

The current recommended therapy for chronic hepatitis C is the combination of peginterferon and ribavirin, which results in a sustained clearance of hepatitis C virus (HCV) in at least half of patients. However, the mechanisms by which interferon alpha (IFN-α) and ribavirin act against HCV are not well defined. The importance of understanding the biological pathways, cellular effects, and antiviral activities of these agents is underscored by clinical observations that nonresponders frequently have little or no decrease in HCV RNA levels during treatment, suggesting intrinsic resistance to IFN-α. Cell culture and animal models of HCV have shown that the infection itself may block IFN-α induction and action. Still other findings suggest that individual innate and adaptive immune responses, host genetic factors, viral genetic diversity, and clinical comorbidities account for part of nonresponse to therapy. To provide an overview of the current knowledge of the mechanisms of action of IFN-α and ribavirin against HCV and offer guidance for future research, the American Association for the Study of Liver Diseases held a single topic conference entitled “Interferon and Ribavirin in Hepatitis C Virus Infection: Mechanisms of Response and Non-Response” on March 1-3, 2007. This article summarizes that conference.
Interferons (IFNs): An Overview
The IFNs
(Dr. Howard Young, National Cancer Institute, National Institutes of Health)
The type 1 IFNs [interferon alpha and beta (IFN-α/β)] comprise a family of distinct proteins1 that are produced by a wide variety of cells, including fibroblasts, epithelial cells, and hepatocytes,2 although plasmacytoid dendritic cells (DCs) are probably the major source in most viral infections. In contrast, type II IFN [interferon gamma (IFN-γ)] is a single gene cytokine unrelated in structure to IFN-α/β that is produced largely by macrophages, natural killer (NK) cells, and T lymphocytes. Both types of IFNs interact with cells via distinct cellular receptors. The details of the signaling mechanisms by which IFN-α/β and IFN-γ induce the transcription of interferon-stimulated genes (ISGs) and depress the transcription of others are still being defined.3 However, it is increasingly clear that the complex transcriptional programs induced differ significantly depending on the IFN type, the cellular target, and the nature of the infection/host challenge.
IFNs and Viral Infection
(Dr. Christine Biron, Brown University)
The outcome of any viral infection depends largely on the accompanying innate and adaptive immune responses. These critical host responses are embedded in human evolution and have been matched by evolutionary changes in the pathogens that allow them to evade these immune mechanisms.4 Immune reactions are first triggered by engagement of pathogen-associated molecular pattern (PAMP) receptors that sense the viral threat and induce an antiviral state through myriad pathways, including limiting cellular translation [via protein kinase R (PKR)], modifying and degrading viral RNA [via RNAspecific adenosine deaminase, 2′,5′-oligoadenylate synthetase (OAS), and ribonuclease L], altering cellular vesicle trafficking (through guanosine triphosphatases such as myxovius resistance [Mx]), and probably many other undiscovered antiviral mechanisms.1
The engagement of PAMP receptors, including Tolllike receptors (TLRs) and the retinoic acid–inducible gene I (RIG-I)–like helicases, following HCV and other RNA viral infections initiates signaling pathways that lead to the synthesis of IFN-α/β, tumor necrosis factor (TNF), and a variety of other cytokines such as interleukin-12 (IL-12) and IL-151 (Fig. 1). These are largely produced by myeloid-derived and especially plasmacytoid DCs that express TLRs in abundance. IFN-α/β produced by DCs activates NK cells, enhancing their cytotoxic potential and stimulating their production of IFN-γ, whereas IL-15 induced by IFN-α/β stimulates the proliferation and accumulation of NK cells.5,6 IFN-α/β produced by DCs also modulates the activation of CD8+ T cells, which produce additional IFN-γ and represent the central players in the pathogen-specific adaptive immune response.7
Fig. 1.

The host innate response to HCV infection. dsRNA indicates double-stranded RNA; HCV, hepatitis C virus; IFN, interferon; IKK-ε, I kappa B kinase ε; IRF, interferon regulatory factor; ISG, interferon-stimulated gene; Jak, Janus kinase; PAMP, pathogen-associated molecular pattern; RIG-I, retinoic acid-inducible gene I; STAT, signal transducer and activator of transcription; TBK1, TANK-binding kinase 1; TLR3, toll-like receptor 3; and Tyk2, tyrosine kinase 2. Adapted from Nature 2005;436:939.
This well-coordinated interaction of PAMP receptors, signaling pathways, cytokines, and effector cells constitutes a continuum between innate responses that occur rapidly and control the pathogen until the slower, highly pathogen-specific, and ultimately more effective T and B cell responses develop. The key concepts are that (1) the integrated and highly regulated nature of these innate and adaptive immune responses and (2) the activation of NK cells, as well as the timing, breadth, and robustness of subsequent antigen-specific T cell immunity, are likely to be substantially shaped by early events in the innate response to pathogens.5,8
IFN-α/β acts to induce antiviral responses in cells far removed from its site of production via interaction with specific cell surface receptors, the type I IFN receptors [interferon alpha receptor 1 (IFNαR1) and IFNαR2; Fig. 1]. These signal to the nucleus through Janus kinase-1 (Jak1) and tyrosine kinase 2 (Tyk2) phosphorylation of the signal transducers and activators of transcription (STATs).9 A total of seven different STATs, 1 through 6 with 5a and 5b, when activated assemble as homodimeric and heterodimeric signaling complexes. The classic IFN-α/β signaling pathways activate STAT1/STAT2 heterodimers, which activate expression of specific subsets of genes controlled by promoters containing interferonstimulated response elements (ISRE; Fig. 1). There is evidence, however, for IFN-α/β activation of each of the STATs under different conditions. This flexibility may provide mechnisms for using cytokines to access a wide range of biological effects as needed.
HCV Interactions with IFN
HCV
(Dr. Stanley Lemon, University of Texas Medical Branch)
HCV strains show extraordinary genetic diversity, which reflects the error-prone HCV RNA replicase, the exceptional replicative capacity of HCV (1012 virions produced daily), and its frequent success in evading both innate and adaptive host immune responses. Most, if not all, of the 10 proteins expressed by HCV have evolved important interactions with multiple host cell proteins that benefit the viral cycle either directly or indirectly and contribute to the pathogenesis of chronic hepatitis C.10 These include interactions such as the cleavage of the TLR3 adaptor protein (Toll-like receptor adaptor protein [TRIF]), as well as the RIG-I adaptor protein (interferon beta promoter stimulator-1, IPS-1; also known as MAVS, Cardif, and VISA), by the HCV nonstructural (NS) 3/4A protease,2,11,12 which results in disruption of the activation of interferon regulatory factor-3 (IRF-3), an essential step in the virus activation of IFN synthesis13 (Fig. 2A). It is likely that these interactions figure prominently in the long-term persistence of HCV, although the recent demonstration of IPS-1 cleavage by the protease of hepatitis A virus, which is not capable to causing persistent infections, argues that disruption of this pathway is not the complete story.14
Fig. 2.

HCV subversion of IFN activation and signal transduction: (A) HCV NS3-4A blocks IFN induction at multiple levels and (B) HCV blocks IFN signal transduction. CBP indicates CREB binding protein; FADD, Fas associated protein with death domain; HCV, hepatitis C virus; IFN, interferon; IKK, I kappa B kinase; IL, interleukin; IPS-1, interferon beta promoter stimulator-1; IRF, interferon regulatory factor; ISG, interferon-stimulated gene; ISRE, interferon-stimulated response element; Jak, Janus kinase; MDA5, melanoma differentiation–associated protein 5; NF-κB, nuclear factor kappa B; NS, nonstructural; PIAS, protein inhibitor of activated signal transducer and activator of transcription; PP2A, protein phosphatase 2A; RIG-I, retinoic acid-inducible gene I; RIP-1, receptor interacting protein 1; SOCS, suppressor of cytokine signaling; STAT, signal transducer and activator of transcription; TBK1, TANK-binding kinase 1; TLR3, toll-like receptor 3; TRAF6, tumor necrosis factor receptor-associated factor 6; TRIF, Toll-like receptor adaptor protein; and Tyk2, tyrosine kinase 2. Adapted from Nature 2005; 436:939.
In addition to disrupting the intracellular pathways that activate IFN synthesis, HCV proteins also effectively block the actions of double-stranded RNA (dsRNA)–induced antiviral molecules and several ISGs. The HCV core protein interferes with STAT signaling and may contribute to resistance to both endogenous and exogenous IFN.15 The NS5A protein binds to the catalytic site of PKR, blocking its ability to phosphorylate eukaryotic initiation factor 2 alpha and thereby inhibiting dsRNA-induced shutdown of host cell protein translation.16
A number of important questions remain concerning the clinical importance of these in vitro observations, particularly as related to the disruption of IRF-3 signaling by the NS3/4A protease. For example, is it possible that genotype-specific differences in the ability of NS3/4A to cleave IPS-1 or TRIF might explain different rates of long-term viral persistence or different rates of response to IFN-α–based therapies? Although the jury is still out on these questions, it is clear that the genotype 3 NS3/4A is capable of cleaving both of these adaptor proteins even though this genotype is quite sensitive to IFN-α therapy. Another important question is whether the observed efficacy of small-molecule inhibitors of NS3/4A may be due in part to a “double-whammy” effect, rescuing IRF-3 activation while also inhibiting HCV replication.17 The answer here seems a little clearer, as in vitro studies demonstrate rescue of IRF-3 signaling only at very high concentrations of NS3/4A inhibitors far greater than those required for significant antiviral effects (Liang et al., in preparation, 2007).
HCV and IFN Induction
(Dr. Michael Gale, Jr., University of Washington)
In cultured hepatocytes, HCV RNA is sufficient to trigger IFN production through processes that signal the activation of IRF-3. This signaling is initiated by RIG-I, which binds to double-stranded regions of the HCV RNA and signals downstream IRF-3 activation. To evade this process and to attenuate the production of IFN during infection, HCV directs a blockade of RIG-I signaling through the actions of the NS3/4A protease.18 Studies in the past year have identified an essential adaptor protein of RIG-I signaling, IPS-1. Remarkably, the HCV NS3/4A protease targets and cleaves IPS-1 early during infection, thus ablating RIG-I signaling of IFN production by the infected cell11 (Fig. 2A). These findings indicate that HCV evades the innate immune system and turns down adaptive immune responses through intracellular alteration in IFN induction and thus allows for persistent infection and perhaps blunting of therapeutic effects of exogenously applied IFN-α.
IPS-1 as the Target of the NS3 Protease
(Dr. Darius Moradpour, University of Lausanne)
The IPS-1 protein is localized to the outer mitochondrial membrane, a location essential for RIG-I signaling.12 The NS3/4A protein is also present on intracellular membranes of the membranous web found in HCV-infected cells. Proper membrane placement of NS3/4A is dependent on the NS4A cofactor as well as the aminoterminal region of the NS3 protease. Thus, NS3/4A most likely targets IPS-1 for proteolysis through localization motifs embedded within both NS3 and NS4A. IPS-1 has been found in its cleaved, inactive form within liver tissue from patients with chronic HCV infection, providing evidence that NS3/4A targets IPS-1 in vivo. NS3/4A cleavage of IPS-1 results in ablation of virus-induced nuclear factor kappa B (NF-κB) activation and function. Thus, HCV cleavage of IPS-1 abolishes virus signaling of IRF-3 and NF-κB actions, suggesting NS3/4A as a major point of HCV control over cellular innate antiviral defenses. However, patients with chronic HCV infection frequently have prominent hepatic ISG expression, and this raises the possibility that other, RIG-I–independent pathways may contribute to hepatic IFN production by infected cells or specific IFN-producing cells.
HCV and IFN Signaling
(Dr. Raymond Chung, Massachusetts General Hospital)
The interaction of NS3/4A with TLR3 and RIG-I mediated pathogen recognition systems provides a rational explanation for how HCV may evade antiviral defenses. Strikingly, several other regions of the HCV genome also appear to participate in the disarming of host antiviral defenses.19 A major role has been suggested for the HCV core protein in impairing IFN-α/β signal transduction. In transient and stably transfected Huh7 cell lines, HCV core led to diminished STAT1 accumulation and promoted its proteasome-dependent degradation (Fig. 2B).20 More importantly, HCV core impaired IFN-α/β–induced STAT1 activation and decreased binding of ISGF3 to nuclear IFN-α ISRE. Structure-function studies have shown that the N-terminal of the HCV core region directly binds to STAT1 at its SH2 domain, which can impair subsequent IFN signaling, suggesting a model in which the direct interaction of HCV core with STAT1 blocks its recruitment and phosphorylation by Jak1 (Fig. 2B).15
HCV and Protein Phosphatase 2A (PP2A)
Dr. Markus Heim, University Hospital, Basel, Switzerland)
HCV replication within hepatocytes may also interfere with IFN-α/β signaling through activation of inhibitors of the pathway. One such inhibitor is PP2A. Extracts of liver tissue from patients with chronic hepatitis C and from HCV transgenic mice demonstrated evidence of impaired binding of STATs to ISREs.21 In cell culture, HCV polyprotein expression induced PP2A, which in turn inhibited protein arginine N-methyltransferase 1, a methyltransferase that arginine-methylates STAT1 and NS3.22,23 This hypomethylation of STAT1 promoted its binding to the protein inhibitor of activated signal transducer and activator of transcription 1 (PIAS1), which led to the inability of phosphorylated STAT1 to bind DNA (Fig. 2B). Interestingly, in vitro S-adenosyl methionine (SAMe; the principal methyl donor for STAT1) combined with betaine was found to reverse HCV blockade of IFN-α/β signaling.24 These findings have led to pilot studies testing whether the addition of SAMe and betaine to peginterferon will improve responses in patients with chronic hepatitis C who are nonresponders to optimal therapy.
Cytokines and IFN Signaling Responses to HCV
(Dr. Stephen Polyak, University of Washington)
HCV may also be able to modulate antiviral responses in the host through interactions with elements of the inflammatory response. Among its many activities, IRF-3 regulates the expression of IL-8, an inflammatory chemokine that inhibits the antiviral actions of IFN-α/β. HCV RNA and infection induced IL-8 transcription via RIG-I and IPS-1, a process that involves recruitment of IRF-3 to the IL-8 promoter. Moreover, RIG-I stabilized IL-8 messenger RNA (mRNA) via AU-rich elements in the 3′ untranslated region of the mRNA.25 A working model proposes that the initial activation of the innate antiviral response during HCV infection also induces expression of inflammatory cytokines and chemokines. During HCV-mediated blockade of dsRNA-induced antiviral responses, inflammatory mediators may still be expressed via activation of other key transcription factors such as NF-κB, which is activated by many signaling pathways. These mediators may act to down-regulate IFN-α/β responses.
HCV Genetic Variability and Antiviral Responses
(Dr. John Tavis, Saint Louis University)
The inherent genetic variability of HCV may have clinical consequences. Evolution from acute to chronic infection may rely in part on mutations occurring within antigenic epitopes by which the virus escapes from immune surveillance. Sequence mutations may also adversely affect viral fitness. Finally, genetic variability may account for differences in response rates to therapy. Resistance mutations are well known to occur in HCV as well as other viruses that permit escape from the antiviral activity of small-molecule therapy. Whether similar genetic variations alter responses to IFN is unknown.
As a part of the investigation of mechanisms of antiviral activity in the Study of Viral Resistance to Antiviral Therapy of Chronic Hepatitis C (Virahep-C) trial, HCV genetic variation was evaluated in 96 patients in whom viral kinetic responses and clinical outcomes were well defined. Patients were stratified evenly by genotype (1a or 1b), race, and virological response to therapy. Virological response was defined by the decrease in serum HCV RNA by 28 days of therapy and categorized as marked (>3.5 log10 decrease or undetectable), intermediate (1.4-3.5 log10 decrease), or poor (<1.4 log10 decrease). HCV RNA from all 96 patients was analyzed, and the whole open reading frame was sequenced from overlapping amplicons.26
A comparison of HCV genomes between individuals found only a few amino acids that differed consistently by response or race. However, viral sequences were significantly more diverse in marked responders than poor responders, and those with an intermediate response were usually intermediate in diversity. The genetic diversity was not uniformly distributed across the HCV genome. The major regions with greater variability among marked responders were NS5A and NS3 for genotype 1a and core and NS3 for genotype 1b. These correlations were observed in both racial groups.
Overall, variability in NS3 was most commonly associated with a marked virological response, and the variability within NS3 was confined to its C-terminal helicase domain.27 Genetic diversity was also noted between the two races, but these were inconsistent and less clear than the differences by response category. These findings are consistent with a more potent immune response imposing greater selective forces on the HCV genome or variations at multiple positions in the HCV genome impairing its ability to resist the antiviral effects of IFN and ribavirin. Either way, these results imply that viral genetic variation is an important determinant of the success of antiviral therapy.
HCV and IFN Priming of the Adaptive Immune Response
DC Function and HCV Infection
(Dr. Gyongyi Szabo, University of Massachusetts)
DC function appears to play a major role in the immune response to HCV infection and may be deficient in patients with chronic hepatitis C. Intracellular expression of HCV core and NS3 proteins has been shown to activate normal monocytes in a TLR2-dependent manner.28 These results may represent one mechanism for increased TNF-α production in chronic HCV. Similarly, in vitro HCV core and NS3 proteins induced monocyte-derived DC differentiation into a phenotype with impaired T cell activation capacity.29 In addition, the overall frequency of plasmacytoid (IFN-α/β–producing) DCs has been found to be reduced in HCV-infected patients. These results suggest that chronic HCV infection is associated with alterations in innate immune responses at multiple levels. Proinflammatory monocyte activation, reduced monocyte DC T cell activation capacity, and impaired IFN-α production by plasmacytoid DCs may each contribute to ongoing liver injury as well as the inefficient elimination of HCV during antiviral therapy.
Complement Actions and HCV Infection
(Dr. Young Hahn, University of Virginia)
Elements of the complement pathway interact at several levels with the innate and adaptive immune response. HCV core protein has been shown to bind to gC1qR30 and inhibit T helper 1 (Th1) differentiation and antiviral IFN-γ production through suppression of DC IL-12 production. Stimulation of monocyte DCs with HCV core protein or monoclonal antibodies to anti-gC1qR selectively decreased TLR-3–induced and TLR4–induced IL-12p70 production but did not affect the production of IL-10, TNF-α, or transforming growth factor-β. Coculture studies of DCs and CD4+ T cells revealed that treatment of monocyte DCs with HCV core or anti-gC1qR strongly and specifically inhibited T cell production of IFN-γ. Moreover, stimulation of allogeneic CD4+ T cells with anti-gC1qR–treated DC resulted in a skewed differentiation from Th1 to T helper 2 (Th2) cells. Suppression of DC IL-12 production strongly correlated with reduced T cell IFN-γ production. Importantly, the addition of IL-12 rescued the suppression of IFN-γ production by CD4+ T cells and restored normal Th1 differentiation. These results suggest that HCV-induced dysregulation of antiviral T cell immunity via DC-mediated induction may also contribute to persistent HCV infection.
IFN and the Adaptive Immune Response During HCV Infection
(Dr. Barbara Rehermann, National Institutes of Health)
Studies in the chimpanzee model of HCV infection have shown that IFN-α/β responses are detectable within the liver during the first week of acute HCV infection. IFN-α/β expression alters the composition and proteolytic function of the immunoproteasome, a major antigen-processing enzyme complex that generates major histocompatibility complex class I–bound peptides recognized by virus-specific CD8+ T cells. This effect was observed not only in primary human hepatocytes and hepatoma cell lines but also in vivo in the HCV-infected chimpanzee model, where the expression kinetics of IFN-α/β in liver correlated closely with changes in proteasome composition.31 Furthermore, IFN-α/β induced chemokines in acute HCV infection, which recruited HCV-specific T cells to the liver. In contrast, in chronic infection, HCV-specific T cell responsiveness was decreased during effective antiviral therapy.32 These findings suggest that exogenous IFN is acting as an antiviral agent in chronic infection and imply that T cell responsiveness in established infection is antigen-driven.
T Cell Immunity to HCV and Outcome of Antiviral Therapy
(Dr. Hugo Rosen, University of Colorado)
Pretreatment levels of T cell immunity to HCV may predict outcome of antiviral therapy in chronic hepatitis C. A large number of patients with chronic hepatitis C, genotype 1, who were being treated with peginterferon and ribavirin as a part of the Virahep-C study were evaluated for the presence and kinetics of HCV-specific CD4+ and CD8+ T cell responses before and during treatment. These studies showed that African Americans displayed relatively abrogated HCV-specific CD4+ T cell responses yet intact responses to other viruses such as cytomegalovirus. Interestingly, a pretreatment enzymelinked immunosorbent spot level was identified that discriminated between responders and nonresponders in both African American and Caucasian cohorts. Strikingly, HCV-specific responses decreased rather than increased during antiviral therapy, whereas cytomegalovirus-specific responses were preserved. Dendritic function may be restored by antiviral therapy (although no longitudinal data exist), and preliminary data indicate that expression of TNF-related apoptosis-inducing ligand on NK cells may be associated with early virologic response. Thus, the level of pretreatment HCV-specific T cell responses appears to correlate with a better outcome of IFN-α–based therapy of chronic hepatitis C; however, the treatment itself may decrease these T cell responses but restore other immune responses that may play an important role in clearance of HCV.
Human Determinants of IFN Efficacy
Therapy of Chronic Hepatitis C in Humans
(Dr. Charles Howell, University of Maryland)
Current best therapy for chronic hepatitis C, the combination of peginterferon and ribavirin given for 48 weeks, yields overall sustained virological response (SVR) rates of 50% to 60%.33,34 Importantly, response rates are higher among patients infected with genotypes 2 and 3 (75% to 80%) than with genotype 1 (46% to 52%) and can be achieved with 24 weeks of therapy and lower doses of ribavirin (800 mg rather than 1000-1200 mg daily).35
Analyses of HCV RNA levels during antiviral therapy have identified important factors associated with response. These factors include both viral (HCV genotype and initial HCV RNA level) and host factors (age, sex, race, body weight, hepatic steatosis, insulin resistance, hepatic fibrosis, immunodeficiency, adherence, and presence of other comorbidities). How these factors are associated with response was the focus of a recent trial of the combination of peginterferon alfa-2a and ribavirin among 196 African American and 205 Caucasian American patients with chronic genotype 1 HCV (Virahep-C).36 In that study, factors found to be most strongly associated with an SVR were Caucasian race, female gender, HCV RNA levels, baseline liver biopsy fibrosis scores, and total amount of peginterferon received. These results confirmed previous studies showing that African Americans were less likely to respond to IFN-α therapy.37-39 Differences between African American and Caucasian American patients were obvious within days of starting therapy, and differences could not be attributed to viral subtype (1a versus 1b), initial viral levels, gender, age, body weight, or hepatic fibrosis. Furthermore, the differences in viral kinetic responses between African Americans and Caucasians were more quantitative than qualitative. Thus, patterns of viral kinetics were similar in responders to treatment regardless of race; African Americans were merely more likely to have a nonresponse. Overall, SVR rates were 52% among Caucasians but only 28% among African Americans.
Thus, the biologic basis for nonresponse to IFN-α–based therapy in African Americans was not clear from clinical features and analysis of viral levels and geno-types.36 Even when researchers controlled for differences in adherence, race was still a strong predictor of response. Similarly, detailed analyses of pharmacokinetics and pharmacodynamics showed only minor differences between the two racial groups.
More careful analysis of the relative effects of body weight, obesity, hepatic steatosis, diabetes, and insulin resistance has suggested that insulin sensitivity has a major role in responses to IFN-α therapy in hepatitis C.40 In multivariate analysis, insulin resistance but not age, gender, body weight, body mass index, or hepatic steatosis was associated with lack of SVR. The association of insulin resistance and nonresponse to IFN therapy is potentially important because insulin sensitivity is modifiable by weight loss or drug therapy. Furthermore, these associations suggest that there may be important intracellular interactions between the signaling pathways of insulin and IFN-α.
Host Genetic Markers and Antiviral Response in Humans
(Dr. Leland Yee, University of Pittsburgh)
Host genetic factors may play an important role in disease outcome and response to therapy in hepatitis C. As part of the Virahep-C study, host genetic markers are being evaluated, with a focus on specific candidate genes thought to be important in modulating disease activity and affecting antiviral responses. The candidates include those involved in the IFN-α/β pathway, including ISGs, genes encoding immunomodulatory cytokines, genes of the major histocompatibility complex regions, and genes involved in cell death and stress responses. In these studies, confounding and spurious associations can affect results, particularly when we are dealing with racially diverse and genetically heterogeneous populations.
In small studies done in patients with hepatitis C receiving antiviral therapy in clinical trials, genes associated with a higher likelihood of response have included major histocompatibility complex alleles, polymorphisms in ISGs such as Mx1,41 and heterozygosity for HFE H63D,42 but these associations need to be reproduced in other populations and independently confirmed. In a cross-sectional study of a large cohort of persons with chronic hepatitis C, a polymorphism in the promoter region of IFN-γ (−764G) was found to be more common in persons who spontaneously recovered from HCV infection compared to those who developed chronic infection (4.2% versus 1.6%) and in patients who had an SVR compared to nonresponse (11.2% versus 4.3%) to IFN-α–based therapy.43 This promoter variant was confirmed in vitro to have increased promoter activity over the wild-type sequence. Thus, candidate gene approaches have demonstrated several promising associations that may have a biological basis. Whole genome studies and admixture mapping using large sample sizes are needed.
Host Gene Expression in Response to HCV Infection and IFN Therapy
Responses to Hepatitis C in the Chimpanzee
(Dr. Francis Chisari, Scripps Research Institute)
The host immune response to acute HCV infection has been studied extensively in the chimpanzee model of infection.44 Onset of infection was accompanied by a rapid and robust innate immune response with expression of multiple ISGs in the liver that paralleled the fluctuating levels of HCV RNA in serum during the acute phase of HCV infection and evolution to chronic infection.44 This immediate response indicated that HCV RNA is likely recognized by PAMP receptors within infected cells, which trigger IFN-β production and ISG expression. In contrast, similar studies of hepatitis B virus (HBV) infection revealed that ISG expression was not induced. The apparent “invisibility” of HBV compared to the high “visibility” of HCV to the host can possibly be explained by distinctions in the replication programs of each virus (HBV DNA replicates within nucleocapsid particles, whereas HCV replicates in membranous webs within the cytoplasm) and perhaps by the greater speed by which HCV replicates and spreads in vivo. In the case of HBV infection, resolution was associated with the hepatic infiltration of T cells and their production of IFN-γ. In contrast, HCV resolution or reduction of serum viral load was associated with onset of the IFN-α/β response and hepatic ISG expression. These studies indicate that HCV triggers innate immune defenses during acute infection, but this virus is capable of resisting and/or controlling this response as infection progresses. The cell source of endogenous IFN production, whether it is the hepatocyte or specific immune cells such as DCs, is not known but could represent an important therapeutic target for enhancement of endogenous IFN production in an effort to control HCV infection.
Insights from Gene Expression Analyses in Chimpanzees
(Dr. Robert Lanford, Southwestern Foundation for Biomedical Research)
The chimpanzee model permits prospective analyses of gene expression responses to HCV infection and antiviral therapy in both the peripheral blood and the liver. With microarray technology, elevated hepatic ISG expression was found during the acute phase of HCV infection in chimpanzees, which disappeared upon resolution of infection.45 Chimpanzees with chronic infection had persistently elevated ISG levels in the liver, which were indicative of continuing endogenous production of IFN-α/β. Microarray analyses of chimpanzees without HCV infection treated with IFN-α reveal up-regulation of more than 1000 genes. The response to IFN-α was tissue-specific, with many genes differentially induced between liver and peripheral blood mononuclear cells (PBMCs), only ~200 genes being commonly induced in both sites. Most ISG mRNAs reached highest levels within 4 hours after peginterferon administration and returned to baseline by 24 hours despite high levels of peginterferon detected in blood, suggesting a rapid negative feedback that shuts off IFN effects.46 In contrast to noninfected animals, HCV-infected chimpanzees had meager increases in mRNAs of typical ISGs in the liver after peginterferon administration. These results indicate that the up-regulation of ISGs during chronic HCV infection may blunt the response to exogenously administered IFN-α (ISGs may already be maximally stimulated) and account for the lack of an apparent response to IFN-α–based antiviral therapy in some patients.
IFN Responses in Peripheral Blood and Liver in the HCV-Infected Chimpanzee
(Dr. T. Jake Liang, National Institutes of Health)
Chimpanzees with and without HCV infection have been studied for intrahepatic and PBMC responses to type I and II IFNs (human IFN-α, IFN-γ, and consensus IFN).47 Quantitative real-time polymerase chain reaction was performed to evaluate the expression of ISGs in both PBMC and liver and to compare the responses to IFNs between naive and HCV-infected chimpanzees. The in vivo responses to all three IFNs were lower in the HCV-infected chimpanzees than uninfected chimpanzees. This defect was particularly evident in the liver, as induction of hepatic ISGs was barely detectable in the infected animals even after maximal doses of IFNs (Fig. 3). Although the basal levels of hepatic ISG expression were higher in HCV-infected than uninfected animals, these increased basal levels did not account entirely for the lack of ISG induction because the absolute levels of ISGs after IFN treatment were substantially lower in infected than uninfected animals. Consistent with the lack of detectable IFN response, there was little or no change in HCV RNA levels of infected animals after treatment. These data indicate a defective response, particularly in the liver, to IFN-based therapies in HCV-infected chimpanzees.
Fig. 3.
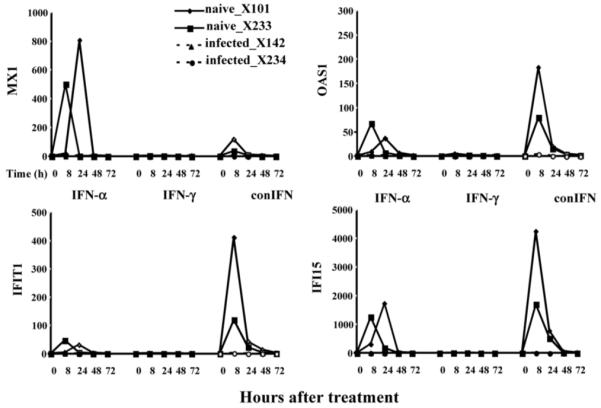
Defective hepatic responses to IFN in HCV-infected chimpanzees. Intrahepatic ISG responses to exogenous IFN treatment were analyzed in chimpanzees. The ISGs studied were Mx1, OAS1, IFIT1, and IFI15. Chimpanzees were administered IFN-α, IFN-γ, and consensus IFN. Fold induction, mRNA responses were determined as a function of time after receipt of IFN in two uninfected animals (solid lines) and two HCV-infected animals (dashed lines). HCV indicates hepatitis C virus; IFN, interferon; ISG, interferon-stimulated gene; mRNA, messenger RNA; and OAS1, 2′,5′-oligoadenylate synthetase 1. Adapted from Gastroenterology 2007;132:733.
In subsequent studies, the hepatic expression of IFN signaling components and inhibitory regulators including suppressor of cytokine signaling 3 (SOCS3) was assessed.47 Following IFN administration, the expression of SOCS3 was significantly up-regulated in the liver of HCV-infected chimpanzees. SOCS3 expression was also evaluated in the liver of HCV-infected patients undergoing IFN-α treatment and showed a similar pattern that was associated with treatment response. The defective hepatic response to IFN in HCV-infected chimpanzees is probably mediated by the activation of SOCS3 and may partly explain the nonresponse of many HCV patients to IFN-α–based therapy.
Hepatic Gene Transcriptional Profiles in Humans with Chronic Hepatitis C
(Dr. Ian McGilvray, University of Toronto)
The liver transcriptional profiles as well as their correlation with response to antiviral therapy in humans with HCV infection have been characterized. Patients with chronic hepatitis C demonstrated a distinct pattern of gene up-regulation, the genes most commonly affected being the ISGs and immune response genes. To evaluate the discriminatory value of gene expression patterns between responders and nonresponders to IFN-α–based therapy, pretreatment liver biopsies were analyzed by microarray.48 In an unsupervised cluster analysis, a set of 18 genes, most of them being well-known ISGs, separated responders from nonresponders (Table 1). Of the 18 genes, 8 were particularly accurate in classifying treatment response in genotype 1 infection. Subsequent analysis of a larger, independent cohort of patients undergoing treatment demonstrated a positive predictive value of 93% for treatment response. Of these 8 genes, ISG15 and ubiquitin specific peptidase 18 (USP18) have been further studied in a tissue culture model of HCV infection.49 USP18 is a cysteine de-ubiquitinase that cleaves ISG15, a ubiquitin-like molecule, from its target. Down-regulation of USP18 was shown to enhance IFN-α/β responses in inhibiting HCV replication in vitro. The mechanism of action appears to be linked to heightened STAT1 activation.
Table 1. Microarray Studies of Liver Tissue in Human HCV Infection: Patterns of ISGs in Pretreatment Liver Tissue Between SVR or RR and NR or SR (Fold Change).
| SVR/NR (Chen et al.48: Toronto: cDNA) |
RR/SR (Feld et al.,50 NIH: Bethesda: Oligo) |
|
|---|---|---|
| ISG | ISGs Up-Regulated in SRs Prior to Treatment | |
| Mx 1 | −1.6 | −3.9 |
| Viperin | −1.8 | −3.3 |
| ISG 15 | −4.4 | −3.1 |
| IFI 6-16 | −2.8 | −3.0 |
| ISG 56 | −2.1 | −2.8 |
| OAS 2 | −3.8 | −2.4 |
| OAS 3 | −2.5 | −2.4 |
| USP 18 | −1.5 | −1.7 |
| Other ISGs with Similar Patterns | ||
| IFITM 1 | −2.3 | |
| G1P3 | −3.0 | |
| IFI 27 | −3.7 | |
| IFI 35 | −2.3 | |
| IFI 44 | −2.7 | |
| IFIT 3 | −2.6 | |
| STAT 1 | −2.0 | |
cDNA indicates complementary DNA; ISG, interferon-stimulated gene; NIH, National Institutes of Health; NR, nonresponder; OAS, 2′,5′-oligoadenylate synthetase; RR, rapid responder; SR, slow responder; STAT 1, signal transducer and activator of transcription 1; SVR, sustained virological response; and USP 18, ubiquitin specific peptidase 18.
Changes in Gene Expression During Peginterferon and Ribavirin Therapy in Humans
(Dr. Jordan Feld, National Institutes of Health)
The changes in hepatic gene expression during antiviral therapy are difficult to study in humans, as they require repeat liver biopsies performed before and during therapy. An alternative is to study separate cohorts of patients undergoing liver biopsy at different times, before or during therapy.50 The latter approach was used to assess intrahepatic gene expression in response to IFN-α therapy in three groups of matched patients with HCV genotype 1 infection: (1) those undergoing routine liver biopsy before therapy, (2) a group that received a single injection of peginterferon 24 hours before liver biopsy, and (3) a group that received 3 days of ribavirin therapy in addition to peginterferon given 24 hours before biopsy. By pooling results from adequate numbers of patients, the separate effects of peginterferon alone and peginterferon with ribavirin could be assessed. Patients from all three groups were then treated in a standard fashion, and their responses were characterized as rapid or slow on the basis of 28-day decreases in HCV RNA levels of less or greater than 2 log10 IU/mL. Known ISGs were induced in all treated patients. In the pretreatment group, slow responders had higher pretreatment ISG expression than rapid responders; these findings are similar to those reported previously (Table 1). On treatment, both slow and rapid responders had similar absolute ISG expression, but when it was corrected for baseline expression with the pretreatment group, rapid responders had a greater increase in ISGs, whereas slow responders exhibited greater increases in IFN-inhibitory pathways. Patients pretreated with ribavirin had heightened induction of IFN-related genes as well as down-regulation of genes involved in IFN inhibition and hepatic stellate cell activation (Table 2). Thus, ISG inducibility appears to be important in determining treatment response, and ribavirin may improve treatment outcome by enhancing hepatic gene responses to IFN-α/β. Collectively, these data indicate that ISGs are induced during chronic HCV infection and suggest that there is a block in the ability of ISGs to effectively clear viral infection. The nature and location(s) of this block remain to be elucidated.
Table 2. Microarray Studies of Liver Tissue in Human HCV Infection: Patterns of IFN-Stimulated Genes or IFN-Inhibitory Genes in Liver Tissue from Ribavirin and Peginterferon Combination-Treated Patients Versus Peginterferon Monotherapy-Treated Patients (Fold Change Comparing On-Treatment Versus Pretreatment).
| IFN-Related ↑ | Ribavirin Versus No Ribavirin |
IFN-Inhibitory ↓ | Ribavirin Versus No Ribavirin |
|---|---|---|---|
| IRF 6 | 1.5 | PP2A | −3.9 |
| IRF 7 | 1.7 | SOCS1 | −1.7 |
| JAK 1 | −2.7 | SUMO1 | −1.7 |
| STAT 1 | 1.5 | SAE1 | −1.7 |
| IFNαR1 | 1.3 |
Abbreviations: IFN interferon; IRF, interferon response factor; JAK 1, Janus kinase 1; PP2A, protein phosphatase 2A; SOCS1, suppressor of cytokine signaling 1; STAT 1, signal transducer and activator of transcription 1; SUMO1, SMT3 suppressor of mif two 3 homolog 1; SAE1, SWMO activating enzyme 1.
Changes in Gene Expression with IFN-α Therapy
(Dr. Milton Taylor, Indiana University)
Studies in the chimpanzee model have shown that HCV-infected animals have a blunted response to peginterferon, particularly in the liver.46,47 There have been few studies of gene array expression with IFN-α therapy in human patients. Optimally, studies of IFN-signaling responses and antiviral action against HCV should be conducted on human liver tissue, but the difficulty of obtaining liver samples on multiple occasions before, during, and after therapy makes this approach impractical.48 For these reasons, in the Virahep-C study, the effects of IFN-α therapy on gene array expression were evaluated in PBMCs.36 A total of 69 patients were studied, stratified by race and peginterferon response (marked, intermediate, or poor viral response at 28 days). Within 1 to 2 days, approximately 300 genes were modified by 2-fold or more (up-regulated or down-regulated).51 No distinction in IFN-induced gene expression was identified by race or gender. However, fewer genes were modified in patients with a poor response in comparison to those with a marked or even intermediate viral response. Classic ISGs increased rapidly and remained elevated for the entire 28-day period in marked responders. In poor viral responders, ISGs were also induced but at significantly lower levels than in marked or intermediate viral responders at all time points. There was no single ISG that correlated with response or nonresponse; the decrease in gene induction among poor responders was global and appeared to affect the major ISGs equally. With mathematical models, a cluster of 36 genes was identified that correlated with changes in viral titer during therapy.
Thus, poor responders to IFN-based therapy of HCV infection appear to have a global defect in intracellular IFN-α/β signaling that is, at least in part, also reflected in gene expression by PBMCs during therapy. The defect is most likely in the induction of IFN-α/β responses and may be due to deficient IFN receptor number or activity, defective Jak-Stat signaling, or decreased TLR activated IRF-7 binding to DNA.
Host Cell Gene Expression During HCV Infection
(Dr. Michael Katze, University of Washington)
Studies of protein expression in patients with chronic hepatitis C reveal increases in many specific ISG products as well as a wide array of other proteins involved in apoptosis, inflammation, lipid metabolism, and cell cycle control. With functional genomics and high-throughput protein profiling of HCV liver transplant patients, specific gene signatures were identified that characterized re-infection of the graft, whereas others were associated with rapid disease progression.52 A “high” ISG response was consistently associated with lesser degrees of fibrosis during follow-up after liver transplantation. These findings suggest that endogenous IFN and ISG production protect against disease progression.
Ribavirin: Mechanism of Action
Clinical Insights
(Dr. Jean-Michel Pawlotsky, University of Paris)
The addition of ribavirin to IFN-α–based regimens produces dramatic improvement in SVR rates among patients with chronic hepatitis C. Clinically, ribavirin appears both to increase the end-of-treatment response rate and to decrease the subsequent relapse rate. The mechanism by which this effect occurs has not been elucidated. Several hypotheses for the mechanism of action of ribavirin have been proposed (Fig. 4): (1) a direct antiviral effect against the HCV RNA–dependent RNA polymerase; (2) depletion of intracellular guanosine triphosphate (GTP) pools through its action as an inhibitor of inosine mono-phosphate dehydrogenase (IMPDH); (3) induction of misincorporation of nucleotides by the viral RNA polymerase, leading to lethal mutagenesis and production of virus with diminished infectivity; and (4) alteration in the T helper cytokine balance from a Th2 profile to a more antiviral Th1 profile.
Fig. 4.

Ribavirin: proposed mechanisms of action. GMP indicates guanosine monophosphate; GTP, guanosine triphosphate; HCV, hepatitis C virus; IFN-γ, interferon gamma; IMPDH, inosine monophosphate dehydrogenase; RDP, ribavirin diphosphate; RdRp, RNA-dependent RNA polymerase; RMP, ribavirin monophosphate; RTP, ribavirin triphosphate; Th1, T helper 1; and Th2, T helper 2. Adapted from Nature 2005;436:967.
In clinical trials of monotherapy, ribavirin produces only a very modest and transient decrease in HCV RNA levels (0.5 log decline at days 2-3). When combined with standard IFN-α, ribavirin appears to increase the second phase decline in HCV RNA levels; however, this effect is slight and is not observed when standard IFN is given daily or peginterferon is used. These results suggest that ribavirin does not have direct antiviral activity against HCV. Although ribavirin is an IMPDH inhibitor, there is little clinical data to support this mechanism of action. Other IMPDH inhibitors have failed to demonstrate antiviral activity in vivo against HCV either alone or in combination with ribavirin.53 The “lethal mutagenesis” hypothesis is frequently cited as the mechanism of action of ribavirin, but human studies have been contradictory in detection of this effect. In a study of 11 patients receiving ribavirin (1000-1200 mg daily) alone or in combination with IFN-α for 28 days, sequencing of multiple clonal isolates from patients revealed no increase in mutation frequency, error generation rate, entropy, or genetic distance between samples over time, suggesting that overt mutagenesis does not occur in vivo at conventional doses. Other human studies have suggested mutagenic effects for ribavirin in vivo but only at early time-points.54,55
Mutagenic Effects
(Dr. Julie Pfeiffer, University of Texas Southwestern)
In studies using HCV replication models, mutagenic effects of ribavirin have been identified by several54,56-60 but not all authors.61 When infectivity was used as a functional readout for mutagenesis, diminished infectivity was found with ribavirin treatment of HCV cultures.58,60 The concentrations of ribavirin needed to achieve mutagenic effects were high (up to 400 uM) and may exceed those achieved in vivo. These concentration differences may help to explain the discordance observed between cell culture and human studies54,55,62-67 (Table 3). The studies done on human samples were often limited by the circumscribed nature of the viral genomic regions sampled and the lack of a functional assay for the sequences identified. Furthermore, nonviable sequences may have been selected out and not identified among those found in the circulation. Only with improved ability to cultivate individual isolates to perform genotype-phenotype correlations can the issue be more completely resolved. In the meantime, further study of ribavirin in the only available infectious HCV model, JFH-1, could address the matter in vitro.
Table 3. Studies of Ribavirin-Related Viral RNA Mutagenesis.
| Evidence for Ribavirin-Induced Mutagenesis |
Evidence Against Ribavirin-Induced Mutagenesis |
|---|---|
| Replicon system | Replicon system |
| Contreras et al.56* | Kato et al.61* |
| Lanford et al.58† | |
| Zhou et al.60† | |
| Tanabe et al.59* | |
| Kanda et al.57* | |
| Hofmann et al.54* | |
| Patient samples | Patient samples |
| Young et al.62‡ | Lee et al.64‡§ |
| Asahina et al.63ठ| Sookoian et al.65 |
| Hofmann et al.54‡§ | Querenghi et al.66‡ |
| Lutchman et al.55‡§ | Schinkel et al.67§ |
Sequencing.
Functional assay.
RBV monotherapy.
Early time point.
Immunomodulatory Effects
(Dr. Gary Levy, University of Toronto)
The immunological effects of ribavirin have been studied in the mouse model of fulminant hepatitis caused by mouse hepatitis virus 3 (MHV-3). In this model, ribavirin therapy attenuates the macrophage inflammatory cyto-kine response to MHV-3.68 More importantly, ribavirin has marked effects on the adaptive immune response by augmenting Th1 cytokine production and inhibiting Th2 IL-4 production. Ribavirin also lowered numbers of T regulatory cells and their associated cytokines, and this suggests another means by which adaptive immunity is promoted. Interestingly, the finding of restoration of STAT-1 patterns after ribavirin therapy back to those seen in MHV-3–resistant animals suggests that ribavirin also alters innate immunity.69 More recently, by the coupling of ribavirin to a macromolecular carrier to enhance hepatocyte delivery, further attenuation of fulminant hepatitis and inhibition of MHV-3 replication were achieved.70 The mechanism by which ribavirin alters Th cytokine responses remains to be clarified, but these data suggest a possible link between ribavirin and IFN-α/β immunomodulatory properties and offer a mechanistically plausible basis for the consistent observation that ribavirin given as monotherapy can improve liver chemistries and when given with IFN-α can decrease clinical relapse rates. Further study to clarify the vigor of cellular immune responses to HCV in the presence of ribavirin will help to address this hypothesis.
IMPDH Inhibitors and HCV Replication
(Dr. Jongdae Lee, University of California, San Diego)
Ribavirin is an IMPDH inhibitor, and some of its effects may be mediated by depletion of intracellular GTP levels through inhibition of the purine salvage pathway. Clinical studies of other IMPDH inhibitors have shown little evidence of antiviral activity. IMPDH inhibitors, however, may have other actions that affect HCV replication and disease. SM360320, a ligand of TLR7, an important intracellular sensing mechanism for pathogens, can induce anti-HCV activity not only through induction of IFN-α/β, a known effect of TLR7 signaling, but also through IFN-independent means. In cell culture studies, IMPDH inhibitors such as mizoribine, although not directly TLR7 ligands, appeared to enhance TLR7 signaling through NF-κB and IRF-7 and did so through depletion of GTP pools because this effect was reversible with restoration of GTP. Furthermore, the addition of SM360230 to mizoribine produced a strong antiviral effect, and this suggests that IMPDH inhibition has beneficial effects on signaling events upstream of IFN-α/β induction. Like SM360230, ribavirin had similar effects on TLR7 signaling in innate immune cells and anti-HCV activity in replicon cells. These results suggest that IM-PDH inhibitors may enhance the antiviral activities of TLRs, provided they are not toxic or immunosuppressive (for example, mycophenolate). These findings may also help to explain the observed induction of ISGs by ribavirin in microarray studies of human liver tissue as well as its observed immunomodulatory effects. More importantly, how these pathways synergize with TLR signaling pathways will provide insights into how ribavirin enhances IFN-α effectiveness in HCV.
Effects of Ribavirin on Viral Kinetics During IFN Therapy
(Dr. Alan Perelson, Los Alamos National Laboratory)
Viral kinetic studies during IFN-α therapy of chronic hepatitis C have defined two phases of viral decline: a rapid, initial first phase, which is believed to reflect antiviral efficacy of IFN-α, and a more delayed and slower second phase, which is believed to reflect eradication of virus-infected hepatocytes. In several studies, ribavirin has been found to have little or no effect on the first phase of viral decline but to enhance the second phase, particularly when IFN-α effectiveness is limited.71 These findings support a theoretical model wherein ribavirin, through its mutagenic actions, lowers HCV fitness, thereby inducing an increased eradication of infected hepatocytes. This mechanism of action would cause a steeper second phase decline, reflecting the balance between de novo infection and loss of infected cells. Diminished fitness in the presence of ribavirin would be particularly apparent when IFN-α effectiveness is low because viral production would be incompletely arrested. Convincing in vivo evidence for this phenomenon is lacking, but there are no good in vitro assays for HCV fitness. Other mechanisms of action of ribavirin do not support the viral kinetic findings or lethal mutagenesis, but the possible effects of ribavirin on TLR7 or IFN-α/β signaling pathways are alternative explanations that merit further evaluation.
Effects of HCV Small-Molecule Inhibitors on IFN Actions
(Dr. John Alam, Vertex Pharmaceuticals)
Recently, several novel small molecules have been developed that demonstrate potent activity against HCV in vitro and in vivo.72 Most promising among these are specific inhibitors of the HCV NS3 protease. Because the NS3/4A protease appears to contribute to HCV’s subversion of IFN activation by inactivating IPS-1, inhibition of NS3/4A may have dual effects.13 In pilot studies on the use of telaprevir (VX950), an HCV NS3/4A protease inhibitor, PBMCs were taken from patients before and during therapy to assess gene expression profiles comparing patients receiving peginterferon alone to those receiving the combination of peginterferon and telaprevir.73 Within 12 hours of starting therapy, expression levels of 532 genes were modified significantly (397 up-regulated and 135 down-regulated) in patients receiving peginterferon alone, and 59% of the genes overlapped with published ISGs. By days 7 and 14 of treatment, at a time when HCV RNA levels had fallen by >4 log10 IU/mL in the combination-treated patients but by only 1 log10 IU/mL in the peginterferon-treated patients, 237 of these genes were differentially expressed in PBMCs. Importantly, many ISGs remained up-regulated to a greater extent with combination therapy than monotherapy (Fig. 5). These preliminary results require confirmation and clinical correlation. They nonetheless suggest that inhibition of the NS3 protease releases the HCV-mediated inhibition of IFN action that contributes to nonresponse.
Fig. 5.
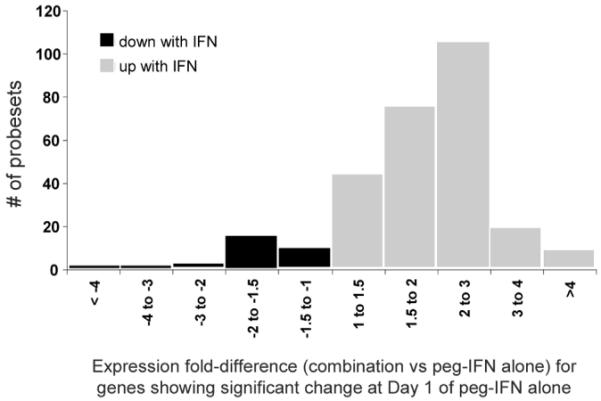
Induction of ISGs by the HCV NS3/4A inhibitor telaprevir. A histogram of the fold changes shows that that most of the genes that are up-regulated on treatment with IFN (gray bars) continue to maintain higher expression levels when treated with telaprevir versus those treated with placebo. A small number of these genes show an almost 4-fold difference. Similarly, a small number of genes are down-regulated with IFN (black bars), and on subsequent treatment with telaprevir, these genes continue to show depressed expression levels in comparison with patients who received the placebo. HCV indicates hepatitis C virus; IFN, interferon; ISG, interferon-stimulated gene; and NS, nonstructural.
Conclusions and Future Directions
The basis for nonresponse of chronic HCV to IFN-α and ribavirin therapy has until recently remained enigmatic. However, a great deal has been learned recently regarding interactions between the virus and host to suggest the bases for nonresponse. The virus has evolved extraordinary mechanisms to counteract both the innate and adaptive immune and proinflammatory responses that likely contribute to both persistence and limitations in responsiveness to exogenous antiviral therapy. Viral subversion of steps both upstream of IFN induction and downstream, including IFN-α/β signaling, ISG function, and DC and T cell responses, has now been well described. Similarly, it appears that there exist host constraints to IFN response as well. Evidence from gene array studies in chimpanzees and humans indicates that the nonresponder patient has a strongly induced intrahepatic repertoire of ISGs, suggesting that this up-regulation may blunt subsequent responsiveness to exogenous IFN and/or that there are downstream blocks to IFN action. SOCS3 and other ISGs involved in suppression of IFN actions may be important mediators of this nonresponse. Groups with limited response rates to IFN-α and ribavirin therapy, such as African Americans, may have both host genetic and environmental (insulin resistance) bases for their inferior response rates. Ribavirin’s mechanisms of action are not well understood. Although some clinical studies suggest transient increases in the HCV RNA mutation rate, convincing evidence for lethal mutagenesis in vivo is thus far lacking. More recent data support an immunomodulatory role for ribavirin, insofar as it appears to enhance sensitivity to IFN-α action.
Further research will be required to address several un-answered questions. Better correlation between in vitro and in vivo findings is necessary. In this regard, further study of IFN signaling and the effects of IFN-α therapy on the only robust infectious HCV tissue culture model (JFH1 clone) is warranted. In addition, further work on gene and protein expression in response to IFN-α treatment is needed to identify differences in ISG repertoires between responder and nonresponder patients. Comparisons between host responses to virus and to treatment in patients with the profoundly IFN-sensitive genotypes 2 and 3 and those with genotype 1 will help to clarify what features define an impaired response. Similarly, comparisons of responses between patients with acute hepatitis C, who also have excellent response rates, and those with chronic infection will also help to address this question. Studies that compare responses of HCV to IFN-α with other viral infections such as HBV will also provide insight into HCV-specific perturbations to the IFN response. Finally, further study of the impact of ribavirin and other HCV-specific agents, including NS3/4A inhibitors, on the IFN response will shed further light on the mechanisms of action of these agents. It is essential that the findings of these studies be rapidly translated into potential means of increasing the efficacy of IFN-α therapy in hepatitis C.
Acknowledgments
Supported by National Institutes of Health grants AI069939 (to R.T.C.), DK078722 (to R.T.C.), AI060389 (to M.G.) DK062187 (to S. J. P.), AI066328 (to S. J. P.), and U19-AI40035 (to S.M.L.) and by the Intramural Division of the National Institute of Diabetes and Digestive and Kidney Diseases (to T.J.L. and J.H.H.).
Abbreviations
- CBP
CREB binding protein
- cDNA
complementary DNA
- DC
dendritic cell
- dsRNA
double-stranded RNA
- FADD
Fas associated protein with death domain
- GMP
guanosine monophosphate
- GTP
guanosine triphosphate
- HBV
hepatitis B virus
- HCV
hepatitis C virus
- IFN
interferon
- IFN-α
interferon alpha
- IFN-α/β
interferon alpha and beta (type 1 interferon)
- IFNαR
interferon alpha receptor
- IFN-γ
interferon gamma (type 2 interferon)
- IKK
I kappa B kinase
- IL
interleukin
- IMPDH
inosine monophosphate dehydrogenase
- IPS-1
interferon beta promoter stimulator-1 (also known as MAVS, Cardif, and VISA)
- IRF
interferon regulatory factor
- ISG
interferon-stimulated gene
- ISRE
interferon-stimulated response element
- Jak
Janus kinase
- MDA5
melanoma differentiation–associated protein 5
- MHV-3
mouse hepatitis virus 3
- mRNA
messenger RNA
- NF-κB
nuclear factor kappa B
- NIDDK
National Institute of Diabetes and Digestive and Kidney Diseases
- NIH
National Institutes of Health
- NK
natural killer
- NR
nonresponder
- NS
nonstructural
- OAS
2′,5′-oligoadenylate synthetase
- PAMP
pathogen-associated molecular pattern
- PBMC
peripheral blood mononuclear cell
- PIAS
protein inhibitor of activated signal transducer and activator of transcription
- PKR
protein kinase R
- PP2A
protein phosphatase 2A
- RDP
ribavirin diphosphate
- RdRp
RNA-dependent RNA polymerase
- RIG-I
retinoic acid–inducible gene I
- RIP-1
receptor interacting protein 1
- RMP
ribavirin monophosphate
- RR
rapid responder
- RTP
ribavirin triphosphate
- SAMe
S-adenosyl methionine
- SOCS
suppressor of cytokine signaling
- SR
slow responder
- STAT
signal transducer and activator of transcription
- SUMO1
SMT3 suppressor of mif two 3 homolog 1
- SVR
sustained virological response
- TBK1
TANK-binding kinase 1
- Th1
T helper 1
- Th2
T helper 2
- TLR
toll-like receptor
- TNF
tumor necrosis factor
- TRAF6
tumor necrosis factor receptor–associated factor 6
- TRIF
Toll-like receptor adaptor protein
- Tyk2
tyrosine kinase 2
- USP18
ubiquitin specific peptidase 18
- Virahep-C
Study of Viral Resistance to Antiviral Therapy of Chronic Hepatitis C
Footnotes
Potential conflict of interest: Dr. Lemon is a consultant for Novartis, GlaxoSmithKline, Hoffman-LaRoche, Genelabs Technology, Abbott, Pharmasett. He also received grants from Schering-Plough and Tibotec. Dr. Chung received grants from Roche and Schering-Plough.
References
- 1.Takaoka A, Yanai H. Interferon signalling network in innate defence. Cell Microbiol. 2006;8:907–922. doi: 10.1111/j.1462-5822.2006.00716.x. [DOI] [PubMed] [Google Scholar]
- 2.Li K, Foy E, Ferreon JC, Nakamura M, Ferreon AC, Ikeda M, et al. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc Natl Acad Sci U S A. 2005;102:2992–2997. doi: 10.1073/pnas.0408824102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Honda K, Takaoka A, Taniguchi T. Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity. 2006;25:349–360. doi: 10.1016/j.immuni.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 4.Garcia-Sastre A, Biron CA. Type 1 interferons and the virus-host relation-ship: a lesson in detente. Science. 2006;312:879–882. doi: 10.1126/science.1125676. [DOI] [PubMed] [Google Scholar]
- 5.Lee SH, Miyagi T, Biron CA. Keeping NK cells in highly regulated antiviral warfare. Trends Immunol. 2007;28:252–259. doi: 10.1016/j.it.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 6.Young HA, Ortaldo J. Cytokines as critical co-stimulatory molecules in modulating the immune response of natural killer cells. Cell Res. 2006;16:20–24. doi: 10.1038/sj.cr.7310004. [DOI] [PubMed] [Google Scholar]
- 7.Gil MP, Salomon R, Louten J, Biron CA. Modulation of STAT1 protein levels: a mechanism shaping CD8 T-cell responses in vivo. Blood. 2006;107:987–993. doi: 10.1182/blood-2005-07-2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kabelitz D, Medzhitov R. Innate immunity—cross-talk with adaptive immunity through pattern recognition receptors and cytokines. Curr Opin Immunol. 2007;19:1–3. doi: 10.1016/j.coi.2006.11.018. [DOI] [PubMed] [Google Scholar]
- 9.Murray PJ. The JAK-STAT signaling pathway: input and output integration. J Immunol. 2007;178:2623–2629. doi: 10.4049/jimmunol.178.5.2623. [DOI] [PubMed] [Google Scholar]
- 10.Lemon SM, Walker C, Alter MJ, Yi M. Hepatitis C viruses. In: Knipe D, Howley P, Griffin DE, Lamb RA, Martin MA, Roizman B, et al., editors. Fields Virology. 5th ed. Lippincott, Williams and Wilkins; Philadelphia, PA: 2007. pp. 1253–1304. [Google Scholar]
- 11.Loo YM, Owen DM, Li K, Erickson AK, Johnson CL, Fish PM, et al. Viral and therapeutic control of IFN-beta promoter stimulator 1 during hepatitis C virus infection. Proc Natl Acad Sci U S A. 2006;103:6001–6006. doi: 10.1073/pnas.0601523103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005;437:1167–1172. doi: 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]
- 13.Gale M, Jr, Foy EM. Evasion of intracellular host defence by hepatitis C virus. Nature. 2005;436:939–945. doi: 10.1038/nature04078. [DOI] [PubMed] [Google Scholar]
- 14.Yang Y, Liang Y, Qu L, Chen Z, Yi M, Li K, et al. Disruption of innate immunity due to mitochondrial targeting of a picornaviral protease pre-cursor. Proc Natl Acad Sci U S A. 2007;104:7253–7258. doi: 10.1073/pnas.0611506104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin W, Kim SS, Yeung E, Kamegaya Y, Blackard JT, Kim KA, et al. Hepatitis C virus core protein blocks interferon signaling by interaction with the STAT1 SH2 domain. J Virol. 2006;80:9226–9235. doi: 10.1128/JVI.00459-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pflugheber J, Fredericksen B, Sumpter R, Jr, Wang C, Ware F, Sodora DL, et al. Regulation of PKR and IRF-1 during hepatitis C virus RNA replication. Proc Natl Acad Sci U S A. 2002;99:4650–4655. doi: 10.1073/pnas.062055699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Foy E, Li K, Wang C, Sumpter R, Jr, Ikeda M, Lemon SM, et al. Regulation of interferon regulatory factor-3 by the hepatitis C virus serine protease. Science. 2003;300:1145–1148. doi: 10.1126/science.1082604. [DOI] [PubMed] [Google Scholar]
- 18.Foy E, Li K, Sumpter R, Jr, Loo YM, Johnson CL, Wang C, et al. Control of antiviral defenses through hepatitis C virus disruption of retinoic acid-inducible gene-I signaling. Proc Natl Acad Sci U S A. 2005;102:2986–2991. doi: 10.1073/pnas.0408707102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Taylor DR, Shi ST, Romano PR, Barber GN, Lai MM. Inhibition of the interferon-inducible protein kinase PKR by HCV E2 protein. Science. 1999;285:107–110. doi: 10.1126/science.285.5424.107. [DOI] [PubMed] [Google Scholar]
- 20.Lin W, Choe WH, Hiasa Y, Kamegaya Y, Blackard JT, Schmidt EV, et al. Hepatitis C virus expression suppresses interferon signaling by degrading STAT1. Gastroenterology. 2005;128:1034–1041. doi: 10.1053/j.gastro.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 21.Heim MH, Moradpour D, Blum HE. Expression of hepatitis C virus proteins inhibits signal transduction through the Jak-STAT pathway. J Virol. 1999;73:8469–8475. doi: 10.1128/jvi.73.10.8469-8475.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Duong FH, Christen V, Berke JM, Penna SH, Moradpour D, Heim MH. Upregulation of protein phosphatase 2Ac by hepatitis C virus modulates NS3 helicase activity through inhibition of protein arginine methyltransferase 1. J Virol. 2005;79:15342–15350. doi: 10.1128/JVI.79.24.15342-15350.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Duong FH, Filipowicz M, Tripodi M, La Monica N, Heim MH. Hepatitis C virus inhibits interferon signaling through up-regulation of protein phosphatase 2A. Gastroenterology. 2004;126:263–277. doi: 10.1053/j.gastro.2003.10.076. [DOI] [PubMed] [Google Scholar]
- 24.Duong FH, Christen V, Filipowicz M, Heim MH. S-Adenosylmethionine and betaine correct hepatitis C virus induced inhibition of interferon signaling in vitro. Hepatology. 2006;43:796–806. doi: 10.1002/hep.21116. [DOI] [PubMed] [Google Scholar]
- 25.Wagoner J, Austin M, Green J, Imaizumi T, Casola A, Brasier A, et al. Regulation of CXCL-8 (interleukin-8) induction by double-stranded RNA signaling pathways during hepatitis C virus infection. J Virol. 2007;81:309–318. doi: 10.1128/JVI.01411-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yao E, Tavis JE. A general method for nested RT-PCR amplification and sequencing the complete HCV genotype 1 open reading frame. Virol J. 2005;2:88. doi: 10.1186/1743-422X-2-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Donlin MJ, Cannon NA, Yao E, Li J, Wahed A, Taylor MW, et al. Pretreatment sequence diversity differences in the full-length hepatitis C virus open reading frame correlate with early response to therapy. J Virol. 2007;81:8211–8224. doi: 10.1128/JVI.00487-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dolganiuc A, Oak S, Kodys K, Golenbock DT, Finberg RW, Kurt-Jones E, et al. Hepatitis C core and nonstructural 3 proteins trigger toll-like receptor 2-mediated pathways and inflammatory activation. Gastroenterology. 2004;127:1513–1524. doi: 10.1053/j.gastro.2004.08.067. [DOI] [PubMed] [Google Scholar]
- 29.Dolganiuc A, Kodys K, Kopasz A, Marshall C, Do T, Romics L, Jr, et al. Hepatitis C virus core and nonstructural protein 3 proteins induce pro- and anti-inflammatory cytokines and inhibit dendritic cell differentiation. J Immunol. 2003;170:5615–5624. doi: 10.4049/jimmunol.170.11.5615. [DOI] [PubMed] [Google Scholar]
- 30.Kittlesen DJ, Chianese-Bullock KA, Yao ZQ, Braciale TJ, Hahn YS. Interaction between complement receptor gC1qR and hepatitis C virus core protein inhibits T-lymphocyte proliferation. J Clin Invest. 2000;106:1239–1249. doi: 10.1172/JCI10323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shin EC, Seifert U, Kato T, Rice CM, Feinstone SM, Kloetzel PM, et al. Virus-induced type I IFN stimulates generation of immunoproteasomes at the site of infection. J Clin Invest. 2006;116:3006–3014. doi: 10.1172/JCI29832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rahman F, Heller T, Sobao Y, Mizukoshi E, Nascimbeni M, Alter H, et al. Effects of antiviral therapy on the cellular immune response in acute hepatitis C. Hepatology. 2004;40:87–97. doi: 10.1002/hep.20253. [DOI] [PubMed] [Google Scholar]
- 33.Fried MW, Shiffman ML, Reddy KR, Smith C, Marinos G, Goncales FL, Jr, et al. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N Engl J Med. 2002;347:975–982. doi: 10.1056/NEJMoa020047. [DOI] [PubMed] [Google Scholar]
- 34.Manns MP, McHutchison JG, Gordon SC, Rustgi VK, Shiffman M, Reindollar R, et al. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomised trial. Lancet. 2001;358:958–965. doi: 10.1016/s0140-6736(01)06102-5. [DOI] [PubMed] [Google Scholar]
- 35.Hadziyannis SJ, Sette H, Jr, Morgan TR, Balan V, Diago M, Marcellin P, et al. Peginterferon-alpha2a and ribavirin combination therapy in chronic hepatitis C: a randomized study of treatment duration and ribavirin dose. Ann Intern Med. 2004;140:346–355. doi: 10.7326/0003-4819-140-5-200403020-00010. [DOI] [PubMed] [Google Scholar]
- 36.Conjeevaram HS, Fried MW, Jeffers LJ, Terrault NA, Wiley-Lucas TE, Afdhal N, et al. Peginterferon and ribavirin treatment in African American and Caucasian American patients with hepatitis C genotype 1. Gastroenterology. 2006;131:470–477. doi: 10.1053/j.gastro.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 37.Howell C, Jeffers L, Hoofnagle JH. Hepatitis C in African Americans: summary of a workshop. Gastroenterology. 2000;119:1385–1396. doi: 10.1053/gast.2000.19582. [DOI] [PubMed] [Google Scholar]
- 38.Jeffers LJ, Cassidy W, Howell CD, Hu S, Reddy KR. Peginterferon alfa-2a (40 kd) and ribavirin for black American patients with chronic HCV genotype 1. Hepatology. 2004;39:1702–1708. doi: 10.1002/hep.20212. [DOI] [PubMed] [Google Scholar]
- 39.Muir AJ, Bornstein JD, Killenberg PG. Peginterferon alfa-2b and ribavirin for the treatment of chronic hepatitis C in blacks and non-Hispanic whites. N Engl J Med. 2004;350:2265–2271. doi: 10.1056/NEJMoa032502. [DOI] [PubMed] [Google Scholar]
- 40.Conjeevaram HS, Kleiner DE, Everhart JE, Hoofnagle JH, Zacks S, Afdhal NH, et al. Race, insulin resistance and hepatic steatosis in chronic hepatitis C. Hepatology. 2007;45:80–87. doi: 10.1002/hep.21455. [DOI] [PubMed] [Google Scholar]
- 41.Yee LJ. Host genetic determinants in hepatitis C virus infection. Genes Immun. 2004;5:237–245. doi: 10.1038/sj.gene.6364090. [DOI] [PubMed] [Google Scholar]
- 42.Bonkovsky HL, Naishadham D, Lambrecht RW, Chung RT, Hoefs JC, Nash SR, et al. Roles of iron and HFE mutations on severity and response to therapy during retreatment of advanced chronic hepatitis C. Gastroenterology. 2006;131:1440–1451. doi: 10.1053/j.gastro.2006.08.036. [DOI] [PubMed] [Google Scholar]
- 43.Huang Y, Yang H, Borg BB, Su X, Rhodes SL, Yang K, et al. A functional SNP of interferon-gamma gene is important for interferon-alpha-induced and spontaneous recovery from hepatitis C virus infection. Proc Natl Acad Sci U S A. 2007;104:985–990. doi: 10.1073/pnas.0609954104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Su AI, Pezacki JP, Wodicka L, Brideau AD, Supekova L, Thimme R, et al. Genomic analysis of the host response to hepatitis C virus infection. Proc Natl Acad Sci U S A. 2002;99:15669–15674. doi: 10.1073/pnas.202608199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bigger CB, Guerra B, Brasky KM, Hubbard G, Beard MR, Luxon BA, et al. Intrahepatic gene expression during chronic hepatitis C virus infection in chimpanzees. J Virol. 2004;78:13779–13792. doi: 10.1128/JVI.78.24.13779-13792.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lanford RE, Guerra B, Lee H, Chavez D, Brasky KM, Bigger CB. Genomic response to interferon-alpha in chimpanzees: implications of rapid downregulation for hepatitis C kinetics. Hepatology. 2006;43:961–972. doi: 10.1002/hep.21167. [DOI] [PubMed] [Google Scholar]
- 47.Huang Y, Feld JJ, Sapp RK, Nanda S, Lin JH, Blatt LM, et al. Defective hepatic response to interferon and activation of suppressor of cytokine signaling 3 in chronic hepatitis C. Gastroenterology. 2007;132:733–744. doi: 10.1053/j.gastro.2006.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen L, Borozan I, Feld J, Sun J, Tannis LL, Coltescu C, et al. Hepatic gene expression discriminates responders and nonresponders in treatment of chronic hepatitis C viral infection. Gastroenterology. 2005;128:1437–1444. doi: 10.1053/j.gastro.2005.01.059. [DOI] [PubMed] [Google Scholar]
- 49.Randall G, Chen L, Panis M, Fischer AK, Lindenbach BD, Sun J, et al. Silencing of USP18 potentiates the antiviral activity of interferon against hepatitis C virus infection. Gastroenterology. 2006;131:1584–1591. doi: 10.1053/j.gastro.2006.08.043. [DOI] [PubMed] [Google Scholar]
- 50.Feld JJ, Nanda S, Huang Y, Cam M, Pusek SN, Schweigler L, et al. Hepatic gene expression during treatment with peginterferon and ribavirin: identifying molecular pathways for treatment response. Hepatology. doi: 10.1002/hep.21853. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Taylor MW, Tsukahara T, Brodsky L, Schaley J, Sanda C, Stephens MJ, et al. Changes in gene expression during pegylated interferon and ribavirin therapy of chronic hepatitis C virus distinguish responders from nonresponders to antiviral therapy. J Virol. 2007;81:3391–3401. doi: 10.1128/JVI.02640-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Smith MW, Walters KA, Korth MJ, Fitzgibbon M, Proll S, Thompson JC, et al. Gene expression patterns that correlate with hepatitis C and early progression to fibrosis in liver transplant recipients. Gastroenterology. 2006;130:179–187. doi: 10.1053/j.gastro.2005.08.015. [DOI] [PubMed] [Google Scholar]
- 53.Hezode A, et al. Hepatology. 2006;44:615A. [Google Scholar]
- 54.Hofmann WP, Polta A, Herrmann E, Mihm U, Kronenberger B, Sonntag T, et al. Mutagenic effect of ribavirin on hepatitis C nonstructural 5B quasispecies in vitro and during antiviral therapy. Gastroenterology. 2007;132:921–930. doi: 10.1053/j.gastro.2006.12.005. [DOI] [PubMed] [Google Scholar]
- 55.Lutchman G, Danehower S, Song BC, Liang TJ, Hoofnagle JH, Thomson M, et al. Mutation rate of the hepatitis C virus NS5B in patients undergoing treatment with ribavirin monotherapy. Gastroenterology. 2007;132:1757–1766. doi: 10.1053/j.gastro.2007.03.035. [DOI] [PubMed] [Google Scholar]
- 56.Contreras AM, Hiasa Y, He W, Terella A, Schmidt EV, Chung RT. Viral RNA mutations are region specific and increased by ribavirin in a full-length hepatitis C virus replication system. J Virol. 2002;76:8505–8517. doi: 10.1128/JVI.76.17.8505-8517.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kanda T, Yokosuka O, Imazeki F, Tanaka M, Shino Y, Shimada H, et al. Inhibition of subgenomic hepatitis C virus RNA in Huh-7 cells: ribavirin induces mutagenesis in HCV RNA. J Viral Hepat. 2004;11:479–487. doi: 10.1111/j.1365-2893.2004.00531.x. [DOI] [PubMed] [Google Scholar]
- 58.Lanford RE, Guerra B, Lee H, Averett DR, Pfeiffer B, Chavez D, et al. Antiviral effect and virus-host interactions in response to alpha interferon, gamma interferon, poly(i)-poly(c), tumor necrosis factor alpha, and ribavirin in hepatitis C virus subgenomic replicons. J Virol. 2003;77:1092–1104. doi: 10.1128/JVI.77.2.1092-1104.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tanabe Y, Sakamoto N, Enomoto N, Kurosaki M, Ueda E, Maekawa S, et al. Synergistic inhibition of intracellular hepatitis C virus replication by combination of ribavirin and interferon-alpha. J Infect Dis. 2004;189:1129–1139. doi: 10.1086/382595. [DOI] [PubMed] [Google Scholar]
- 60.Zhou S, Liu R, Baroudy BM, Malcolm BA, Reyes GR. The effect of ribavirin and IMPDH inhibitors on hepatitis C virus subgenomic replicon RNA. Virology. 2003;310:333–342. doi: 10.1016/s0042-6822(03)00152-1. [DOI] [PubMed] [Google Scholar]
- 61.Kato T, Date T, Miyamoto M, Sugiyama M, Tanaka Y, Orito E, et al. Detection of anti-hepatitis C virus effects of interferon and ribavirin by a sensitive replicon system. J Clin Microbiol. 2005;43:5679–5684. doi: 10.1128/JCM.43.11.5679-5684.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Young KC, Lindsay KL, Lee KJ, Liu WC, He JW, Milstein SL, et al. Identification of a ribavirin-resistant NS5B mutation of hepatitis C virus during ribavirin monotherapy. Hepatology. 2003;38:869–878. doi: 10.1053/jhep.2003.50445. [DOI] [PubMed] [Google Scholar]
- 63.Asahina Y, Izumi N, Enomoto N, Uchihara M, Kurosaki M, Onuki Y, et al. Mutagenic effects of ribavirin and response to interferon/ribavirin combination therapy in chronic hepatitis C. J Hepatol. 2005;43:623–629. doi: 10.1016/j.jhep.2005.05.032. [DOI] [PubMed] [Google Scholar]
- 64.Lee JH, von Wagner M, Roth WK, Teuber G, Sarrazin C, Zeuzem S. Effect of ribavirin on virus load and quasispecies distribution in patients infected with hepatitis C virus. J Hepatol. 1998;29:29–35. doi: 10.1016/s0168-8278(98)80175-x. [DOI] [PubMed] [Google Scholar]
- 65.Sookoian S, Castano G, Frider B, Cello J, Campos R, Flichman D. Combined therapy with interferon and ribavirin in chronic hepatitis C does not affect serum quasispecies diversity. Dig Dis Sci. 2001;46:1067–1071. doi: 10.1023/a:1010718213584. [DOI] [PubMed] [Google Scholar]
- 66.Querenghi F, Yu Q, Billaud G, Maertens G, Trepo C, Zoulim F. Evolution of hepatitis C virus genome in chronically infected patients receiving ribavirin monotherapy. J Viral Hepat. 2001;8:120–131. doi: 10.1046/j.1365-2893.2001.00265.x. [DOI] [PubMed] [Google Scholar]
- 67.Schinkel J, de Jong MD, Bruning B, van Hoek B, Spaan WJ, Kroes AC. The potentiating effect of ribavirin on interferon in the treatment of hepatitis C: lack of evidence for ribavirin-induced viral mutagenesis. Antivir Ther. 2003;8:535–540. [PubMed] [Google Scholar]
- 68.Ning Q, Brown D, Parodo J, Cattral M, Gorczynski R, Cole E, et al. Ribavirin inhibits viral-induced macrophage production of TNF, IL-1, the procoagulant fgl2 prothrombinase and preserves Th1 cytokine production but inhibits Th2 cytokine response. J Immunol. 1998;160:3487–3493. [PubMed] [Google Scholar]
- 69.Ning Q, Berger L, Luo X, Yan W, Gong F, Dennis J, et al. STAT1 and STAT3 alpha/beta splice form activation predicts host responses in mouse hepatitis virus type 3 infection. J Med Virol. 2003;69:306–312. doi: 10.1002/jmv.10290. [DOI] [PubMed] [Google Scholar]
- 70.Levy GA, Adamson G, Phillips MJ, Scrocchi LA, Fung L, Biessels P, et al. Targeted delivery of ribavirin improves outcome of murine viral fulminant hepatitis via enhanced anti-viral activity. Hepatology. 2006;43:581–591. doi: 10.1002/hep.21072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dixit NM, Layden-Almer JE, Layden TJ, Perelson AS. Modelling how ribavirin improves interferon response rates in hepatitis C virus infection. Nature. 2004;432:922–924. doi: 10.1038/nature03153. [DOI] [PubMed] [Google Scholar]
- 72.Sulkowski MS. Specific targeted antiviral therapy for hepatitis C. Curr Gastroenterol Rep. 2007;9:5–13. doi: 10.1007/s11894-008-0015-x. [DOI] [PubMed] [Google Scholar]
- 73.Forestier N, Weegink C, Purdy S, McNair L, Jansen P, Zeuzem S, et al. Current status of subjects receiving peginterferon alfa-2a (PEG-IFN) and ribavirin (RBV) after a 14-day study of the hepatitis C protease inhibitor telaprevir (VX-950) with PEG-IFN. Hepatology. 2006;44:614A. [Google Scholar]