Concentration (a.u.) | T1 (s) | SNR at TR = 8s | |
| NAA singlet (3 equivalent protons) | 7.5 | 1.55 | 2 | 9 | 1.45 | 2 |
| Cho (9 equivalent protons) | 1.8 | 1.2 | 1.44 | 1.5 | 1.2 | 1 |
| Cr-CH3 (3 equivalent protons) | 5.2 | 1.3 | 1.39 | 7.7 | 1.4 | 1.71 |
| mI | 3.8 | 1.1 | 0.91 (at 3.56ppm) | 5 | 1.1 | 0.99 |
| Glu (T1 not estimated) | 7 | 1.3 | / | 9 | 1.3 | / |
| Background signal | WM/GM | ||
|---|---|---|---|
| α | ω (ppm) | SNR at TR = 8s | |
| Nine gaussian components, time domain model: Σ9i=1ai exp(jωt)exp(−β2t2)T1 = 0.2s and β = 50 Hz for all the components | 2.5 | 1.88 | 0.21 |
| 15 | 2.08 | 1.29 | |
| 2.5 | 2.39 | ||
| 2.5 | 2.55 | ||
| 2.5 | 2.71 | ||
| 10 | 3.09 | 0.86 | |
| 5 | 3.5 | 0.43 | |
| 5 | 3.66 | ||
| 10 | 3.91 | ||
The macromolecular background components were chosen to make the background signal resemble the patterns shown in Ref. [3] a.u. arbitrary unit, WM white matter, GM gray matter
Study 1: Effects of averaging
In the first Monte Carlo study, we evaluated the statistical performance of the method in terms of bias and standard deviation for SNR = 2 along with an increasing number of voxels in an average, Navg = 2, 4 and 6, and the macromolecular background signal (see Table 1) added to the simulated short echo time signals. For each TR, Ngen = 120 voxels are created by the proposed averaging method from the original MRSI grid. We also tested our method in the case of no averaging. In this case, designated by Navg = 0, only the 48 voxels (Ngen = Ntotal) in the MRSI grid contribute to the fitting method.
Study 2: Macromolecular background effect
We tested the macromolecular background effect on the T1 estimation in the second Monte Carlo study. We compared the statistical results obtained with Navg = 6 with and without a macromolecule signal in the simulation. The weighted average using the bootstrapped standard error estimation is tested in these two cases.
Results
Monte Carlo studies
Results of the Monte Carlo simulation are shown in Figs. 6, 7 and Table 2. SE, bootstrap estimates of the SE and bias are expressed as a percentage of the true T1 value.
Fig. 6.

From Monte Carlo simulations, gold standard standard errors of metabolite T1s compared against standard errors estimated by the bootstrap technique with varying numbers of voxels used for signal averaging Navg. The sign (*) indicates cases where the standard deviation of the bootstrap estimate is larger than its mean value or where the bootstrap estimate is biased by more than 100% from the actual value. Note the general decrease of the SE with Navg
Fig. 7.

The bias (b) on metabolite T1 and the bias for a weighted average (bw) calculated with Eq. 3. b and bw are displayed as percentage of the true metabolite T1 value
Table 2.
From Monte Carlo simulations (NMC = 100), gold standard standard errors (SE), standard error (SEw) corresponding to the weighted average and calculated with Eq. 4, bias (b) and bias for a weighted average (bw) calculated with Eq. 3
| Metabolite | SE (%) | SEw(%) | b(%) | bw(%) | ||||
|---|---|---|---|---|---|---|---|---|
| w/o | w/ | w/o | w/ | w/o | w/ | w/o | w/ | |
| NAA-WM | 7.51 | 12.84 | 6.94 | 11.58 | 1.96 | 5.33 | 0.54 | 0.41 |
| NAA-GM | 9.28 | 17.65 | 9.12 | 15.96 | 1.95 | 0.82 | 2.02 | 0.73 |
| tCho-WM | 12.14 | 14.99 | 10.18 | 14.97 | 1.83 | −5.260 | 0.14 | −8.20 |
| tCho-GM | 25.44 | 42.47 | 24.60 | 37.31 | 6.21 | 13.82 | 3.37 | 9.74 |
| tCr-WM | 12.84 | 14.08 | 12.06 | 13.32 | 2.31 | −8.041 | 1.93 | −7.63 |
| tCr-GM | 14.83 | 18.38 | 13.46 | 17.28 | 2.74 | −3.01 | −0.70 | −6.83 |
| mI-WM | 14.57 | 34.25 | 13.51 | 30.14 | 2.63 | 9.95 | 0.93 | 3.26 |
| mI-GM | 25.18 | 59.20 | 20.95 | 42.59 | 3.97 | 15.30 | −1.50 | −1.75 |
Results shown with (“w/”) and without (“w/o”) a background contamination, SNRNAA = 2, Navg = 6
Study 1: Effects of averaging
Figure 6 shows the mean and the standard deviation of the bootstrap estimates, as well as the gold standard of the SE for each metabolite, for a range of Navg, SNRNAA equals 2, and the set of 5 TRs. (*) indicates cases where the bootstrap estimate is biased by more than 100 %from the actual value.
For the four metabolites of interest (NAA, Cr-CH3, Cho, mI), higher SE were found in the GM than in WM consistent with the discrepancy between the number of GM voxels versus the number of WM voxels available in the masks we used in the simulation. The SE globally decreases with the number of voxels used in an average Navg. For Navg = 6, the gold standard SE is below 20% of the true T1 for NAA, Cho and Cre in the WM.
For all of the considered metabolites in the WM and the GM and for Navg ≥ 4, the SE estimated by the bootstrap technique was within 50% of the actual SE value. Less biased bootstrap estimates were generally obtained for Navg = 2 and the bias between the bootstrap estimates and the actual SE value increased with Navg. For mI, the bootstrap approach successfully estimated the SE with a large standard deviation in the WM and only for Navg = 6 in the GM. For Navg = 0, the bootstrap estimates have reasonable values in WM for NAA, Cho and Cre and failed to estimate the SE in the other cases.
Figure 7 shows the bias (denoted by b) on T1 values for the four metabolites of interest and for a weighted average calculated by Eq. 3 and denoted by bw. b and bw are displayed as percentages of the true metabolite T1 value. For (WM/GM)-T1,NAA (WM/GM)-T1,Cr-CH3 and WM-T1,Cho the bias is below ±10% of the true T1 value. We note that, in the case of our simulation, increasing the Navg did not reduce the bias for WM-T1,Cr-CH3. As seen in the next study, we think that this bias is more due to the interaction with the macromolecules than to a lack of SNR. Increasing Navg seems to reduce the bias for (WM/GM) T1,NAA, GM-T1,Cho, GM-T1,Cre, and (WM/GM) T1,mI. For WM and GM-T1,mI, the weighted average makes the bias below 10% of the actual value for Navg ≥ 2.
The best bias and standard deviation trade-off is obtained for Navg = 6 at the expense of slightly biased bootstrap estimates.
Study 2: Macromolecular background effect
Table 2 shows the statistical results (regular SE, SEw calculated with Eq. 4, b and bw) of the T1 estimation procedure for Navg = 6, SNR = 2, with and without a background contamination in the signals to process. All the metabolite T1 estimations show the same trend with a larger standard deviation and a bigger bias in the case of macromolecular contamination as compared to the absence of a background signal. In the absence of a macromolecular background, the weighted average that takes into account the bootstrapped standard error successfully reduces the bias (bw as compared to b) on the metabolite T1.
In the presence of the simulated background signal, the SE of the T1 estimate increased by 10%(for WM-T1,Cr-CH3) and more than doubled for the T1,mI. The bias was also affected (but usually reduced) by using a weighted average. The GMT1,Cr-CH3 and WM-T1,Cho estimation were more downward biased in the presence of a background signal when using a weighted average than when using the standard mean calculation.
Metabolite T1 estimation on in vivo data
For the eight subjects, the anterior-posterior center of the PRESS Box (12 × 12, 1 cc) was centered in the anterior-posterior middle of the corpus callosum body visualized in a sagittal plane. Voxels anterior to the center of the PRESS Box were analyzed as part of the anterior brain region (Ant.) and the rest of the voxels were evaluated as posterior voxels (Post.).
The bootstrap estimate of standard error on in vivo data
Figure 8 shows, for each subject of the study, a histogram of B = 200 bootstrap replications of ^TWM1,NAA calculated in the posterior part of the PRESS box. For each subject i(i = 1,…, 8), these replications are used to estimate standard error SEi. In this example, subject 3 presents a small standard error and thus will have a larger weighting in the proposed weighted average in Eq. 3 while the results from subject 2 or 5 will have smaller weighting due to their bigger bootstrap estimated standard error. This bootstrap standard error estimate gives good insight about the reliability of the fitted metabolite T1. In the case of subject 8, the bootstrap histogram reveals a peak quite distinct from the average when fitting ^TWM1,NAA from the selected voxels. The dotted line indicates the estimated T1 when all the selected voxels are considered while the bootstrap histogram is calculated with the bootknife approach that randomly removes one voxel at each calculation. Therefore, this case shows that some voxels were essential in determining the T1.
Fig. 8.

Histograms of B = 200 bootstrap replications of WM-T1,NAA, calculated from in vivo data (posterior region) of eight healthy subjects. A broken line is drawn at the parameter T1 estimate
Anterior versus posterior metabolite T1
The estimated in vivo T1 relaxation times (mean±SD) for “pure” WM and “pure” GM at 1.5 T for Navg = 6 and computed from the anterior region (Ant.) or from the posterior region (Post.) are given in Table 3. These results are calculated with Eqs. 3 and 4 using the bootstrap standard error estimates and are given as weighted means ± the corresponding standard deviations. For statistical analysis, a two-tailed paired t-test was used. See Appendix.
Table 3.
From eight healthy volunteers, estimated T1-relaxation times (in seconds) of NAA, Cho, Cr-CH3, mI at 1.5 T, in pure WM and pure GM, using MRSI data, (Navg = 6)
| Metabolite | T1, Navg = 6 | |
|---|---|---|
| Ant. | Post. | |
| NAA | ||
| WM | 1.38 ± 0.15 | 1.28 ± 0.10** |
| GM | 1.23 ± 0.36 | 1.31 ± 0.20 |
| Cho | ||
| WM | 1.20 ± 0.13 | 1.20 ± 0.05 |
| GM | 1.18 ± 0.20 | 1.20 ± 0.10 |
| Cr-CH3 | ||
| WM | 1.26 ± 0.09 | 1.23 ± 0.08 |
| GM | 1.19 ± 0.12 | 1.22 ± 0.06 |
| mI | ||
| WM | 1.29 ± 0.09 | 1.23 ± 0.10 |
| GM | 1.19 ± 0.12 | 1.22 ± 0.09 |
Note the anterior and posterior difference (P < 0.05) found with a paired t-test in NAA T1 in the WM as well as a trend, not statistically significant, of a WM/GM difference for NAA, Cr-CH3 and mI T1 in the anterior part
** Anterior WM-T1 > posterior WM T1 with P < 0.05
The standard deviation in the GM was larger than in the WM, for most of the time, as in the Monte Carlo simulations. From these results, no significant differences were found in metabolite T1s between GM and WM in the posterior region. In anterior region WM-T1,NAA, 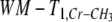 , and WM-T1,mI tend to be higher than GM-T1s but this trend did not reach statistical significance (0.1 < P < 0.2). The T1 of NAA in the anterior part of the WM was significantly longer than in the posterior part of the WM (P < 0.05).
, and WM-T1,mI tend to be higher than GM-T1s but this trend did not reach statistical significance (0.1 < P < 0.2). The T1 of NAA in the anterior part of the WM was significantly longer than in the posterior part of the WM (P < 0.05).
Discussion
This work presents a novel method for estimating metabolite T1s using MRSI data, enabling estimates within tissue types (GM and WM) and across different regions (anterior and posterior were demonstrated here). The smaller voxel size of the MRSI data versus previous single voxel studies partially addresses the issue of large partial volume artifacts between gray matter and white matter. Additionally, incorporating the information regarding tissue type composition obtained from higher resolution MR images and the multiple voxel data obtained with MRSI allows better correction of partial volume artifacts than possible with the previous single voxel data. This method has been validated and tested on simulations and applied in vivo. We also proposed assessing a confidence interval in the fitted T1 results by introducing a bootstrap approach. We showed that a weighting average that takes into account the confidence assessment can reduce the bias on the estimated T1 value for a metabolite with a small SNR. This method relies upon a group mean metabolite T1 approach. The proposed algorithm yielded results that are in agreement with the literature and support the hypothesis of regional differences in T1 in the brain.
Methodology
The important parameters in the proposed method are the SNR of the considered metabolite signal, the number of voxels used in an average, Navg, and the number of available, mostly GM or mostly WM voxels in the MRSI grid.
The SNR, as expected, appeared to clearly play a role both on the bias and the standard deviation of the T1 estimation. In Figs 6 and 7 the results for NAA and Cr-CH3 which have the greatest SNR in our simulation, present good biases and standard deviations on T1 estimation compared to the ones for Cho (especially in GM) and mI. In order to realize a robust four-parameter fit with low SNR, non-reliable voxel quantification results should be rejected from the analysis. The use of criteria, such as Cramér-Rao lower bounds [18] or linewidth thresholds, is necessary to determine the quality of the metabolite amplitude quantification and to perform voxel selection.
Increasing the number of voxels used in an average Navg, improves SNR and so typically reduces both bias and standard deviation. Note that, by averaging the voxels, the independence between the averaged voxels is reduced and the bootstrap technique can tend to underestimate the real standard error. Also note that this averaging is performed while taking into account the WM-GM distribution and the introduced dependence tracks the initial tissue content. It was also shown by the Monte Carlo simulation results that, for an original SNR of two, the use of six voxels in an average corresponds to a good trade-off between the SNR gain and the lost of voxel independence and leads to reliable metabolite T1 estimations. Furthermore, this voxel averaging introduces some partial voluming with CSF to the generated voxels, especially for mostly GM voxels originating from the thin cortical ribbon at the midline. Nevertheless, as the percentage of GM and WM (and thus, CSF) in the voxel are explicitly taken into account during the fitting procedure, the regression presented in Eq. 1 enables a correction of this partial voluming effect.
The number of voxels available for a specific tissue type influences the standard deviation of the T1 estimation. Consequently, the dispersion of the results is larger for the GM than for the WM values from both our simulation and the in vivo data.
The first Monte Carlo simulations showed that for an original SNRNAA of 2 in the MRSI data, an almost unbiased estimation of the T1,NAA, T1,Cho, T1,Cr-CH3 is possible. A weighted average, taking into account the bootstrap estimates of the uncertainty on T1 could also yield to an unbiased estimation of T1,mI. This approach tends to reduce the dispersion due to bad data points and makes a group mean value more accurate.
We conclude from the second Monte Carlo study that the presence of a macromolecular background signal has an important effect on the dispersion and the bias of the results and should be considered as another effect, besides the set and number of TRs and the type and parameters of the sequence used [25], leading to the discrepancy between the different published T1 values. The bootstrapped standard error estimation is also hampered by the macromolecular contribution. In the proposed simulation, all the voxels were contaminated in the same way and the background signal was arbitrary and particularly elevated under the mI making its T1 estimation harder.
Finally, the proposed bootstrap procedure is a novel method to estimate metabolite T1 standard error and is enabled in this study by the use of MRSI data. The bootstrap technique is a non-parametric method that does not require a complex implementation. This approach may be beneficial in future uncertainty and/or bias estimation studies of spectroscopic quantification.
In vivo results
The range of our results for the four metabolites is in good agreement with existing literature [1–4], when using the weighted average and Navg = 6. The method gives reliable results, for an original SNR around 2, when processing equivalent 6 cc voxels, which corresponds to an SNR around 4.9 for NAA. Voxel averaging combined with the quantification procedure, which constrained the additional damping factor allowed for each metabolite, was required to achieve the accuracy of the results.
We found significantly greater WM-T1,NAA values in the anterior (frontal) part (1.38 ± 0.15, mean ± SD) than in the posterior part of the slice (1.28 ± 0.10), but we were not able to see this result for the GM or for the other metabolites. Brief et al. [1] also reported similar results between WM frontal (1.59 ± 0.10), and WM parietal (1.35) regions. In the literature, WM-T1,NAA values can differ to some extent. Our anterior WM T1,NAA is lower than the one reported by Brief et al. or Kreis et al. [3] (1.88 ± 0.09), but still higher than others, as for example the value reported by Ethofer [2] (1.19±0.09 in the fronto-parietal region). This discrepancy may be partly due to the way the macromolecular background signal was fitted.
The effect of regional variation in metabolite T1 values may be necessary to take into account in the estimated metabolite concentration, depending on the ratio of TR/T1 used, on the ratio of the regional T1s, and on the accuracy of the metabolite amplitude estimation. In our study, a regional T1 variation for NAA of 7.8% would result in a difference of only 5% in the NAA signal amplitude for a short TR of 1 s. Considering the biological variability and the accuracy achievable for the NAA signal amplitude estimation, this difference might be negligible for a concentration estimation point of view. In the case of healthy versus diseased brain, the metabolite T1 difference may be larger than 7%. The measured NAA signal amplitudes could have important differences (greater than 5%), due solely to the T1 variation and not to a tissue concentration difference. Conversely, the T1 differences could mask the concentration changes due to the disease. Of course, when the repetition time exceeds three expected T1(TR > 3T1), the difference in regional T1 can range from 0 to as much as 50%, and the difference (due to the T1 weight) in metabolite signal amplitude will remain below 5% of the actual concentration value. Then the regional T1 variation will have effectively no effect on the estimated concentrations. In practice, the use of a long TR increases scan time, especially for MRSI acquisition and is therefore avoided.
The T1 value found for NAA in the WM of the posterior part of the slice (1.28 ± 0.10) is close to the values reported by Rutgers et al. [5,26] (1.30 ± 0.14) in the centrum semiovale. The T1 relaxation times found for the other metabolites Cho, Cr and mI, are also essentially the same as other reported values [2,5,26].
Especially in gray matter, where the glutamate signal is higher, the macromolecular signal, the NAA and the glutamate signals and some contribution from metabolite present at low concentration such as GABA are unknowingly entangled at 2 ppm. Moreover, the amount of macromolecule signal compared to the metabolite signal differs at each TR, as macromolecules have a shorter T1 than metabolites. As a consequence, the variability of the quantification results increases. We think that the higher standard error found for the GM-T1,NAA is partly due to this variability and partly due to the few number of available gray matter voxels.
We also observed without reaching statistical significance that, as opposed to water T1, the T1 for NAA, Cr-CH3 and mI could be greater in the white matter than in the gray matter in the anterior part while no difference was seen in the posterior part. Although a difference of WM/GM voxel distribution in the anterior and posterior regions (see Fig. 1) might have influenced this result, this observation supports the assumption of different underlying mechanisms for water and metabolite relaxation times. While tissue composition and difference in anisotropy may be involved in water T1 relaxation process, the intra-cellular metabolite T1, may be more dependent, as suggested by Ethofer et al., on micro-structural characteristics and viscosity properties.
Conclusion
Brain metabolite T1 measurements were calculated using a novel MRSI voxel averaging and bootstrapping approach. The proposed method takes advantage of the multi-voxel acquisition provided by MRSI and enables the investigation of regional variations in metabolite T1 values. It also introduces a bootstrap technique for estimating a standard error on metabolite T1s. Significant differences were found between anterior WM-T1,NAA and posterior WM-T,1,NAA. This result emphasizes the need to take into account tissue and regional T1 differences in MRSI metabolite quantification. Finally, the method only requires a multi-WM/GM voxel acquisition and is not restricted to short echo time 2D MRSI acquisition. The presence of a macromolecular background made the metabolite T1 estimation less accurate and substantially increased the dispersion. The principle of the method can be applied and extended to other field strengths (to increase the SNR) or to other types of data acquisition that present less macromolecular contamination such as TE-averaging [27], longer TE acquisition or to using localization in a third spatial dimension (3D-CSI).
Acknowledgement
We thank Dr. Ying Lu, Associate Professor in Residence of Radiology and Biostatistics at UCSF, for his expert assistance in the statistical aspects of this project and are grateful to Sung Won Chung and Yan Li for helpful discussions. This study was supported in part by Research Grant RG-3517A2 from the National Multiple Sclerosis Society (PI: Daniel Pelletier). Dr. Daniel Pelletier is a recipient of the Harry Weaver Neuroscholar Award from the National Multiple Sclerosis Society (JF-2122-A).
Appendix
Paired t-test in the case of weighted combinations
To compare the WM versus GM, or anterior versus posterior metabolite T1s, we used a paired t-test analysis that took into account the uncertainties estimated by bootstrapping. To perform the statistical analysis, we modified the usual paired t-test with the following steps:
1. Calculate the weighted sample mean of the variable “difference”
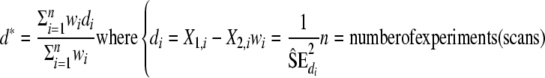 |
,
X1,i and X2,i are either the estimated T1 in the WM versus in the GM or the estimated T1 in the anterior region versus in the posterior region for a given metabolite.
wi should reflect the confidence in the difference value and is set to the inverse of the bootstrap estimate of the variance of di.
For comparisons between WM and GM T1 values:
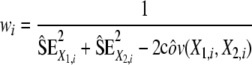 |
.
Indeed, as WM and GM T1 values were estimated on the same set of data, they can be correlated and the covariance between the two variables has to be taken into account.  are estimated using the proposed bootstrap approach. For comparison between WM anterior versus WM posterior T1, we assume no correlation (cov(X1,i, X2,i) = 0) as the values are calculated from a different data set.
are estimated using the proposed bootstrap approach. For comparison between WM anterior versus WM posterior T1, we assume no correlation (cov(X1,i, X2,i) = 0) as the values are calculated from a different data set.
2. Calculate the standard error of the weighted sample mean
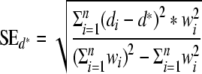 |
.
3. Get the current value of the statistic t and its degree of freedom ν
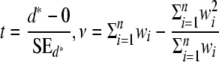 |
.
Note that if wi = 1 for i = 1, …, n, we retrieve the usual t statistic with  .
.
4. Determine the two-tailed P value from the Student’s t cumulative distribution function f(t, ν)
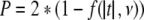 |
.
References
- 1.Brief EE, Whittall KP, Li DK, MacKay A. Proton T1 relaxation times of cerebral metabolites differ within and between regions of normal human brain. NMR Biomed. 2003;16:503–509. doi: 10.1002/nbm.857. [DOI] [PubMed] [Google Scholar]
- 2.Ethofer T, Mader I, Seeger U, Helms G, Erb M, Grodd W, Ludolph A, Klose U. Comparison of longitudinal metabolite relaxation times in different regions of the human brain at 1.5 and 3 Tesla. Magn Reson Med. 2003;50:1296–1301. doi: 10.1002/mrm.10640. [DOI] [PubMed] [Google Scholar]
- 3.Kreis R, Slotboom J, Hofmann L, Boesch C. Integrated data acquisition and processing to determine metabolite contents, relaxation times, and macromolecule baseline in single examinations of individual subjects. Magn Reson Med. 2005;54:761–768. doi: 10.1002/mrm.20673. [DOI] [PubMed] [Google Scholar]
- 4.Mlynarik V, Gruber S, Moser E. Proton T (1) and T (2) relaxation times of human brain metabolites at 3 Tesla. NMR Biomed. 2001;14:325–331. doi: 10.1002/nbm.713. [DOI] [PubMed] [Google Scholar]
- 5.Rutgers DR, Grond J. Relaxation times of choline, creatine and N-acetyl aspartate in human cerebral white matter at 1.5 T. NMR Biomed. 2002;15:215–221. doi: 10.1002/nbm.762. [DOI] [PubMed] [Google Scholar]
- 6.Traber F, Block W, Lamerichs R, Gieseke J, Schild HH. 1H metabolite relaxation times at 3.0 tesla: measurements of T1 and T2 values in normal brain and determination of regional differences in transverse relaxation. J Magn Reson Imaging. 2004;19:537–545. doi: 10.1002/jmri.20053. [DOI] [PubMed] [Google Scholar]
- 7.Hetherington HP, Mason GF, Pan JW, Ponder SL, Vaughan JT, Twieg DB, Pohost GM. Evaluation of cerebral gray and white matter metabolite differences by spectroscopic imaging at 4.1 T. Magn Reson Med. 1994;32:565–571. doi: 10.1002/mrm.1910320504. [DOI] [PubMed] [Google Scholar]
- 8.Maudsley AA, Darkazanli A, Alger JR, Hall LO, Schuff N, Studholme C, Yu Y, Ebel A, Frew A, Goldgof D, Gu Y, Pagare R, Rousseau F, Sivasankaran K, Soher BJ, Weber P, Young K, Zhu X. Comprehensive processing, display and analysis for in vivo MR spectroscopic imaging. NMR Biomed. 2006;19:492–503. doi: 10.1002/nbm.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Noworolski SM, Nelson SJ, Henry RG, Day MR, Wald LL, Star-Lack J, Vigneron DB. High spatial resolution 1H-MRSI and segmented MRI of cortical gray matter and subcortical white matter in three regions of the human brain. Magn Reson Med. 1999;41:21–29. doi: 10.1002/(SICI)1522-2594(199901)41:1<21::AID-MRM5>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 10.Schuff N, Ezekiel F, Gamst AC, Amend DL, Capizzano AA, Maudsley AA, Weiner MW (2001) Region and tissue differences of metabolites in normally aged brain using multislice 1H magnetic resonance spectroscopic imaging, pp 899–07 [DOI] [PMC free article] [PubMed]
- 11.Zhang YMB, Smith S. Segmentation of brain MR images through a hidden Markov random field model and the expectation maximization algorithm. IEEE Trans Med Imaging. 2001;20(1):45–57. doi: 10.1109/42.906424. [DOI] [PubMed] [Google Scholar]
- 12.Tran TK, Vigneron DB, Sailasuta N, Tropp J, Le Roux P, Kurhanewicz J, Nelson S, Hurd R (2000) Very selective suppression pulses for clinical MRSI studies of brain and prostate cancer, pp 23–3 [DOI] [PubMed]
- 13.Ratiney H, Sdika M, Coenradie Y, Cavassila S, Ormondt D, Graveron-Demilly D. Time-domain semi-parametric estimation based on a metabolite basis set. NMR Biomed. 2005;18:1–13. doi: 10.1002/nbm.895. [DOI] [PubMed] [Google Scholar]
- 14.Pijnappel WWF, Boogaart A, Beer R, Ormondt D. SVD-based quantification of magnetic resonance signals. J Magn Reson. 1992;97(1):122–134. [Google Scholar]
- 15.Graveron-Demilly DAD, Briguet A, Fenet B. Product-operator algebra for strongly coupled spin systems. J Magn Reson Ser A. 1993;101(3):233–239. doi: 10.1006/jmra.1993.1038. [DOI] [Google Scholar]
- 16.Naressi A, Couturier C, Devos JM, Janssen M, Mangeat C, Beer R, Graveron-Demilly D. Java-based graphical user interface for the MRUI quantitation package. Magn Reson Mater Phy. 2001;12:141–152. doi: 10.1007/BF02668096. [DOI] [PubMed] [Google Scholar]
- 17.Ratiney H, Coenradie Y, Cavassila S, Ormondt D, Graveron-Demilly D. Time-domain quantitation of 1H short echo-time signals: background accommodation. Magn Reson Mater Phy. 2004;16(6):284–296. doi: 10.1007/s10334-004-0037-9. [DOI] [PubMed] [Google Scholar]
- 18.Cavassila S, Deval S, Huegen C, Ormondt D, Graveron-Demilly D. Cramer-Rao bounds: an evaluation tool for quantitation. NMR Biomed. 2001;14:278–283. doi: 10.1002/nbm.701. [DOI] [PubMed] [Google Scholar]
- 19.Auffermann WF, Ngan SC, Hu X. Cluster significance testing using the bootstrap. Neuroimage. 2002;17:583–591. doi: 10.1016/S1053-8119(02)91223-1. [DOI] [PubMed] [Google Scholar]
- 20.Chung S, Lu Y, Henry RG. Comparison of bootstrap approaches for estimation of uncertainties of DTI parameters. Neuroimage. 2006;24(2):531–541. doi: 10.1016/j.neuroimage.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 21.Bolan PJAW, Henry P-G, Garwood M (2004) Feasibility of computer-intensive methods for estimating the variance of spectral fitting parameters. In: Proceedings of the 12th annual meeting ISMRM (abstract 304). Kyoto
- 22.Efron B, Tibshirani R. An introduction to the bootstrap. New York: Chapman and Hall; 1993. [Google Scholar]
- 23.Hesterberg T (2004) Unbiasing the bootstrap-boot knife sampling vs. smoothing. Section on Statistics and the Environment: American Statistical Association, pp 2924–930
- 24.Nelson SJ. Analysis of volume MRI and MR spectroscopic imaging data for the evaluation of patients with brain tumors. Magn Reson Med. 2001;46:228–239. doi: 10.1002/mrm.1183. [DOI] [PubMed] [Google Scholar]
- 25.Knight-Scott J, Li SJ. Effect of long TE on T1 measurement in STEAM progressive saturation experiment. J Magn Reson. 1997;126:266–269. doi: 10.1006/jmre.1997.1171. [DOI] [PubMed] [Google Scholar]
- 26.Rutgers DR, Kingsley PB, Grond J. Saturation-corrected T 1 and T 2 relaxation times of choline, creatine and N-acetyl aspartate in human cerebral white matter at 1.5 T. NMR Biomed. 2003;16:286–288. doi: 10.1002/nbm.828. [DOI] [PubMed] [Google Scholar]
- 27.Srinivasan R, Cunningham C, Chen A, Vigneron D, Hurd R, Nelson S, Pelletier D. TE-averaged two-dimensional proton spectroscopic imaging of glutamate at 3 T. Neuroimage. 2006;30:1171–1178. doi: 10.1016/j.neuroimage.2005.10.048. [DOI] [PubMed] [Google Scholar]