

Abstract
Gel electrophoresis is a standard biochemical technique used for separating biomolecules on the basis of size and charge. Despite the use of gels in early single-molecule experiments, gel electrophoresis has not been widely adopted for single-molecule fluorescence spectroscopy. We present a novel method that combines gel electrophoresis and single-molecule fluorescence spectroscopy to simultaneously purify and analyze biomolecules in a gel matrix. Our method, in-gel ALEX, uses non-denaturing gels to purify biomolecular complexes of interest from free components, aggregates, and non-specific complexes. The gel matrix also slows down translational diffusion of molecules, giving rise to long, high-resolution time traces without surface immobilization, which allow extended observations of conformational dynamics in a biologically friendly environment. We demonstrated the compatibility of this method with different types of single molecule spectroscopy techniques, including confocal detection and fluorescence-correlation spectroscopy. We demonstrated that in-gel ALEX can be used to study conformational dynamics at the millisecond timescale; by studying a DNA hairpin in gels, we directly observed fluorescence fluctuations due to conformational interconversion between folded and unfolded states. Our method is amenable to the addition of small molecules that can alter the equilibrium and dynamic properties of the system. In-gel ALEX will be a versatile tool for studying structures and dynamics of complex biomolecules and their assemblies.
Keywords: Gel electrophoresis, single molecule FRET, ALEX, FCS, conformational dynamics, DNA hairpin
Biological mechanisms depend on multi-component protein machines and dynamic complexes of biomolecules. A popular separation technique used for purification and characterization of such biomolecules is gel electrophoresis1-3, during which molecules are separated in gel matrices based on properties such as size and net electrical charge. In denaturing gel electrophoresis, non-covalent interactions are disrupted by chaotropic agents such as urea or SDS1,3; in contrast, non-denaturing (native) gel electrophoresis preserves the native 3D structure and functionality of biomolecules4-6.
Quantitative analysis of electrophoresis gels mainly depends on ensemble measurements of radioactivity or fluorescence5,7-11, which report on observables averaged over many molecules. This averaging can be eliminated using single-molecule methods, which directly observe and analyze sources of static and dynamic heterogeneity. Early works combining gels and single-molecule fluorescence explored the behavior of single phage DNA molecules (labeled with a large number of DNA-intercalating dyes) during electrophoresis12,13. Single-molecule detection of individual fluorophores in gels was first reported by Dickson et al14, and was soon followed by several other studies15-25 in which the gels were used mainly to slow down diffusion. These methods offer important advantages for single-molecule detection such as the elimination of surface-immobilization artifacts. However, these approaches relied on co-polymerizing the sample with the gel matrix, (either in polyacrylamide or agarose) and were hampered by several limitations, including low signal-to-noise ratio26, large reduction in the number of detected fluorescent molecules compared to the amount supplied in the gel18,26, changes in the photophysical properties of chromophores22,26, as well as complete immobilization of molecules even when the average pore size of the gel was much larger than the molecules14,19,27. More recently, our group28 reported single-molecule FRET studies of purified complexes in polyacrylamide gel slices. While this approach seems straightforward, single molecule measurements in gel slices have several practical shortcomings. The need for many preparatory steps (for running, imaging, and slicing the gel) introduces higher background fluorescence and lengthens the preparation time (that includes the gel-running time plus >30 min for gel imaging and gel-slice excision), making it unsuitable for complexes with short half-life. Moreover, it is difficult to “tune” the in-gel concentration to the 50-100 pM level needed for single-molecule detection using confocal optics. Here, we use novel combinations of gel electrophoresis with alternating-laser excitation spectroscopy (ALEX; Ref. 28-30) to overcome most of these problems and take advantage of the benefits of single-molecule detection in gels. Using DNA standards, we demonstrated that in-gel ALEX combines excellent detection sensitivity with the remarkable separation capability provided by polyacrylamide gels. We exploited the extension of molecular diffusion in gels to study real-time FRET fluctuations in DNA-hairpin molecules diffusing in 6% polyacrylamide (PA) and recovered kinetic parameters in agreement with published reports. We expect that in-gel ALEX will be a useful tool for studying structures and dynamics of complex biomolecules and their assemblies.
Experimental Section
Reagents
The quality and purity of the buffers and gel reagents were important for achieving an optimal signal-to-noise ratio for single-molecule detection in gel; if available, chemicals of “luminescence-grade” were employed. All reagents are listed in Table S-1.
DNA
Amino-modified oligonucleotides (IBA, Germany) were labeled with NHS-conjugated fluorophores according to the manufacturer's instructions and purified on a reverse-phase C18 FPLC column (μRPC C2/C18, GE Healthcare, UK) on a liquid chromatography system (AKTA, GE Healthcare, UK). The nomenclature for linear DNA is “TX-D,BY-A”, where T is top strand, B is bottom strand, X & Y indicate the labelling positions as the number of bases counted from the 5′ end of the top strand, and D & A indicate the fluorophores used. The DNA hairpin was annealed in hybridization buffer (50 mM Tris pH 8.0, 1 mM EDTA, 500 mM NaCl) from labelled and gel-purified single-stranded DNA. All DNA sequences are listed in Table S-2.
RNA polymerase (RNAP) open complex
The open complex of Escherichia coli RNA polymerase (RNAP) with promoter DNA was formed as described31. A reaction mixture (20 μl) of 40 nM holoenzyme (Epicentre, WI) and 10 nM T25-Cy3B,B54-Alexa647 promoter DNA in transcription buffer [40 mM HEPES-NaOH, pH 7.0, 100 mM potassium-L-glutamate, 10 mM MgCl2, 1 mM DTT, 100 μg/ml bovine serum albumin, 5% (v/v) glycerol] was incubated at 37°C for 15 min. Heparin Sepharose (GE Healthcare, UK; 1 mg/ml) was added to disrupt non-specific RNAP-DNA complexes and remove free RNAP. After 1 min at 37°C, samples were centrifuged, and 13 μl of supernatant were transferred to tubes containing 0.5 μl of 10 mM ApA (RiboMed, CA) at 37°C.
Gel electrophoresis
We prepared gels of different 37:1 acrylamide:bis-acrylamide concentrations using 0.1% (w/v) ammonium persulfate and 0.4 μl/ml TEMED. Unless specified, gels were formed and run in Tris-Glycine (TG) buffer (25 mM Tris, 200 mM glycine, pH 8.0). One hairpin sample (Fig. 5, right most column) was measured in Mg2+-containing buffer (25 mM Tris, 200 mM glycine, 5 mM MgCl2, pH 8.0) and Mg2+-containing gel compositions [acrylamide mixture (to make 6%), 25 mM Tris, 200 mM glycine, 5 mM MgCl2]. Since cations move towards the cathode during electrophoresis, extended runs reduce the in-gel Mg2+ concentration; thus, we kept the Mg2+ concentration constant by replacing the running buffer every 10 min. Slab-gel electrophoresis (Fig. 3E) was run on a Mini-Protean gel (BioRad, CA) at 120 V for 45 min and imaged using a gel scanner (PharosFX, BioRad, CA).
FIGURE 5.

Studying DNA hairpin conformations using in-gel ALEX.
Double-stranded DNA (T1-Cy3B,B18-ATTO647N; leftmost column) and DNA hairpin (middle and rightmost columns) were measured in 6% PA (A-C) and in solution (D). The samples in the left and middle columns were measured in TG buffer, while the hairpin in the rightmost column was measured in buffer and gel containing 5 mM MgCl2.
(A) Representative time traces of the above samples in 6% PA (burst selection criteria: L=60, M=10, T=1 ms, S>0.5, burst length>8 ms; per-0.5ms bin filter: S>0.5, FDex>10; see Data analysis). The upper panels show the photon counts due to ALEX partitioned into 0.5-ms time bins (green: FDex,Dem; red: FDex,Aem; FAex,Aem not shown). Notice that strong anti-correlation between FDex,Dem and FDex,Aem is visible, especially in the hairpin sample with 5 mM MgCl2.
(B) Two-dimensional plots of E* and standard deviation of E* (σE) for bursts detected using the selection criteria given in (A). Top subpanel in each 2D plot: E* histogram; right subpanel: σE histogram. For relatively static samples (DNA and hairpin in TG buffer), the σE distributions are biased towards low σE value (<0.2). For the dynamic sample (hairpin in 5 mM MgCl2), the σE distribution is shifted towards higher value (σE>0.2) indicating the higher variability of FRET values within a burst. Note that the FRET distribution for this sample is shifted towards intermediate FRET value as compared to that measured in solution (Fig. 4B); this is caused by an increased FRET averaging within the fluorescence bursts, as individual hairpin molecules sample both the folded and unfolded conformations many times during their transit from the confocal spot.
(C) Determination of interconversion rates using the FCS-ratio method35. Top subpanels: donor autocorrelation [GDD(τ), black lines] and donor-acceptor cross correlation [GDA(τ), grey lines] curves normalized such that GDD(τ) curves fall between 0 and 1. The amplitude of the cross correlation curve is proportional to the relative concentration of the doubly labeled species, therefore samples with differing donor-acceptor concentrations will have different maximum correlation amplitudes. Bottom subpanels: ratios of GDD(τ) and GDA(τ) (black lines) and the fitted function (red line). The parts of the curves above 20 ms are dominated by noise due to the low amplitudes of the correlation curves, and consequently were not considered during fitting. The DNA and hairpin sample in the absence of MgCl2 showed little dynamics; however, the DNA-hairpin sample in 5 mM MgCl2 exhibited a large fluctuation that can be fitted with a stretched exponential function with mean relaxation time (<τ>) of 0.92 ± 0.23 ms (with the standard deviation calculated from 6 replicates).
(D) Determination of interconversion rates for samples in solution using the FCS-ratio method. Similar to (C), the DNA and hairpin samples showed no dynamics in the absence of MgCl2; in contrast the hairpin sample in 5 mM MgCl2 exhibited large fluctuations with a mean relaxation time (<τ>) of 1.08 ± 0.25 ms (standard deviation from 6 replicates). The similarity of interconversion rates in solution and in gel shows that the gel matrix largely preserves the dynamics of the DNA-hairpin.
FIGURE 3.
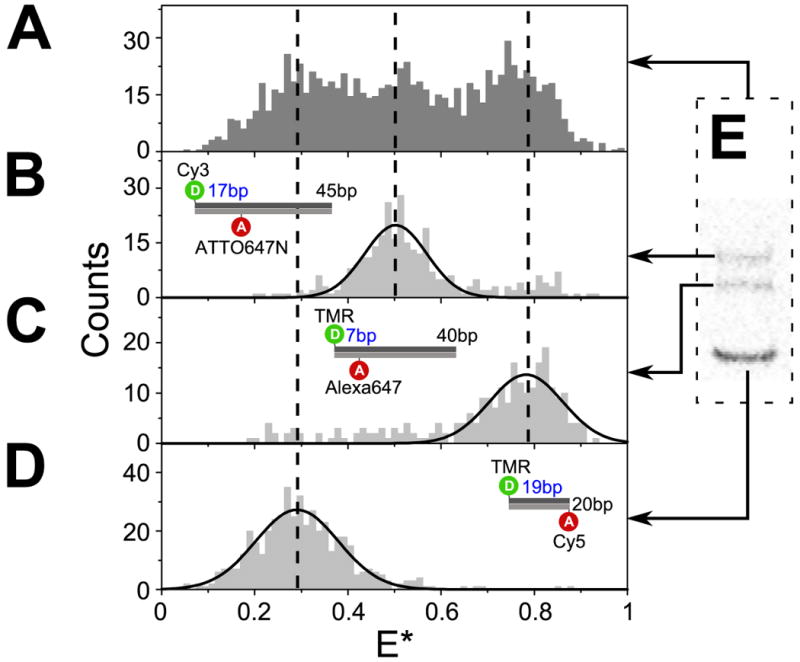
In-gel ALEX separates subpopulations with different electrophoretic mobility.
(A-D) ALEX-based FRET distributions for 3 DNA standards and their mixture. (A) Examination of the mixture of three DNA of different lengths and mean E* using in-solution ALEX could not clearly resolve the individual species. In contrast, in-gel ALEX examination of the same mixture after electrophoretic separation in 10% PA recovered E* distributions for the individual species: (B) T1-Cy3,B18-ATTO647N (intermediate FRET, 45 bp;), (C) T1-TMR,B8-Alexa647 (high FRET, 40 bp), and (D) T1-TMR,B20-Cy5 (low FRET, 20 bp). The dashed vertical lines indicate the mean E* values of the individual subpopulations as measured in gel. The mean E* values measured in gel were consistent with the mean E* values measured separately for each type of DNA in solution.
(E) Fluorescence image (FAex,Aem) of an analytical gel (10% PA) showing the separation of the DNA mixture described in (A).
Mini-gel chambers (Fig. 1A) were formed by placing a 2-mm silicone gasket (Grace Bio Labs, OR) between a large and a small glass coverslip (22 × 50 mm and 22 × 20 mm, respectively; 0.13-0.16 mm thick, VWR, PA) and sealing one end with a Blu-Tack dam. To form the gel, the chamber was filled with ≈500 μl of acrylamide mixture and sealed with a comb (Fig. 1A). After polymerization, the comb and dam were removed and the chamber was filled with running buffer. The electric field is supplied through two Pt electrodes (WPI, FL) connected to a power supply (Wolf Labs, UK). Extensive coverslip cleaning was unnecessary for confocal experiments on diffusing molecules, but useful for wide-field or TIRF imaging. The gels were pre-run at 5 V/cm for 30 min and the running buffer was replenished to remove any free radicals and acrylamide1,2. Five μl of 1 nM sample was loaded into the well using gel-loading tips (Gel-Saver II, USA Scientific, FL) with the voltage at 5 V/cm. Joule heating (i.e., excess heat due to electrical-current passage through the media) was <1°C at 5 V/cm. Higher voltage will accelerate electrophoresis, but could lead to overheating and worse band separation.
FIGURE 1.

Schematic of a mini-gel assembly for in-gel ALEX. (A) Top view of the gel apparatus. Polyacrylamide (PA) is polymerized between two glass coverslips spaced by a 2-mm silicone gasket. The electric field is supplied through two platinum electrodes immersed in the running buffer. Sample is loaded in the well (formed by leaving a small comb during polymerization process). Negatively charged biomolecules (e.g., DNA) migrate towards the positive electrode (migration direction denoted by the bold arrow). The migration rate is determined by the charge and size of the biomolecules.
(B) Cross-section of the mini-gel assembly. The confocal volume (green ellipse; not to scale) is positioned halfway in the gel along the direction of electrophoresis and ≈30 μm above the top surface of the bottom coverslip.
(C) Fluorescence intensity traces from a stationary confocal spot show the appearance of fluorescence bands as labeled molecules undergo electrophoresis through the confocal volume. Here, an RNA polymerase (RNAP) open complex mixture was separated in 5% PA and observed under ALEX. The time traces were binned with 1-s resolution. Green: fluorescence in the donor-emission channel due to donor excitation (FDex,Dem); blue: fluorescence in the acceptor-emission channel due to donor excitation (FDex,Aem); red: fluorescence in the acceptor channel due to acceptor excitation (FAex,Aem). The burst centered at ≈3.5 min corresponds to the free doubly labeled DNA; the burst starting at ≈10 min is the (RNAP)-DNA open complex.
Single-molecule observations were performed by placing the gel-containing coverslip sandwich (with the large coverslip on the bottom) on the top of a microscope objective (water immersion, 1.2 NA, 60×, Olympus, Japan). Systematic in-gel fluorescence correlation spectroscopy (FCS) measurements on DNA standards at varying distances (10 to 60 μm) of the confocal volume from the bottom coverslip showed extended diffusion in all cases (not shown), indicating the presence and integrity of the gel matrix for the studied range of distances from the coverslip. Unless otherwise noted, our in-gel ALEX measurements were performed with the confocal volumes placed at ≈30 μm above the top surface of the bottom coverslip.
For reagent delivery, the gel apparatus was modified (Fig. 6A) by replacing the top coverslip with two small coverslips attached together using tape (Scotch® tape, 3M, MN). After gel polymerization, one of the top coverslips and the tape were removed to expose the top surface of the gel, while the other small coverslip kept the gel in place. With the voltage off, the running buffer was drained and the reagent was deposited directly on the exposed gel surface closest to location of the confocal spot.
FIGURE 6.

Reagent delivery for in-gel ALEX measurements.
(A) Modified mini-gel assembly for reagent delivery in gels. Instead of one small coverslip, two small coverslips (attached with tape) are used to seal the top of the chamber during gel polymerization. Once the gel is polymerized, one coverslip is left in place to prevent the gel from floating in the buffer, while the other one is removed to expose the top surface of the gel for reagent delivery.
(B) E* histogram of RNAP open complex in 5% PA (burst selection criteria: L=40, M=10, T=1 ms, S>0.6) shows a FRET distribution centered on E*≈0.3 due to unwinding and bending of the promoter DNA, conformational changes that result in a decreased interfluorophore distance.
(C) E* histograms [prepared using the same burst selection criteria as in (B)] measured 10 min after the addition of 100 μL of 10% SDS on the gel shows complete denaturation of the open complex. The marked decrease in the mean E*, to ≈0.2, reflects the efficient denaturation of the RNAP molecules, leaving the double-stranded promoter DNA in its linear B-DNA conformation.
Optical setup
Single-molecule fluorescence experiments were performed using a home-built confocal microscope with excitation wavelengths of 532 nm (Samba, Cobolt, Sweden) and 638 nm (Cube, Coherent, CA) with alternating-laser excitation (ALEX) as described29,31,32. The two lasers were alternated with a modulation frequency of 10 kHz and coupled into an inverted confocal microscope (IX71, Olympus, Japan). Data acquisitions were performed with custom software written in LabVIEW (National Instruments, TX). Fluorescence arrival times were recorded on two spectrally separated detectors (SPQR-14, Perkin Elmer, CA) and processed using custom software written in LabVIEW, MATLAB (MathWorks, MA), and Python (Python Software Foundation). Experiments requiring high photon counts to achieve high temporal resolution (Fig. 5) were conducted with excitation power of 400 μW (donor excitation, Dex) and 60 μW (acceptor excitation, Aex); excitation powers were measured at continuous-wave mode before the dichroic. The rest of in-gel ALEX experiments were performed using 200 μW (for Dex) and 50 μW (for Aex).
FCS experiments were performed with continuous excitation of either 532 nm (150 μW) or 638 nm (60 μW). Photon arrival times in the donor and acceptor channels were directly correlated using hardware correlator (Flex02-02D, Correlator.com, NJ).
ALEX data analysis
ALEX-based experiments produce four photon streams: FDex,Dem, FDex,Aem, FAex,Dem, FAex,Aem, where FXex,Yem is the photon count detected in Y-emission wavelength upon excitation with the X-excitation laser. Fluorescence bursts corresponding to molecules diffusing through the confocal volume were identified by applying a search algorithm33,34 that searches for L photons, each having M neighboring photons within a time interval of T ms. We performed burst search on FAex,Aem to find molecules with the acceptor present. Stoichiometry, S, and apparent FRET efficiency, E*, were calculated for each burst29,30, yielding two-dimensional E*-S histograms that allow for sorting and identification of subpopulations.
Once bursts were identified, photon arrival times within each burst were further binned to provide information about temporal E* and S fluctuations within bursts. Since extended exposure of fluorophores to laser excitation increases photobleaching, we used burst-selection criteria (i.e., a search threshold on the FAex,Aem channel) to remove bursts without an active acceptor (e.g., due to photobleaching). In some experiments (indicated in the relevant figure legends), further per-bin filtering was applied to remove spurious changes in fluorescence intensity within the bursts, such as donor-blinking (indicated by a sudden drop in the donor intensity relative to the acceptor intensity) and sudden change in relative brightness between donor and acceptor (indicated by a large change in S)34.
Burst-selection strategies are different for in-solution and in-gel measurements, due to the different durations of the bursts. Large T and L values coupled with small values of M would preferentially detect continuous large bursts while including time intervals when the molecules transiently exit the confocal volume. On the other hand, decreasing T and increasing M would dissect large bursts into many smaller bursts as the intensity transiently dips below the prescribed threshold. For within-burst time-trace analysis (Fig. 5), we adopted the former strategy where transient drops in intensity within the bursts were further screened using per-bin filter. The latter strategy is simpler to implement and is suitable in cases where maximization of burst duration is not important.
The standard deviation of FRET values within one burst, σE, was calculated from the 0.5-ms binned E* trace (see Fig. 5A). The mean FRET value, E*, and its standard deviation, σE, were plotted into 2D histograms (Fig. 5B). Artificial “spikes” and “voids” due to binning artifact were removed by applying Gaussian smoothing to the 2D histograms.
Ensemble FRET data analysis
Ensemble FRET values from in-gel ALEX measurements were calculated using the expression: E*=FDex,Aem/(FDex,Aem+FDex,Dem), where FDex,Dem and FDex,Aem, were the ALEX-based fluorescence intensities integrated for a period of 1 s within the band of interest. To improve the accuracy of the calculation, we corrected each channel for background counts measured from the same gel sample at the start of the experiment (e.g., at t≈0 in Fig. 1C).
FCS data analysis
To compare the diffusion properties of molecules in solution and in gel, we used autocorrelation of the acceptor channel due to acceptor excitation to avoid any FRET-induced donor intensity fluctuations. The autocorrelation curves of the acceptor channel, GAA(τ), were fitted with a simple expression describing 3D diffusion in the absence any photophysical changes:
| (1) |
where N is the average number of molecules diffusing inside the confocal volume, τD is the characteristic time spent inside the confocal volume, and γ is the ratio of axial to radial radii of the confocal volume.
FRET dynamics were analyzed by taking the ratio of donor autocorrelation GDD(τ) and donor-acceptor cross correlation GDA(τ) due to donor excitation35. The correlation curves contain terms describing diffusion, Diff(τ), as well as kinetic reaction, RXY(τ):
| (2) |
In the ideal case without spectral leakage between the donor and acceptor channels, the reaction terms for the donor autocorrelation and donor-acceptor cross correlation are given as follow
| (3) |
| (4) |
where Ex is the FRET value of state x and kx is the rate of forming state x. Note that due to the anti-correlated nature of the donor-acceptor emissions in FRET, the reaction term for the cross correlation curve increases monotonically with longer time lags. Taking the ratio of GDD(τ) and GDA(τ) removes the diffusion terms, leaving only the reaction terms defined by equations (3) and (4). In non-ideal conditions, equations (3) and (4) become very complex once corrections terms are introduced. Since we were only interested in recovering the timescale of folding-unfolding process, we fit the curves to a simple stretched exponential function
| (5) |
where C is a proportionality constant related to the relative concentration of the doubly labeled species, K is the equilibrium constant between the folded and unfolded states, β is the stretch parameter, and τR is the timescale of reaction. The stretch parameter, β, can vary between 1 (where the system displays normal two-state Arrhenius kinetics, with one discrete energy barrier) and 0 (where there is a continuum of equal energy barriers and the system shows power-law kinetics)36. The value of β between 0 and 1 indicates that the reaction is heterogeneous with multiple overlapping pathways. The mean relaxation time from such reaction can be obtained by considering
| (6) |
where Γ(β-1) is the gamma function. The low amplitudes of correlation curves at longer time lags caused their ratio to be noise-dominated (Fig. 5C); consequently, we limit our fitting to data points up to 20 ms.
Results and Discussion
Identification of electrophoretic bands
We first positioned the confocal spot halfway in the gel (≈30 μm above the coverslip surface, Fig. 1B) and observed the fluorescence intensities due to molecules electrophoresed through the confocal volume. Migration of electrophoretic bands was observed as clustering of fluorescence bursts. Binning the fluorescence intensity traces in 1-s bins removed fast intensity fluctuations and facilitated the identification of individual bands (Fig. 1C). Once the band of interest was identified, electrophoretic migration was stopped by switching off the power supply, followed by single-molecule detection of diffusing molecules.
The concentration gradient within a gel band allows for ensemble, FCS, and single-molecule measurements on the same sample. By moving the confocal volume towards the edges of the band, we enter the concentration regime suitable for single-molecule detection using confocal microscopy (10-100 pM). Measuring towards the middle of the band provides the concentration regime suitable for FCS and ensemble experiments. The physical separation of samples in gel allows easy examination of all electrophoretic species in a single gel lane by moving the detection volume from one band to another.
Ensemble FRET measurements using in-gel ALEX offer several advantages over ensemble fluorescence techniques such as laser-based gel scanning7,8 or in-gel fluorescence measurements in cuvettes10,11. Firstly, the use of avalanche photodiodes detectors offers higher sensitivity in the red spectral region compared to photomultiplier tubes used in gel scanners and commercial fluorimeters. Secondly, FRET measurements using in-gel ALEX do not require singly labeled control samples. Finally, in-gel ALEX improves the accuracy of ensemble FRET measurements by facilitating background correction, corrections for leakage, direct excitation, and difference in detection efficiencies between the donor and acceptor channels (since such correction factors can be easily obtained from single-molecule measurements30, and by excluding signals from the edges of the band where noise dominates.
We demonstrated the ability to obtain ensemble FRET values from purified species by studying the DNA-RNAP reaction mixture (Fig. 1C, see RNA polymerase (RNAP) open complex). The ensemble FRET values (calculated from 1-s time-bins from the middle of each band and corrected for background) obtained for free DNA (E*≈0.17) and DNA-bound RNA polymerase open complex (E*≈0.24) are consistent with the expected FRET changes during open complex formation37.
Comparison between in-solution and in-gel measurements
Previous observations of single-molecule FRET in polyacrylamide26 reported lower signal-to-background ratio (SBR) for in-gel experiments (SBR≈5) compared to measurements in solution (SBR≈11). To validate our approach, we compared single-molecule measurements of double-stranded DNA standards using in-gel and in-solution ALEX. Fluorescence-intensity traces in gel (Fig. 2B) showed that single molecules are detected with a slightly better SBR (SBR≈25) than in solution (Fig. 2A; SBR≈20), and with a substantially better SBR than the previous reports; although we cannot directly compare our SBR values with the one obtained in other labs, our results indicate that our method for gel preparation and in-gel spectroscopy enables sensitive single-molecule detection in the gel matrix. This improvement in SBR was likely due to the longer transit through the confocal volume, which increased the number of emitted photons per-burst (not shown). Importantly, the mean E* and S values in gel were essentially identical to those in solution (Fig. 2A-B, right panels). The in-gel S distribution was wider due to more opportunities for blinking and photobleaching during the longer time spent inside the confocal volume. We have also observed that at high excitation intensity (Dex > 500 μW, Aex > 80 μW) the background photon counts in gel increased more rapidly than in solution, causing a slight shift in E* and S relative to the values obtained in solution (not shown). We attribute this to higher impurities or scattering from the polyacrylamide gel matrix. If necessary, the effect of increasing background counts can be accounted for by applying simple corrections during the calculation of E* and S for each burst30.
FIGURE 2.

Comparison between in-solution and in-gel measurements.
(A) Fluorescence-intensity time traces (left, binned with 1-ms resolution) and the resulting E*-S histograms (right) of doubly labeled DNA (T1-Cy3B,B18-ATTO647N) freely diffusing in solution. Only molecules containing both the green and red fluorophores are displayed in the 2D E*-S histogram (using the threshold criterion of 0.5 < S < 0.9). Above and on the right of the 2D histogram are the 1D projections of the E* and S axis, respectively. The 1D histograms were fitted with Gaussian distributions (black solid lines); the mean values and standard deviations of the fits are given as insets.
(B) Time traces (left) and the resulting E*-S histograms (right) of the same DNA construct measured in 5% PA gel; the mean E* and S values measured in the gel are essentially identical to the ones in solution. The S histogram in gel is wider due to increased blinking and photobleaching during the longer transit time through the confocal volume.
(C) Extended diffusion of DNA in 5% PA is illustrated by FCS. The autocorrelation curve (FAex,Aem) obtained in-gel (gray line) is shifted towards a slower timescale when compared to the curve measured in solution (black line). Specifically, the average molecular transit time inside the confocal volume (τD) measured in gel (≈7.7 ms) is ≈ 4-fold longer than that measured in solution (≈2.1 ms).
(D) Comparison of the distributions of burst duration (obtained using the burst selection criteria: L=50, M=10, T=2 ms, S>0.5; this maximizes the length of detected bursts) shows a marked increase in the probability of having longer bursts in gel. The probability of observing a burst longer than 10 ms increased ≈3-fold in gel (50% of the selected bursts) compared to in solution (16%).
The longer fluorescence bursts found in Fig. 2B indicate that molecules in gel spent, on average, longer time in the confocal volume compared to those in solution. This was anticipated, since cross-linked polyacrylamide forms a water-filled web of pores, with pore size decreasing non-linearly with increasing concentration of PA38,39. For example, 5% PA has an average pore diameter of ≈100 nm (Ref. 2), while 8% PA has an average pore diameter of ≈2 nm (Ref. 40). The caging effect of the pores slowed down the translational diffusion of molecules inside the gel4 (Fig. 2C-D, Fig. 4C), permitting direct observation of fluorescence fluctuations within slowly diffusing bursts without surface immobilization. The extended molecular diffusion in gels was conclusively confirmed after comparing the temporal autocorrelation curves for the same DNA in solution and in gel (Fig. 2C). The average molecular transit time through the confocal volume (τD) measured in 5% PA (τD≈ 7.7 ms) was ≈ 4-fold longer than the value obtained in solution (τD ≈ 2.1 ms). This effect was also reflected in the distributions of burst duration (Fig. 2D), which showed a shift towards longer burst durations in gel (note that the burst length is influenced by the burst-search process; see ALEX data analysis).
FIGURE 4.

Using a DNA hairpin as a model for studying conformational dynamics.
(A) Schematic of a DNA hairpin interconverting between folded and unfolded conformations. In the folded conformation (top subpanel), the donor and acceptor fluorophores are brought close by the zipping action of the hairpin stem, producing a high FRET value. In the unfolded conformation (bottom subpanel), the hairpin stem is unzipped, creating a disordered single stranded DNA region that results in a lower FRET value due to the longer average interfluorophore distance.
(B) The equilibrium between the folded and unfolded conformations is influenced by the presence of Mg2+ ions. The unfolded conformation (low FRET) is favored at 0 mM MgCl2 (top subpanel), while at saturating concentration of MgCl2 (100 mM, middle subpanel), the folded conformation of the hairpin is favored (high FRET). Non-saturating (5 mM) MgCl2 concentration produced a bimodal FRET distribution with a substantial number of bursts having intermediate FRET values. This is consistent with the hairpins undergoing interconversion between the folded and unfolded states around the timescale of diffusion. Burst selection criteria: L=50, M=6, T=1 ms, S>0.6.
(C) The presence of gel matrix extended the diffusion of hairpin ≈4.5-fold as shown by the τD increase between freely diffusing molecules in buffer (≈2.2 ms) and in 6% PA gel (≈9.7 ms). Autocorrelation of FAex,Aem was used in order to avoid complications introduced by FRET-induced donor intensity fluctuations.
The results above signify that in-gel ALEX can extend molecular diffusion without perturbing the E* and S observables. In contrast, when we embedded fluorescent DNA-hairpin molecules in polyacrylamide gel using co-polymerization protocol as described14,17, we were not able to detect any fluorescence bursts using confocal optics (Fig. S-1C). This implied that the molecules were not diffusing, consistent with previous reports of molecule immobilization when co-polymerized with PA14,16-18,20-22,26. On the other hand, when we brought the confocal volume close to the coverslip surface (<3 μm), we were able to see diffusing fluorescent molecules (Fig. S-1D); however, this is most likely due to molecules diffusing in the aqueous layer between the gel and the coverslip. Co-polymerization with polyacrylamide is expected to be problematic because it exposes biological molecules to free radicals, which can damage biomolecules and fluorophores, as well as to bis-acrylamide, an effective cross-linking agent that might cause the observed immobilization.
Electrophoretic separation of charged biomolecules
The capability to separate molecular species using gel electrophoresis on top of a microscope objective and perform immediate ALEX measurements in the same gel is vital for studying the structure and dynamics of complexes separated in the gel, since it bypasses the need for additional preparatory steps (separate gel electrophoresis, gel disassembly, imaging, gel-band excision and coverslip mounting) which can lead to dissociation of the complexes of interest. As a proof-of-principle of the ability to separate molecular species using gels and perform immediate in-gel ALEX measurements, we recovered FRET distributions for a mixture of three DNAs of different lengths and FRET efficiencies (Fig. 3). DNA (a negatively charged molecule) migrates towards the positive electrode during electrophoresis, with its mobility being a complex interplay between its net electronic charge and molecular size. In the absence of any higher-order structure, shorter double-stranded DNAs are expected to migrate faster than longer double-stranded DNAs in non-denaturing gels.
Figure 3E shows the three DNAs (20, 40, and 45 bp in length) separated in 10% non-denaturing PA gel. This sample simulates a mixture of molecules with different conformational states (represented by different FRET values) and different electrophoretic mobility. Examination of the mixture using in-solution ALEX produced a broad E* histogram that could not be conclusively resolved into its individual sub-populations (Fig. 3A). In contrast, in-gel ALEX separated the three DNAs on the basis of electrophoretic mobility (seen as three gel bands in Fig. 3E) and produced “purified” E* distributions that correspond to each DNA component of the mix (Fig. 3B-D). Our results showed that size differences as small as 5 bp (cf. Fig. 3B and 3C, where >95% of the molecules correspond to the main DNA population) could be resolved given the right gel concentration (here, 10% PA). In fact, a separation of 1 bp is routinely done in many biochemical studies involving DNA and RNA3,41,42. As a result, in-gel ALEX should be very useful for analyzing complex multi-component mixtures with multiple conformational states and stoichiometries.
Conformational changes in a DNA hairpin
Information about conformational dynamics and kinetics provides an important link between biomolecular structure and function. To achieve direct observation of conformational dynamics, we used gels to slow down the translational diffusion of biomolecules and observed FRET fluctuations within diffusing molecules. As a test case, we examined a DNA hairpin with a 5 bp stem and poly-A (30 nt) loop.
DNA and RNA hairpins are important models for understanding the conformational dynamics of biopolymers. Their folding rates and equilibria are influenced by external factors, such as temperature43-45 and concentration of cations36,45 (see ref. 46 for a recent review). Figure 4A shows a simple schematic of a two-state reaction describing the folding and unfolding of the hairpin. Although it has been suggested that hairpin folding is a complex process that involves several intermediate states44,46, for the purpose of demonstrating the capability of in-gel ALEX, we model the hairpin as a two-state system.
Metal cations, such as Mg2+, are known to stabilize the folded state of hairpins45. Figure 4B shows the FRET distributions for freely diffusing hairpin molecules in buffers with different MgCl2 concentrations. At 0 mM MgCl2, the hairpin is found primarily in the unfolded state where the donor-acceptor distance is large (low FRET). At 100 mM MgCl2, the folded state (characterized by high FRET efficiency) is more prominent. At 5 mM MgCl2, we observe a large population of bursts with intermediate FRET efficiencies (E* from 0.3 to 0.7), suggesting the presence of interconversion between the folded and unfolded states.
To test whether the intermediate FRET efficiencies are due to the dynamic nature of the hairpin, we compared a short double-stranded DNA (which can be treated as a rigid rod), with the DNA hairpin measured at 0 mM MgCl2 and at 5 mM MgCl2. Using in-gel ALEX, we were able to extend the diffusion of the hairpin by ≈4.5-fold (Fig. 4C; τD in buffer and in 6% PA gel are 2.2 and 9.7 ms respectively). We then utilized the extended diffusion to study real-time FRET fluctuations within long bursts (Fig. 5A). Both the DNA and the hairpin sample without MgCl2 showed relatively small FRET fluctuations (Fig. 5A, left and middle panels). However, the hairpin at 5 mM MgCl2 exhibited clear, multiple, large-scale FRET fluctuations accompanied by anti-correlated donor and acceptor signals (Fig. 5A, right panel). In contrast, measurements of hairpin dynamics in solution and in the presence of 5 mM MgCl2 (Fig. S-2C) resulted in time-traces that were relatively short compared to the timescale of dynamics, making it difficult to draw conclusions regarding dynamics solely from time-traces.
The dynamic nature of the hairpin at 5 mM MgCl2 was confirmed by examining the distribution of FRET standard deviation (σE; Fig. 5B). Due to the non-linear nature of FRET and the photon counts involved in the calculation of E*, bursts with intermediate FRET values (≈0.5) exhibit larger fluctuations (hence, they are characterized by larger standard deviations) than bursts with low or high FRET values34. To separate dynamics from shot-noise-induced variability, we used a static dsDNA sample with E*≈0.5 as a reference point for determining the upper limit of σE on the basis of shot-noise. Essentially all DNA molecules showed σE values lower than 0.2; hence, any bursts with σE higher than the experimentally determined threshold of 0.2 are classified as dynamic. Using these criteria, most bursts obtained in 5 mM MgCl2 (Fig. 5B, right panel) are dynamic. In contrast, most bursts obtained in the absence of MgCl2 (Fig. 5B, middle) display low σE values, consistent with that fact that they are mostly static. An extensive discussion of the standard-deviation analysis for static and dynamic samples will be presented elsewhere (Joseph P. Torella, Y.S. and A.N.K. in preparation).
To arrive at quantitative estimates of the folding-unfolding reaction rates, we used FCS, a sensitive method for detecting fluorescence fluctuations and their timescales. In theory, the reaction timescale can be obtained by directly fitting equation (2) to the donor-autocorrelation curves; however, in practice, this requires precise knowledge of the diffusion term Diff(τ) which means that the confocal volume has to be well characterized. This can be circumvented if information about the diffusion term can be obtained independently, e.g., by measuring the diffusion of donor-only control sample under the same experimental conditions47. An alternative approach that does not require separate measurement is proposed by Torres et al35, where the donor-acceptor cross-correlation from the same sample is used for characterizing diffusion instead of the autocorrelation curve from donor-only sample.
Using the method of Torres et al35, we compared reaction rates for the three different samples in 6% gels and in solution. Both the dsDNA and the hairpin-DNA in the absence of MgCl2 produced essentially constant ratio curves when measured either in gel or in solution (Fig. 5C-D); this implied that the original correlation curves are composed primarily of the diffusion term. However, the hairpin sample measured in 6% PA containing 5 mM MgCl2 exhibited an exponential decay that can be fitted to equation (5) to produce τR = 0.71 ± 0.15 ms, β = 0.69 ± 0.07, and mean relaxation time <τ> = 0.92 ± 0.23 ms (Fig. 5C). The value of β suggests that the reaction consists of multiple reaction pathways over a complex energy landscape34,44. The results in gel are comparable to those obtained for the same hairpin in a solution containing 5 mM MgCl2 (Fig. 5D), where τR = 0.97 ± 0.21 ms, β = 0.83 ± 0.08, and <τ> = 1.08 ± 0.25 ms, indicating that the dynamics of the hairpin are largely preserved in the gel. Additionally, the mean relaxation times obtained in solution and in gel are in good agreement with the 0.45 – 0.70 ms mean relaxation times obtained in ref. 36 for a similar hairpin using slightly different buffer conditions and a different method of analysis.
While it is tempting to increase the concentration of polyacrylamide to extend molecular diffusion even further, at high concentrations, the pores of polyacrylamide gel may not be large enough to avoid perturbations to the system studied. For example, we have observed “trapping” of DNA Polymerase I in one conformation at 8% PA; this was in contrast to the situation in a 5% PA, where conformational dynamics can be clearly seen (Santoso and Kapanidis, in preparation). The result is not surprising, since the average size of gel pores in 8% PA (≈2 nm; Ref. 40), a size smaller than the smallest dimension of the polymerase used (≈6 nm; PDB file 1KFS, Ref. 48). The important observation of the shift in the equilibrium behavior between 5% and 8% PA highlights the trade off between prolonging translational diffusion and maintaining the native conformational dynamics. While similar concern is also valid for other methods for extending single-molecule observation (e.g., surface immobilization), in-gel ALEX allows for this effect to be systematically studied by using varying gel concentrations.
Delivering small molecules using in-gel ALEX
The ability to deliver reagents, such as nucleotides, amino acids, metal ions, or other small molecules, is crucial for many single-molecule FRET experiments11,19,49. Modification of the in-gel ALEX apparatus (Fig. 6A) allows for rapid and efficient on-site delivery of small molecules on the top surface of the gel close to the confocal spot; this capability is vital for studying the effect of small molecules on the structure and dynamics of rare, transient or unstable molecular complexes since it bypasses the need for several additional preparatory steps which can lead to dissociation of these complexes (see also Results' section on Electrophoretic separation of charged biomolecules).
Figure 6B is a FRET histogram of RNAP-DNA open complex. In the open complex, the promoter DNA within the transcription bubble is bent and unwound37, causing a decrease in donor-acceptor distance and consequently an increase in the FRET value (E*≈0.3). Within 10 min of delivering denaturant (Sodium dodecyl sulfate, SDS), most of the open complexes (>90%) were destroyed, leaving the promoter DNA in its linear B-DNA confirmation, which exhibits a reduced FRET efficiency due to the increase in the mean separation of the donor and acceptor. The incubation time can be shortened by increasing the amount and the concentration of the reagents as well as by decreasing the gel thickness by using thinner spacer. We also note that when a specific concentration of a certain analyte is required in the gel (e.g., the concentration of Mg2+ in Fig. 5), it is best to pre-soak the gel with buffer in the right concentration for 1 hour before loading the sample.
Conclusions
Our work introduces simple, general, and robust ways to combine the use of gels with single-molecule FRET/ALEX spectroscopy and FCS. Our results clearly establish that excellent signal-to-background ratio (≈25) can be obtained in polyacrylamide gels, in contrast to previous report26. We attributed this to the complete gel polymerization prior to the addition of fluorescent molecules; this ensures removal of free radicals (present during acrylamide polymerization), which are known to induce cross linking1,27 and to change the photophysical properties of fluorophores18,22,26. We further demonstrated simultaneous separation and extended diffusion of biomolecules using in-gel ALEX, and recovered FRET distributions of individual DNA populations differing by only 5 bp in length; these capabilities should be instrumental for studying complex molecular assemblies, such as transcription8,10,11,32, recombination7 and nucleosomal complexes9,50. We further exploited extended molecular diffusion in gel to observe real-time FRET fluctuations within diffusing hairpin without the need for surface immobilization. Consistent to a previous report44, we showed that DNA hairpin with 5 bp stem and (A)30 loop fluctuates between the folded and unfolded conformations at 0.5-1 ms timescale. The ability to extend diffusion time without affecting the inherent dynamics enabled us to study FRET dynamics using FCS where the timescale of reaction (τR) is similar or faster than the timescale of diffusion (τD)50.
In-gel ALEX can be extended further by borrowing from established techniques in gel electrophoresis and single molecule fluorescence. For instance, ALEX spectroscopy is fully compatible with agarose gels or use of stacking layers (where a thin gel layer of different composition gel is added on top of the main gel matrix) for improving band resolution1. Moreover, preliminary in-gel ALEX observations (YS and AK, unpublished) in the presence of an electric field resulted in reduced photobleaching, faster sampling of molecules, and improvement in photon statistics, benefits similar to ones observed for single-molecule detection in nanopipettes51.
Supplementary Material
Acknowledgments
Y. S. and A. N. K. were supported by a UK Bionanotechnology IRC grant, EPSRC grant EP/D058775, NIH grant GM069709-01, and European Community's Seventh Framework Programme (FP7/2007-2013) grant HEALTH-F4-2008-201418 (entitled READNA). Y.S. was partially supported by an EPA Cephalosporin Scholarship (Linacre College, University of Oxford). The authors thank Joe Torella for help with the DNA-hairpin preparation, and Cathy Joyce (Yale University) for discussions.
Footnotes
Supporting Information Available Additional information as noted in text. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Chrambach A, Rodbard D. Science. 1971;172:440–451. doi: 10.1126/science.172.3982.440. [DOI] [PubMed] [Google Scholar]
- 2.Stellwagen N, Stellwagen E. J Chromatogr A. 2008;1216:1917–1929. doi: 10.1016/j.chroma.2008.11.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sambrook J, Russell D. Molecular cloning: a laboratory manual. CSHL press; 2001. [Google Scholar]
- 4.Fried M, Crothers D. Nucleic Acids Res. 1981;9:6505–6525. doi: 10.1093/nar/9.23.6505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chelm B, Geiduschek E. Nucleic Acids Res. 1979;7:1851. doi: 10.1093/nar/7.7.1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Garner M, Revzin A. Nucleic Acids Res. 1981;9:3047. doi: 10.1093/nar/9.13.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Radman-Livaja M, Biswas T, Mierke D, Landy A. Proc Natl Acad Sci U S A. 2005;102:3913–3920. doi: 10.1073/pnas.0500844102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen C, Chang C, Yen C, Chiu M, Chang W. Proc Natl Acad Sci U S A. 2009;106:127. doi: 10.1073/pnas.0811689106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kelbauskas L, Chan N, Bash R, Yodh J, Woodbury N, Lohr D. Biochemistry. 2007;46:2239–2248. doi: 10.1021/bi061289l. [DOI] [PubMed] [Google Scholar]
- 10.Mekler V, Kortkhonjia E, Mukhopadhyay J, Knight J, Revyakin A, Kapanidis A, Niu W, Ebright Y, Levy R, Ebright R. Cell. 2002;108:599–614. doi: 10.1016/s0092-8674(02)00667-0. [DOI] [PubMed] [Google Scholar]
- 11.Mukhopadhyay J, Kapanidis A, Mekler V, Kortkhonjia E, Ebright Y, Ebright R. Cell. 2001;106:453–463. doi: 10.1016/s0092-8674(01)00464-0. [DOI] [PubMed] [Google Scholar]
- 12.Oana H, Masubuchi Y, Matsumoto M, Doi M, Matsuzawa Y, Yoshikawa K. Macromolecules. 1994;27:6061–6067. [Google Scholar]
- 13.Ueda M, Oana H, Baba Y, Doi M, Yoshikawa K. Biophys Chem. 1998;71:113–123. doi: 10.1016/s0301-4622(98)00093-3. [DOI] [PubMed] [Google Scholar]
- 14.Dickson R, Norris D, Tzeng Y, Moerner W. Science. 1996;274:966–969. doi: 10.1126/science.274.5289.966. [DOI] [PubMed] [Google Scholar]
- 15.Brasselet S, Peterman E, Miyawaki A, Moerner W. J Phys Chem B. 2000;104:3676–3682. [Google Scholar]
- 16.Dickson R, Cubitt A, Tsien R, Moerner W. Nature. 1997;388:355–358. doi: 10.1038/41048. [DOI] [PubMed] [Google Scholar]
- 17.Dickson R, Norris D, Tzeng Y, Sakowicz R, Goldstein L, Moerner W. Mol Cryst Liq Cryst. 1996;291:31–39. [Google Scholar]
- 18.Kubitscheck U, Kückmann O, Kues T, Peters R. Biophys J. 2000;78:2170–2179. doi: 10.1016/S0006-3495(00)76764-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lu H, Xun L, Xie X. Science. 1998;282:1877. doi: 10.1126/science.282.5395.1877. [DOI] [PubMed] [Google Scholar]
- 20.Moerner W, Peterman E, Brasselet S, Kummer S, Dickson R. Cytometry. 1999;36 doi: 10.1002/(sici)1097-0320(19990701)36:3<232::aid-cyto13>3.0.co;2-l. year. [DOI] [PubMed] [Google Scholar]
- 21.Peterman E, Brasselet S, Moerner W. J Phys Chem A. 1999;103:10553–10560. [Google Scholar]
- 22.Schafer S, Dittrich P, Petrov E, Schwille P. Microsc Res Tech. 2006;69:210–219. doi: 10.1002/jemt.20283. [DOI] [PubMed] [Google Scholar]
- 23.Segers-Nolten G, Wyman C, Wijgers N, Vermeulen W, Lenferink A, Hoeijmakers J, Greve J, Otto C. Nucleic Acids Res. 2002;30:4720–4727. doi: 10.1093/nar/gkf599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi J, Dertouzos J, Gafni A, Steel D, Palfey B. Proc Natl Acad Sci U S A. 2006;103:5775–5780. doi: 10.1073/pnas.0510482103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shi J, Palfey B, Dertouzos J, Jensen K, Gafni A, Steel D. J Am Chem Soc. 2004;126:6914–6922. doi: 10.1021/ja038902y. [DOI] [PubMed] [Google Scholar]
- 26.Koopmans W, Schmidt T, van Noort J. ChemPhysChem. 2008;9:2002–2009. doi: 10.1002/cphc.200800370. [DOI] [PubMed] [Google Scholar]
- 27.Gai H, Griess G, Demeler B, Weintraub S, Serwer P. J Microsc. 2007;226:256–262. doi: 10.1111/j.1365-2818.2007.01776.x. [DOI] [PubMed] [Google Scholar]
- 28.Santoso Y, Hwang L, Le Reste L, Kapanidis A. Biochem Soc Trans. 2008;36:738–744. doi: 10.1042/BST0360738. [DOI] [PubMed] [Google Scholar]
- 29.Kapanidis A, Lee N, Laurence T, Doose S, Margeat E, Weiss S. Proc Natl Acad Sci U S A. 2004;101:8936–8941. doi: 10.1073/pnas.0401690101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee N, Kapanidis A, Wang Y, Michalet X, Mukhopadhyay J, Ebright R, Weiss S. Biophys J. 2005;88:2939–2953. doi: 10.1529/biophysj.104.054114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heilemann M, Hwang L, Lymperopoulos K, Kapanidis A. In: DNA-Protein Interactions: Principles and Protocols, Third Edition. 3rd. Moss T, Leblanc B, editors. Vol. 543. Humana Press; 2009. pp. 503–521. Chapter 9. [Google Scholar]
- 32.Heilemann M, Lymperopoulos K, Wigneshweraraj S, Buck M, Kapanidis A. Studying sigma 54-dependent transcription at the single-molecule level using alternating-laser excitation (ALEX) spectroscopy. Biophotonics 2007: Optics in Life Science Proceedings of the SPIE. 2007:66332K. [Google Scholar]
- 33.Eggeling C, Berger S, Brand L, Fries J, Schaffer J, Volkmer A, Seidel C. J Biotechnol. 2001;86:163–180. doi: 10.1016/s0168-1656(00)00412-0. [DOI] [PubMed] [Google Scholar]
- 34.Nir E, Michalet X, Hamadani K, Laurence T, Neuhauser D, Kovchegov Y, Weiss S. J Phys Chem B. 2006;110:22103–22124. doi: 10.1021/jp063483n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Torres T, Levitus M. J Phys Chem B. 2007;111:7392–7400. doi: 10.1021/jp070659s. [DOI] [PubMed] [Google Scholar]
- 36.Wallace M, Ying L, Balasubramanian S, Klenerman D. J Phys Chem B. 2000;104:11551–11555. [Google Scholar]
- 37.Kapanidis A, Margeat E, Ho S, Kortkhonjia E, Weiss S, Ebright R. Science. 2006;314:1144–1147. doi: 10.1126/science.1131399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rüchel R, Steere R, Erbe E. J Chromatogr. 1978;166:563–75. [Google Scholar]
- 39.Rüchel R, Brager M. Anal Biochem. 1975;68:415–428. doi: 10.1016/0003-2697(75)90637-5. [DOI] [PubMed] [Google Scholar]
- 40.Zimm B, Levene S. Q Rev Biophys. 1992;25:171. doi: 10.1017/s0033583500004662. [DOI] [PubMed] [Google Scholar]
- 41.Rodrguez S, Zapata C. Mol Biotechnol. 2002;21:117–122. doi: 10.1385/MB:21:2:117. [DOI] [PubMed] [Google Scholar]
- 42.Sanger F, Nicklen S, Coulson A. Proc Natl Acad Sci U S A. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grunwell J, Glass J, Lacoste T, Deniz A, Chemla D, Schultz P. Nat Med. 1998;4:350–353. [Google Scholar]
- 44.Wallace M, Ying L, Balasubramanian S, Klenerman D. Proc Natl Acad Sci U S A. 2001;98:5584–5589. doi: 10.1073/pnas.101523498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ha T. Biochemistry. 2004;43:4055–4063. doi: 10.1021/bi049973s. [DOI] [PubMed] [Google Scholar]
- 46.Van Orden A, Jung J. Biopolymers. 2008;89:1–16. doi: 10.1002/bip.20826. [DOI] [PubMed] [Google Scholar]
- 47.Bonnet G, Krichevsky O, Libchaber A. Proc Natl Acad Sci U S A. 1998;95:8602–8606. doi: 10.1073/pnas.95.15.8602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brautigam C, Steitz T. J Mol Biol. 1998;277:363–377. doi: 10.1006/jmbi.1997.1586. [DOI] [PubMed] [Google Scholar]
- 49.Margeat E, Kapanidis A, Tinnefeld P, Wang Y, Mukhopadhyay J, Ebright R, Weiss S. Biophys J. 2006;90:1419–1431. doi: 10.1529/biophysj.105.069252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kelbauskas L, Chan N, Bash R, DeBartolo P, Sun J, Woodbury N, Lohr D. Biophys J. 2008;94:147–158. doi: 10.1529/biophysj.107.111906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vogelsang J, Doose S, Sauer M, Tinnefeld P. Anal Chem. 2007;79:7367–7375. doi: 10.1021/ac071176n. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.