

Abstract
Inherited mutations in voltage-gated sodium channels (VGSCs; or Nav) cause many disorders of excitability, including epilepsy, chronic pain, myotonia, and cardiac arrhythmias. Understanding the functional consequences of the disease-causing mutations is likely to provide invaluable insight into the roles that VGSCs play in normal and abnormal excitability. Here, we sought to test the hypothesis that disease-causing mutations lead to increased resurgent currents, unusual sodium currents that have not previously been implicated in disorders of excitability. We demonstrated that a paroxysmal extreme pain disorder (PEPD) mutation in the human peripheral neuronal sodium channel Nav1.7, a paramyotonia congenita (PMC) mutation in the human skeletal muscle sodium channel Nav1.4, and a long-QT3/SIDS mutation in the human cardiac sodium channel Nav1.5 all substantially increased the amplitude of resurgent sodium currents in an optimized adult rat–derived dorsal root ganglion neuronal expression system. Computer simulations indicated that resurgent currents associated with the Nav1.7 mutation could induce high-frequency action potential firing in nociceptive neurons and that resurgent currents associated with the Nav1.5 mutation could broaden the action potential in cardiac myocytes. These effects are consistent with the pathophysiology associated with the respective channelopathies. Our results indicate that resurgent currents are associated with multiple channelopathies and are likely to be important contributors to neuronal and muscle disorders of excitability.
Introduction
Voltage-gated sodium channels (VGSCs; or Nav) are crucial to the generation and propagation of the all-or-none action potentials (APs) in excitable cells, such as neurons and muscle. More than 200 different missense mutations in 7 VGSCs have been identified as causing disorders of excitability, or channelopathies, in humans (1, 2). Although these channelopathies are relatively rare, understanding the functional consequences of the disease mutations provides invaluable insight into the roles that VGSCs play in normal and abnormal excitability. Mutant channels have been extensively studied in nonexcitable heterologous expression systems, providing substantial knowledge; however, a major concern is that the functional properties of VGSCs in neurons and muscle cells are not always accurately reproduced in nonexcitable cells. A prime example of this phenomenon is VGSC resurgent currents. Although resurgent VGSC currents have been recorded from neurons, it has not been possible to record resurgent currents in nonexcitable heterologous expression systems (3–5), and it is not known whether VGSC mutations that cause channelopathies alter resurgent currents.
Resurgent sodium currents were first described in cerebellar Purkinje neurons (3) and more recently have been observed in dorsal root ganglion (DRG) neurons (4). Normally, VGSCs open, and subsequently rapidly inactivate, upon membrane depolarization. Inactivated channels cannot reopen until they have been substantially hyperpolarized for many milliseconds; this is the basis for the AP refractory period (6). In contrast, resurgent currents reopen during the repolarization of the membrane potential and therefore are likely to contribute to spontaneous and high-frequency firing (7). These unusual sodium currents are proposed to result from a putative intracellular blocking factor that binds to open VGSCs and prevents classical VGSC inactivation, but unbinds during repolarization at potentials at which channels normally remain inactivated (8). In both cerebellar Purkinje and DRG neurons isolated from Nav1.6 knockout mice, resurgent currents are greatly reduced, which indicates that Nav1.6 is normally the predominant generator of resurgent currents in neurons (3, 4). However, artificial slowing of VGSC inactivation via toxin application can induce resurgent currents in Purkinje neurons isolated from Nav1.6 knockout mice, which indicates that VGSCs other than Nav1.6 may have the capacity to generate resurgent sodium currents (9). In this study, we hypothesized that disease mutations that slow or destabilize sodium channel inactivation lead to increased resurgent currents. We have previously shown that recombinant Nav1.6 channels expressed in DRG neurons can generate resurgent currents in approximately 60% of the transfected neurons (4), which indicates that DRG neurons should be an ideal cell background for testing our hypothesis. Although DRG neurons express multiple VGSCs, it is worth noting that not all isoforms are natively expressed in DRG neurons (10). Adult DRG sensory neurons can express combinations of Nav1.1, Nav1.6, Nav1.7, Nav1.8, and Nav1.9. In addition, Nav1.2, Nav1.3, and Nav1.5 are expressed, at least at low levels, in embryonic rat DRG. However, because the cellular background of DRG neurons, which may include expression of particular kinases and auxiliary subunits, is appropriate to generate resurgent sodium currents in Nav1.6, we proposed that cultured DRG neurons might provide an opportune expression system for investigating the capability of other wild-type and mutant VGSCs to generate resurgent current. Using this expression system, we showed for the first time to our knowledge that disease mutations in 3 different VGSC isoforms, associated with 3 distinct disorders of excitability, increased resurgent sodium currents. Based on our data, we propose that resurgent sodium currents play important and previously unrecognized roles in disorders of excitability.
Results
A Nav1.7 inactivation gate mutation implicated in PEPD increases resurgent sodium currents.
We first asked whether a mutation in Nav1.7 that slows inactivation and causes paroxysmal extreme pain disorder (PEPD; ref. 11) generates resurgent sodium currents. Highly expressed in DRG neurons, Nav1.7 channels are essential in nociception, as evidenced by single-point missense mutations causing a spectrum of pain syndromes, including PEPD, and by nonsense mutations resulting in human insensitivity to pain (12). Although Nav1.7 channels have not previously been shown to produce resurgent currents, PEPD mutations destabilize inactivation, shifting voltage dependence and decreasing the rate of inactivation (11, 13). Therefore, we hypothesized that PEPD mutations induce resurgent currents. We studied the Nav1.7 PEPD mutation I1461T, located within the highly conserved D3-D4 inactivation particle (IFMT) critical for VGSC inactivation (14, 15). Modified wild-type Nav1.7 and Nav1.7-I1461T (referred to herein as Nav1.7r and Nav1.7r-I1461T, respectively) channels that generate currents that can be pharmacologically isolated were expressed in adult rat DRG neurons (Figure 1, A and B). In addition to the recombinant channel of interest, neurons were also cotransfected with a second plasmid encoding for both EGFP, to help identify transfected neurons, and a specific Nav1.8 shRNA, to minimize endogenous Nav1.8 currents (see Methods for details). In DRG neurons, the I1461T PEPD mutation impaired inactivation (Figure 1, C and D, and Table 1), but did not alter activation (data not shown), of Nav1.7r channels. The persistent component, measured at the end of a 50-ms pulse to –10 mV, was also significantly larger for Nav1.7r-I1461T channels than for Nav1.7r channels (Nav1.7r, 0.2% ± 0.2 % of peak transient current, n = 13; Nav1.7r-I1461T, 1.0% ± 0.3%, n = 22; P < 0.05). These changes were identical to those observed for wild-type and I1461T Nav1.7 channels expressed in HEK293 cells (13). However, in DRG neurons, both Nav1.7r and Nav1.7r-I1461T channels also generated resurgent sodium currents. Resurgent sodium currents were observed in 5 of 21 neurons transfected with Nav1.7r (Figure 2, A and B), with an average amplitude — expressed as a percentage of the peak transient sodium current elicited with a test pulse to –10 mV — of 1.0% ± 0.5% for these 5 neurons. Conversely, 20 of 30 neurons expressing Nav1.7r-I1461T channels produced resurgent sodium current (Figure 2, C and D). The frequency of resurgent current with Nav1.7r-I1461T channels was significantly increased (P < 0.05, χ2 test) compared with Nav1.7r channels. Moreover, the relative amplitude of the resurgent current (2.0% ± 0.1%) was also significantly greater for Nav1.7r-I1461T than for Nav1.7r channels (P < 0.05). Because all PEPD mutations characterized to date result in slower inactivation of Nav1.7 (11, 13), we predict that all PEPD mutations are likely to increase resurgent current generation.
Figure 1. Currents generated by recombinant Nav1.7 channels expressed in DRG neurons.

(A) Representative Nav1.7r current traces recorded from a transfected DRG neuron. (B) Representative Nav1.7r-I1461T current traces recorded from a transfected DRG neuron. Currents were elicited with step depolarizations to voltages ranging from –80 to +40 mV from a holding potential of –100 mV. (C) The painful mutation I1461T slowed the rate of inactivation of Nav1.7r. Black trace, Nav1.7r; red trace, Nav1.7r-I1461T. Currents were elicited with a step depolarization to +10 mV. (D) Steady-state inactivation curves for Nav1.7r (black) and Nav1.7r-I1461T (red) channels expressed in DRG neurons. Cultured adult rat DRG neurons were transfected with the recombinant VGSC construct and Nav1.8 shRNA, and recordings were done in the presence of 500 nM TTX.
Table 1 .
Biophysical properties of wild-type and mutant sodium channels

Figure 2. Resurgent currents are produced by recombinant Nav1.7 channels expressed in DRG neurons.

(A and B) Representative current traces recorded from DRG neurons expressing Nav1.7r that did not (A) and that did (B) generate resurgent currents. (C and D) Representative current traces recorded from DRG neurons expressing Nav1.7r-I1461T channels that did not (C) and that did (D) generate resurgent currents. Resurgent currents were larger on average for Nav1.7r-I1461T than for Nav1.7 channels. Currents are magnified ×30 relative to the peak transient current (elicited with a pulse to –10 mV) to emphasize the resurgent current components. (E) Resurgent current voltage protocol. (F) Voltage dependence of resurgent current shown in D.
A PEPD mutation that increases resurgent currents increases AP repetitive firing in simulated neurons.
Based on work done in cerebellar Purkinje neurons (3), we expected increased resurgent sodium currents generated by PEPD mutations to result in spontaneous and/or repetitive APs in DRG neurons. To test this, we performed computer simulations of DRG neuron excitability. We used an established model of DRG neuron excitability (16) that had been implemented in the NEURON simulation environment (17) and, with modifications only to the appropriate sodium channel formulation, simulated and evaluated the impact of the I1461T mutation and resurgent currents on AP firing. Mathematical models of Nav1.7 and Nav1.7-I1461T currents with and without a resurgent current blocking factor were developed using a multistate Markov-type model of Nav1.7 (Figure 3). The resurgent current blocking factor was implemented using the strategy developed by Khaliq et al. (18). The forward and reverse transition rate expressions for the blocking factor were adjusted (see Supplemental Table 2; supplemental material available online with this article; doi: 10.1172/JCI40801DS1) in order to match the relative amplitude of the experimentally observed resurgent currents. In our model, the blocking factor–induced resurgent current was 1.0% of the peak transient current in the modeled Nav1.7 conductance and 2.0% of the peak transient current in the modeled Nav1.7-I1461T conductance (Figure 3C). The computer simulations of AP firing in DRG neurons indicated that, whereas the destabilization of inactivation caused by the I1461T mutation was sufficient to decrease the threshold for eliciting an AP, inclusion of the resurgent current blocking factor led to high-frequency trains of APs (Figure 3D). It is important to note that even when the transition rate expressions for the blocking factor were adjusted so that the wild-type resurgent current was doubled (2% of peak), a 70-pA stimulus still only elicited a single AP in the model neuron. These results indicate the resurgent current blocking factor and the I1461T mutations act synergistically to increase neuronal excitability and that resurgent currents probably contribute to the extreme pain sensations associated with PEPD mutations.
Figure 3. Computer simulation of sodium conductances and DRG neuron excitability.

(A) Diagram of Markov models for VGSC conductances. An 8-state Markov model was used for simulation of VGSC conductances without resurgent currents. C1–C3, closed (nonconducting) states; O, open (conducting) state; I1–I4, inactivated (also nonconducting) states. A 9-state Markov model incorporated the resurgent current blocking factor. The OB state (boxed) represents channels blocked by this factor. (B) Simulated Nav1.7 (black trace) and Nav1.7-I1461T (red trace) currents elicited by a voltage step from –100 mV to +10 mV. (C) Simulated resurgent currents generated by model Nav1.7 (black trace) and Nav1.7-I1461T (red trace) conductances. Model currents were elicited with the standard resurgent current voltage protocol shown in Figure 2E. (D) In simulated DRG neurons with Nav1.7 channels, 70 pA depolarizing current is required to elicit an AP with or without resurgent current simulation. Conversely, only 40 pA is needed to elicit an AP in a simulated neuron with Nav1.7-I1461T channels, and a train of high-frequency APs is generated when the modeled Nav1.7-I1461T channels generate resurgent currents.
A cardiac long-QT3/sudden infant death syndrome mutation that slows inactivation increases resurgent Nav1.5 current and broadens AP waveform in a modeled myocyte.
Disease mutations that impair inactivation have also been identified in several other VGSCs, including Nav1.1 and Nav1.3 mutations associated with epilepsies, Nav1.4 mutations associated with skeletal muscle myotonias, and Nav1.5 mutations associated with cardiac arrhythmias (1, 2). More than 50 different disease mutations that impair inactivation have been characterized (see Supplemental Table 1 for a partial listing). However, these mutations have all been characterized in heterologous expression systems that do not support the generation of resurgent sodium currents. Therefore, we next used the DRG expression system to determine whether the Nav1.5 mutation F1486L (19), associated with long-QT3/sudden infant death syndrome (LQT3/SIDS), generates increased resurgent currents. As was previously shown in HEK293 cells (19), the F1486L mutation, which is also located in the IFMT inactivation particle, slowed the rate of inactivation, increased the fraction of persistent currents, and shifted the voltage dependence of activation in the depolarizing direction (Table 1). However, 9 of 18 DRG neurons expressing wild-type Nav1.5 generated resurgent currents (Figure 4, A and B). In our expression system, the Nav1.5-F1486L LQT3/SIDS mutation significantly increased the relative amplitude of the resurgent currents (Table 1). Of 17 neurons expressing Nav1.5-F1486L channels, 8 generated resurgent currents, with an average relative amplitude of 2.0% ± 0.4% (Figure 4, C and D). As resurgent sodium currents are activated during repolarization, we expected increased resurgent currents in Nav1.5 to broaden the AP, increase the QT interval, and thus contribute to the potentially lethal cardiac arrhythmias associated with LQT3/SIDS mutations. Mathematical modeling of resurgent sodium currents and computer simulations of a cardiac myocyte (20) indicated that this may indeed be the case (Figure 5).
Figure 4. Resurgent currents are produced by Nav1.5 channels.

(A and B) Representative current traces recorded from DRG neurons expressing Nav1.5 that did not (A) and that did (B) generate resurgent currents. (C and D) Representative current traces recorded from DRG neurons expressing Nav1.5-F1486L channels that did not (C) and that did (D) generate resurgent currents. Resurgent currents were larger on average for Nav1.5-F1486L than for Nav1.5 channels. Currents were elicited with the standard resurgent current protocol shown in Figure 2E and are magnified ×30 relative to the peak current amplitude.
Figure 5. Computer simulations of Nav1.5 currents and cardiac myocyte APs.

(A) Simulated Nav1.5 (black trace) and Nav1.5-F1486L (blue trace) currents elicited by a voltage step from –100 mV to +10 mV. (B) Simulated resurgent currents generated by model Nav1.5 (black trace) and Nav1.5-F1486L (blue trace) conductances. Model currents were elicited with the standard resurgent current voltage protocol shown in Figure 2E. The modeled resurgent current was 0.9% of the peak current for Nav1.5 and 1.7% of the peak current for Nav1.5-F1486L. (C) Simulated APs from a modeled cardiac myocyte. There was little difference between the APs of a model cell with Nav1.5 that did not include resurgent currents (black trace) and those of a cell with Nav1.5 that did include resurgent currents (green trace). Nav1.5-F1486L simulated without resurgent current generation broadened the AP (red trace), and this effect was exacerbated in the simulation of Nav1.5-F1486L with resurgent current generation (blue trace).
A D4/S4 paramyotonia congenita mutation in Nav1.4 that replaces a charged residue and uncouples fast inactivation generates resurgent sodium currents.
We next asked whether a mutation that slows inactivation of Nav1.4 and causes paramyotonia congenita (PMC) induces resurgent sodium currents. We studied the R1448P mutation (21), which alters the outermost extracellular charged residue in the sodium channel voltage sensor that couples channel activation and inactivation (22). Interestingly, although this mutation slows the inactivation of Nav1.4 currents by approximately 10-fold (Figure 6A), it causes a hyperpolarizing shift in the voltage dependence of inactivation (Table 1). In a previous study, resurgent sodium currents were not detected in any of 41 DRG neurons transfected with wild-type skeletal muscle sodium channel Nav1.4r (4). Again, we did not detect resurgent currents in any of 11 neurons expressing Nav1.4r (Figure 6B); however, 13 of 20 neurons expressing Nav1.4r-R1448P channels generated resurgent currents (Figure 6C), with an average relative amplitude of 4.8% ± 0.7% of peak. Thus, at least for Nav1.4, slowing the rate of inactivation seemed to be crucial to the production of resurgent sodium currents, and the impact of the mutation on the voltage dependence of inactivation may be less important. It should be noted that although the R1448P mutation does not alter the voltage dependence of activation, it slows deactivation (the rate at which open channels transit to the closed state; refs. 21, 23), and slower deactivation might also contribute to enhanced resurgent currents, especially in combination with slower inactivation. Resurgent currents generated by Nav1.4 are likely to increase repetitive AP firing in skeletal muscle, which is one of the hallmarks of PMC. However, it should be noted that patients with PMC can also experience episodes of muscle weakness or paralysis in addition to myotonia (24). Although resurgent currents could clearly contribute to myotonic activity associated with PMC, incomplete inactivation observed with mutant PMC channels is likely to be an important factor in muscle weakness associated with PMC (2).
Figure 6. A PMC mutation induces resurgent currents in Nav1.4.

(A) The paramyotonia R1448P mutation caused a pronounced slowing of the rate of Nav1.4r inactivation. Currents were elicited with a step depolarization to +10 mV. (B) Resurgent currents were not detectable in any of the neurons expressing Nav1.4r channels. (C) Conversely, the majority of neurons expressing Nav1.4r-R1448P channels generated robust resurgent currents. Resurgent currents were elicited with the protocol shown in Figure 2E and are magnified ×20 relative to the peak current amplitude.
A homologous inactivation gate mutation engineered into Nav1.6 channels dramatically increases resurgent currents and destabilizes transition to an inactivated state.
Because Nav1.6 appears to be the predominant generator of resurgent currents in cerebellar Purkinje neurons and DRG neurons (3, 4), we next compared the amplitude of resurgent currents generated by Nav1.6 channels with those produced by the Nav1.4, Nav1.5, and Nav1.7 disease mutations. Resurgent currents were detected in 8 of 14 DRG neurons expressing wild-type Nav1.6r channels, with an average relative amplitude of 2.4% ± 0.3% of the peak current (Figure 7A). This relative amplitude is similar to that produced by the Nav1.7r-I1461T PEPD mutant and the Nav1.5-F1486L LQT3/SIDS mutant, but slightly smaller than that produced by the Nav1.4r-R1448P PMC mutant (Figure 7C). This indicates that the resurgent currents produced by the disease mutants are indeed likely to have important impact on cellular excitability. Remarkably, gain-of-function disease mutations, to date, have not been identified in Nav1.6 channels. To determine the potential impact of mutations in Nav1.6 that impair inactivation, we next asked if a mutation in Nav1.6 (I1477T) corresponding to the I1461T mutation in Nav1.7 could increase resurgent sodium currents generated by Nav1.6. The Nav1.6r-I1477T mutation shifted the voltage dependence of inactivation in the depolarizing direction, slowed the rate of inactivation and significantly increased the persistent sodium current (Table 1). Resurgent currents were observed in 7 of 14 neurons expressing Nav1.6r-I1477T channels. Notably, these currents were approximately 8-fold larger than those produced by either Nav1.6r or Nav1.7r-I1461T channels, with an average relative amplitude of 15.3% ± 3.4% (Figure 7C). These data suggest that mutations impairing inactivation of Nav1.6 may be lethal as a result of the proclivity of Nav1.6 to generate resurgent currents.
Figure 7. Nav1.6 channels generate large resurgent currents.

(A and B) Representative resurgent currents recorded from a neuron expressing Nav1.6r (A) and Nav1.6r-I1477T (B) channels. Current traces are magnified ×20 and ×5, respectively, relative to the peak current. (C) Comparison of the relative resurgent current amplitude, expressed as a percentage of peak transient current, for wild-type and mutant VGSCs. *P < 0.05 versus the wild-type channel for the given isoform.
Discussion
Our data show, for the first time to our knowledge, that Nav1.4, Nav1.5, and Nav1.7 channels have the capability to generate resurgent currents, and that the relative resurgent sodium current amplitudes observed with the disease mutations that impair inactivation are of approximately the same magnitude as the resurgent currents generated by Nav1.6 under control conditions in cerebellar Purkinje neurons and DRG neurons (3, 4). An extensive series of studies on cerebellar Purkinje neurons indicates that resurgent currents have a major impact on excitability, contributing to spontaneous firing and accelerating the rate of repetitive firing of APs (3, 7, 8, 18, 25). As a consequence, it is predicted that the resurgent currents associated with the inherited neuronal and muscle channelopathies could markedly affect AP firing in neurons and muscle and contribute to disease pathophysiology. Indeed, our computer simulations of a DRG neuron and cardiac myocyte indicated that resurgent currents could substantially exacerbate the effects of the disease mutations on cellular excitability.
Our data provided further evidence that the sodium channel isoforms differ in their proclivity to produce resurgent sodium currents. We showed that Nav1.6 was more inclined to generate resurgent currents than Nav1.4, Nav1.5, or Nav1.7. Furthermore, the identical mutation produced a greater increase of resurgent currents in Nav1.6 than in Nav1.7 (compare Nav1.6r-I1477T data with that of Nav1.7r-I1461T; Table 1 and Figures 2 and 7). Although studies of Nav1.6 knockout mice strongly indicate that Nav1.6 is the predominant generator of resurgent currents in cerebellar Purkinje and DRG neurons (3, 4), resurgent currents have been detected in Purkinje and other central nervous system neurons from Nav1.6 knockout mice (26). Nav1.2 channels have been shown to produce resurgent currents when expressed in DRG neurons, albeit in only 2 of 25 transfected cells (27). Although it is not known whether Nav1.1 and Nav1.3 channels produce resurgent currents, mutations that slow the rate of inactivation have been identified in these isoforms in patients with epilepsy (28, 29), and it is therefore conceivable that resurgent currents could contribute to the pathophysiology of some inherited epilepsies. In our study, we showed that a mutation in the voltage-sensing segment 4 of the fourth domain, as well as mutations in the IFMT inactivation particle, induced enhanced resurgent currents, which suggests that any mutation that slows the rate of open channel fast inactivation might be able to induce resurgent currents. However, it is important to note that the rate of inactivation cannot be the only determinant of resurgent current production. Nav1.8 currents are slower to inactivate than are Nav1.7r-I1461T channels. Regardless of this, cultured rat DRG neurons treated with tetrodotoxin (TTX) that produced large endogenous Nav1.8 currents, but no recombinant currents, did not produce resurgent currents (n = 11; see Supplemental Figure 1). Thus, other factors must also contribute to the inclination of specific VGSC isoforms to generate resurgent currents.
The data presented here, in conjunction with the findings of our previous study (4), clearly show that DRG neurons have the appropriate cellular environment for production of resurgent currents. Conversely, adult DRG sensory neurons are not the native tissue for expression of Nav1.4 and Nav1.5, and it is not known whether cardiac and skeletal muscle cells have the appropriate cellular environment for the production of resurgent currents. Data from cerebellar Purkinje neurons indicate that the auxiliary sodium channel β4 subunit may be the putative blocking factor crucial for generation of resurgent currents with Nav1.6 channels (30). Previous work has demonstrated that DRG neurons and cardiac and skeletal muscle all express high levels of β4 (31). However, although the β4 subunit may be necessary, it does not appear to be sufficient. Phosphorylation of the sodium channel, the β4 subunit, or possibly an unidentified protein also seems to be required (32), at least in cerebellar Purkinje neurons. This indicates that changes in kinase and phosphatase activity are likely to affect the generation of resurgent currents. Therefore, even if the conditions in normal cardiac and skeletal muscle are not appropriate for generation of resurgent currents, under specific, possibly pathological, conditions, these cells might express the appropriate accessory subunits and kinases. Because our optimized expression system used adult DRG neurons, in which Nav1.4 and Nav1.5 are not normally expressed, it would be of great interest to determine whether Nav1.4 and Nav1.5 wild-type and mutant channels produce resurgent sodium currents in skeletal and cardiac muscle, respectively, under either normal or pathophysiological conditions.
It is important to note that not all of the DRG neurons expressing recombinant channels capable of generating resurgent currents produced detectable resurgent currents. This further indicates that the cellular environment is a crucial determinant of resurgent current production. Regional differences in the properties of sensory afferents that affect resurgent current generation might contribute to the phenotypical association of pain with particular body regions in PEPD. Interestingly, cells that generated detectable resurgent currents generally generated larger peak transient currents (Supplemental Table 4). Although this difference was only significant for 3 of the 7 channel constructs that generated resurgent currents, it might suggest that cellular factors underlying resurgent current generation could also affect peak current expression. However, more than 40 cells with peak transient current amplitudes greater than 20 nA were recorded that lacked detectable resurgent currents. Furthermore, the relative amplitude of resurgent currents is poorly correlated with peak transient current amplitude (Supplemental Figure 3). At this time, we do not know how DRG neurons that do and do not produce resurgent currents differ in terms of β subunit expression and/or kinase activity. Our optimized DRG expression system could be useful in identifying cellular factors that modulate resurgent currents.
Our data clearly show that resurgent sodium currents are likely to play a role in the functional consequences of inherited neuronal and muscle channelopathies. In addition, our data — in conjunction with the previous study indicating that β-pompilidotoxin can artificially induce resurgent currents in cerebellar Purkinje neurons (9) — indicate that any manipulation that slows or destabilizes inactivation has the potential to induce resurgent currents. Many posttranslational modifications have been reported to slow the rate of inactivation or increase the amplitude of persistent sodium currents, including hypoxia (33), phosphorylation (34), altered calcium signaling (35), G protein activation (36), and oxidation (37). We propose that these alterations could also result in abnormal resurgent current generation. The induction of resurgent sodium currents likely contributes to the more extreme electrophysiological changes and disease sequelae that can be associated with both inherited and acquired disorders of neuronal and muscle excitability.
Methods
Sodium channel constructs and mutagenesis
These studies used cDNA constructs encoding the human Nav1.4 open reading frame (38), the human Nav1.5 open reading frame (39), the mouse Nav1.6 open reading frame (40), and the human Nav1.7 open reading frame (41). To aid in isolation and characterization of sodium currents generated in DRG neurons by recombinant VGSCs, cDNA constructs for Nav1.4, Nav1.6, and Nav1.7 were modified with a single point mutation to confer high resistance to TTX (referred to herein as Nav1.4r, Nav1.6r, and Nav1.7r, respectively; Ki, approximately 100 μM; ref. 42) using the QuikChange XL (Stratagene) mutagenesis kit, as previously described (35, 43). Nav1.5 channels are natively resistant to TTX (Ki, about 2 μM); therefore, their sensitivity to TTX was not modified. Additional channelopathy constructs (Nav1.4r-R1448P, Nav1.5-F1486L, Nav1.6r-I1477T, and Nav1.7r-I1461T) were made by inserting the respective mutation into the TTX-resistant cDNA constructs using the QuikChange XL mutagenesis kit following the manufacturer’s instructions (Stratagene). Mutations were confirmed by sequencing.
cDNA and cell culture and transfection
Adult rat DRG neurons were harvested and cultured as previously described (43, 44). Animal procedures were approved by the Indiana University School of Medicine Institutional Animal Care and Use Committee. Briefly, rats were rendered unconscious by exposure to CO2 and decapitated. Cells were treated with collagenase (1 mg/ml) and papain (1 mg/ml), dissociated in DMEM supplemented with 10% fetal bovine serum, and plated on glass coverslips coated with polyornithine and laminin. Cultures were maintained at 37°C in a 5% CO2 incubator, and media was changed every 2 days during experimental incubation periods. The Helios Gene Gun (Bio-Rad Laboratories) was used to transiently cotransfect rat DRG neurons, as described previously (35, 43, 45, 46). Cells were cotransfected with a plasmid encoding the recombinant VGSC and an internal ribosome entry site–EGFP (IRES-EGFP) vector plasmid that also encoded a Nav1.8 shRNA (see below for details).
Electrophysiology
DRG recordings were obtained from cells 20–72 hours after transfection. Whole-cell patch-clamp recordings were conducted under voltage-clamp mode at room temperature (about 22°C) after obtaining a Giga-ohm seal (1–20 GΩ) using a HEKA EPC-10 amplifier (47). Data were acquired on a Windows-based Pentium IV computer using the Pulse program (version 8.65; HEKA Elektronik). Fire-polished electrodes (0.9–2.5 MΩ) were fabricated from 1.7-mm capillary glass using a Sutter P-97 puller (Novato), and the tips were coated with sticky wax (KerrLab) to minimize capacitive artifacts and increase series resistance compensation. The standard electrode solution consisted of 140 mM CsF, 10 mM NaCl, 1.1 mM EGTA, and 10 mM HEPES. The standard extracellular bathing solution contained 130 mM NaCl, 30 mM TEA Chloride, 1 mM MgCl2, 3 mM KCl, 1 mM CaCl2, 0.05 mM CdCl2, 10 mM HEPES, and 10 mM d-glucose. Recording solutions were adjusted using d-glucose to maintain physiological pH and osmolarity values. Cells on glass coverslips were transferred to a recording chamber containing 250 μl bathing solution. Transfected cells were selected based on their ability to express EGFP. Cells not expressing EGFP were also used for nontransfected control experiments and analysis. Voltage-clamp protocols were used to assay the biophysical properties of the recombinant sodium currents in the presence of 500 nM TTX to knock down native TTX-sensitive sodium currents. Offset potential was zeroed before patching. Capacitive artifacts were canceled using the computer-controlled circuitry of the patch clamp amplifier. Series resistance errors were always compensated with 75%–90% series resistance compensation and were typically less than 5 mV during voltage-clamp recordings. Leak currents were linearly canceled by digital P/–5 subtraction, whereby currents elicited by 5 pulses that are one-fifth of the test pulse are subtracted from the test pulse. Cells were held at a membrane potential of –100 mV. Membrane currents were filtered at 5 kHz, sampled at 20 kHz. Whole-cell patch recordings did not last more than 45 minutes, and cells were not held in the standard bathing solution for more than 1 hour. Inward sodium currents had a reversal potential of approximately +65 mV, corresponding closely to the calculated Nernst potential observed during the standard current-voltage (I/V) protocol. Data were not recorded before 3 minutes after whole-cell configuration had been established to allow adequate time for the electrode to equilibrate. Once recording started, cells underwent a series of 60 pulses of 20 ms each to –10 mV, to ensure rundown of any residual endogenous Nav1.9 current (42). I/V relationships were determined by an incremental depolarizing step protocol, testing every +5 mV for 50 ms, from –80 to +50 mV. To determine the fraction of channels transitioning to a fast-inactivated state, a double-pulse protocol (h∞/V) was used that incrementally conditioned the channels from –120 to –10 mV for 500 ms before testing for the fraction of channels available at –10 mV. Resurgent currents were assayed with a 2-step protocol that initially depolarized the membrane to +30 mV for 20 ms before testing for inward resurgent sodium currents by hyperpolarizing the membrane potential in –5-mV increments from 0 mV to –80 mV, for 100 ms, before returning to the holding potential. Experimental voltage-clamp data were analyzed using Pulsefit (version 8.65; HEKA Elektronik), Origin (version 7.0; OriginLab Corp.), and Microsoft Excel software programs. Data from individual steady-state recording conditions were fit using a standard single-phase Boltzmann distribution for voltage-dependent activation (m∞) and steady-state fast-inactivation (h∞) data. Midpoint (V1/2) and slope factors (Z) were calculated using a standard single-phase Boltzmann distribution fit according to Equation 1.
Equation 1
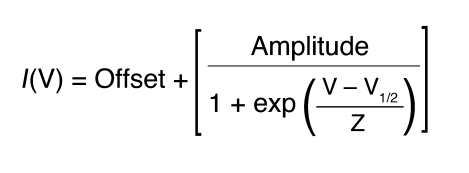
Resurgent sodium current quantification and analysis
Cells were assayed for the ability to produce resurgent sodium currents using a step protocol (Figure 2E) that initially conditioned the cells to +30 mV for 20 ms, from a holding potential of –100 mV, before repolarizing the membrane potential from 0 to –80 mV (in –5-mV increments) to test for resurgent current; cells were then returned to their holding potential. Resurgent sodium currents display distinct features (25), and these were used to determine whether a cell generated resurgent currents. For example, resurgent currents display a unique voltage dependence in which the peak resurgent currents are elicited by moderately hyperpolarized potentials relative to the holding potentials during the repolarizing steps. For all cells identified with resurgent current in the current study, maximal peak currents during the repolarizing pulses were produced within a window of potentials from –35 to –50 mV (Figure 2F) and were first observed around –10 mV. Additionally, these currents displayed unique gating kinetics within the window of potentials with a noticeably slower onset and decay phase. This is in contrast with classic VGSC tail currents, which are observed instantaneously following hyperpolarizing steps and decay within a few milliseconds. In approximately 50% of the cells, resurgent currents were not detected (for example, see Figures 2, 4, and 6). Currents were analyzed with leak subtraction in PulseFit and were filtered at 1,000 Hz to reduce noise but maintain the current waveform. The resurgent current amplitude was measured relative to the leak-subtracted baseline (Supplemental Figure 2). The relative amplitude of the resurgent current was calculated as a percentage of the peak transient current by multiplying the peak resurgent current by 100 and then dividing by the peak transient current generated during a test pulse to –10 mV from a holding potential of –100 mV. The average resurgent current amplitude for each VGSC construct was only calculated using data from those cells in which resurgent current was detected. Recordings contaminated with endogenous TTX-resistant currents, determined from steady-state inactivation plots (see below), were not used for further analysis. Cells that expressed recombinant currents with peak transient sodium current amplitudes that were less than 5 nA were also excluded from the overall analysis because of uncertainties associated with measuring resurgent current amplitude in cells that had small peak current amplitudes.
Isolation of recombinant VGSC currents in DRG neurons
The goal of these experiments was to isolate and record the sodium currents generated by the recombinant VGSCs. As described above, the recombinant channels either were naturally resistant (Nav1.5), or were mutated to be resistant (Nav1.4, Nav1.6, Nav1.7), to TTX. Endogenous DRG TTX-sensitive channels were blocked with 500 nM TTX. DRG neurons can also express endogenous Nav1.8 and Nav1.9 currents, which are resistant to TTX. Nav1.9 currents are not observed under the culture and recording conditions we used (45, 46, 48, 49), and were therefore not an issue. Although Nav1.8 currents are substantially decreased with time in culture (50), we used additional measures to minimize contamination of the recordings by Nav1.8 currents. Nav1.8 currents were knocked down using a targeted shRNA plasmid (pLL3.7; targeting sequence, GATGAGGTCGCTGCTAAGG) to silence native rat NaV1.8 gene expression via RNAi (51). Additionally, the Nav1.8 shRNA was in an IRES-EGFP vector, allowing transfected cells to be identified by their green fluorescence. The shRNA plasmid and recombinant sodium channel constructs were transfected at a ratio of 2:1. Under control conditions (less than 48 hours in culture), Nav1.8 current amplitude averaged 34.9 ± 4.8 nA (n = 42). To determine the efficiency of Nav1.8 shRNA knockdown, neurons were transfected with TTX-sensitive Nav1.7 plasmid plus the Nav1.8 shRNA. Cells with green fluorescence were recorded in the presence of TTX (to block both endogenous and recombinant TTX-sensitive currents in this control experiment), and the remaining TTX-resistant current, which must be generated by endogenous TTX-resistant channels, was measured (n = 17). Nav1.8 and Nav1.9 produce currents with distinctive kinetic and voltage-dependent properties that can be readily distinguished from each other and from TTX-sensitive currents (10, 48). In the 17 cells examined, Nav1.9 currents were not observed, and the Nav1.8 current amplitude averaged 0.5 ± 0.3 nA. Thus, endogenous Nav1.8 currents were reduced by greater than 98% under our experimental conditions (Supplemental Figure 1, A and B). In addition, because Nav1.8 currents have distinctive kinetic and voltage-dependent properties, contamination by Nav1.8 current can be determined for each individual cell expressing recombinant current. The midpoint of the voltage dependence of inactivation for Nav1.8 currents is –34.7 ± 2.0 mV, substantially more depolarized than any of the recombinant constructs investigated in the present study (see Table 1). Analysis of the voltage dependence of inactivation curve can therefore be used to determine the absolute and relative amplitude of the recombinant VGSC current and the endogenous Nav1.8 current for each individual cell (Supplemental Figure 1C). Cells that expressed Nav1.8 currents with amplitudes greater than 15% of the recombinant current amplitude were excluded from the final data analysis. In the 150 cells expressing TTX-resistant recombinant VGSCs that were used in the final analysis, the peak recombinant current amplitude averaged 36.2 ± 2.1 nA, and the peak residual Nav1.8 current amplitude averaged 0.3 nA. It is important to note that resurgent sodium currents were not observed in a control group of neurons transfected with Nav1.8 shRNA and treated with TTX that did not express recombinant TTX-resistant VGSC current (n = 11; Supplemental Figure 1E). Furthermore, in another control group of cultured rat DRG neurons treated with TTX that produced large endogenous Nav1.8 currents (average peak amplitude, 32 ± 8 nA; n = 11), resurgent currents were not observed (Supplemental Figure 1D). These data confirmed that (a) the use of TTX and the Nav1.8 shRNA allowed effective isolation of the current produced by recombinant VGSCs in transfected DRG neurons and (b) the resurgent currents recorded from DRG neurons transfected with Nav1.8 shRNA and TTX-resistant recombinant channels in the presence of 500 nM TTX were indeed generated by the recombinant channels.
Computational simulations
Simulations were performed to explore the impact that resurgent currents generated by VGSCs and disease mutants might have on AP firing. The basic approach was to use established models of DRG neuron and cardiac myocyte excitability that had been implemented in the NEURON simulation environment (17) and, with modifications only to the appropriate sodium channel formulation, simulate and evaluate the impact of the disease mutation and/or the resurgent current blocking factor.
DRG neuron simulation
The DRG neuron model used was developed previously (16) and included the following voltage-dependent currents: a delayed rectifier potassium current (IKDR), an A-type potassium current (IKA), and Nav1.8, slowly inactivating TTX-R current. The only changes made to the model were to the Nav1.7 voltage-dependent sodium current in the model. The sodium current changes were implemented in a Markov model based on the Hodgkin-Huxley formulation of Nav1.7 previously used (16). The Markov formulation is more amenable to implementation of both the I1461T effects and the resurgent blocking factor (18). Simulations were run with (a) 100% Nav1.7, (b) 100% Nav1.7 with resurgent current, (c) 50% Nav1.7 and 50% Nav1.7-I1461T, and (d) 50% Nav1.7 and 50% Nav1.7-I1461T, both with resurgent current. Mutant channels were simulated with 50% wild-type channels because the PEPD mutations displayed autosomal dominance.
Nav1.7.
The diagram for the Markov model used for the simulated voltage-gated sodium conductances is shown in Figure 3A. The model includes 3 closed states, a conducting open state, and 3 inactivated states. The Markov model used for the simulated voltage-gated sodium conductance with resurgent current included 1 additional state, the open-blocked state (Figure 3A, boxed region). The resurgent current factor was implemented as done for simulation of resurgent currents in cerebellar Purkinje neurons by Khaliq et al. (18), with slight modifications to the transition rate expressions (see Supplemental Table 2).
Nav1.7-I1461T.
Characterization of the functional impact of the I1461T mutation in HEK293 cells (13) and DRG neurons (present study) showed that this mutation destabilized inactivation, shifting the voltage dependence of inactivation in the depolarizing direction and slowing the rate of open-state inactivation. The measured values of channel availability and time constants of inactivation from these studies were used to reformulate expressions for the vertical transitions in Figure 3A (between the inactivated states and the closed and open states). The horizontal transitions were unchanged (Supplemental Table 2). Figure 3B compares the modeled Nav1.7 and Nav1.7-I1461T currents elicited with a step depolarization to +10 mV. The resurgent current was added to the Nav1.7-I1461T conductance model in the exact way that it was added to the wild-type Nav1.7 conductance model. Figure 3C compares the resurgent currents generated by Nav1.7and Nav1.7-I1461T simulated conductances.
Cardiac myocyte simulation
We modified mathematical models of cardiac AP firing (20, 52) to simulate the impact of the F1486L LQT3/SIDS mutation and resurgent currents. The cardiac myocyte model (see http://senselab.med.yale.edu/ModelDB/ShowModel.asp?model=3800) was previously implemented by I. Jacobson in the NEURON simulation environment (17). The only changes made to the model were to the voltage-dependent sodium current (Naf in the original model). Simulations were run with (a) 100% Nav1.5, (b) 100% Nav1.5 with resurgent current, (c) 50% Nav1.5 and 50% Nav1.5-F1486L, and (d) 50% Nav1.5 and 50% Nav1.5-F1486L, both with resurgent current. Mutant channels were simulated at with 50% wild-type channels because the LQT3/SIDS mutations displayed autosomal dominance.
Nav1.5.
We developed a Markov model based on the Hodgkin-Huxley formulation of Nav1.5 in the original model (20, 52). The Markov formulation was more amenable to implementation of both the F1486L effects and the resurgent blocking factor. Intermediate inactivation states were not included in this formulation, as it was unclear how to model potential interactions between the resurgent current blocking factor and intermediate inactivation states. This is reasonable, as the F1486L mutation may have little or no impact on intermediate inactivation (19). The diagram for the Markov model used for the simulated voltage-gated sodium conductances is shown in Figure 3A, and the transition rate expressions for Nav1.5 are provided in Supplemental Table 3.
Nav1.5 F1486L.
Our data on the F1486L mutation indicate that it produces moderate destabilization of inactivation, shifting the voltage dependence of inactivation by approximately 8 mV in the depolarizing direction, slowing the rate of inactivation, and slightly increasing persistent currents. We modeled these effects by altering the transition rates from the closed and open state into the inactivated states (see Supplemental Table 3). Simulated Nav1.5 and Nav1.5-F1486L currents are shown in Figure 5A.
Nav1.5 and Nav1.5 F1486L with resurgent current.
The resurgent current factor was modeled to be similar to that of Nav1.7, but with slight modifications to the overall expression to account for the relative resurgent current amplitude observed with Nav1.5 and Nav1.5-F1486L channels in our experiments (Supplemental Table 3). Simulated resurgent currents are shown in Figure 5B.
Statistics
All data are mean ± SEM. Comparison of frequency was determined using χ2 test. Statistical significance was assessed with Microsoft Excel using 2-tailed Student’s unpaired t tests. Statistical significance of difference was accepted at P values less than 0.05.
Supplementary Material
Acknowledgments
We thank E. Thompson and M. Vasko for assistance with the Nav1.8 shRNA. B.W. Jarecki was supported by an Indiana Clinical and Translational Sciences Institute Career Development Award (PHS grant 5TL1RR025759), and A.D. Piekarz was partially supported by NIH training fellowship NS066663. This work was supported by NIH research grant NS053422 (to T.R. Cummins).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J. Clin. Invest. 120:369–378 (2010). doi:10.1172/JCI40801.
See the related Commentary beginning on page 80.
References
- 1.George AL Jr. Inherited disorders of voltage-gated sodium channels. J Clin Invest. 2005;115(8):1990–1999. doi: 10.1172/JCI25505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cannon SC. Pathomechanisms in channelopathies of skeletal muscle and brain. Annu Rev Neurosci. 2006;29:387–415. doi: 10.1146/annurev.neuro.29.051605.112815. [DOI] [PubMed] [Google Scholar]
- 3.Raman IM, Sprunger LK, Meisler MH, Bean BP. Altered subthreshold sodium currents and disrupted firing patterns in Purkinje neurons of Scn8a mutant mice. Neuron. 1997;19(4):881–891. doi: 10.1016/S0896-6273(00)80969-1. [DOI] [PubMed] [Google Scholar]
- 4.Cummins TR, Dib-Hajj SD, Herzog RI, Waxman SG. Nav1.6 channels generate resurgent sodium currents in spinal sensory neurons. FEBS Lett. 2005;579(10):2166–2170. doi: 10.1016/j.febslet.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 5.Chen Y, Yu FH, Sharp EM, Beacham D, Scheuer T, Catterall WA. Functional properties and differential neuromodulation of Na(v)1.6 channels. Mol Cell Neurosci. 2008;38(4):607–615. doi: 10.1016/j.mcn.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hodgkin AL, Huxley AF. The dual effect of membrane potential on sodium conductance in the giant axon of Loligo. J Physiol. 1952;116(4):497–506. doi: 10.1113/jphysiol.1952.sp004719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Raman IM, Bean BP. Ionic currents underlying spontaneous action potentials in isolated cerebellar Purkinje neurons. J Neurosci. 1999;19(5):1663–1674. doi: 10.1523/JNEUROSCI.19-05-01663.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Raman IM, Bean BP. Inactivation and recovery of sodium currents in cerebellar Purkinje neurons: evidence for two mechanisms. Biophys J. 2001;80(2):729–737. doi: 10.1016/S0006-3495(01)76052-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grieco TM, Raman IM. Production of resurgent current in NaV1.6-null Purkinje neurons by slowing sodium channel inactivation with beta-pompilidotoxin. J Neurosci. 2004;24(1):35–42. doi: 10.1523/JNEUROSCI.3807-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rush AM, Cummins TR, Waxman SG. Multiple sodium channels and their roles in electrogenesis within dorsal root ganglion neurons. J Physiol. 2007;579(Pt 1):1–14. doi: 10.1113/jphysiol.2006.121483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fertleman CR, et al. SCN9A mutations in paroxysmal extreme pain disorder: allelic variants underlie distinct channel defects and phenotypes. Neuron. 2006;52(5):767–774. doi: 10.1016/j.neuron.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 12.Dib-Hajj SD, Cummins TR, Black JA, Waxman SG. From genes to pain: Na v 1.7 and human pain disorders. Trends Neurosci. 2007;30(11):555–563. doi: 10.1016/j.tins.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 13.Jarecki BW, Sheets PL, Jackson JO, II, Cummins TR. Paroxysmal extreme pain disorder mutations within the D3/S4-S5 linker of Nav1.7 cause moderate destabilization of fast inactivation. J Physiol. 2008;586(Pt 17):4137–4153. doi: 10.1113/jphysiol.2008.154906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Armstrong CM, Bezanilla F, Rojas E. Destruction of sodium conductance inactivation in squid axons perfused with pronase. J Gen Physiol. 1973;62(4):375–391. doi: 10.1085/jgp.62.4.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.West JW, et al. A cluster of hydrophobic amino acid residues required for fast Na(+)-channel inactivation. Proc Natl Acad Sci U S A. 1992;89(22):10910–10914. doi: 10.1073/pnas.89.22.10910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sheets PL, Jackson JO, II, Waxman SG, Dib-Hajj SD, Cummins TR. A Nav1.7 channel mutation associated with hereditary erythromelalgia contributes to neuronal hyperexcitability and displays reduced lidocaine sensitivity. J Physiol. 2007;581(Pt 3):1019–1031. doi: 10.1113/jphysiol.2006.127027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hines ML, Carnevale NT. The NEURON simulation environment. Neural Comput. 1997;9(6):1179–1209. doi: 10.1162/neco.1997.9.6.1179. [DOI] [PubMed] [Google Scholar]
- 18.Khaliq ZM, Gouwens NW, Raman IM. The contribution of resurgent sodium current to high-frequency firing in Purkinje neurons: an experimental and modeling study. J Neurosci. 2003;23(12):4899–4912. doi: 10.1523/JNEUROSCI.23-12-04899.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang DW, et al. Cardiac sodium channel dysfunction in sudden infant death syndrome. Circulation. 2007;115(3):368–376. doi: 10.1161/CIRCULATIONAHA.106.646513. [DOI] [PubMed] [Google Scholar]
- 20.Courtemanche M, Ramirez RJ, Nattel S. Ionic mechanisms underlying human atrial action potential properties: insights from a mathematical model. Am J Physiol. 1998;275(1 Pt 2):H301–H321. doi: 10.1152/ajpheart.1998.275.1.H301. [DOI] [PubMed] [Google Scholar]
- 21.Featherstone DE, Fujimoto E, Ruben PC. A defect in skeletal muscle sodium channel deactivation exacerbates hyperexcitability in human paramyotonia congenita. J Physiol. 1998;506(Pt 3):627–638. doi: 10.1111/j.1469-7793.1998.627bv.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chahine M, et al. Sodium channel mutations in paramyotonia congenita uncouple inactivation from activation. Neuron. 1994;12(2):281–294. doi: 10.1016/0896-6273(94)90271-2. [DOI] [PubMed] [Google Scholar]
- 23.Oxford GS. Some kinetic and steady-state properties of sodium channels after removal of inactivation. J Gen Physiol. 1981;77(1):1–22. doi: 10.1085/jgp.77.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Neurotherapeutics. 2007;4(2):238–251. doi: 10.1016/j.nurt.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 25.Raman IM, Bean BP. Resurgent sodium current and action potential formation in dissociated cerebellar Purkinje neurons. J Neurosci. 1997;17(12):4517–4526. doi: 10.1523/JNEUROSCI.17-12-04517.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Do MT, Bean BP. Sodium currents in subthalamic nucleus neurons from Nav1.6-null mice. J Neurophysiol. 2004;92(2):726–733. doi: 10.1152/jn.00186.2004. [DOI] [PubMed] [Google Scholar]
- 27.Rush AM, Dib-Hajj SD, Waxman SG. Electrophysiological properties of two axonal sodium channels, Nav1.2 and Nav1.6, expressed in mouse spinal sensory neurones. J Physiol. 2005;564(Pt 3):803–815. doi: 10.1113/jphysiol.2005.083089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Holland KD, et al. Mutation of sodium channel SCN3A in a patient with cryptogenic pediatric partial epilepsy. Neurosci Lett. 2008;433(1):65–70. doi: 10.1016/j.neulet.2007.12.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lossin C, Wang DW, Rhodes TH, Vanoye CG, George AL., Jr Molecular basis of an inherited epilepsy. Neuron. 2002;34(6):877–884. doi: 10.1016/S0896-6273(02)00714-6. [DOI] [PubMed] [Google Scholar]
- 30.Grieco TM, Malhotra JD, Chen C, Isom LL, Raman IM. Open-channel block by the cytoplasmic tail of sodium channel beta4 as a mechanism for resurgent sodium current. Neuron. 2005;45(2):233–244. doi: 10.1016/j.neuron.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 31.Yu FH, et al. Sodium channel beta4, a new disulfide-linked auxiliary subunit with similarity to beta2. J Neurosci. 2003;23(20):7577–7585. doi: 10.1523/JNEUROSCI.23-20-07577.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grieco TM, Afshari FS, Raman IM. A role for phosphorylation in the maintenance of resurgent sodium current in cerebellar purkinje neurons. . J Neurosci. 2002;22(8):3100–3107. doi: 10.1523/JNEUROSCI.22-08-03100.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hammarstrom AK, Gage PW. Hypoxia and persistent sodium current. Eur Biophys J. 2002;31(5):323–330. doi: 10.1007/s00249-002-0218-2. [DOI] [PubMed] [Google Scholar]
- 34.Numann R, Catterall WA, Scheuer T. Functional modulation of brain sodium channels by protein kinase C phosphorylation. Science. 1991;254(5028):115–118. doi: 10.1126/science.1656525. [DOI] [PubMed] [Google Scholar]
- 35.Herzog RI, Liu C, Waxman SG, Cummins TR. Calmodulin binds to the C terminus of sodium channels Nav1.4 and Nav1.6 and differentially modulates their functional properties. J Neurosci. 2003;23(23):8261–8270. doi: 10.1523/JNEUROSCI.23-23-08261.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ma JY, Catterall WA, Scheuer T. Persistent sodium currents through brain sodium channels induced by G protein betagamma subunits. Neuron. 1997;19(2):443–452. doi: 10.1016/s0896-6273(00)80952-6. [DOI] [PubMed] [Google Scholar]
- 37.Kassmann M, et al. Oxidation of multiple methionine residues impairs rapid sodium channel inactivation. Pflugers Arch. 2008;456(6):1085–1095. doi: 10.1007/s00424-008-0477-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.George AL, Jr, Komisarof J, Kallen RG, Barchi RL. Primary structure of the adult human skeletal muscle voltage-dependent sodium channel. Ann Neurol. 1992;31(2):131–137. doi: 10.1002/ana.410310203. [DOI] [PubMed] [Google Scholar]
- 39.Gellens ME, et al. Primary structure and functional expression of the human cardiac tetrodotoxin-insensitive voltage-dependent sodium channel. Proc Natl Acad Sci U S A. 1992;89(2):554–558. doi: 10.1073/pnas.89.2.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smith MR, Smith RD, Plummer NW, Meisler MH, Goldin AL. Functional analysis of the mouse Scn8a sodium channel. J Neurosci. 1998;18(16):6093–6102. doi: 10.1523/JNEUROSCI.18-16-06093.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Klugbauer N, Lacinova L, Flockerzi V, Hofmann F. Structure and functional expression of a new member of the tetrodotoxin-sensitive voltage-activated sodium channel family from human neuroendocrine cells. EMBO J. 1995;14(6):1084–1090. doi: 10.1002/j.1460-2075.1995.tb07091.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Leffler A, Herzog RI, Dib-Hajj SD, Waxman SG, Cummins TR. Pharmacological properties of neuronal TTX-resistant sodium channels and the role of a critical serine pore residue. Pflugers Arch. 2005;451(3):454–463. doi: 10.1007/s00424-005-1463-x. [DOI] [PubMed] [Google Scholar]
- 43.Herzog RI, Cummins TR, Ghassemi F, Dib-Hajj SD, Waxman SG. Distinct repriming and closed-state inactivation kinetics of Nav1.6 and Nav1.7 sodium channels in mouse spinal sensory neurons. J Physiol. 2003;551(Pt 3):741–750. doi: 10.1113/jphysiol.2003.047357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Caffrey JM, Eng DL, Black JA, Waxman SG, Kocsis JD. Three types of sodium channels in adult rat dorsal root ganglion neurons. Brain Res. 1992;592(1–2):283–297. doi: 10.1016/0006-8993(92)91687-a. [DOI] [PubMed] [Google Scholar]
- 45.Cummins TR, et al. Nav1.3 sodium channels: rapid repriming and slow closed-state inactivation display quantitative differences after expression in a mammalian cell line and in spinal sensory neurons. J Neurosci. 2001;21(16):5952–5961. doi: 10.1523/JNEUROSCI.21-16-05952.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dib-Hajj SD, et al. Transfection of rat or mouse neurons by biolistics or electroporation. Nat Protoc. 2009;4(8):1118–1126. doi: 10.1038/nprot.2009.90. [DOI] [PubMed] [Google Scholar]
- 47.Cummins TR, Rush AM, Estacion M, Dib-Hajj SD, Waxman SG. Voltage-clamp and current-clamp recordings from mammalian DRG neurons. Nat Protoc. 2009;4(8):1103–1112. doi: 10.1038/nprot.2009.91. [DOI] [PubMed] [Google Scholar]
- 48.Cummins TR, Dib-Hajj SD, Black JA, Akopian AN, Wood JN, Waxman SG. A novel persistent tetrodotoxin-resistant sodium current in SNS-null and wild-type small primary sensory neurons. J Neurosci. 1999;19(24):RC43. doi: 10.1523/JNEUROSCI.19-24-j0001.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dib-Hajj S, Black JA, Cummins TR, Waxman SG. NaN/Nav1.9: a sodium channel with unique properties. Trends Neurosci. 2002;25(5):253–259. doi: 10.1016/S0166-2236(02)02150-1. [DOI] [PubMed] [Google Scholar]
- 50.Fjell J, Cummins TR, Dib-Hajj SD, Fried K, Black JA, Waxman SG. Differential role of GDNF and NGF in the maintenance of two TTX-resistant sodium channels in adult DRG neurons. Brain Res Mol Brain Res. 1999;67(2):267–282. doi: 10.1016/S0169-328X(99)00070-4. [DOI] [PubMed] [Google Scholar]
- 51.Mikami M, Yang J. Short hairpin RNA-mediated selective knockdown of NaV1.8 tetrodotoxin-resistant voltage-gated sodium channel in dorsal root ganglion neurons. Anesthesiology. 2005;103(4):828–836. doi: 10.1097/00000542-200510000-00022. [DOI] [PubMed] [Google Scholar]
- 52.Luo CH, Rudy Y.A dynamic model of the cardiac ventricular action potential. I. Simulations of ionic currents and concentration changes. Circ Res. 19947461071–1096. . [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.