

Abstract
Brain abnormality in surviving premature infants is associated with an enormous amount of neurodevelopmental disability, manifested principally by cognitive, behavioral, attentional, and socialization deficits, most commonly with only relatively modest motor deficits. The most recognized contributing neuropathology is cerebral white matter injury. The thesis of this review is that acquired cerebellar abnormality is a relatively less recognized but likely important cause of neurodevelopmental disability in small premature infants. The cerebellar disease may be primarily destructive (eg, hemorrhage, infarction) or primarily underdevelopment. The latter appears to be especially common and relates to a particular vulnerability of the cerebellum of the small premature infant. Central to this vulnerability are the extraordinarily rapid and complex developmental events occurring in the cerebellum. The disturbance of development can be caused either by direct adverse effects on the cerebellum, especially the distinctive transient external granular layer, or by indirect remote trans-synaptic effects. This review describes the fascinating details of cerebellar development, with an emphasis on events in the premature period, the major types of cerebellar abnormality acquired during the premature period, their likely mechanisms of occurrence, and new insights into the relation of cerebellar disease in early life to subsequent cognitive/behavioral/attentional/socialization deficits.
Keywords: premature infants, cerebellum, injury, underdevelopment, external granule cells, clinical sequelae
Brain abnormality in surviving premature infants is manifested by an enormous amount of neurodevelopmental disability. This disability consists of significant cognitive, behavioral, attentional, and socialization deficits in approximately 25% to 50% and serious motor deficits (ie, cerebral palsy) in 5% to 10%.1–7 Notably, therefore, contrary to many writings, the principal subsequent neurological syndrome in the large majority of premature infants involves cognitive deficits without major motor deficits.
The overall magnitude of this problem of neurodevelopmental disability relates to the large number of affected infants. Thus, approximately 63 000 infants are born yearly in the United States with a birth weight <1500 g (very low birth weight).8 This group represents 1.5% of all live births, a proportion that has increased gradually over the past decade. With current survival rates at approximately 90%, the yearly number of new cases of significant cognitive disturbances is at least 10 000 to 20 000, and the number of infants with cerebral palsy approaches 5000. Of particular importance in this context is the increasing number of extremely low-birth-weight infants, including those born between 24 weeks’ and 28 weeks’ gestation, that is, at the interface of the second and third trimesters of gestation. Because of sharply increased survival rates of these infants (50%–70%) and rates of disability of over 50% in most series, their contribution to the overall burden of neurodevelopmental disturbance is increasing.4,9–14
The neuropathology of the brain abnormality in surviving premature infants involves both white matter and gray matter disease.7 For decades, the principal disturbance has been considered to involve cerebral white matter. Indeed, periventricular leukomalacia, a primarily nonhemorrhagic cerebral white matter lesion, occurs to varying degrees in about 50% of very low-birth-weight infants. The major hemorrhagic lesion, that is, germinal matrix-intraventricular hemorrhage, is quantitatively less frequent (approximately 20%), and the clinically most important variety of intraventricular hemorrhage, associated with periventricular hemorrhagic infarction, is still less frequent (approximately 5%). However, in the smallest infants (eg, <750 g birth weight), this severe type of intraventricular hemorrhage is more common, with incidence of 20% or more.15,16 Moreover, in these very immature infants, other lesions, for example, cerebellar hemorrhage, become prominent (see later). In recent years, a diverse spectrum of neuronal/axonal disturbances involving thalamus, basal ganglia, and cerebral cortex has been shown to be frequent in premature infants, usually though not exclusively in association with periventricular leukomalacia.7 It is now apparent that an additional and clinically important component of this neuronal/axonal constellation is the involvement of the cerebellum and the cerebellar relay nuclei in brain stem (ie, base of the pons and inferior olivary nuclei).
Thus, the focus of this review is the cerebellar abnormalities of the premature infant, particularly the extremely low-birth-weight infant. The discussion will emphasize the cerebellar disorders characteristic of prematurity and acquired during the premature period and of course will not include dysgenetic, infectious, and other cerebellar lesions seen in infants of any gestational age. The principal themes of this review will be that the cerebellum (1) is an extraordinarily rapidly developing structure during the premature period, (2) because of this rapidity of development, it is vulnerable to the multiple insults to which the premature infant is exposed, and (3) because of this vulnerability, it sustains distinctive structural abnormalities likely to be of major clinical importance. In the following, first I review cerebellar development, with an emphasis on events occurring in the premature period, then the principal cerebellar abnormalities acquired during this maturational stage, and finally, the likely later clinical correlates.
Cerebellar Development
Overall Growth
From 24 weeks to 40 weeks of gestation, the cerebellum undergoes a rate of growth nearly unparalleled elsewhere in the brain. When assessed in vivo by 3-dimensional volumetric ultrasound, the cerebellar volume increases 5-fold from 24 weeks to 40 weeks postconception (Figure 1).17 When assessed in vivo by 3-dimensional volumetric magnetic resonance imaging (MRI), cerebellar volume increases approximately 3.5-fold from 28 weeks to 40 weeks postconception.18 Decades ago, the rapidity of cerebellar growth during this period of human brain development was described postmortem in terms of increase in weight and DNA content, by Dobbing and coworkers19–21 as well as by many others.22 The structure also shows exponential growth in foliation during this period (Figure 2), and as a consequence, the surface area of the cerebellar cortex increases more than 30-fold (from 448 mm2 to 15 200 mm2) from 24 weeks of gestation to term.22
Figure 1.
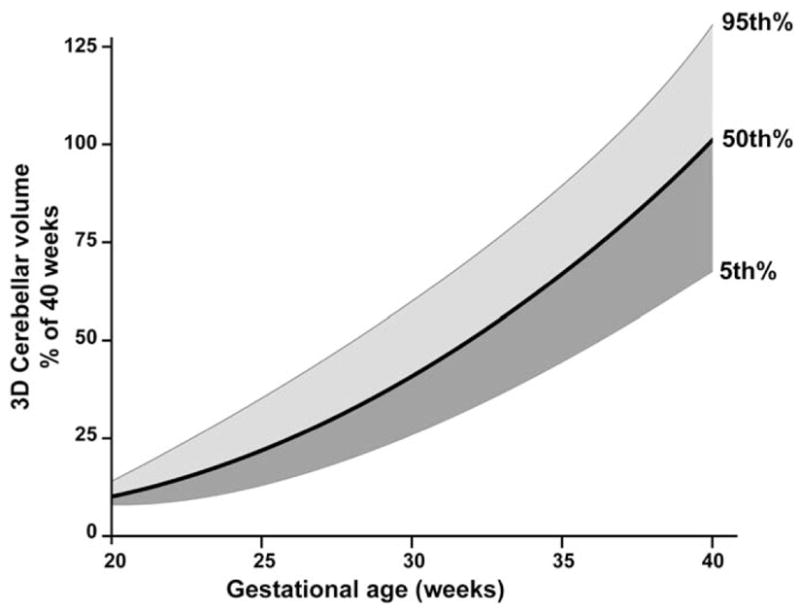
Cerebellar volume as a function of gestational age. The three curves are the 5th, 50th, and 95th percentile values of cerebellar volume obtained by 3-dimensional volumetric ultrasonography. Data obtained from 231 studies of fetuses from 20 to 40 weeks’ gestation and expressed as percentage of cerebellar volume at 40 weeks’ gestation. The 100% value is the 50th percentile value at 40 weeks’ gestation (absolute value, 17.6 mL). Note the dramatic increase in cerebellar volume from 24 to 40 weeks’ gestation.
SOURCE: Adapted from Ultrasound Med Biol. 2000;26(6):981–988.17
Figure 2.
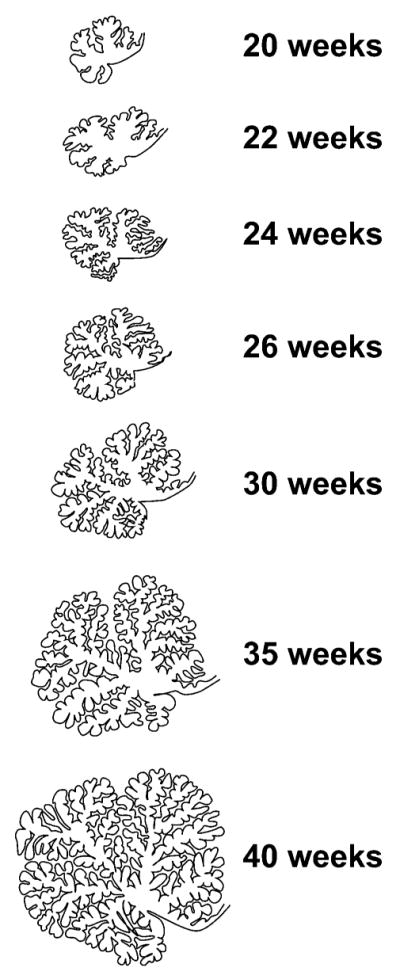
Growth of the cerebellar surface from 24 to 40 weeks. Drawings were made in the mid-sagittal plane. Note the extraordinary increase from 24 weeks to 40 weeks in cerebellar surface area, related primarily to increased foliation but also to increased overall cerebellar growth.
SOURCE: Adapted from J Comp Neurol. 1970;139(4):473–500.23
Histogenetic/Cytogenetic Events
The major histogenetic and cytogenetic manifestations of human cerebellar development provide substance to the gross measurements described above (Table 1). Prior to 20 weeks, the earliest events occur in the first month and involve the development of the cerebellar primordia and related pontine structures from the rostral hindbrain by the action of the isthmus organizer at the midbrain–hindbrain boundary.24,25 The major initial events in development of the cerebellum per se involve the establishment of 2 proliferative zones (Figure 3; Table 1).23–27 The first of these is the dorsomedial ventricular zone that gives rise, by radial migration, to interneurons of the deep (roof) nuclei and to the Purkinje cells. Neurons originating in the ventricular zone are γ-aminobutyric acid (GABA)ergic. The ventricular zone later gives rise to other GABAergic neurons, that is, Golgi cells of the internal granular layer and basket and stellate cells of the molecular layer. The second proliferative zone is the dorsolateral subventricular zone of the rhombic lip (Figure 3). Neurons originating in the rhombic lip are glutamatergic and those in the upper portion of the rhombic lip give rise to granule precursor cells and some neurons of the deep nuclei. The granule cell precursors initially migrate tangentially over the surface of the cerebellum to form the external granular layer (Figure 4). It is noteworthy in this regard that at their site of origin in the rhombic lip, the neuronal precursors have close access to the cerebellar surface because the pial and ventricular surfaces are nearly apposed (Figure 3). Later, the external granular layer cells migrate radially inward along Bergmann radial glial fibers to form the dense internal granular layer (see later). Additionally, rhombic lip precursors give rise to the projection (glutamatergic) neurons of the deep nuclei.28 These cells migrate initially tangentially beneath the pial surface before migrating to the deep nuclei (Figure 3). The precursors in the lower portion of the rhombic lip also migrate tangentially, here along the ventrolateral surface of the pons and rostral medulla to form the pontine nuclei (basis pontis) and inferior olivary nuclei, critical cerebellar relay nuclei. The latter 2 later provide crucial excitatory afferent input to the cerebellum, the former via mossy fibers to internal granule cells and the latter via climbing fibers to Purkinje cells (see later).
Table 1.
Major Features of Human Cerebellar Development
| <20 weeks |
| Two proliferative zones established: dorsomedial ventricular zone and dorsolateral subventricular zone of the rhombic lip |
| Ventricular zone gives rise, by radial migration, to the deep (eg, dentate) nuclei and Purkinje cells, which are γ-aminobutyric acid (GABA)ergic |
| Rhombic lip (rostral portion) gives rise, by tangential migration, to the external granular layer (and ultimately the internal granular layer by inward radial migration), neurons of which are glutamatergic |
| Rhombic lip (caudal portion) gives rise, by tangential migration, to the pontine and inferior olivary nuclei, which are glutamatergic |
| 20 to 30 weeks |
| External cortical surface of the cerebellum begins rapid expansion and foliation |
| External granular layer reaches peak thickness, approximately 6 to 9 cells thick and highly proliferative, and can be divided into inner and outer halves: proliferative neurons in outer half and neurons preparing to migrate in inner half |
| External granular layer neurons begin inward radial migration along radial glial (Bergmann glia) fibers to begin formation of the internal granule cell layer |
| Purkinje cell neurons are differentiating and secrete Sonic hedgehog; granule precursor cells of the external granular layer express molecules of Sonic hedgehog–signaling pathway; Sonic hedgehog strongly stimulates proliferation of granule precursor cells of the external granular layer |
| 30 to 40 weeks |
| External granular layer proliferation and inward migration to form the internal granular layer continues at a peak |
| External granular layer continues rapid expansion as thickness remains essentially constant, while cortical surface area increases markedly, requiring a remarkable increase in number of external granule cells |
| >40 weeks |
| External granular layer gradually dissipates throughout first postnatal year and internal granule cell layer becomes greatly compacted |
| Purkinje cells enlarge and differentiate further as major outflow to dentate nuclei |
| Molecular layer enlarges several-fold with Purkinje cell differentiation and elaboration of granule cell input |
Figure 3.
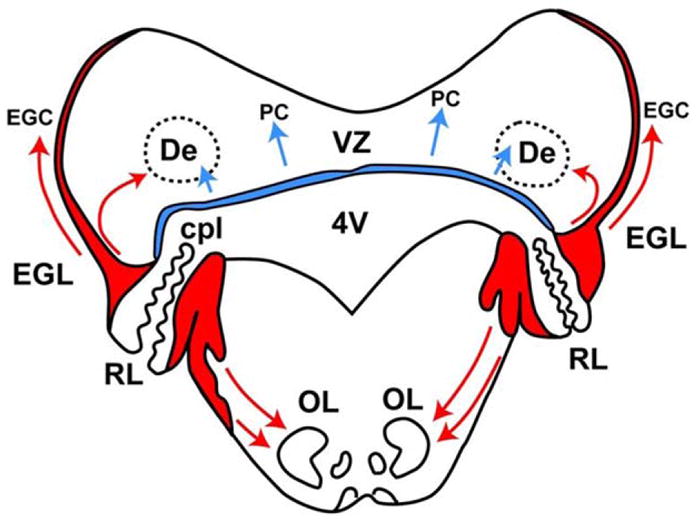
Two proliferative zones in cerebellar development. Transverse section of the rhombencephalon at approximately 14 weeks’ gestation. Note the dorsally placed ventricular zone (VZ, blue) and the dorsolaterally placed rhombic lip (RL, red). Arrows indicate directions of migration. The VZ gives rise to interneurons of the deep nuclei, for example dentate (De), and to the Purkinje cells. The RL has 2 portions divided by the choroid plexus (cpl) of the fourth ventricle (4V); the upper portion gives rise to granule precursor cells of the external granular layer (EGL) and to projection neurons of the deep nuclei, and the lower portion gives rise to neurons of the basis pontis and inferior olive (OL).
SOURCE: Adapted from Brain Res. 1973;62(1):1–35.26
Figure 4.

Major events in the histogenesis of the cerebellum in 4 major time periods from 9 weeks of gestation to 7 months postnatal (pn). The 2 zones of proliferation are the ventricular zone (VZ) and the external granule cell layer (EGL). Three directions of migration are indicated by arrows, that is radial from the VZ, tangential over the surface of the cerebellum to form the EGL, and later, inward to form the internal granular layer (IGL). Proliferation in the outer half of the EGL is under positive control by Sonic hedgehog (Shh) secreted by Purkinje cells (P-cells). Note the markedly active proliferation and migration of the granule precursor cells of the EGL during the premature period. Not shown is the marked increase in size of the molecular layer (ML) during the postnatal period. De, dentate; IZ, intermediate zone; pn, postnatal; WM, white matter.
SOURCE: Adapted from J Comp Neurol. 1970;139(4):473–50023; Brain Res. 1973;62(1):1–3526; and J Neurol. 2003;250(9):1025–1036.24
From 20 weeks to 30 weeks, cerebellar development accelerates. This acceleration is apparent grossly by the prominent increase in surface foliation (Figure 2).22,23,26 At the microscopic level, the external granular layer reaches its peak thickness by 25 weeks, approximately 6 to 9 cells thick and is highly proliferative. The latter conclusion is based in part on supravital autoradiography studies performed by Rakic and Sidman.23 Proliferation in the external granular layer occurs principally in the outer half, and thus these rapidly proliferating cells are directly exposed to the cerebrospinal fluid (Figure 4; see later).23,27 Proliferation of external granular layer neuronal precursors is under potent positive control by Sonic hedgehog, which is secreted by the differentiating Purkinje cells (Figure 4).27
From 30 weeks to 40 weeks, cerebellar growth and differentiation continues at a peak (Figure 4).19,20,22–25 Indeed, proliferation of granule precursor cells of the external granular layer (followed by inward migration to form the internal granular layer) is by far the dominant cellular determinant of cerebellar growth. During this period, because the external granular layer maintains its vertical thickness essentially constant but expands horizontally to accommodate the more than 30-fold increase in hemispheral surface area, the increase in number of cells must be extraordinary.
The importance of this proliferative phase for the ultimate growth of the cerebellum is perhaps best understood by considering that in the adult cerebellum, the total number of granule cells accounts for more than 95% of all neurons in the structure.29 Indeed, the ratio of Purkinje cells to granule cells is 1:3300 and of dentate nuclear cells to granule cells, 1:21 000. Moreover, the number of granule cells in the mature cerebellum, about 1011, exceeds the total number of neurons in the entire cerebral cortex by 4-fold and the total number of all neurons in the human body in aggregate. Thus, occurring on the surface of the cerebellum during the premature period, especially at the interface of the second and third trimesters of gestation, is a remarkably important series of events essential for the structural and functional integrity of the cerebellum. Not only must the granule precursor cells proliferate and migrate inward but on their way through the molecular layer to their final placement in the internal granular layer they make critical initial contacts to establish cerebellar circuitry. Thus, prior to the granule cell migration, Purkinje cell dendrites have begun elaboration in the molecular layer. During their migration through the molecular layer, guided by the Bergmann radial glial fibers, the granule cells extend horizontal parallel fibers that contact Purkinje cell dendrites. The deepest parallel fibers derive from the first granule cells to migrate and those closest to the surface from the last.26 Shortly after their arrival in the internal granular layer, these cells are contacted by mossy fiber input from the pons.
After 40 weeks’ gestation, cerebellar growth decelerates and the external granular layer gradually dissipates (Figure 4). However, this deceleration is not abrupt and a thinned external granular layer is apparent throughout the first postnatal year. During this period, neuronal differentiation throughout the cerebellar cortex is active, and axonal input (via the inferior cerebellar peduncle [climbing fibers] from dorsal spinocerebellar, vestibulocerebellar, and olivocerebellar tracts, and via the middle cerebellar peduncle [mossy fibers] from the pontine nuclei) and output (from the roof nuclei) increase. Thus, it is not surprising that in the first postnatal year as the external granular layer dissipates, the internal granule cell layer continues to increase in size, and with the associated elaboration of granule cell axons, that is, parallel fibers, and Purkinje cell dendrites, the molecular layer enlarges several-fold.22 Indeed, the events during the premature period are essential for the occurrence of this subsequent maturation.
Cerebellar Abnormalities of Prematurity
Cerebellar abnormalities acquired during the premature period and leading to structural cerebellar deficits on follow-up include disorders that appear to be primarily destructive or primarily impaired development (underdevelopment; Table 2). It is highly likely that there is overlap between these 2 categories but currently the distinction seems most rational. The overt destructive disorders are hemorrhagic or ischemic in basic nature. The disorders characterized by underdevelopment likely are much more common and relate primarily either to direct effects on cerebellum and relay nuclei or to remote trophic effects (see later).
Table 2.
Cerebellar Abnormalities of Prematurity
| Primarily destructive |
| Hemorrhage |
| Infarction |
| Primarily impaired development |
| Direct effects on cerebellum (hemosiderin-blood products; hypoxia-ischemia/infection-inflammation; glucocorticoids; undernutrition) |
| Remote effects on cerebellum (impaired trans-synaptic trophic effects) |
Destructive Cerebellar Lesions
Hemorrhage
Cerebellar hemorrhage is the best studied destructive cerebellar lesion of prematurity. Several reports used ultrasonographic imaging through the posterolateral fontanel (“mastoid window”) or MRI to show that cerebellar hemorrhage in premature infants is more common than previously revealed by ultrasonographic imaging through the anterior fontanel.30–33 Moreover, the incidence is strikingly dependent on the degree of prematurity. Thus, in the last 3 years of study (2000–2002) of the largest reported series, the incidence in infants <750 g birth weight was 17%, and in those >750 to 1499 g, only 2%.32
Cerebellar hemorrhage is usually unilateral, hemispheric in location (71%) and associated with ultrasonographically detectable supratentorial lesions, primarily intraventricular hemorrhage (77%). The hemorrhages are followed by cerebellar atrophy, detectable about 2 months later, in 37%; atrophy is focal in the unilateral lesions and more generalized in the minority of hemorrhages that are bilateral. Notably, reduction in contralateral cerebral volume has been defined and may reflect impaired remote trans-synaptic trophic effects (see later).
The likely origin and pathogenesis of cerebellar hemorrhage have been reviewed.7 The common focal unilateral hemispheric lesions may originate in the external granular layer, a germinal matrix, whereas the less common vermian hemorrhages may originate in the residual germinal matrix of the ventricular zone in the roof of the fourth ventricle. Etiology is likely multifactorial, as with supratentorial germinal matrix-intraventricular hemorrhage.7 Circulatory factors related to impaired cerebrovascular autoregulation, large patent ductus arteriosus, and other parameters of a compromised cerebral circulation seem important in pathogenesis, as in supratentorial germinal matrix-intraventricular hemorrhage.7,32
Infarction
Cerebellar infarction is a recognized complication of prematurity, especially extreme prematurity. Mercuri and colleagues initially described 9 cases of focal, unilateral cerebellar atrophy evident on MRI, among 73 premature infants, months to several years following birth; at birth, the affected infants weighed 600 to 1200 g.34 Although several infants were studied in the first postnatal year, no obvious evidence for previous hemorrhage was reported. The authors offered the reasonable proposal that the lesions represented infarction, and thus 10%to 15%of this relatively unselected premature population was affected.
A series of subsequent seminal reports by Johnsen, Bodensteiner, and colleagues described a high frequency of MRI evidence of cerebellar parenchymal loss in a selected population of former premature infants, that is, children with cerebral palsy whose birth weight was <1000 g and gestational age <28 weeks.35–38 In their most recent series of such children (n = 50), 64% had MRI evidence for cerebellar parenchymal loss. Of these, the loci involved (ie, bilateral inferior in approximately 60% and focal/unilateral in 10%) suggested vascular distributions, especially involving inferior cerebellar arteries.38 Although no clear evidence of ischemic events could be found, the scans were done several years after the neonatal period. The nearly invariable association of the cerebellar lesions with supratentorial white matter injury, that is, cystic periventricular leukomalacia and/or cerebral white matter volume loss, raised the possibility that the pathogenesis of the cerebellar and cerebral lesions is similar. The relation of cerebral white matter injury to ischemia and infection/inflammation has been reviewed elsewhere.7,39 Thus, it is possible that at least some of these cases were related to a generalized ischemic insult or insults rather than to a vascular occlusive event. Finally, the likelihood that infants with this combined cerebellar and cerebral lesion represent the severe end of the spectrum of cerebellar parenchymal destructive disease is suggested by the severity of the clinical features, that is, severe mixed spastic-dystonic-ataxic cerebral palsy (100%), microcephaly (63%), epilepsy (42%), and mental retardation (68%). The subsequent observation of a bilaterally small cerebellum could reflect an initial destructive ischemic event followed by cerebellar underdevelopment, as described in the next section.
The relation of the bilateral cerebellar parenchymal loss discussed in this context to the cerebellar underdevelopment discussed later is an important topic for future research. Most critically needed is a large-scale prospective analysis from the first days of life, with a focus on the cerebellum that is comparable in detail to the usual emphasis on supratentorial structures. Such an approach is required to elucidate evolution and mechanisms and allow distinction between the relative roles of destructive and dysmaturational influences.
Cerebellar Underdevelopment
Cerebellar underdevelopment, unrelated to overt destructive parenchymal disease from hemorrhage or infarction, may represent the most common type of cerebellar abnormality of the premature infant (Table 2). The considerable prevalence of this abnormality has been illustrated by numerous neuroimaging studies, principally MRI, carried out generally either weeks later at term equivalent age or months or years later.18,40–50 The cerebellar involvement has consisted most often of bilateral, generally symmetric deficits in cerebellar hemispheric volumes (Figure 5). The abnormality is most characteristic of infants within the gestational age range of 24 to 32 weeks, generally with the mean of 26 to 28 weeks. In the largest reported series (n = 75), decreased cerebellar volume at term equivalent age correlated with decreasing gestational age.18 A strong association of the cerebellar underdevelopment with supratentorial lesions, especially periventricular leukomalacia and intraventricular hemorrhage, and with MRI evidence for hemosiderin over the surface of the cerebellum is apparent (see later). These findings raise important possibilities concerning the mechanism of the cerebellar underdevelopment, as discussed later.
Figure 5.

Cerebellar underdevelopment as evidenced by magnetic resonance imaging at 7 months of age in an infant born at 29 gestational weeks. Sagittal image (A) shows markedly small vermis and pons below the inclined tentorium. Coronal image (B) shows small cerebellar hemispheres immediately below the tentorium.
SOURCE: Reproduced with permission from AJNR Am J Neuroradiol. 2005;26(7):1659–1667.44
The neuropathological basis of the decreased cerebellar volume remains to be elucidated. The largest autopsy study of modern-day premature infants (n = 41) involved infants with mean gestational age of approximately 32 weeks, that is, just beyond the period of greatest cerebellar vulnerability.51 Neuronal loss was noted in dentate nucleus, cerebellar cortex or the brain stem cerebellar relay nuclei, basis pontis, and inferior olive, in only 5% to 15%of infants.51 However, the neuronal loss was essentially confined to those infants with periventricular leukomalacia (n = 17), in whom the neuronal deficit occurred in 25% to 30% of infants. Similarly, gliosis in these structures was more common in the presence of periventricular leukomalacia and, in this group, the incidences were 43% in dentate, 29% in cerebellar cortex, 100% in basis pontis, and 92% in inferior olive. The pathological findings could be secondary to direct insults or a trophic effect or both (see below). Most importantly, specific studies of the external granular layer, quantitative study of cerebellar cortical layers, or more detailed mechanistic study (eg, caspase or other staining for apoptosis) were not carried out. More detailed neuropathological study of the cytological characteristics of the cerebellum of the premature infant is needed.
Mechanisms
Two broad mechanisms appear most plausible to explain the disturbance of cerebellar development after premature birth (Table 2). The first of these involves direct effects on the rapidly growing cerebellum, and the second, remote effects operating via altered trophic trans-synaptic interactions. Although these mechanisms are not mutually exclusive, and indeed may operate simultaneously, they will be discussed separately for clarity.
Direct effects
More than 3 decades ago, Dobbing and coworkers postulated that brain structures undergoing most rapid growth (ie, during a “growth spurt”) were most vulnerable to the negative effects of an insult.19,52 Cerebellum was considered particularly vulnerable in the developing animal because of its very rapid growth at that time, a period comparable to the perinatal human. The concept of a particular vulnerability of the cerebellum during its phase of rapid growth was documented in the late 1960s and early 1970s in experimental models of undernutrition, glucocorticoid exposure, and x-irradiation.19,20,52–54 Diminished DNA content, indicative of diminished cell number, was the principal outcome assessed. Glucocorticoid exposure and undernutrition could play an important contributory role in the human premature infant (see later).
Blood products, hemosiderin
The possibility that the cerebellar underdevelopment in premature infants may be related to adverse effects of blood products has been raised principally by the observations of Messerschmidt and coworkers, who have described the severe end of the spectrum of the acquired cerebellar growth failure.44,49,50 In their series of 35 infants (mean gestational age, 27 weeks; mean birth weight, 900 g), after an initially normal cerebellar ultrasonographic examination in the first week of life, subsequent ultrasound and then MRI scans identified a gradual deficit in volume, without any apparent injury pattern, over the ensuing weeks (Figure 6). The pons and medulla also were found to be small subsequently. Using MRI sequences optimal for detection of hemosiderin, they identified infratentorial hemosiderin deposition in 70% of infants.50 The deposition was particularly prominent on the cerebellar surface but was also noted on the surface of the brain stem and in the fourth ventricular region (Figure 6). Hemosiderin in the posterior fossa conveyed a sensitivity of 0.70 (95% confidence interval [CI], 0.48–0.86) and a specificity of 0.95 (CI, 0.84–0.99), with a positive predictive value of 0.88 and a negative predictive value of 0.87, for cerebellar underdevelopment.50 Nearly all infants had experienced intraventricular hemorrhage and 69% had posthemorrhagic hydrocephalus. In other reports, which included a less severe portion of the spectrum of cerebellar underdevelopment, intraventricular hemorrhage occurred in only a minority of infants and, as an isolated factor, was not clearly correlated with cerebellar volume.18,45,46 However, it is important to recognize that subarachnoid hemorrhage, in the absence of intraventricular hemorrhage, is common in premature infants.7 Indeed, in one older series of premature infants studied by computed tomography (CT) after the finding of bloody cerebrospinal fluid by lumbar puncture, 29% had subarachnoid hemorrhage without evidence for intraventricular hemorrhage7 (an additional 63% had intraventricular hemorrhage with associated subarachnoid blood). Thus, the presence of blood products in the cerebrospinal fluid of premature infants is not uncommon, even in the absence of overt intraventricular hemorrhage.
Figure 6.

Cerebellar underdevelopment and hemosiderin deposition by magnetic resonance imaging (MRI) in a newborn at term equivalent age. Sagittal T2-weighted MRI (A) from a 13-week-old infant born at 26 weeks’ gestation shows small vermis, enlarged fourth ventricle, reduced dimensions of the brain stem, and inclined tentorium; hemosiderin deposition is apparent on the surface of the pons and the lining of the fourth ventricle (black arrows). Horizontal MRI (B) shows reduced volume of cerebellar hemispheres with hemosiderin deposition in both hemispheres (black arrows).
SOURCE: Reproduced with permission from Eur J Paediatr Neurol. 2008;12(6):455–460.50
A relation between hemosiderin deposition and cerebellar atrophy is well documented in adults.55,56 With subarachnoid hemorrhage, hemosiderin deposits, as well as nonheme iron, are deposited particularly over the cerebellar surface, and infiltration of the cerebellar cortex is especially severe.56 A predilection for cerebellar cortex and brain stem may relate in part to cerebrospinal fluid flow patterns. In adult brain, the cellular structures disturbed are axons in the molecular layer and Purkinje cells. A related phenomenon probably accounts for the unexpected subsequent cerebellar atrophy described in children with traumatic brain injury.57 Similarly, older premature infants with posthemorrhagic hydrocephalus have been shown at autopsy to have subarachnoid blood products over the cerebellar surface, ferritin-positive glia in the molecular layer, and loss of Purkinje cells and inferior olivary neurons.58 Studies of small premature infants at the interface of the second and third trimesters of gestation are needed.
Nevertheless, taken together, the data raise the strong possibility that the key targets for the adverse effects of blood over the surface of the cerebellum of the small premature infant are the granule precursor cells of the external granular layer. The proliferating cells of the external granular layer are located directly at the interface with the subarachnoid space (see Figure 4). Impairment of the survival or proliferation, or both, of these cells could result in the cerebellar underdevelopment as evidenced by MRI. The effect on the external granular layer would result not only in deficient generation of internal granule cells but also in disturbance of the granular excitatory input to Purkinje cells and other cells of the molecular layer. The result would be deficient development of the full spectrum of cerebellar circuitry (Figure 7).
Figure 7.

Likely mechanisms by which direct adverse effects on the external granule cell layer lead to diminished volumetric development of cerebellum and pontine and olivary nuclei. See text for details.
The mechanisms of disturbance to the external granular layer in the context of hemosiderin deposition almost certainly would relate to the generation of free radicals, especially reactive oxygen species. Hemosiderin is derived from blood by the following sequential steps: hemolysis of red blood cells, formation of heme, conversion of heme to free iron (and biliverdin, carbon monoxide) by heme oxygenase, and formation of ferritin and then hemosiderin.59 Free iron is toxic because it leads to the generation of reactive oxygen species, especially the hydroxyl radical by the Fenton reaction.7 In one adult study of brain with hemosiderin deposits, free iron was increased 2.5-fold in cerebellar cortex and 14.5-fold in medulla.56 In experimental models, intracortical injections of free iron lead to lipid peroxidation products and epileptogenic necrotic foci.60–62 Additionally, hemosiderin, although a storage form of iron, may also release iron from its protein matrix.56 The central nervous system has limited ability to discharge iron, and thus the accumulated iron can produce a chronic deleterious effect. Notably, studies of cerebrospinal fluid of infants with posthemorrhagic hydrocephalus show persistence of copious amounts of nonprotein-bound iron for weeks after intraventricular hemorrhage.63
Other mechanisms could contribute to the cerebellar disturbance with subarachnoid blood. Thus, neuropathological studies of cerebellar subarachnoid hemorrhage in preterm infants show a decrease in the glutamate transporters, excitatory amino acid transporter 4 (EAAT4) in Purkinje cell dendrites, and glutamate/aspartate transporter (GLAST) in Bergmann glia.64 The effects of these 2 transporter disturbances could lead to an increase in extracellular glutamate and excitotoxic Purkinje cell death. Additionally, subarachnoid blood in piglets has been shown to lead to enhanced pial vasoconstrictor responses and thus the potential for local ischemia at the cerebellar surface.65
Hypoxia-ischemia and infection/inflammation
A possible relation of the cerebellar underdevelopment to hypoxia-ischemia or infection/inflammation or both is suggested by the strong correlation in vivo of the cerebellar abnormality with MRI-demonstrated periventricular leukomalacia.18,41,43,45–47 As noted earlier, neuronal loss or gliosis or both in the cerebellum were noted at postmortem examination almost exclusively in premature infants with periventricular leukomalacia.51 These correlations raise the possibility that the cerebellar underdevelopment could be secondary to the action of one or more of the pathogenetic insults operative in periventricular leukomalacia, that is, hypoxia-ischemia and infection/inflammation.7 In this context, it is noteworthy that there is ample experimental evidence that either hypoxia-ischemia or infection/inflammation can lead to deleterious effects on the developing cerebellum.66–68
Regarding hypoxia-ischemia, in studies of fetal sheep early in gestation so that the cerebellum contains an external granular layer roughly comparable to that in the human infant at the interface of the second and third trimesters, hypoxemia (induced by restricting uteroplacental blood flow) resulted in a 25% decline in mitotic cells in the external granular layer and a 3-fold increase in pyknotic (apoptotic) cells.68 Slightly later in development, Purkinje cell injury was identified66; this finding raises the possibility that the production and secretion of Sonic hedgehog by Purkinje cells, an event critical for proliferation in the external granular layer (see earlier), could be impaired. The result would be deficient proliferation in the external granular layer and a failure of development of the internal granule cell layer, with its critical connections for cerebellar circuitry (Figure 7).
The possibility that hypoxia-ischemia is operative in the human premature infant to lead to cerebellar abnormality is suggested indirectly in studies of premature infants by the correlation with diminished cerebellar volume of clinical factors consistent with hypoxia-ischemia. In 1 large study (n = 145), these factors included duration and intensity of mechanical ventilation and presence of patent ductus arteriosus.18 In another report (n = 35), the need for “early intubation” and “catecholamine support” was significantly associated with cerebellar underdevelopment.50 Additionally, direct exposure of proliferating cells in the outer half of the external granular layer to free radicals in cerebrospinal fluid could occur, in view of the observation by Inder and colleagues of elevated free radical products in the cerebrospinal fluid of very low-birth-weight infants with MRI evidence of periventricular leukomalacia.69
A strong relation of maternal intrauterine infection with systemic fetal inflammation or of postnatal neonatal infection with systemic inflammation and the occurrence of periventricular leukomalacia is well documented.7,70,71 Potential relevance to cerebellar underdevelopment is suggested by experimental studies of systemic inflammation in fetal sheep that show apoptosis of Purkinje cells and injury to their dendrites.66 These effects appeared to be mediated by attack by free radicals, perhaps principally generated by microglia, a mechanism operative in human cerebral white matter in periventricular leukomalacia.7 The experimental studies were carried out later in cerebellar development than occurs in the human very low-birth-weight infant, although some of the findings may be relevant. Currently, there are no direct data on the role of infection/inflammation in the genesis of cerebellar abnormality of the human premature infant.
Glucocorticoid exposure
Concerning glucocorticoid exposure, none of the aforementioned reports of premature infants with cerebellar underdevelopment has shown a clear relation of cerebellar volume to antenatal or postnatal glucocorticoid exposure. However, the prevalence of these exposures is so high, often 85% to 100%, that “control” comparison groups are difficult to define with sufficient statistical power for analysis. It is well known that postnatal dexamethasone administration to premature infants for prevention or treatment of bronchopulmonary dysplasia is associated with adverse neurologic outcome.72–76 Moreover, this therapy is associated with diminished cerebral cortical gray matter volume at term.77 Specific study of the cerebellum in this context would be of particular interest.
Indeed, a recent experimental study is especially noteworthy and potentially relevant to the human infant.78 Thus, in the developing mouse, studied at an age when the external granular layer is prominent (postnatal day 7), as in the human premature infant, a single injection of dexamethasone or corticosterone led to rapid (within 4 hrs) and selective, caspase-3-dependent apoptotic death of granule precursor cells in the external granular layer and permanent reductions in neuronal cell counts of their progeny, the internal granular layer neurons.78 This effect was lost after postnatal day 10 when the external granular layer dissipates in the mouse, as in the human infant (see earlier). Importantly, immunocytochemical studies showed that glucocorticoid receptor immunoreactivity is dramatically upregulated in the granule precursor cells at this time (Figure 8). Activation of the receptor activates a death pathway regulated by the proapoptotic molecule, Bcl-2 family member Puma.78 The experimental data define a previously unknown window of vulnerability when a single glucocorticoid exposure at clinically relevant doses leads to granule precursor cell apoptosis and a permanent cerebellar deficit.78 The findings raise the possibility that glucocorticoid administration could play a role in cerebellar underdevelopment in the human. Of additional importance in this context are the findings (1) that in premature infants, basal and peak serum cortisol responses in the first 2 weeks of life are highly variable, (2) that high serum cortisol levels documented in certain premature infants probably represent “continuing stress” from a variety of respiratory and related disorders, and (3) that such high levels are associated with neonatal morbidity (chronic lung disease, retinopathy of prematurity).79 Taken together, the data suggest that the cerebellum of the sick small premature infant may be exposed to high glucocorticoid levels from a variety of sources, endogenous and exogenous, and that these compounds may play a critical additive and/or central role in impaired proliferation of the external granular layer and thereby cerebellar underdevelopment.
Figure 8.
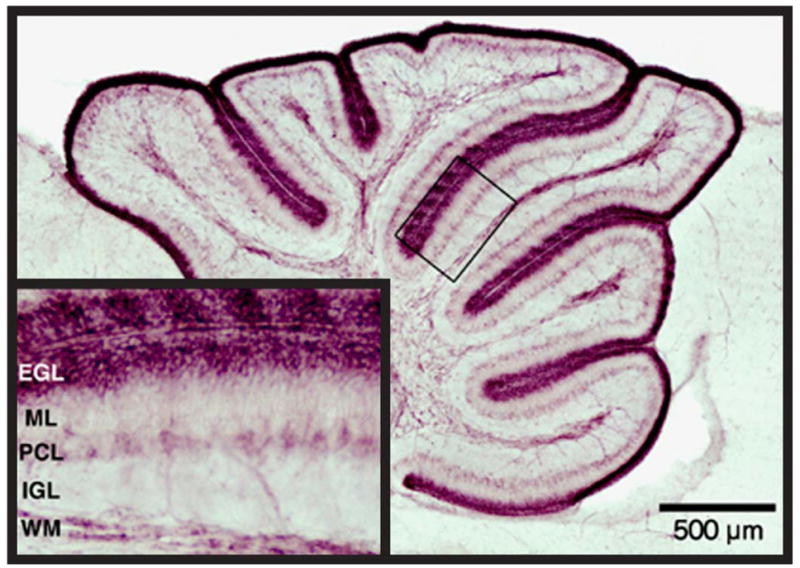
Glucocorticoid (GC) receptor immunoreactivity in the external granular layer (EGL) of the developing cerebellum. Glucocorticoid receptor immunohistochemistry in the postnatal day 7 mouse pup shows specific, dramatically elevated levels of immunoreactivity in the EGL. At this specific time point, a single GC injection in vivo leads to marked degeneration. IGL, internal granular layer; ML, molecular layer; PCL, Purkinje cell layer; WM, white matter.
SOURCE: Reproduced with permission from Cell Death Differ. 2008;15(10):1582–1592.78
Undernutrition
Concerning undernutrition, both earlier and recent reports indicate a strong need to address a potential relation of cerebellar underdevelopment in premature infants to undernutrition. As noted earlier, abundant experimental data show that during its phase of rapid growth, the cerebellum is especially vulnerable to undernutrition.19,52 Current information indicates a clear relationship between undernutrition and disturbed structural and functional human brain development (see below). Earlier reports demonstrated that early postnatal undernutrition in term infants may lead to subsequent cognitive deficits and disturbances of total brain growth.80–94 A seminal later study by Lucas and coworkers showed that just a 4-week exposure to a “standard-“ or “high-nutrient” diet in preterm infants (≤30 weeks’ gestational age) was followed by more rapid somatic and brain growth, subsequent cognitive performance, and caudate volumes.95–97 In another prospective study, Hayakawa and colleagues showed in 21 infants of 24 to 27 weeks’ gestational age that those with early-established enteral feeding (<3 weeks) had at 15 postnatal weeks improved somatic growth, brain growth (head growth), and electroencephalography (EEG) maturation compared with infants with delayed establishment of enteral feeding (>3 weeks).98 Although detailed studies of the relationship of nutrition and cerebellar development are needed, notably Limperopoulos and coworkers in their study of 169 preterm infants did show that cerebellar volume correlated significantly both with weight percentile and head circumference percentile at term equivalent age.18 Thus, this important initial observation is consistent with the notion that postnatal undernutrition could play a role in the cerebellar underdevelopment of prematurity.
Further supportive of a deleterious effect of undernutrition on brain development in the premature infant is recent work concerning structural and functional outcome in small-for-gestational age very low-birth-weight infants. Small-for-gestational age premature infants are of considerable interest in this context because intrauterine undernutrition likely occurs particularly during phases of rapid brain development, including especially cerebellar development, in the last one half of gestation. Outcome studies have documented impaired cognitive development in childhood among small-for-gestational age premature infants,99–102 and volumetric MRI research has shown diminished overall brain volume, as well as decreased cerebral cortical gray matter and hippocampal volumes in similar infants.103,104 Cerebellum was not studied separately in the MRI studies. A potential relation of intrauterine undernutrition to the adverse effects of excessive glucocorticoid, described above, seems possible because undernutrition during pregnancy has been shown to alter expression of the placental steroid dehydrogenase that metabolizes maternal cortisol, thereby exposing the fetus to excess cortisol.105–107 In this context, it is noteworthy that studies of placental insufficiency and intrauterine growth restriction in fetal sheep and guinea pigs showed restricted cerebellar growth and differentiation.108–113 In the models in which cerebellar histologic development appeared to be most closely comparable to the human cerebellum of approximately 24 weeks, a subsequent 25% reduction in overall cerebellar growth and a 22% reduction in volume of the granule cell layer were documented.109 The external granular layer was not specifically studied, but in view of the ultimate decrease in volume of the internal granule cell layer, an impairment of proliferation in the external granular layer is a likely possibility.
Remote effects
A second mechanism potentially involved in the cerebellar underdevelopment of the small premature infant involves remote trans-synaptic effects primarily involving neuronal connections between cerebrum and cerebellum (Figure 9). This mechanism would represent a maturation-distinctive form of diaschisis, a term initially used by Von Monakow114 and subsequently by others to characterize a loss of function in a brain area neurally connected to but remote from a separate lesioned brain area. The evidence supporting a role for diaschisis in cerebellar underdevelopment includes the strong relation of the abnormality with periventricular leukomalacia and other cerebral lesions in recent MRI volumetric studies in premature infants, as outlined in the next section.
Figure 9.
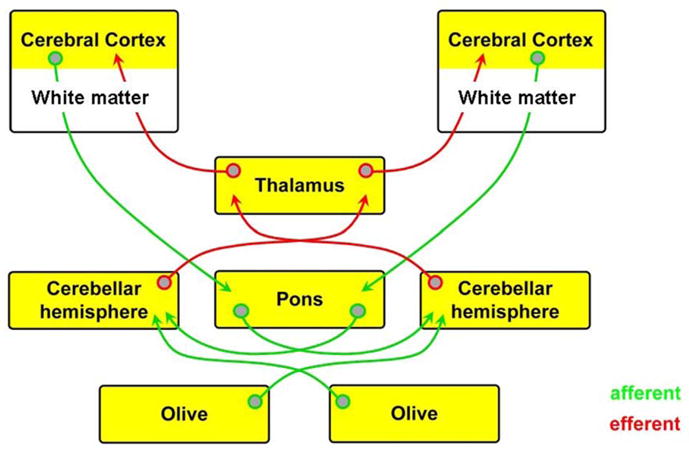
Major afferent and efferent connections between cerebrum and cerebellum. In premature infants, the most likely disturbances of afferent connections occur at the level of the cerebrum (cerebral white matter and cortex) and of efferent connections at the levels of the cerebellum and thalamus. See text for details.
Association with cerebral lesions
A strong relation of cerebellar underdevelopment with cerebral white matter lesions is supported by several large-scale studies using volumetric MRI.18,43,45,46,50 Although most often the MRI abnormalities have been consistent with diffuse noncystic periventricular leukomalacia, such other lesions as periventricular hemorrhagic infarction and posthemorrhagic hydrocephalus have been observed. In one study in which severity of white matter injury was evaluated relative to cerebellar volume deficit, a direct correlation was observed.46 Notably also concerning remote effects (see later), in 2 MRI studies that analyzed pontine size, pontine diameter as well as cerebellar volume was found to be reduced in premature infants with periventricular leukomalacia.41,47
Crossed cerebral and cerebellar effects
The association of cerebellar underdevelopment with supratentorial abnormalities may involve multiple remote trophic transneuronal interactions (Figure 9). The most detailed and systematic study of the structural relations of cerebrum and cerebellum in very low-birth-weight infants (n = 74) used 3-dimensional volumetric MRI (Table 3).43 Notable findings were first that infants with bilateral diffuse periventricular leukomalacia had bilateral symmetric decreases not only in cerebral volumes but also in cerebellar volumes, raising the possibility that the cerebral lesions had an adverse effect on cerebellum. This possibility is supported further by the finding that with unilateral periventricular hemorrhagic infarction, there was not only the expected prominent loss of ipsilateral cerebral volume but also a marked loss of contralateral cerebellar volume (Table 3). The finding of crossed cerebellar “atrophy” in children who had been born prematurely and had had unilateral cerebral lesions was made initially by qualitative imaging of 7 former premature infants (gestational age 25 to 28 weeks) by Rollins and colleagues.115 Additionally, however, in the study of Limperopoulos and colleagues,43 with bilateral cerebellar hemorrhage, there was not only a symmetric decrease in cerebellar volume but also a symmetric decrease in cerebral volume, suggesting that the cerebellar lesions had an adverse effect on the cerebrum (Table 3). Further supportive of this possibility, with unilateral cerebellar hemorrhage, in addition to the expected sharp decrease in ipsilateral cerebellar volume, there was a statistically significant decrease in contralateral cerebral volume.
Table 3.
Relationship Between Cerebellar and Cerebral Hemispheric Volumes Among Premature Infants at Term Equivalent Age
| Cerebellar Volumea |
Cerebral Volumea |
|||||
|---|---|---|---|---|---|---|
| Ipsilateral (%) | Contralateral (%) | P | Ipsilateral(%) | Contralateral (%) | P | |
| Normal MRI (n = 30) | 100 | 100 | NS | 100 | 100 | NS |
| Bilateral dPVL (n = 14) | 81 | 80 | NS | 84 | 85 | NS |
| Unilateral PHI (n = 14) | 83 | 66 | <.001 | 74 | 85 | <.001 |
| Bilateral CBH (n = 4) | 64 | 63 | NS | 89 | 90 | NS |
| Unilateral CBH (n = 6) | 46 | 83 | <.001 | 92 | 87 | <.01 |
SOURCE: Adapted from Pediatrics. 2005;116(4):844–850.43
Note: CBH, cerebellar hemorrhage; dPVL, diffuse noncystic periventricular leukomalacia; MRI, magnetic resonance imaging; NS, nonsignificant; PHI, periventricular hemorrhagic infarction.
Values are expressed as percentages, with values for infants with normal MRI assigned 100%. See original report for absolute values and standard deviations. Absolute values of 100% for cerebellum were 12.4 mL (ipsilateral) and 12.3 mL (contralateral) and for cerebrum 181.2 mL (ipsilateral) and 179.8 mL (contralateral). P values refer to ipsilateral-contralateral comparisons.
Maturation-distinctive diaschisis
As noted above, diaschisis is a term most commonly used in adult neurology to refer to a loss of function in a neurally connected region remote from a brain lesion. The remote disturbance of function has generally been a decrease in blood flow or metabolism or appearance of neurologic signs referable to the remote region.116–123 Most commonly, the primary lesion has been in cerebral cortex (eg, infarction), and the remote effect, in contralateral cerebellum, leading to the term crossed cerebellar diaschisis (or crossed cerebrocerebellar diaschisis). In the adult, a structural consequence is unusual. Thus, mild cerebellar atrophy has been documented several years after the primary lesion in only about 30% of cases.118 The crossed cerebellar diaschisis has been attributed to the loss of excitatory input from cerebrum via corticopontine tracts, which synapse on pontine nuclei, the fibers of which cross the pons and ascend in the contralateral cerebellar peduncle to the contralateral cerebellar hemisphere (Figure 9). Notably, crossed cerebellocerebral diaschisis has been shown in adults with cerebellar lesions; the contralateral cerebral abnormality has been manifested by a decrease in perfusion or development of relevant cerebral neurological signs.119–123 Cerebellocerebral diaschisis has been attributed to loss of feedback from the cerebellar hemisphere via the crossed dentato-rubro-thalamic-cortical pathway to the contralateral cerebral cortex (Figure 9).
The findings of Limperopoulos and colleagues (Table 3)43 are consistent with both crossed cerebrocerebellar diaschisis and crossed cerebellocerebral diaschisis. The structural volumetric growth deficits in small premature infants appear to be considerably greater and more frequent than the mild atrophy observed only in a minority of adults. A possible reason for a maturation-distinctive diaschisis in the small premature infant may relate to the phase of rapid cerebellar development; thus, careful studies of the developing rat (14-day old) show that unilateral forebrain injury is followed after hours by increased apoptosis in cerebellar granule cells.67 (The external granular layer could not be studied because at 14 postnatal days in the rat, the layer has disappeared.) Moreover, the fact that apoptosis is a much more active process in developing than in adult neurons suggests that the apoptotic stimulus could have more profound effects on the rapidly developing cerebellum of the small premature infant than on the adult cerebellum.
Potential mechanisms underlying the cerebrocerebellar and cerebellocerebral diaschisis in premature infants are apparent in Figure 9. In the crossed cerebrocerebellar deficits, the loss of corticopontine input could relate to cerebral white matter disease or associated cerebral cortical abnormality or both. Loss of excitatory pontocerebellar input to the contralateral cerebellar hemisphere would follow, although a role for possible primary injury to pontine nuclei should also be considered (see earlier regarding pontine pathology in premature infants at autopsy). In the crossed cerebellocerebral deficits, the loss of cortical input could relate to cerebellar disease and disturbance of the dentato-rubro-thalamic-cortical pathway (Figure 9). Additionally, the high frequency of diffuse thalamic disease in the premature infant, as recently described,51,124 could further interrupt the reverberating cerebral cortical input from cerebellum at the level of the thalamus (Figure 9).
Conclusions
To conclude, cerebellar underdevelopment in the small premature infant exists on a continuum of severity from a mild volumetric deficit to an extraordinary cerebellar deficiency. The origin of the disorder appears to be primarily at the interface of the second and third trimesters of gestation and to be acquired postnatally. The most vulnerable site appears to be the external granular layer, located on the surface of the cerebellum, although other elements of cerebellar cortex may be affected. The 2 most likely operative pathogenetic mechanisms involve either direct effects on the cerebellum, especially the external granular layer, or remote trans-synaptic trophic effects. Concerning the direct effects, leading negative effectors are hemosiderin deposition, as a marker of iron accumulation and free radical attack, hypoxia-ischemia or infection/inflammation or both, glucocorticoids, and undernutrition. The first of these effectors may be the most important, but each may be operative, with the degree depending on the gestational age of the infant, postnatal illness, drug exposures, and so on. Importantly, 2 or more of the effectors may be synergistic or at least act in concert. Concerning remote trans-synaptic effects, it appears that cerebellar growth and development can be impaired by deficits in excitatory input from cerebrum and also, importantly, by failure of cerebellar output to cerebrum. An intact reverberating circuit between cerebrum and cerebellum and vice versa may be especially critical for normal growth during this critical phase of development. Again, the remote trans-synaptic effects may operate in concert with the direct effects in an additive or synergistic manner. Both the direct and remote effects may operate postnatally over a prolonged period, possibly sufficiently prolonged for deleterious effects to be interrupted by appropriate interventions and prevention thereby effected.
Clinicopathological Correlations
Clinicopathological correlations in cerebellar disease of the premature infant are especially difficult to make because of the frequent lack of detailed neurodevelopmental follow-up and the frequent confounding presence of associated lesions of supratentorial structures, especially periventricular leukomalacia, severe intraventricular hemorrhage, and periventricular hemorrhagic infarction, all of which can lead independently to subsequent neurological deficits. Additionally, many of the studies of cerebellar abnormality have not included MRI in the neonatal period or subsequently, and thus the presence of supratentorial lesions, especially diffuse noncystic periventricular leukomalacia, cannot be assessed. Nevertheless, some tentative conclusions seem warranted, and the initial data suggest that cerebellar abnormality acquired in the premature period is associated with clinically diverse and somewhat unexpected sequelae. In the following sections, clinicopathological correlations are discussed separately for the 2 categories of cerebellar abnormality (see earlier), that is, primarily destructive disease and primarily impaired development.
Primarily Destructive Disease
Cerebellar Hemorrhage
The most detailed study of the neurodevelopmental correlates of cerebellar hemorrhage involved 35 premature infants (mean gestational age, 26 weeks), who were compared with 35 age-matched premature infants without hemorrhage and 16 infants with cerebellar hemorrhage plus supratentorial parenchymal injury.125 The clinical correlates of isolated cerebellar hemorrhage were serious. Thus, 66% exhibited subsequent neurological abnormalities, including significant neuromotor disabilities, some expected as a consequence of cerebellar affection, that is, hypotonia, gait abnormalities, and extraocular disturbances. Other deficits broadened the scope of apparent cerebellar-related sequelae, that is, impaired receptive (37%) and expressive (42%) language, cognitive deficits (40%), socialization-behavioral deficits (34%), and abnormal results on autism screener measures (37%). These deficits were uncommon or absent in the infants without cerebellar hemorrhage and could not be clearly accounted for by associated cerebral injury. Deficits were distinctly worse in those with bilateral or unilateral hemispheral involvement, and notably infants with vermian involvement almost exclusively accounted for those with socialization difficulties and abnormal autism screening. Cerebellar lesions, particularly in the vermis, have been described in neuropathological and neuroimaging studies of children with autism.126–129 Overall, the observations implicate the cerebellum in a broad spectrum of cognitive-behavioral-socialization functions (see later).
Infarction
For clinicopathological correlations, cerebellar infarction in premature infants is best considered in terms of unilateral focal and bilateral lesions. Limited information is available on unilateral focal lesions that appear most clearly to be infarctions, but in general, focal ataxia and hypotonia have been observed.34 Accompanying supratentorial parenchymal injury has been associated with bilateral neurological signs. Clinicopathological correlations attributable specifically to the cerebellar disease in these generally severe cases are particularly difficult in bilateral cerebellar lesions, because of the frequent presence of major supratentorial disease (periventricular leukomalacia, periventricular hemorrhagic infarction, white matter “volume loss,” posthemorrhagic hydrocephalus). Thus, in the interesting series of Bodensteiner, Johnsen, and colleagues35–38 selected from children with cerebral palsy whose birth weight was less than 1000 g (mean, approximately 750 g) and gestational age less than 28 weeks (mean, approximately 26 weeks), 85%had supratentorial abnormalities. Approximately 65% later exhibited microcephaly, consistent with severe bilateral cerebral disease. The clinical outcome was poor—40% had epilepsy and 70% had mental retardation.38 The pattern of cerebral palsy was distinctive and consisted of a mixed motor deficit with spasticity, dystonia, and ataxia, unlike the predominantly “pure” spasticity in infants without cerebellar disease. Per the authors’ experience, this distinctive motor pattern and cerebellar disease accounted for 15% of the patients with cerebral palsy in a large regional rehabilitation facility from which the children were derived.38 The principal anatomic pattern of cerebellar injury was generally symmetric with preferential involvement of inferior cerebellar hemispheres. It is likely that the cerebellar involvement is crucial in determination of the mixed type of cerebral palsy, in view of its role in generation of appropriate patterns of limb movements and adjustment of locomotor output.130 However, the role of the cerebellum in the genesis of the other deficits, separate from the role of the supratentorial disease, is unclear.
Primarily Impaired Cerebellar Development
Those disorders with primarily impaired cerebellar development, that is, underdevelopment, occur on a spectrum of severity. The most severely affected infants, for example those described by Messerschmidt and coworkers,44,49,50 exhibited severe cerebellar underdevelopment by term equivalent age (with no intervening evidence of injury) and subsequently had severe neurological features, comparable to those described above by Bodensteiner, Johnsen, and colleagues (see above). Thus, all infants (n = 31) exhibited a distinctive mixed pattern of cerebral palsy (spastic-ataxic-dyskinetic), 94% had severe cognitive deficits, 90% were microcephalic, and 55% had epilepsy.49 Concerning accompanying cerebral injury, posthemorrhagic hydrocephalus occurred in approximately 70%, but noncystic periventricular leukomalacia was not included in their analysis.49,50 Thus, distinguishing the relative roles of cerebral white matter injury versus cerebellar abnormality vis-á-vis the neurological sequelae is difficult. It is likely that the cerebellar abnormality contributes importantly to the distinctive cerebral palsy pattern. The potential role of cerebellar deficit in the cognitive deficits could be considerable but is difficult to dissect conclusively in the absence of more detailed analysis of accompanying cerebral involvement.
The less severely affected infants with cerebellar underdevelopment, that is, the large population of infants described in the aforementioned MRI volumetric studies,18,40,45,46,48 may be those particularly likely to exhibit deficits related to cerebellar abnormality per se. Thus, these infants in general do not have as severe confounding cerebral disease as observed in those with the severe cerebellar underdevelopment described above. The cerebellar deficits may be “unmasked” as a result. Concerning the clinical correlates of the less severe cerebellar underdevelopment, the studies of human infants are still confounded to some extent by associated cerebral white matter disease, albeit less so than for those with severe cerebellar underdevelopment. In one report of 83 premature infants, cerebellar volume correlated with Bayley Psychomotor Developmental Index scores, but, as the authors acknowledge, the test is unlikely to be sensitive enough to detect cerebellar-related deficits in higher level cognitive function.46 In a study of premature infants at adolescence, there were significant associations between cerebellar volume and various cognitive scores, including deficits in executive and visual-spatial functions and language.40 As noted earlier, follow-up studies of infants with cerebellar hemorrhage without cerebral disease suggest that disturbances of language, cognition, socialization, and affect may occur.125 Comparable follow-up studies of the cerebellar underdevelopment discussed here are limited. Thus, more detailed data in premature infant populations studied by MRI, to assess cerebral as well as cerebellar disease, and by careful neuropsychological analysis, to evaluate the full cognitive spectrum, are needed.
The potential clinical correlates of cerebellar abnormality in premature infants can be derived from studies of children and adults with cerebellar disease (Table 4).131–137 The topographic aspects of the role of the cerebellum in these functions have been elucidated by neuroanatomical and functional neuroimaging studies that emphasize the reverberating connections of cerebellar outflow, via the dentato-rubro-thalamic circuit, to the prefrontal cortex (executive functions), posterior parietal cortex (spatial cognition), superior temporal cortex (language and complex auditory and visual processing), and cerebellar input from these cortical regions via the corticopontine tracts and pontocerebellar fibers (Figure 9). Recent careful neuropsychological study of children with cerebellar malformations support the correlation of cerebellar abnormality with cognitive and affective disturbances, such as those noted in Table 4.132 Of particular note, malformations of cerebellar hemispheres are especially associated with selective neuropsychological deficits involving mainly executive functions and visuospatial and linguistic abilities, whereas malformations affecting the cerebellar vermis are especially related to affective and social disorders and often an autistic syndrome.132 In some ways, these observations are reminiscent of the so-called cerebellar cognitive affective syndrome well described in adults with acquired cerebellar lesions.138 Relevance to the premature newborn is suggested by the aforementioned findings of Limperopoulos and colleagues.125 On follow-up of premature infants with cerebellar hemorrhage, it was found that those infants with socialization difficulties and positive autism screening were almost exclusively the infants with involvement of the vermis. Additionally noteworthy is that among the infants with cerebellar malformations, motor deficits are less severe than cognitive deficits and, importantly, improve progressively.132 This constellation is similar on follow-up of premature infants (see earlier).
Table 4.
Potential Clinical Correlates of Cerebellar Abnormality in Premature Infantsa
| Motor disturbances |
| Spectrum from incoordination to overt ataxia (“mixed cerebral palsy”) |
| Deficits in motor planning and execution |
| Cognitive deficits |
| Involving visual-spatial abilities, verbal fluency, reading, memory, and learning |
| Attentional deficits |
| Involving regulation of shifts in attention |
| Social/affective disturbances |
| Socialization deficits |
| Mood abnormalities |
| Autistic behavior |
See text for references.
Clearly, the stage is now set for detailed follow-up studies of small premature infants from the perspective of cognitive/linguistic/affective/socialization functions, as a function of cerebellar volume and other structural manifestations of underdevelopment. There are sufficient data to hypothesize that cerebellar abnormality, especially acquired underdevelopment, is an important determinant of higher level cognitive outcomes in surviving small premature infants.
Acknowledgments
The study was supported by grants from the National Institutes of Health (5R13NS040925-09), the Cerebral Palsy International Research Foundation, the Kennedy Krieger Institute, and the Child Neurology Society.
Footnotes
The online version of this article can be found at: http://jcn.sagepub.com/cgi/content/abstract/24/9/1085
The authors have no conflicts of interest to disclose with regard to this article.
The study was presented at the Neurobiology of Disease in Children Conference: Symposium on Injury to the Preterm Brain and Cerebral Palsy, in conjunction with the 37th Annual Meeting of the Child Neurology Society, Santa Clara, California, November 5, 2008.
For reprints and permissions queries, please visit SAGE’s Web site at http://www.sagepub.com/journalsPermissions.nav
References
- 1.Woodward LJ, Edgin JO, Thompson D, Inder TE. Object working memory deficits predicted by early brain injury and development in the preterm infant. Brain. 2005;128(pt 11):2578–2587. doi: 10.1093/brain/awh618. [DOI] [PubMed] [Google Scholar]
- 2.Bayless S, Stevenson J. Executive functions in school-age children born very prematurely. Early Hum Dev. 2007;83(4):247–254. doi: 10.1016/j.earlhumdev.2006.05.021. [DOI] [PubMed] [Google Scholar]
- 3.Platt MJ, Cans C, Johnson A, et al. Trends in cerebral palsy among infants of very low birthweight (<1500 g) or born prematurely (<32 weeks) in 16 European centres: a database study. Lancet. 2007;369(9555):43–50. doi: 10.1016/S0140-6736(07)60030-0. [DOI] [PubMed] [Google Scholar]
- 4.Larroque B, Ancel PY, Marret S, et al. Neurodevelopmental disabilities and special care of 5-year-old children born before 33 weeks of gestation (the EPIPAGE study): a longitudinal cohort study. Lancet. 2008;371(9615):813–820. doi: 10.1016/S0140-6736(08)60380-3. [DOI] [PubMed] [Google Scholar]
- 5.Kobaly K, Schluchter M, Minich N, et al. Outcomes of extremely low birth weight (<1 kg) and extremely low gestational age (<28 weeks) infants with bronchopulmonary dysplasia: effects of practice changes in 2000 to 2003. Pediatrics. 2008;121(1):73–81. doi: 10.1542/peds.2007-1444. [DOI] [PubMed] [Google Scholar]
- 6.Allin M, Walshe M, Fern A, et al. Cognitive maturation in preterm and term born adolescents. J Neurol Neurosurg Psychiatr. 2008;79(4):381–386. doi: 10.1136/jnnp.2006.110858. [DOI] [PubMed] [Google Scholar]
- 7.Volpe JJ. Neurology of the Newborn. 5. Philadelphia, PA: Elsevier; 2008. [Google Scholar]
- 8.Martin JA, Kung HC, Mathews TJ, et al. Annual summary of vital statistics: 2006. Pediatrics. 2008;121(4):788–801. doi: 10.1542/peds.2007-3753. [DOI] [PubMed] [Google Scholar]
- 9.Marlow N, Wolke D, Bracewell MA, Samara M for the EPICure Study Group. Neurologic and developmental disability at six years of age after extremely preterm birth. N Engl J Med. 2005;352(1):9–19. doi: 10.1056/NEJMoa041367. [DOI] [PubMed] [Google Scholar]
- 10.Wood NS, Costeloe K, Gibson AT, et al. The EPICure study: associations and antecedents of neurological and developmental disability at 30 months of age following extremely preterm birth. Arch Dis Child. 2005;90(2):F134–F140. doi: 10.1136/adc.2004.052407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marlow N, Hennessy EM, Bracewell MA, Wolke D for the EPICure Study Group. Motor and executive function at 6 years of age after extremely preterm birth. Pediatrics. 2007;120(4):793–804. doi: 10.1542/peds.2007-0440. [DOI] [PubMed] [Google Scholar]
- 12.Wolke D, Samara M, Bracewell M, Marlow N for the EPICure Study Group. Specific language difficulties and school achievement in children born at 25 weeks of gestation or less. J Pediatr. 2008;152(2):256–262. doi: 10.1016/j.jpeds.2007.06.043. [DOI] [PubMed] [Google Scholar]
- 13.Tyson JE, Parikh NA, Langer J, Green C, Higgins RD for the National Institute of Child Health and Human Development Neonatal Research Network. Intensive care for extreme prematurity—moving beyond gestational age. N Engl J Med. 2008;358(16):1672–1681. doi: 10.1056/NEJMoa073059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Steinmacher J, Pohlandt F, Bode H, Sander S, Kron M, Franz AR. J Pediatr. 6. Vol. 152. 2008. Neurodevelopmental follow-up of very preterm infants after proactive treatment at a gestational age of > or = 23 weeks; pp. 771–776.pp. 776.e771–e772. [DOI] [PubMed] [Google Scholar]
- 15.Bassan H, Feldman HA, Limperopoulos C, et al. Periventricular hemorrhagic infarction: risk factors and neonatal outcome. Pediatr Neurol. 2006;35(2):85–92. doi: 10.1016/j.pediatrneurol.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 16.Qiu X, Lee SK, Tan K, Piedboeuf B, Canning R for the Canadian Neonatal Network. Comparison of singleton and multiple-birth outcomes of infants born at or before 32 weeks of gestation. Obstet Gynecol. 2008;111(2 pt 1):365–371. doi: 10.1097/AOG.0b013e318162688f. [DOI] [PubMed] [Google Scholar]
- 17.Chang CH, Chang FM, Yu CH, Ko HC, Chen HY. Assessment of fetal cerebellar volume using three-dimensional ultrasound. Ultrasound Med Biol. 2000;26(6):981–988. doi: 10.1016/s0301-5629(00)00225-8. [DOI] [PubMed] [Google Scholar]
- 18.Limperopoulos C, Soul JS, Gauvreau K, et al. Late gestation cerebellar growth is rapid and impeded by premature birth. Pediatrics. 2005;115(3):688–695. doi: 10.1542/peds.2004-1169. [DOI] [PubMed] [Google Scholar]
- 19.Dobbing J. The later growth of the brain and its vulnerability. Pediatrics. 1974;53(1):2–6. [PubMed] [Google Scholar]
- 20.Dobbing J, Sands J. Quantitative growth and development of human brain. Arch Dis Child. 1973;48(10):757–767. doi: 10.1136/adc.48.10.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Friede RL. Developmental Neuropathology. 2. New York: Springer-Verlag; 1989. [Google Scholar]
- 22.Lemire RJ, Loeser JD, Leech RW, Alvord EC., Jr . Normal and Abnormal Development of the Human Nervous System. Hagerstown: Harper & Row; 1975. [Google Scholar]
- 23.Rakic P, Sidman RL. Histogenesis of cortical layers in human cerebellum, particularly the lamina dissecans. J Comp Neurol. 1970;139(4):473–500. doi: 10.1002/cne.901390407. [DOI] [PubMed] [Google Scholar]
- 24.ten-Donkelaar HJ, Lammens M, Wesseling P, Thijssen HOM, Renier WO. Development and developmental disorders of the human cerebellum. J Neurol. 2003;250(9):1025–1036. doi: 10.1007/s00415-003-0199-9. [DOI] [PubMed] [Google Scholar]
- 25.ten-Donkelaar HJ, Lammens M, Hori A. Clinical Neuroembryology: Development and Developmental Disorders of the Human Central Nervous System. Berlin: Springer-Verlag; 2006. [Google Scholar]
- 26.Sidman RL, Rakic P. Neuronal migration, with special reference to developing human brain: a review. Brain Res. 1973;62(1):1–35. doi: 10.1016/0006-8993(73)90617-3. [DOI] [PubMed] [Google Scholar]
- 27.Carletti B, Rossi F. Neurogenesis in the cerebellum. Neuroscientist. 2008;14(1):91–100. doi: 10.1177/1073858407304629. [DOI] [PubMed] [Google Scholar]
- 28.Fink AJ, Englund C, Daza RA, et al. Development of the deep cerebellar nuclei: transcription factors and cell migration from the rhombic lip. J Neurosci. 2006;26(11):3066–3076. doi: 10.1523/JNEUROSCI.5203-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Andersen BB, Korbo L, Pakkenberg B. A quantitative study of the human cerebellum with unbiased stereological techniques. J Comp Neurol. 1992;326(4):549–560. doi: 10.1002/cne.903260405. [DOI] [PubMed] [Google Scholar]
- 30.Merrill JD, Piecuch RE, Fell SC, Barkovich AJ, Goldstein RB. A new pattern of cerebellar hemorrhages in preterm infants. Pediatrics. 1998;102(6):E62. doi: 10.1542/peds.102.6.e62. [DOI] [PubMed] [Google Scholar]
- 31.Miall LS, Cornette LG, Tanner SF, Arthur RJ, Levene MI. Posterior fossa abnormalities seen on magnetic resonance brain imaging in a cohort of newborn infants. J Perinatol. 2003;23(5):396–403. doi: 10.1038/sj.jp.7210941. [DOI] [PubMed] [Google Scholar]
- 32.Limperopoulos C, Benson CB, Bassan H, et al. Cerebellar hemorrhage in the preterm infant: ultrasonographic findings and risk factors. Pediatrics. 2005;116(3):717–724. doi: 10.1542/peds.2005-0556. [DOI] [PubMed] [Google Scholar]
- 33.Bednarek N, Akhavi A, Pietrement C, Mesmin F, Loron G, Morville P. Outcome of cerebellar injury in very low birth-weight infants: 6 case reports. J Child Neurol. 2008;23(8):906–911. doi: 10.1177/0883073808318063. [DOI] [PubMed] [Google Scholar]
- 34.Mercuri E, He J, Curati WL, Dubowitz LM, Cowan FM, Bydder GM. Cerebellar infarction and atrophy in infants and children with a history of premature birth. Pediatr Radiol. 1997;27(2):139–143. doi: 10.1007/s002470050085. [DOI] [PubMed] [Google Scholar]
- 35.Johnsen SD, Tarby TJ, Lewis KS, Bird R, Prenger E. Cerebellar infarction: an unrecognized complication of very low birthweight. J Child Neurol. 2002;17(5):320–324. doi: 10.1177/088307380201700502. [DOI] [PubMed] [Google Scholar]
- 36.Bodensteiner JB, Johnsen SD. Cerebellar injury in the extremely premature infant: newly recognized but relatively common outcome. J Child Neurol. 2005;20(2):139–142. doi: 10.1177/08830738050200021101. [DOI] [PubMed] [Google Scholar]
- 37.Johnsen SD, Bodensteiner JB, Lotze TE. Frequency and nature of cerebellar injury in the extremely premature survivor with cerebral palsy. J Child Neurol. 2005;20(1):60–64. doi: 10.1177/08830738050200011001. [DOI] [PubMed] [Google Scholar]
- 38.Bodensteiner JB, Johnsen SD. Magnetic resonance imaging (MRI) findings in children surviving extremely premature delivery and extremely low birthweight with cerebral palsy. J Child Neurol. 2006;21(9):743–747. doi: 10.1177/08830738060210091101. [DOI] [PubMed] [Google Scholar]
- 39.Khwaja O, Volpe JJ. Pathogenesis of cerebral white matter injury of prematurity. Arch Dis Child Fetal Neonatal Ed. 2008;93(2):F153–F161. doi: 10.1136/adc.2006.108837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Allin M, Matsumoto H, Santhouse AM, et al. Cognitive and motor function and the size of the cerebellum in adolescents born very pre-term. Brain. 2001;124(pt 2):60–66. doi: 10.1093/brain/124.1.60. [DOI] [PubMed] [Google Scholar]
- 41.Argyropoulou MI, Xydis V, Drougia A, et al. MRI measurements of the pons and cerebellum in children born preterm; associations with the severity of periventricular leukomalacia and perinatal risk factors. Neuroradiology. 2003;45(10):730–734. doi: 10.1007/s00234-003-1067-0. [DOI] [PubMed] [Google Scholar]
- 42.Peterson BS, Vohr B, Staib LH, et al. Regional brain volume abnormalities and long-term cognitive outcome in preterm infants. JAMA. 2000;284(15):1939–1947. doi: 10.1001/jama.284.15.1939. [DOI] [PubMed] [Google Scholar]
- 43.Limperopoulos C, Soul JS, Haidar H, et al. Impaired trophic interactions between the cerebellum and the cerebrum among preterm infants. Pediatrics. 2005;116(4):844–850. doi: 10.1542/peds.2004-2282. [DOI] [PubMed] [Google Scholar]
- 44.Messerschmidt A, Brugger PC, Boltshauser E, et al. Disruption of cerebellar development: potential complication of extreme prematurity. AJNR Am J Neuroradiol. 2005;26(7):1659–1667. [PMC free article] [PubMed] [Google Scholar]
- 45.Srinivasan L, Allsop J, Counsell SJ, Boardman JP, Edwards AD, Rutherford M. Smaller cerebellar volumes in very preterm infants at term-equivalent age are associated with the presence of supratentorial lesions. AJNR Am J Neuroradiol. 2006;27(3):573–579. [PMC free article] [PubMed] [Google Scholar]
- 46.Shah DK, Anderson PJ, Carlin JB, et al. Reduction in cerebellar volumes in preterm infants: relationship to white matter injury and neurodevelopment at two years of age. Pediatr Res. 2006;60(1):97–102. doi: 10.1203/01.pdr.0000220324.27597.f0. [DOI] [PubMed] [Google Scholar]
- 47.Yoshida S, Hayakawa K, Yamamoto A, Kanda T, Yamori Y. Pontine hypoplasia in children with periventricular leukomalacia. AJNR Am J Neuroradiol. 2008;29(3):425–430. doi: 10.3174/ajnr.A0873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Parker J, Mitchell AA, Kalpakidou A, et al. Cerebellar growth and behavioural and neuropsychological outcome in preterm adolescents. Brain. 2008;131(pt 5):1344–1351. doi: 10.1093/brain/awn062. [DOI] [PubMed] [Google Scholar]
- 49.Messerschmidt A, Fuiko R, Prayer D, et al. Disrupted cerebellar development in preterm infants is associated with impaired neurodevelopmental outcome. Eur J Pediatr. 2008;167(10):1141–1147. doi: 10.1007/s00431-007-0647-0. [DOI] [PubMed] [Google Scholar]
- 50.Messerschmidt A, Prayer D, Brugger PC, et al. Preterm birth and disruptive cerebellar development: assessment of perinatal risk factors. Eur J Paediatr Neurol. 2008;12(6):455–460. doi: 10.1016/j.ejpn.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 51.Pierson CR, Folkerth RD, Billards SS, et al. Gray matter injury associated with periventricular leukomalacia in the premature infant. Acta Neuropathol. 2007;114(6):619–631. doi: 10.1007/s00401-007-0295-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dobbing J, Hopewell JW, Lynch A, Sands J. Vulnerability of developing brain. I. Some lasting effects of x-irradiation. Exp Neurol. 1970;28(3):442–449. doi: 10.1016/0014-4886(70)90181-0. [DOI] [PubMed] [Google Scholar]
- 53.Chase HP, Lindsley WF, Jr, O’Brien D. Undernutrition and cerebellar development. Nature. 1969;221(5180):554–555. doi: 10.1038/221554a0. [DOI] [PubMed] [Google Scholar]
- 54.Howard E. Reductions in size and total DNA of cerebrum and cerebellum in adult mice after corticosterone treatment in infancy. Exp Neurol. 1968;22(2):191–208. doi: 10.1016/0014-4886(68)90051-4. [DOI] [PubMed] [Google Scholar]
- 55.Fearnley JM, Stevens JM, Rudge P. Superficial siderosis of the central nervous system. Brain. 1995;118(pt 4):1051–1066. doi: 10.1093/brain/118.4.1051. [DOI] [PubMed] [Google Scholar]
- 56.Koeppen AH, Michael SC, Li D, et al. The pathology of superficial siderosis of the central nervous system. Acta Neuropathol. 2008;116(4):371–382. doi: 10.1007/s00401-008-0421-z. [DOI] [PubMed] [Google Scholar]
- 57.Soto-Ares G, Vinchon M, Delmaire C, Abecidan E, Dhellemes P, Pruvo JP. Cerebellar atrophy after severe traumatic head injury in children. Childs Nerv Syst. 2001;17(4–5):263–269. doi: 10.1007/s003810000411. [DOI] [PubMed] [Google Scholar]
- 58.Fukumizu M, Takashima S, Becker LE. Neonatal posthemorrhagic hydrocephalus: neuropathologic and immunohistochemical studies. Pediatr Neurol. 1995;13(3):230–234. doi: 10.1016/0887-8994(95)00183-g. [DOI] [PubMed] [Google Scholar]
- 59.Thompson KJ, Shoham S, Connor JR. Iron and neurodegenerative disorders. Brain Res Bull. 2001;55(2):155–164. doi: 10.1016/s0361-9230(01)00510-x. [DOI] [PubMed] [Google Scholar]
- 60.Singh R, Pathak DN. Lipid peroxidation and glutathione peroxidase, glutathione reductase, superoxide dismutase, catalase, and glucose-6-phosphate dehydrogenase activities in FeC13-induced epileptogenic foci in the rat brain. Epilepsia. 1990;31(1):15–26. doi: 10.1111/j.1528-1157.1990.tb05354.x. [DOI] [PubMed] [Google Scholar]
- 61.Triggs WJ, Willmore LJ. In vivo lipid peroxidation in rat brain following intracortical Fe2+ injection. J Neurochem. 1984;42(4):976–980. doi: 10.1111/j.1471-4159.1984.tb12699.x. [DOI] [PubMed] [Google Scholar]
- 62.Willmore LJ, Hiramatsu M, Kochi H, Mori A. Formation of superoxide radicals after FeC13 injection into rat isocortex. Brain Res. 1983;277(2):393–396. doi: 10.1016/0006-8993(83)90954-x. [DOI] [PubMed] [Google Scholar]
- 63.Savman K, Nilsson UA, Blennow M, Kjellmer I, Whitelaw A. Non-protein-bound iron is elevated in cerebrospinal fluid from preterm infants with posthemorrhagic ventricular dilation. Pediatr Res. 2001;49(2):208–212. doi: 10.1203/00006450-200102000-00013. [DOI] [PubMed] [Google Scholar]
- 64.Inage YW, Itoh M, Wada K, Hoshika A, Takashima S. Glutamate transporters in neonatal cerebellar subarachnoid hemorrhage. Pediatr Neurol. 2000;23(1):42–48. doi: 10.1016/s0887-8994(00)00142-9. [DOI] [PubMed] [Google Scholar]
- 65.Yakubu MA, Leffler CW. 5-Hydroxtryptamine-induced vasoconstriction after cerebral hematoma in piglets. Pediatr Res. 1997;41(3):317–320. doi: 10.1203/00006450-199703000-00002. [DOI] [PubMed] [Google Scholar]
- 66.Hutton LC, Yan E, Yawno T, Castillo-Melendez M, Hirst JJ, Walker DW. Injury of the developing cerebellum: a brief review of the effects of endotoxin and asphyxial challenges in the late gestation sheep fetus. Cerebellum. 2007:1–10. doi: 10.1007/s12311-014-0602-3. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 67.Joashi UC, Greenwood K, Taylor DL, et al. Poly(ADP ribose) polymerase cleavage precedes neuronal death in the hippocampus and cerebellum following injury to the developing rat forebrain. Eur J Neurosci. 1999;11(1):91–100. doi: 10.1046/j.1460-9568.1999.00409.x. [DOI] [PubMed] [Google Scholar]
- 68.Rees S, Stringer M, Just Y, Hooper SB, Harding R. The vulnerability of the fetal sheep brain to hypoxemia at mid-gestation. Brain Res Dev Brain Res. 1997;103(2):103–118. doi: 10.1016/s0165-3806(97)81787-7. [DOI] [PubMed] [Google Scholar]
- 69.Inder TE, Mocatta T, Darlow B, Spencer C, Volpe JJ, Winterbourn C. Elevated free radical products in the cerebrospinal fluid of VLBW infants with cerebral white matter injury. Pediatr Res. 2002;52(2):213–218. doi: 10.1203/00006450-200208000-00013. [DOI] [PubMed] [Google Scholar]
- 70.Volpe JJ. Postnatal sepsis, necrotizing entercolitis, and the critical role of systemic inflammation in white matter injury in premature infants. J Pediatr. 2008;153(2):160–163. doi: 10.1016/j.jpeds.2008.04.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shah DK, Doyle LW, Anderson PJ, et al. Adverse neurodevelopment in preterm infants with postnatal sepsis or necrotizing enterocolitis is mediated by white matter abnormalities on magnetic resonance imaging at term. J Pediatr. 2008;153(2):170–175. 175.e1. doi: 10.1016/j.jpeds.2008.02.033. [DOI] [PubMed] [Google Scholar]
- 72.Volpe JJ. Encephalopathy of prematurity includes neuronal abnormalities. Pediatrics. 2005;116(1):221–225. doi: 10.1542/peds.2005-0191. [DOI] [PubMed] [Google Scholar]
- 73.Yeh TF, Lin YJ, Lin HC, et al. Outcomes at school age after postnatal dexamethasone therapy for lung disease of prematurity. N Engl J Med. 2004;350(13):1304–1313. doi: 10.1056/NEJMoa032089. [DOI] [PubMed] [Google Scholar]
- 74.Barrington KJ. The adverse neuro-developmental effects of postnatal steroids in the preterm infant: a systematic review of RCTs. BMC Pediatr. 2001;1:1. doi: 10.1186/1471-2431-1-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Short EJ, Klein NK, Lewis BA, et al. Cognitive and academic consequences of bronchopulmonary dysplasia and very low birth weight: 8-year-old outcomes. Pediatrics. 2003;112(5):e359. doi: 10.1542/peds.112.5.e359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Needelman H, Evans M, Roberts H, Sweney M, Bodensteiner JB. Effects of postnatal dexamethasone exposure on the developmental outcome of premature infants. J Child Neurol. 2008;23(4):421–424. doi: 10.1177/0883073807309232. [DOI] [PubMed] [Google Scholar]
- 77.Murphy BP, Inder TE, Huppi PS, et al. Impaired cerebral cortical gray matter growth after treatment with dexamethasone for neonatal chronic lung disease. Pediatrics. 2001;107(2):217–221. doi: 10.1542/peds.107.2.217. [DOI] [PubMed] [Google Scholar]
- 78.Noguchi KK, Walls KC, Wozniak DF, Olney JW, Roth KA, Farber NB. Acute neonatal glucocorticoid exposure produces selective and rapid cerebellar neural progenitor cell apoptotic death. Cell Death Differ. 2008;15(10):1582–1592. doi: 10.1038/cdd.2008.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ng PC. Is there a “normal” range of serum cortisol concentration for preterm infants? Pediatrics. 2008;122(4):873–875. doi: 10.1542/peds.2008-0516. [DOI] [PubMed] [Google Scholar]
- 80.Cordero ME, D’Acuna E, Benveniste S, Prado R, Nuñez JA, Colombo M. Dendritic development in neocortex of infants with early postnatal life undernutrition. Pediatr Neurol. 1993;9(6):457–464. doi: 10.1016/0887-8994(93)90025-8. [DOI] [PubMed] [Google Scholar]
- 81.Lanting CI, Fidler V, Huisman M, Touwen B, Boersma ER. Neurological differences between 9-year-old children fed breast-milk or formula-milk as babies. Lancet. 1994;344(8933):1319–1322. doi: 10.1016/s0140-6736(94)90692-0. [DOI] [PubMed] [Google Scholar]
- 82.Pollock JI. Long-term associations with infant feeding in a clinically advantages population of babies. Dev Med Child Neurol. 1994;36(5):429–440. doi: 10.1111/j.1469-8749.1994.tb11869.x. [DOI] [PubMed] [Google Scholar]
- 83.Donma MM, Donma O. The influence of feeding patterns on head circumference among Turkish infants during the first six months of life. Brain Dev. 1997;19(6):393–397. doi: 10.1016/s0387-7604(97)00041-7. [DOI] [PubMed] [Google Scholar]
- 84.Golding J, Rogers IS, Emmett PM. Association between breast feeding, child development and behavior. Early Hum Dev. 1997;49(suppl):S175–S184. doi: 10.1016/s0378-3782(97)00062-5. [DOI] [PubMed] [Google Scholar]
- 85.Gordon N. Nutrition and cognitive function. Brain Dev. 1997;19(3):165–170. doi: 10.1016/s0387-7604(96)00560-8. [DOI] [PubMed] [Google Scholar]
- 86.Richards M, Wardsworth M, Rahimi-Foroushani A, Hardy R, Kuh D, Paul A. Infant nutrition and cognitive development in the first offspring of a national UK birth cohort. Dev Med Child Neurol. 1998;40(3):163–167. [PubMed] [Google Scholar]
- 87.Gordon N. Some influences on cognition in early life: a short review of recent opinions. Eur J Paediatr Neurol. 1998;2(1):1–5. doi: 10.1016/1090-3798(98)01000-1. [DOI] [PubMed] [Google Scholar]
- 88.Lanting CI, Patandin S, Weisglas-Kuperus N, Touwen BC, Boersma ER. Breastfeeding and neurological outcome at 42 months. Acta Paediatrica. 1998;87(12):1224–1229. doi: 10.1080/080352598750030870. [DOI] [PubMed] [Google Scholar]
- 89.Chase HP, Martin HP. Undernutrition and child development. N Engl J Med. 1970;282(12):933–939. doi: 10.1056/NEJM197004232821701. [DOI] [PubMed] [Google Scholar]
- 90.Stoch MB, Smythe PM, Moodie AD, Bradshaw D. Psychosocial outcome and CT findings after gross undernourishment during infancy: a 20-year developmental study. Dev Med Child Neurol. 1982;24(4):419–436. doi: 10.1111/j.1469-8749.1982.tb13647.x. [DOI] [PubMed] [Google Scholar]
- 91.Galler JR, Ramsey F, Solimano G. The influence of early malnutrition on subsequent behavioral development III. Learning disabilities as a sequel to malnutrition. Pediatr Res. 1984;18(4):309–313. doi: 10.1203/00006450-198404000-00001. [DOI] [PubMed] [Google Scholar]
- 92.Galler JR, Ramsey FC, Morley DS, Archer E, Salt P. The long-term effects of early kwashiorkor compared with marasmus. IV. Performance on the national high school entrance examination. Pediatr Res. 1990;28(3):235–239. doi: 10.1203/00006450-199009000-00018. [DOI] [PubMed] [Google Scholar]
- 93.Dobbing J. Maternal nutrition in pregnancy and later achievement of offspring: a personal interpretation. Early Hum Dev. 1985;12(1):1–8. doi: 10.1016/0378-3782(85)90130-6. [DOI] [PubMed] [Google Scholar]
- 94.Dobbing J. Infant nutrition and later achievement. Am J Clin Nutr. 1985;41(2 suppl):477–484. doi: 10.1093/ajcn/41.2.477. [DOI] [PubMed] [Google Scholar]
- 95.Isaacs EB, Gadian DG, Sabatini S, et al. The effect of early human diet on caudate volumes and IQ. Pediatr Res. 2008;63(3):308–314. doi: 10.1203/PDR.0b013e318163a271. [DOI] [PubMed] [Google Scholar]
- 96.Morley R, Lucas A. Early diet and outcome in prematurely born children. Clin Nutr. 1993;12(suppl):S6–S11. [Google Scholar]
- 97.Lucas A, Morley R, Cole TJ. Randomised trial of early diet in preterm babies and later intelligence quotient. Br Med J. 1998;317(7171):1481–1487. doi: 10.1136/bmj.317.7171.1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hayakawa M, Okumura A, Hayakawa F, Kato Y, et al. Nutritional state and growth and functional maturation of the brain in extremely low birth weight infants. Pediatrics. 2003;111(5 pt 1):991–995. doi: 10.1542/peds.111.5.991. [DOI] [PubMed] [Google Scholar]
- 99.Kok JH, den Ouden AL, Verloove-Vanhorick SP, Brand R. Outcome of very preterm small for gestational age infants: the first nine years of life. Br J Obstet Gynaecol. 1998;105(2):162–168. doi: 10.1111/j.1471-0528.1998.tb10046.x. [DOI] [PubMed] [Google Scholar]
- 100.Scherjon S, Briët J, Oosting H, Kok J. The discrepancy between maturation of visual-evoked potentials and cognitive outcome at five years in very preterm infants with and without hemodynamic signs of fetal brain-sparing. Pediatrics. 2000;105(2):385–391. doi: 10.1542/peds.105.2.385. [DOI] [PubMed] [Google Scholar]
- 101.Ley D, Laurin J, Bjerre I, Marsal K. Abnormal fetal aortic velocity waveform and minor neurological dysfunction at 7 years of age. Ultrasound Obstet Gynecol. 1996;8(3):152–159. doi: 10.1046/j.1469-0705.1996.08030152.x. [DOI] [PubMed] [Google Scholar]
- 102.Schreuder AM, McDonnell M, Gaffney G, Johnson A, Hope PL. Outcome at school age following antenatal detection of absent or reversed end diastolic flow velocity in the umbilical artery. Arch Dis Child. 2002;86(2):F108–F114. doi: 10.1136/fn.86.2.F108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tolsa CB, Zimine S, Warfield SK, et al. Early alteration of structural and functional brain development in premature infants born with intrauterine growth restriction. Pediatr Res. 2004;56(1):132–138. doi: 10.1203/01.PDR.0000128983.54614.7E. [DOI] [PubMed] [Google Scholar]
- 104.Lodygensky GA, Seghier ML, Warfield SK, et al. Intrauterine growth restriction affects the preterm infant’s hippocampus. Pediatr Res. 2008;63(4):438–443. doi: 10.1203/PDR.0b013e318165c005. [DOI] [PubMed] [Google Scholar]
- 105.Uno H, Eisele S, Sakai A, et al. Neurotoxicity of glucocorticoids in the primate brain. Horm Behav. 1994;28(4):336–348. doi: 10.1006/hbeh.1994.1030. [DOI] [PubMed] [Google Scholar]
- 106.Seckl JR. Glucocorticoids, feto-placental 11 betahydroxysteroid dehydrogenase type 2, and the early life origins of adult disease. Steroids. 1997;62(1):89–94. doi: 10.1016/s0039-128x(96)00165-1. [DOI] [PubMed] [Google Scholar]
- 107.Goland RS, Jozak S, Warren WB, Conwell IM, Stark RI, Tropper PJ. Elevated levels of umbilical cord plasma corticotropin-releasing hormone in growth-retarded fetuses. J Clin Endocrinol Metab. 1993;77(5):1174–1179. doi: 10.1210/jcem.77.5.8077309. [DOI] [PubMed] [Google Scholar]
- 108.Mallard EC, Rees S, Stringer M, Cock MI, Harding R. Effects of chronic placental insufficiency on brain development in fetal sheep. Pediatr Res. 1998;43(2):262–270. doi: 10.1203/00006450-199802000-00018. [DOI] [PubMed] [Google Scholar]
- 109.Mallard C, Loeliger M, Copolov D, Rees S. Reduced number of neurons in the hippocampus and the cerebellum in the postnatal guinea-pig following intrauterine growth-restriction. Neuroscience. 2000;100(2):327–333. doi: 10.1016/s0306-4522(00)00271-2. [DOI] [PubMed] [Google Scholar]
- 110.Duncan JR, Cock ML, Loeliger M, Louey S, Harding R, Rees SM. Effects of exposure to chronic placental insufficiency on the postnatal brain and retina in sheep. J Neuropathol Exp Neurol. 2004;63(11):1131–1143. doi: 10.1093/jnen/63.11.1131. [DOI] [PubMed] [Google Scholar]
- 111.Rees S, Bocking AD, Harding R. Structure of the fetal sheep brain in experimental growth retardation. J Dev Physiol. 1988;10(3):211–225. [PubMed] [Google Scholar]
- 112.Bisignano M, Rees S. The effects of intrauterine growth retardation on synaptogenesis and mitochondrial formation in the cerebral and cerebellar cortices of fetal sheep. Int J Dev Neurosci. 1988;6(5):453–460. doi: 10.1016/0736-5748(88)90051-2. [DOI] [PubMed] [Google Scholar]
- 113.Rees S, Harding R. The effects of intrauterine growth retardation on the development of the Purkinje cell dendritic tree in the cerebellar cortex of fetal sheep: a note on the ontogeny of the Purkinje cell. Int J Dev Neurosci. 1988;6(5):461–469. doi: 10.1016/0736-5748(88)90052-4. [DOI] [PubMed] [Google Scholar]
- 114.Von Monakow C. Brain and behavior I: mood states and mind. In: Pibram KH, editor. Diaschisis. Baltimore: Penguin; 1969. pp. 27–36. [Google Scholar]
- 115.Rollins NK, Wen TS, Dominguez R. Crossed cerebellar atrophy in children: a neurologic sequela of extreme prematurity. Pediatr Radiol. 1995;25(suppl 1):S20–S25. [PubMed] [Google Scholar]
- 116.Hausen HS, Lachmann EA, Nagler W. Cerebral diaschisis following cerebellar hemorrhage. Arch Phys Med Rehabil. 1997;78(5):546–548. doi: 10.1016/s0003-9993(97)90175-1. [DOI] [PubMed] [Google Scholar]
- 117.Komaba Y, Mishina M, Utsumi K, Katayama Y, Kobayashi S, Mori O. Crossed cerebellar diaschisis in patients with cortical infarction: logistic regression analysis to control for confounding effects. Stroke. 2004;35(2):472–476. doi: 10.1161/01.STR.0000109771.56160.F5. [DOI] [PubMed] [Google Scholar]
- 118.Tien RD, Ashdown BC. Crossed cerebellar diaschisis and crossed cerebellar atrophy: correlation of MR findings, clinical symptoms, and supratentorial diseases in 26 patients. AJR Am J Roentgenol. 1992;158(5):1155–1159. doi: 10.2214/ajr.158.5.1566683. [DOI] [PubMed] [Google Scholar]
- 119.Broich K, Hartmann A, Biersack HJ, Horn R. Crossed cerebello-cerebral diaschisis in a patient with cerebellar infarction. Neurosci Lett. 1987;83(1–2):7–12. doi: 10.1016/0304-3940(87)90207-2. [DOI] [PubMed] [Google Scholar]
- 120.Rousseaux M, Steinling M. Crossed hemispheric diaschisis in unilateral cerebellar lesions. Stroke. 1992;23(4):511–514. doi: 10.1161/01.str.23.4.511. [DOI] [PubMed] [Google Scholar]
- 121.Komaba Y, Osono E, Kitamura S, Katayama Y. Crossed cerebellocerebral diaschisis in patients with cerebellar stroke. Acta Neurol Scand. 2000;101(1):8–12. doi: 10.1034/j.1600-0404.2000.00002.x. [DOI] [PubMed] [Google Scholar]
- 122.Botez-Marquard T, Bard C, Léveillé J, Botez MI. A severe frontal-parietal lobe syndrome following cerebellar damage. Eur J Neurol. 2001;8(4):347–353. doi: 10.1046/j.1468-1331.2001.00204.x. [DOI] [PubMed] [Google Scholar]
- 123.Sagiuchi T, Ishii K, Aoki Y, et al. Bilateral crossed cerebello-cerebral diaschisis and mutism after surgery for cerebellar medulloblastoma. Ann Nucl Med. 2001;15(2):157–160. doi: 10.1007/BF02988609. [DOI] [PubMed] [Google Scholar]
- 124.Ligam P, Haynes RL, Forkerth RD, et al. Thalamic damage in periventricular leukomalacia: novel pathologic observations relevant to cognitive deficits in survivors of prematurity. Pediatr Res. 2008 doi: 10.1203/PDR.0b013e3181998baf. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Limperopoulos C, Bassan H, Gauvreau K, et al. Does cerebellar injury in premature infants contribute to the high prevalence of long-term cognitive, learning, and behavioral disability in survivors? Pediatrics. 2007;120(3):584–593. doi: 10.1542/peds.2007-1041. [DOI] [PubMed] [Google Scholar]
- 126.Courchesne E, Karns CM, Davis HR, et al. Unusual brain growth patterns in early life in patients with autistic disorder: an MRI study. Neurology. 2001;57(2):245–254. doi: 10.1212/wnl.57.2.245. [DOI] [PubMed] [Google Scholar]
- 127.Courchesne E. Brain development in autism: early overgrowth followed by premature arrest of growth. Ment Retard Dev Disabil Res Rev. 2004;10(2):106–111. doi: 10.1002/mrdd.20020. [DOI] [PubMed] [Google Scholar]
- 128.Hashimoto T, Tayama M, Murakawa K, et al. Development of the brainstem and cerebellum in autistic patients. J Autism Dev Disord. 1995;25(1):1–18. doi: 10.1007/BF02178163. [DOI] [PubMed] [Google Scholar]
- 129.Bauman ML, Kemper TL. Neuroanatomic observations of the brain in autism: a review and future directions. Int J Dev Neurosci. 2005;23(2–3):183–187. doi: 10.1016/j.ijdevneu.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 130.Morton SM, Bastian AJ. Mechanisms of cerebellar gait ataxia. Cerebellum. 2007;6(1):79–86. doi: 10.1080/14734220601187741. [DOI] [PubMed] [Google Scholar]
- 131.Ravizza SM, McCormick CA, Schlerf JE, Justus T, Ivry RB, Fiez JA. Cerebellar damage produces selective deficits in verbal working memory. Brain. 2006;129(pt 2):306–320. doi: 10.1093/brain/awh685. [DOI] [PubMed] [Google Scholar]
- 132.Tavano A, Grasso R, Gagliardi C, et al. Disorders of cognitive and affective development in cerebellar malformations. Brain. 2007;130(pt 10):2646–2660. doi: 10.1093/brain/awm201. [DOI] [PubMed] [Google Scholar]
- 133.Gordon N. The cerebellum and cognition. Eur J Paediatr Neurol. 2007;11(4):b232–234. doi: 10.1016/j.ejpn.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 134.Steinlin M. Cerebellar disorders in childhood: cognitive problems. Cerebellum. 2008;7(4):607–610. doi: 10.1007/s12311-008-0083-3. [DOI] [PubMed] [Google Scholar]
- 135.Lidzba K, Wilke M, Staudt M, Krägeloh-Mann I, Grodd W. Reorganization of the cerebro-cerebellar network of language production in patients with congenital left-hemispheric brain lesions. Brain Lang. 2008;106(3):204–210. doi: 10.1016/j.bandl.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 136.Ito M. Control of mental activities by internal models in the cerebellum. Nat Rev Neurosci. 2008;9(4):304–313. doi: 10.1038/nrn2332. [DOI] [PubMed] [Google Scholar]
- 137.Baillieux H, De Smet HJ, Paquier PF, De Deyn PP, Mariën P. Cerebellar neurocognition: insights into the bottom of the brain. Clin Neurol Neurosurg. 2008;110(8):763–773. doi: 10.1016/j.clineuro.2008.05.013. [DOI] [PubMed] [Google Scholar]
- 138.Schmahmann JD, Sherman JC. The cerebellar cognitive affective syndrome. Brain. 1998;121(pt 4):561–579. doi: 10.1093/brain/121.4.561. [DOI] [PubMed] [Google Scholar]