

Abstract
A vital quest in biology is comprehensible visualization and interpretation of correlation relationships on a genome scale. Such relationships may be represented in the form of networks, which usually require disassembly into smaller manageable units, or clusters, to facilitate interpretation. Several graph-clustering algorithms that may be used to visualize biological networks are available. However, only some of these support weighted edges, and none provides good control of cluster sizes, which is crucial for comprehensible visualization of large networks. We constructed an interactive coexpression network for the Arabidopsis (Arabidopsis thaliana) genome using a novel Heuristic Cluster Chiseling Algorithm (HCCA) that supports weighted edges and that may control average cluster sizes. Comparative clustering analyses demonstrated that the HCCA performed as well as, or better than, the commonly used Markov, MCODE, and k-means clustering algorithms. We mapped MapMan ontology terms onto coexpressed node vicinities of the network, which revealed transcriptional organization of previously unrelated cellular processes. We further explored the predictive power of this network through mutant analyses and identified six new genes that are essential to plant growth. We show that the HCCA-partitioned network constitutes an ideal “cartographic” platform for visualization of correlation networks. This approach rapidly provides network partitions with relative uniform cluster sizes on a genome-scale level and may thus be used for correlation network layouts also for other species.
The complete, or partial, genome sequences from a vast number of organisms have increased our understanding of the design principles for biological systems (Kitano, 2002). The sequence availability has also provided platforms for various omics technologies, including transcriptomics, interactomics, and proteomics (Schena et al., 1995; Li et al., 2004; Baerenfaller et al., 2008). Such techniques have generated an immense amount of data that for the most part are publicly available. One of the central ideas behind the concept of systems biology is to utilize these types of data sets to reveal functional relationships between genes, proteins, and other molecules (Kitano, 2002).
Transcriptional coordination, or coexpression, of genes may uncover groups of functionally related genes (DeRisi et al., 1997; Ihmels et al., 2004; Brown et al., 2005; Persson et al., 2005; Wei et al., 2006; Usadel et al., 2009). Such relationships were initially utilized to reveal functional gene modules in yeast and mammals (Ihmels et al., 2004) and to explore orthologous gene functions between different species and kingdoms (Stuart et al., 2003; Bergmann et al., 2004). Comparable studies have also been undertaken in plants (Brown et al., 2005; Persson et al., 2005; Hirai et al., 2007). In addition, several Web-based tools for plants offer various forms of coexpression analyses. These include CressExpress (Srinivasasainagendra et al., 2008), ATTED-II (Obayashi et al., 2009), Arabidopsis Coexpression Data Mining Tools (Manfield et al., 2006), Genevestigator (Zimmermann et al., 2004), GeneCAT (Mutwil et al., 2008), CSB.DB (Steinhauser et al., 2004), CoreCarb (Mutwil et al., 2009), and Expression Angler of the Bio-Array Resource (Toufighi et al., 2005). These tools can provide coexpressed gene lists for user-specified query genes and thus represent user-friendly Web resources for biologists.
While it appears useful for scientists to examine these types of coexpression lists, more information is generally acquired by visualizing the relationships in the form of networks (Jupiter and VanBuren, 2008). Several studies have explored the properties of such network assemblies (Barabási and Oltvai, 2004; Ihmels et al., 2004; Ma et al., 2007; Mentzen and Wurtele, 2008). The distribution of connections in the networks may generally be described by power-law-related relationships (i.e. a small number of nodes appear to have a large number of connections, while most nodes have very few connections; Albert, 2005). Another apparent feature is that essentiality correlates with high connectivity in both coexpression and protein-protein interaction networks in several species (Jeong et al., 2001; Bergmann et al., 2004; Carlson et al., 2006), although this relationship is less clear in mammalian protein-protein interaction networks (Gandhi et al., 2006; Zotenko et al., 2008).
Although features of coexpression and protein-protein interaction networks have been investigated, the output is generally not very useful for visual inspection and interpretation. One major task, therefore, is to make the networks more accessible to biologists (i.e. to produce visualizations of networks that may easily be interpreted; Aoki et al., 2007). For genome-scale networks, this requires dividing the network into smaller manageable units, or clusters. Such clustering, however, may artificially assign genes to certain clusters and therefore skew the output of the biologically “correct” network. It is important, therefore, to maintain as many relevant biological relationships as possible despite division. The ideal number, or size, of clusters to maintain these relationships is very rarely known and is generally very difficult to predict for biological networks. On the other hand, biological networks may also be viewed as clusters within clusters (i.e. as a hierarchical structure that can be viewed on different levels). For example, genes associated with photosynthesis may be viewed as a cluster that belongs to a supercluster of genes associated with functions in the chloroplast. Thus, the ideal clustering algorithm, and subsequent visualization scheme, should generate partitions of manageable sizes that can be readily reconnected into a whole network to be used for manual inspection.
Several graph-clustering algorithms are available, for example Markov Clustering (MCL; van Dongen, 2000), Restricted Neighborhood Search Clustering (King et al., 2004), MCODE (Bader and Hogue, 2003), and others, such as the recently published CAST algorithm (Huttenhower et al., 2007; Vandepoele et al., 2009), but none of these may efficiently control cluster sizes. While these partitioning methods provide useful layouts for global biological and clustering interpretations, they are not particularly useful for visual inspection. To overcome this problem, we developed a novel Heuristic Cluster Chiseling Algorithm (HCCA) and employed it to construct an interactive correlation network for the Arabidopsis (Arabidopsis thaliana) genome (Arabidopsis Gene Network [AraGenNet]; http://aranet.mpimp-golm.mpg.de/aranet). We show that the HCCA-generated cluster solutions were as good as, or better than, the commonly used partition algorithms Markov, MCODE, and k-means using real-world data. We also show that this type of visualization may reveal biological relationships that are not apparent from single gene coexpression approaches. Finally, we explored the network surroundings to identify essential Arabidopsis genes and present six new genes that are essential for plant growth through mutant analyses.
RESULTS AND DISCUSSION
Calculation of Pearson-Based Correlation Networks
To generate a starting network for the HCCA, we calculated the degree of transcriptional coordination between all the genes present on the Arabidopsis ATH1 array (22,810 probe sets) using 351 Robust Multi-array Average (RMA)-normalized microarray data sets from The Arabidopsis Information Resource (TAIR). Prior to choosing these data sets, we removed data sets that displayed poor replication between arrays (Mutwil et al., 2008). Since it is rather difficult to assess whether lowly expressed genes represent noise or real data, we chose to include all probe sets in the analysis. We then calculated an all-versus-all coexpression network matrix using a Pearson correlation coefficient cutoff of 0.8. In contrast to Spearman correlation, Pearson correlations only capture linear relationships between any two given components. However, it is anticipated that most linked expression profiles will adhere to a linear relationship (Daub et al., 2004).
The distribution of connections in Pearson correlation-based biological networks may generally be described by power-law-related relationships (i.e. a small number of nodes appear to have a large number of connections, while most nodes have very few connections; Barabási and Oltvai, 2004). To assess whether the topology of the obtained Pearson correlation network for Arabidopsis also followed such a relationship, we calculated the node degree distribution of all individual nodes in the network. Figure 1A shows that the node degree distribution is best described by a truncated power-law behavior. We also observed similar deviations from classical power-law behavior in Pearson correlation networks generated for yeast (Saccharomyces cerevisiae) and to a lesser degree for Escherichia coli (Fig. 1B), in agreement with recent reports (van Noort et al., 2004).
Figure 1.

Network characteristics and mutant analyses. A, Log-log plot of node degree distribution for 261 essential genes (red points), 1,224 nonessential genes (green points), and all genes (22,810; blue points) in the Pearson correlation network (r ≥ 0.8) for Arabidopsis. B, Log-log plot of node degree distribution for Pearson correlation networks (r ≥ 0.8) from E. coli (blue), yeast (red), and Arabidopsis (green). The x axis represents the node degree (i.e. the number of connections a node holds), and the y axis displays the frequency (i.e. the number of genes [B]) or the normalized frequency (i.e. the normalized number of genes [A]) showing this degree.
Centrality Versus Essentiality
Another apparent feature in biological networks is that essentiality typically correlates positively with high node degree (i.e. mutations in highly connected nodes tend to result in more severe phenotypes compared with less well-connected nodes; Jeong et al., 2001; Albert, 2005; Carlson et al., 2006; Zotenko et al., 2008). To assess if this type of relationship also is evident in our Pearson correlation network, we analyzed gene connectivity versus embryo lethality. We did this by linking phenotypic data from TAIR (www.arabidopsis.org) to the genes in our Pearson-based network (r = 0.8). Figure 1A shows the node degree distribution of embryo-lethal genes, genes associated with any type of phenotype, and all genes included on the ATH1 microarray. Whereas the node degree distribution for genes associated with nonlethal phenotypes did not deviate significantly compared with all genes present on the ATH1 gene chips (Fig. 1A), genes corresponding to embryo lethality were significantly more connected compared with nonessential genes (Fig. 1A; Supplemental Fig. S4B; P < 0.05). Similar observations have also been reported for coexpression and protein-protein interaction networks in yeast (Albert, 2005; Carlson et al., 2006).
Construction of a Highest Reciprocal Rank-Based Correlation Network in Arabidopsis
Several studies have used r value cutoffs ranging between 0.6 and 0.8 to depict coexpression correlations (van Noort et al., 2004). However, different genes have different distributions of r values (i.e. at a given cutoff, some genes may correlate significantly with hundreds of genes while other genes may not correlate with any). Despite this, it is still possible that the latter may hold biologically relevant relationships. For example, the two transcription factors MYB33 (At5g06100) and MYB65 (At3g11440) regulate pollen and anther development, are expressed similarly, and are functionally redundant (Millar and Gubler, 2005). However, an r value cutoff of 0.8 did not associate these genes transcriptionally (r = 0.7; data not shown; Mutwil et al., 2008). To minimize this problem, we chose to normalize the r value distributions in the calculated Pearson correlation networks using highest reciprocal rank (HRR) as they define the mutual coexpression relationship between two genes of interest. Using this approach, MYB33 and MYB65 were readily transcriptionally linked (mutual average rank = 2 using GeneCAT; Mutwil et al., 2008). With this approach, we were also able to define a connection cutoff, or maximum number of connections, for a given gene. The importance of defining such a cutoff is apparent when looking at the distribution of r values among the data. For example, approximately 1,500 genes are only expressed in pollen (estimated from GeneCAT; Mutwil et al., 2008). All of these genes are correlated with each other with an r value of 0.8 and therefore should be connected to each other in a Pearson-based correlation network (Mentzen and Wurtele, 2008). However, it is virtually impossible to retain any information from such a network structure through manual inspection. Instead, we argue that displaying these genes in close network vicinities, which is achieved by the HRR-based network, is more useful. In addition, recent results indicate that correlation-ranked networks produce sounder results than networks based on correlation coefficients (Obayashi and Kinoshita, 2009).
We set the HRR limit to 30, thus capping the maximum number of edges per node to 30. The resulting HRR network seemed a reasonable compromise between readability and richness of information. In addition, we defined three degrees of coexpression weights using highest reciprocal ranks of 10, 20, and 30 (Mutwil et al., 2008). Similar approaches have also been used by several coexpression Web tools, such as GeneCAT and ATTED-II (Mutwil et al., 2008; Obayashi et al., 2009). The resulting weighted HRR network contained 103,587 edges between 20,785 nodes and was used as the starting network for the HCCA. As anticipated, not all the probe sets shared strong correlation with other probe sets, resulting in 2,025 nodes that were not included in the network (data not shown). The HRR-based network shared 29,956 edges and 6,942 nodes with the Pearson-based coexpression network using r ≥ 0.8 as cutoff (total of 231,882 edges and 7,178 nodes).
Designing the HCCA
Genome-scale coexpression networks, like other networks, consist of nodes and edges that may form a continuous structure or separate islands of clusters, depending on what cutoff one uses. While the smaller structures in such networks may be suitable for visual inspection, other regions may not be due to the number of nodes and edges in these regions. To make such regions more accessible, it is necessary to partition the network into smaller units, or clusters. Obviously, such partitioning will lead to a division of network structures that may, or may not, reflect the “real” network properties. Most biological networks do not contain sufficient data to assess whether the divisions are justifiable or not. However, the flaws in network divisions may be overcome if the different partitions can be reassembled into the structures they were initiated from. We argue that if we can visualize individual network partitions, or clusters, and put these into context with other clusters, then the connectivity between the individual clusters may reflect the larger structures that were partitioned.
Many graph-clustering algorithms do not support weighted edges and do not yield cluster sizes that readily allow visual interpretations. In addition, many graph-clustering algorithms do not allow clustering of large networks (i.e. networks consisting of several thousand nodes). Therefore, we developed a novel graph-clustering algorithm (Fig. 2) referred to as HCCA. The HCCA algorithm takes step size (n) and desired cluster size range as parameters. The HCCA accepts a network as starting point (Fig. 2). For each node in the network, the algorithm generates node vicinity networks (NVNs) by collecting all nodes within n steps away from the seed node. Nodes with higher connectivity to the outside of the NVN are iteratively removed. The resulting clusters are then ranked by outside-to-inside connectivity ratio and filtered according to desired cluster size range. Nonoverlapping clusters are retained by the algorithm, and nodes in these clusters are removed from the network. Nodes associated with rejected clusters are returned to the network and reevaluated. The HCCA recursively creates nonoverlapping clusters until no nodes are left in the network or no more stable clusters can be obtained (Fig. 2). In the latter case, remaining nodes are associated with clusters to which they display the highest connectivity.
Figure 2.

Schematic work flow of the HCCA (n = 3). The HCCA accepts a network as input. Step 1, Each of the nodes in the network is used to generate NVNs by taking n steps away from a seed node (indicated as a star). Step 2, Each NVN is then “chiseled” by recursively removing nodes that have higher connectivity to nodes outside of an NVN than to nodes inside the NVN. In this example, squared and triangular nodes are removed in the first and second rounds of chiseling, respectively. Step 3, The chiseling either completely depletes a NVN of nodes or produces a stable putative cluster (SPC). Step 4, Nonoverlapping SPCs with highest cSPC values are extracted and accepted as clusters. Step 5, Nodes that were accepted as clusters in step 4 are removed from the network. The remaining network is then transferred to step 1 and rechiseled (steps 2–5).
Visual Inspection of the Network Solutions
To partition the network, we used the HCCA with different steps (n) away from the seed node (Fig. 2) and desired cluster sizes ranging from 40 to 400. For example, for n = 3, the HCCA generated 181 clusters that contained approximately 40 to 300 genes per cluster (Fig. 3A). To assess the biological relevance of the partitioned network, we initially compared obtained connections with known biological data through visual inspection. For example, the secondary cell wall cellulose synthase genes CESA4, CESA7, and CESA8 have been used extensively for coexpression analyses (Brown et al., 2005; Persson et al., 2005; Ma et al., 2007). In agreement with these analyses, we obtained genes associated with secondary cell wall synthesis, including IRX6, IRX8, IRX9, IRX12, and several transcription factors that recently have been implicated in secondary cell wall regulation (Zhong and Ye, 2007), in the network vicinity of the three CESA genes (Supplemental Fig. S1).
Figure 3.

Cluster comparison of HCCA, MCL, k-means, and MCODE. A, Graph displaying the cluster size range (x axis) versus number of clusters (y axis; observations) for selected HCCA, MCL, k-means, and MCODE partitions of the HRR network (HRR cutoff = 30). B, Modularity scores for different settings for the HCCA, MCL, k-means, and MCODE algorithms. k-means 100, 200, and 400 represent desired cluster number parameters for k-means; MCL 1.15, 1.5, and 2.0 represent different inflation degrees for the MCL; HCCA n = 2, 3, and 4 represent different step size (n) as described in Figure 2; MCODE (A, B, C, and D) represent degree cutoff, node score cutoff, k-core, and maximum depth, respectively. High modularity values represent better clustering. C, Davies-Bouldin score, or index, for different settings for the HCCA, MCL, k-means, and MCODE. The settings are in accordance with B. Low Davies-Bouldin score represents better clustering. D, ClusterJudge scores of the clustering generated by HCCA, MCL, k-means, and MCODE, respectively. The settings are in accordance with B. High ClusterJudge score represents better clustering.
Estimates of Clustering Solutions
A few other graph-clustering algorithms also support weighted edge graphs, such as the commonly used MCL (van Dongen, 2000; Enright et al., 2002; Mentzen and Wurtele, 2008). To estimate the quality of the clustering solution obtained by HCCA, we clustered the HRR network using the MCL algorithm with a range of different inflation values (Supplemental Table S1). In addition, we included clustering solutions for MCODE (Bader and Hogue, 2003; Prieto et al., 2008), performed clustering using k-means with different settings (Hartigan and Wong, 1979), and then compared the results obtained from the HCCA with the different clustering solutions for the other algorithms (Fig. 3; Supplemental Table S1). We used two different metrics to evaluate the clustering efficiency: the commonly used quantity modularity (Newman and Girvan, 2004), which judges partitions by comparing inside-to-outside connectivity ratios, and the Davies-Bouldin index, which measures the compactness and separation of the obtained clusters (Davies and Bouldin, 1979). Our HCCA approach yielded better cluster partitioning compared with the MCL, k-means, and MCODE in terms of modularity (Fig. 3B; Supplemental Table S1). In addition, the HCCA solutions were clearly better than all the k-means partitions in terms of the Davies-Bouldin index (Fig. 3C; Supplemental Table S1). However, the MCL and MCODE partitions rendered better Davies-Bouldin scores compared with the HCCA (Fig. 3C; Supplemental Table S1). While the best overall MCL solution was the MCL 1.15 (inflation value 1.15), it is important to point out that this partition contains cluster sizes in the range of two to 2,500 genes per cluster (Fig. 3A; Supplemental Table S2) and therefore is not useful for our purposes. These results show that the HCCA performed better than k-means in terms of modularity and Davies-Bouldin index and scored comparable index numbers as MCL and MCODE in terms of modularity.
When considering modular networks, it is generally expected that neighboring nodes fulfill related functions, which also has been recognized in social networks (Wasserman and Faust, 1994). Hence, ideally, one coexpressed gene cluster should contain genes associated with similar biological functions. Therefore, we also tested the overlap of MapMan ontology classes with the clusters generated by the HCCA, MCL, MCODE, and k-means. We used an approach similar to ClusterJudge (Gibbons and Roth, 2002), which uses mutual information between clusters and MapMan ontology terms to score clustering quality (Steuer et al., 2006). In brief, this approach scores the overlap between the ontological terms and the clusters, then subtracts the mean score obtained for randomly assigned clusters, and divides this by the sd of the random clustering solutions. Therefore, a score of 0 (or even negative scores) would indicate random biological categories and clusters, whereas higher scores (which have no upper bound) indicate better concordance between biological categories and clusters. Using this assessment, the HCCA-partitioned networks scored better than all of the MCL and MCODE partitions and scored nearly as well as the solutions generated by k-means (Fig. 3D; Supplemental Table S1). It is important to note that the latter commonly used algorithm cannot generate clusters based on graphs but must use the original expression data, which has an inherent advantage compared with the HCCA, MCODE, and MCL.
We also investigated how HCCA performs on unweighted HRR networks. The HCCA-generated partitions performed slightly better in terms of modularity and ClusterJudge score and much better in terms of Davies-Bouldin score compared with the other clustering algorithms (Supplemental Table S1). However, it is important to note that the HCCA partitions of unweighted networks produced several clusters exceeding the desired maximum cluster size of 400 (Supplemental Table S2). This is most likely due to the more detailed information retained in the weighted network. It should be noted that by lowering the cSPC cutoff value (see Fig. 2 legend), it should still be possible to generate clusters within the desired cluster range using HCCA. Also, the number of clusters obtained from the unweighted network was smaller than for the weighted network (Supplemental Table S2).
Taken together, these tests show that the HCCA partitions scored better than k-means, MCL, and MCODE in terms of modularity and Davies-Bouldin index and outperformed the MCL and MCODE solutions in terms of biologically relevant associations.
Comparisons of Partition Similarities
While the above results show that HCCA generated cluster solutions that are as good as, or better than, MCL, MCODE, and k-means, the HCCA also produced clusters with relative uniform size (Fig. 3A; Supplemental Table S2) and therefore is well suited for cluster visualization for manual inspection. In contrast, the best performing MCL partitions resulted in cluster sizes between two and 2,500 genes (Fig. 3A; Supplemental Table S2), which is in good agreement with what has recently been reported (Mentzen and Wurtele, 2008). Although the cluster size distribution between the different algorithms varied, we anticipated a relatively high overlap in cluster content between the different solutions. Therefore, we compared the overlap of genes associated with certain clusters for the HCCA, MCL, MCODE, and k-means solutions by adjusted Rand indices, which measure similarities between two clustering solutions (Supplemental Table S3; Hubert and Arabie, 1985). Interestingly, each of the algorithms appeared to have generated clusters with different contents. For example, comparison of the HCCA (n = 3) and MCL 1.2 (inflation value = 1.2) solutions resulted in an adjusted Rand index of 0.2495 (identical partitions result in an index of 1; Supplemental Table S3). However, these solutions contain different cluster sizes, which influence the outcome of the adjusted Rand index. Comparing 1,000 k-means-partitioned networks, each featuring 100 cluster centers, with a reference k-means network resulted in an average adjusted Rand index of 0.4, which is considerably lower than the index of 1 for identical partitions. Therefore, it appears that the seemingly low average adjusted Rand indices for the different solutions may in fact signify rather good agreement in cluster contents. The rather low values may be explained by unequal cluster size distributions and by uncertain cluster partitioning for some of the genes.
Robustness of Clustering toward Node Removal and Different HRR Cutoffs
The ATH1 microarray chip contains 22,810 probe sets covering roughly 80% of the genes in the Arabidopsis genome. This means that approximately 5,000 genes are omitted from the chip and, therefore, from our analysis. To assess whether omission of such a number of genes may significantly skew the connections in the HRR network, we randomly removed approximately 20% of the genes from our data sets and reclustered the network using HCCA. We repeated this 20 times and then assessed whether the clusters were significantly different by estimating the average adjusted Rand index. Supplemental Table S3 shows that the average score for HCCA (n = 3) was 0.3818, with only 4% sd. This value is similar to the value obtained for the comparison of 1,000 k-means clustering solution using 100 cluster centers. These data show that the network outline and HCCA clustering are robust against removal of a significant portion of randomly selected genes and therefore also should display biologically meaningful correlations despite the absence of some genes on the ATH1 chip.
To test how different HRR cutoffs influence the clustering by HCCA, we calculated adjusted Rand indices between networks generated using HRR of 10, 20, 30, 40, and 50. Supplemental Table S4 shows that the adjusted Rand index is relatively high (>0.4) for networks generated by similar HRR cutoffs (HRR20 versus HRR30, HRR30 versus HRR40, and HRR40 versus HRR50), despite the fact that the networks differ dramatically in the number of edges (Supplemental Table S4). Taken together, these results indicate that clusters obtained by HCCA are robust against the parameters used to generate the coexpression networks.
Construction of an Interactive Correlation Network for the Arabidopsis Genome
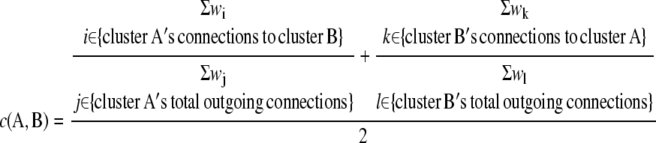
To illustrate the usefulness of the network partition obtained from the HCCA, we implemented an interactive coexpression network browser, which we named AraGenNet (http://aranet.mpimp-golm.mpg.de/aranet). Since the aim of the visualization scheme was to reassemble the partitioned HRR network for manual inspection, the network works on two levels: on the assembled cluster level (meta-network) and on the gene level (Figs. 4 and 5). The cluster-level network (Fig. 4) represents an overview of the interactions between different partitions, or clusters, and therefore depicts the coexpressed context for individual clusters. Therefore, we refer to this network as a meta-network. Any two clusters in the meta-network are connected if the combined weight of edges between them is larger than a certain threshold. We set this linkage threshold, or connectivity score, to 0.02, as this value produced a connection-rich but readable meta-network (Fig. 4, A and B). A node in the meta-network consists of a cluster of coexpressed genes generated from the HCCA (n = 3; Fig. 5). This gene-level network becomes visible by clicking on a cluster node in the meta-network. All connections in the gene-level network are based on HRR and are weighted accordingly (i.e. HRR below 10, 20, and 30 are color coded green, orange, and red, respectively; Fig. 5). These visualization schemes prove the capability and functionality of the HCCA clustering approach.
Figure 4.

Meta-network of coexpressed gene clusters generated by HCCA (n = 3). A, Nodes in the meta-network, or assembled cluster-level network, represent clusters generated by HCCA. Edges between any two nodes represent interconnectivity between the nodes above threshold 0.02 (according to C). B, Enlarged region depicts part of the meta-network presumably associated with photosynthesis. Cluster annotations were inferred by MapMan terms, phenotypic, and expression data (http://aranet.mpimp-golm.mpg.de/aranet). C, Connectivity cutoff values [c(A,B)] for edges in the meta-network. We used a cutoff of 0.02 for visualization purposes.
Figure 5.

Features of HCCA (n = 3) gene cluster 59. Nodes in this cluster, or gene-level network, represent genes, while edges and edge coloration depict the HRR values between any two nodes. Red, yellow, and green node colors depict gene mutants displaying embryo-lethal, gametophyte-lethal, and other described phenotypes, respectively. Gray nodes represent genes with no described phenotype.
Phenotype and Ontology Mapping onto the Network
Since coexpressed genes often tend to be functionally related (DeRisi et al., 1997; Ihmels et al., 2004; Brown et al., 2005; Persson et al., 2005; Wei et al., 2006), we anticipated that connected clusters in the meta-network would share a certain degree of functional commonalities (Freeman et al., 2007). To assess this, we analyzed the genes in each cluster for MapMan ontology term enrichments. We also mapped phenotypic data (http://www.arabidopsis.org/) and tissue-dependent expression profiling for the individual genes. By combining these analyses, we then attempted to describe what biological functions are associated with the individual clusters. For example, mutations in genes associated with cluster 59 (Fig. 5) often result in embryo lethality or pale green plants. The dominant expression profile of genes in this cluster shows high expression in aerial tissues and low expression in roots, pollen, and seeds. MapMan ontology analysis revealed that the most significantly enriched term is amino acid metabolism (P ≤ 10−9). Taken together, these data suggest that cluster 59 is overrepresented for genes involved in amino acid metabolism in the chloroplast and that this function is important for chloroplast development, photosynthesis, and embryo development. This conclusion is supported by the fact that cluster 59 was highly enriched for genes with plastidic localization (P < 0.001; data not shown).
Prediction and Verification of Essential Genes in the Network
To expand the visual features of the network, we color coded the severity of the phenotypic traits using red (embryo lethality), yellow (gametophyte lethality), and green (other phenotypes) nodes in the network (Fig. 5). Interestingly, we observed an uneven distribution of embryo-lethal genes per cluster compared with genes associated with nonlethal phenotypes (Fig. 6A). For example, the chloroplast-associated clusters 21, 59, and 137 showed strong enrichment for essential genes (P < 10−5; Supplemental Table S5). This suggests that nodes in clusters associated with certain biological processes are more essential. For example, of the 111 genes associated with cluster 59, 12 are known to be essential for embryo development (Fig. 6A; Supplemental Table S5). As described above, this cluster may be associated with amino acid activation in the chloroplast.
Figure 6.

Essentiality distribution and mutant phenotypes in the HCCA (n = 3) partitioned network. A, The graph displays the relative distribution of essential genes per any given cluster in the network (HRR cutoff = 30). Black bars depict clusters significantly enriched (P ≤ 0.05) for essential genes. B, Distribution of single-copy genes from 1,000 samplings of 152 random nodes from the HRR network (black bars). Any given gene was referred to as being single copy if no close homolog was detected (score coverage threshold of 30 and length coverage of the protein of 70%). The observed 152 essential, single-copy genes are denoted by the red line. C, Siliques from a plant heterozygous for mutation in At3g14900 (cluster 137). Red arrows indicate chlorotic embryos. Bar = 3 mm. D, Mutant seedlings (At1g15510) from cluster 137 exhibiting pale cotyledons (indicated by arrows). Bar = 3 mm. E, Chlorotic dwarfed mutant (At3g57180; indicated by the arrow) from cluster 21. Bar = 1 cm.
We also investigated how the essentiality of a gene is determined by the number and the distances of its homologs in the network. Figure 6B shows that embryo-lethal genes are clearly overrepresented by single-copy genes (P < 0.001; Supplemental Fig. S2A). Furthermore, essential genes tend to be underrepresented for genes with family members in the network vicinity (i.e. in the node vicinity network; P < 0.05; Supplemental Fig. S2, B and C). Conversely, nonessential genes tend to be neighbors to their family members (P < 0.05; Supplemental Fig. S2, E and F). Taken together, the probability of essentiality for a given gene appears to depend not only on the connectivity of the gene (Fig. 1A) but also on its functional uniqueness in the network vicinity and on its biological role. Interestingly, similar results have recently also been observed in protein-protein interaction studies in yeast (Zotenko et al., 2008). This study convincingly showed that essentiality corresponded to gene products that are well connected and that are associated with certain biological functions.
To explore the prediction of essentiality, we chose 20 genes associated with clusters that harbor numerous essential genes (i.e. the connected clusters 21, 59, and 137; Fig. 6A; Supplemental Fig. S3) and that are well connected to other essential genes in the network. We ordered T-DNA mutant lines corresponding to these genes and analyzed them for mutant phenotypes (Table I). Out of the 20 mutant lines, two resulted in embryo lethality, one in seedling lethality, two in male gametophyte lethality, and one in dwarfed pale green plants (Fig. 6, C–E; Table I). Chlorotic cotyledon phenotypes are typically associated with chloroplastic functions (Flores-Pérez et al., 2008), supporting our prediction that genes belonging to these clusters (i.e. 21, 59, and 137) are functionally associated with the chloroplast. These results illustrate how a coherent and easy-to-navigate data visualization scheme, such as the AraGenNet, can predict biologically meaningful relationships. Recently, the pollen-deficient mutant corresponding to the gene At1g74260 was confirmed by another study (Berthomé et al., 2008).
Table I.
Characteristics of mutants
Family size and family members in vicinity indicate the size of a gene family as defined by Clusters of Orthologous Groups of proteins and the number of family members in the gene network vicinity (n = 2), respectively.
| Gene | T-DNA Line | Phenotype | Family Size | Family Members in Vicinity |
|---|---|---|---|---|
| At3g23940 | SALK_069706 | Gametophyte lethal | 0 | 0 |
| At1g74260 | SALK_050980 | Gametophyte lethal | 0 | 0 |
| At5g64580 | SAIL_74_G12 | Embryo lethal | 0 | 0 |
| At3g14900 | SALK_123989 | Embryo lethal | 0 | 0 |
| At1g15510 | SALK_112251 | Seedling lethal | 182 | 38 |
| At3g57180 |
SALK_068713 |
Pale green, dwarf |
0 |
0 |
Associations of Functional Annotations Using MapMan Ontology
Although the visualization of coexpressed genes may give insight into functional gene patterns and arrangements, an equally relevant quest is to understand how these patterns and arrangements are organized to fulfill cellular functions. To investigate this, we explored the notion that coexpressed genes, and therefore network vicinities, often are functionally related (Ihmels et al., 2004; Brown et al., 2005; Persson et al., 2005; Wei et al., 2006). To assess how different ontological terms are transcriptionally connected, we used the nonclustered HRR network (HRR cutoff = 30) and calculated whether certain MapMan ontology terms were overrepresented in nonoverlapping node vicinities (NVNs in Fig. 2). We then identified terms that co-occurred more often than expected by chance (P ≤ 0.05). These significantly associated terms were connected, and the resulting ontological network was visualized as an interactive network browser (Fig. 7; http://aranet.mpimp-golm.mpg.de/aranet/Mapman_network). To get a more complete network, we also retained connections representing parent-child relationships, which are trivial due to their mutual overlap. From this visualization, it became evident that terms that represent related processes tend to be connected; for example, photosynthesis-related processes (dark green) were connected to plastidial protein synthesis (light blue) and to “protein assembly and cofactor ligation,” which comprises many proteins involved in the assembly of the plastidial apparatus (light blue). Furthermore, the chloroplast cluster (dark green) is closely associated with genes related to tetrapyrrole biosynthesis (light green; Fig. 7). These processes most likely reflect parts of the basal plastidial photosynthetic activity program. Other examples were mitochondrial processes linked to the tricarboxylic acid cycle as well as polyamine synthesis being coupled to Arg degradation more than would be expected by the trivial link of Arg decarboxylase, which is present in both processes. Also, arabinogalactan proteins were linked to abiotic stress, which is in line with their up-regulation upon salt stress (Lamport et al., 2006).
Figure 7.

Network of coexpressed MapMan ontology terms. Nodes in this network represent biological processes as defined by MapMan ontology terms. Node colors and numbers depict the different MapMan terms (legend at left), while edges represent significant (P ≤ 0.001) associations between the terms based on coexpression. OPP, Oxidative pentose pathway; PS, photosynthesis; CHO, carbohydrate.
Since biologically relevant associations were confirmed in the MapMan ontology network, we also investigated associations between other biological processes, which were previously unrelated MapMan terms and which might help to generate new functional insights. Interestingly, plant defensins were connected to sphingolipid biosynthesis in planta. As often the mode of action of plant defensins seems to be mediated by sphingolipids of the attacking pathogen (Thevissen et al., 2000, 2005; Ramamoorthy et al., 2009), it could be speculated that plant sphingolipids might play a role in this mechanism as well. Furthermore, it might be interesting to investigate what caused the link introduced between aromatic amino acid degradation and starch breakdown (Fig. 7, bottom left corner). Thus, the combination of coexpressed gene vicinities and ontology terms may similarly reveal new associations between different processes in the cell.
CONCLUSION
We have constructed an interactive correlation network for Arabidopsis using a novel HCCA. The cluster solutions obtained from this clustering algorithm performed as well as, or better than, the commonly used clustering algorithms MCL, MCODE, and k-means. More importantly, by visualizing the portioned clusters, we could reassemble the network; therefore, we were able to place the obtained partitions into larger biological contexts. We predicted that unique, well-connected genes with certain biological functions tend to be more essential than other genes and confirmed this by mutant analyses. The presented data, therefore, show that comprehensible visualization of genome-scale correlation networks may render new insights into the wiring of biological systems. We propose that this type of network visualization constitutes an easy-to-navigate framework for biologists to prioritize genes for functional analyses.
MATERIALS AND METHODS
Microarray Data
All calculations for this work were done using python and java scripts. Databases for Arabidopsis (Arabidopsis thaliana), yeast, and Escherichia coli use Affymetrix ATH1 (22,810 probe sets), Affymetrix Yeast Genome S98 (9,335 probe sets), and Affymetrix Ecoli_ASv2 (7,312 probe sets) GeneChips, respectively. Arabidopsis microarray data sets consisting of 1,428 ATH1 microarrays were obtained from TAIR (http://www.arabidopsis.org/). Separate Arabidopsis tissue atlas data sets containing 121 microarrays, which were used for plotting the gene expression across Arabidopsis tissues, were generated by the AtGenExpress project (Schmid et al., 2005) and were obtained from TAIR. The data were quality controlled by visual inspection of box plots of raw positive match data and RMA residuals of RMA-normalized data using the RMA express program. Cel files showing artifacts on RMA residual plots or visibly deviating from the majority on the positive match box plots were removed from further analysis. In addition, we removed experiments representing very similar transcriptomic snapshots by iteratively discarding microarrays that displayed Pearson correlation [r(A,B) ≥ 0.95] to more than three other microarrays. From these analyses, we retained 351 microarrays, which subsequently were normalized using R package simpleAffy. The 244 E. coli and 789 yeast microarray data sets used to generate Figure 1 were downloaded from Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/), RMA normalized, and quality controlled as for the arrays for Arabidopsis. Names of the cel files used to construct the Arabidopsis HRR networks are downloadable from the AraGenNet home page.
Phenotypic Data for Arabidopsis
Phenotypic data for Arabidopsis were requested and obtained from TAIR curators and were divided into essential, gametophyte lethal, and nonlethal sets. All the expression data, coexpression network, and phenotypic data presented in this work are downloadable from the AraGenNet home page (http://aranet.mpimp-golm.mpg.de/aranet).
Construction of Coexpression Networks
Pearson-based coexpression networks were used for the centrality-versus-essentiality study and for generating log-log plots. These networks were created using the 351 ATH1 microarrays described above. An edge in the network represents two genes with Pearson correlation [r(A,B) ≥ 0.8]. All subsequent analyses were done on HRR-based networks, including the visualized interactive coexpression network used on the AraGenNet home page. The HRR score between genes A and B is calculated according to:
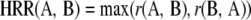 |
where r(A,B) is correlation rank of gene B in gene A's coexpression list. Any two genes that were present in each other's top 10, 20, or 30 correlation lists were connected by green, orange, or red connections, respectively. Edges representing HRR values of 10, 20, and 30 were assigned weights of 1/5, 1/15, and 1/25, respectively. Any two clusters in the meta-network were connected if the connectivity score exceeded 0.02 according to:
 |
where
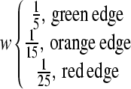 |
We used c(A,B) ≥ 0.02, which connects clusters A and B, if the average mutual weights of edges between the two clusters exceed 0.02. The connectivity score can range from 0 (no edges between the clusters) to 1 (all outgoing connections from cluster A are connected to cluster B and vice versa).
Comparison of a Pearson Correlation Network and a Graphical Gaussian Network
Our Pearson correlation network (r = 0.8) was compared with data sets from a recently published Graphical Gaussian (GGM) network (Ma et al., 2007), and common edges were identified by set comparisons (Supplemental Fig. S4A). Approximately one-third of the edges in the GGM network were also present in our network, consistent with a previous comparison made between the GGM and a Pearson correlation network (Ma et al., 2007).
To assess the association of node degree (number of nodes a node is connected to) with phenotype characteristics (essential or nonessential), a node degree of genes showing a phenotype versus those not showing any phenotype was compared. This was done across 20 coexpression networks generated using Pearson r values ranging from 0.9 to −0.9 (steps of 0.1). The median node degree of genes showing a phenotype was compared with the median node degree of genes not showing any phenotype at a given r value cutoff. Significant differences (Wilcoxon test; P < 0.05) in the median node degree between these two classes were used to indicate significant differences between the two classes.
HCCA Clustering Algorithm
The HCCA can be implemented by a pseudocode available from the AraGenNet home page, and the full source code is available upon request from the authors. A simplified description of the algorithm is depicted in Figure 2 and in “Results and Discussion.” Python implementation of HCCA, together with sample networks, is available from the AraGenNet home page.
MCL
We used the available C code (http://micans.org/mcl/; van Dongen, 2000) for MCL calculations. The method simulates random walks on the graph, with the walking probability respecting the weight (i.e. HRR values) of the edges (HRR value of 10 received weight 1/5, 20 received 1/15, and 30 received 1/25). We used different inflation values, which are the Hadamard power of a stochastic matrix that gives the probabilities for the random walk. Low inflations result in slower random walks and vice versa. The inflation parameter may range from >1 to 5, where small values generate fewer but larger clusters.
k-Means Clustering
To partition probe sets based on the original data, the expression values for each probe set were centered, scaled, and then subjected to the k-means clustering procedure provided by R using the default algorithm of Hartigan and Wong (1979).
MCODE Clustering
The MCODE plugin for Cytoscape (http://baderlab.org/Software/MCODE; Bader and Hogue, 2003) calculates the local density of nodes in a network. Based on this score, a seed node is chosen as a starting point to collect nodes as long as their scores deviate from the seed node within a certain range. After clustering, it allows postprocessing single clusters without changing the rest of the network. Since MCODE has the option to vary six or seven parameters, we attempted to make the output comparable to the HCCA, MCL, and k-means cluster solutions; therefore, we emphasized the solutions that cluster a large portion of nodes (Bader and Hogue, 2003).
Comparison of Clustering Solutions
The clustering solutions were judged by modularity (Newman and Girvan, 2004), which evaluates the graph partitioning by comparing the sum of edge weights within clusters with edge weights linking different clusters. This value is subsequently subtracted by the value that one expects for random partitions. The obtained modularity score ranges between −1 and 1, where 1 represents perfect modularity, 0 represents value expected by chance, and −1 represents a value worse than expected by chance.
The partitions were also evaluated by the Davies-Bouldin (DB) index (Davies and Bouldin, 1979) using the clusterSim R package. It is defined as:
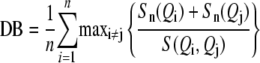 |
with n number of clusters, Sn average distance of all objects from the cluster to their cluster center, and S(QiQj)distance between two cluster centers. Davies-Bouldin score can range from 0 to infinity. Values close to 0 are achieved by good (distant) clustering. However, the value of 0 is gained by just one big cluster.
We used adjusted Rand indices to compare two clustering solutions by pairwise affiliation of nodes (Hubert and Arabie, 1985).
The scores for biological significance of clusters were calculated using the approximate mutual information between the clustering and MapMan categories (Usadel et al., 2006) having at least 10 members. In the case where the clustering solution did not assign all genes to clusters, only those that could be assigned were considered. To make the HCCA clustering comparable to k-means, genes not assigned to any cluster by HCCA were not subjected to k-means, as these genes are most likely difficult to cluster. From this mutual information value, the mean mutual information from 1,000 random assignments (denoted by 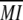 ) with preserved cluster sizes was subtracted, and the result was divided by the sd (denoted by σ) of these random mutual information values according to:
) with preserved cluster sizes was subtracted, and the result was divided by the sd (denoted by σ) of these random mutual information values according to:
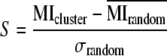 |
Overrepresentation Analysis
To identify terms that might be associated, we randomly sampled approximately 700 nonoverlapping NVNs from the whole network and tested for a significant overrepresentation of MapMan terms within these clusters using a Fisher exact test (P < 0.05 after Benjamini-Hochberg correction). This was repeated several times to exclude random effects. Subsequently, we tested for significant co-occurrence of overrepresented terms using the Fisher exact test.
Uniqueness-Versus-Essentiality Estimates
To group Arabidopsis genes into gene families, a BLASTCLUST analysis on Arabidopsis protein sequences obtained from TAIR was performed. Length coverage threshold of 70% and score coverage threshold were used as parameters.
We used random sampling to investigate whether there is correspondence between a gene having essential or nonessential characteristics and its uniqueness in the genome or node vicinity network. So far, 261 genes are characterized as being essential (phenotypic data from TAIR), and 152 of these are single-copy genes based on the settings above. To investigate whether essential genes tend to be single copy, we sampled 261 random nodes 1,000 times and counted the number of single-copy genes acquired in each sampling. To investigate whether essential genes that do belong to a gene family tend to be unique in the network vicinity, we sampled 109 (261 total − 152 single copy) random nodes 1,000 times. The number of genes unique or nonunique in the network vicinity was then counted and represented as a histogram. The same was done for nonessential genes with characterized nonlethal phenotypes (1,224 total, 422 single copy).
Plant Cultivation and Mutant Analysis
T-DNA knockout lines (Supplemental Table S6) were obtained from the Nottingham Arabidopsis Stock Centre (Alonso et al., 2003). The seeds were surface sterilized, sown on plates containing Murashige and Skoog medium (1× Murashige and Skoog salts, 8 g L−1 agar, 1× B5 vitamins, and 10.8 g L−1 Suc), and incubated for 48 h at 4°C in the dark. The plates were then incubated for 7 d at 21°C with a 16-h photoperiod. T-DNA insertions were confirmed using PCR (Supplemental Table S6). Images of seedlings and siliques were made using a Leica MZ 16 FA stereomicroscope.
Sequence data from this article can be found in the GenBank/EMBL data libraries under accession numbers NC_003074.8, NC_003070.9, NC_003076.8, NC_003074.8, NC_003070.9, and NC_003074.8.
Supplemental Data
The following materials are available in the online version of this article.
Supplemental Figure S1. Cluster 20 containing genes involved in secondary cell wall cellulose synthesis.
Supplemental Figure S2. Distribution of 1,000 random samplings of essential and nonessential genes from the mutual rank network.
Supplemental Figure S3. Clusters 21, 59, and 137.
Supplemental Figure S4. Comparison of a Pearson network and a GGM-generated network.
Supplemental Table S1. ClusterJudge, Modularity, and Davies-Bouldin scores for HCCA, k-means, MCL, and MCODE clustering solutions.
Supplemental Table S2. Cluster size distributions for HCCA, k-means, MCL, and MCODE clustering solutions.
Supplemental Table S3. Adjusted Rand index analysis of clustering solutions generated by the MCL, k-means, and HCCA algorithms.
Supplemental Table S4. Adjusted Rand index analysis of clustering solutions generated by HCCA using HRR cutoffs.
Supplemental Table S5. Fisher's exact test for enrichment of characterized and essential genes in HCCA n = 3 obtained clusters.
Supplemental Table S6. T-DNA knockout lines and primers used.
Supplementary Material
Acknowledgments
We thank Ms. Christy Hipsley and Drs. Chris Somerville, Alisdair Fernie, and Lothar Willmitzer for useful comments on the manuscript. We also thank Mrs. Anja Fröhlich and Mrs. Anett Döring for technical assistance.
This work was supported by the German Ministry of Education and Research (grant no. 0313924 to O.E.) and the German Research Foundation (grant no. IRTG 1360 to M.S.).
The author responsible for distribution of materials integral to the findings presented in this article in accordance with the policy described in the Instructions for Authors (www.plantphysiol.org) is: Staffan Persson (persson@mpimp-golm.mpg.de).
The online version of this article contains Web-only data.
References
- Albert R (2005) Scale-free networks in cell biology. J Cell Sci 118 4947–4957 [DOI] [PubMed] [Google Scholar]
- Alonso JM, Stepanova AN, Leisse TJ, Kim CJ, Chen H, Shinn P, Stevenson DK, Zimmerman J, Barajas P, Cheuk R, et al (2003) Genome-wide insertional mutagenesis of Arabidopsis thaliana. Science 301 653–657 [DOI] [PubMed] [Google Scholar]
- Aoki K, Ogata Y, Shibata D (2007) Approaches for extracting practical information from gene co-expression networks in plant biology. Plant Cell Physiol 48 381–390 [DOI] [PubMed] [Google Scholar]
- Bader GD, Hogue CW (2003) An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics 4 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baerenfaller K, Grossmann J, Grobei MA, Hull R, Hirsch-Hoffmann M, Yalovsky S, Zimmermann P, Grossniklaus U, Gruissem W, Baginsky S (2008) Genome-scale proteomics reveals Arabidopsis thaliana gene models and proteome dynamics. Science 320 938–941 [DOI] [PubMed] [Google Scholar]
- Barabási AL, Oltvai ZN (2004) Network biology: understanding the cell's functional organization. Nat Rev Genet 5 101–113 [DOI] [PubMed] [Google Scholar]
- Bergmann S, Ihmels J, Barkai N (2004) Similarities and differences in genome-wide expression data of six organisms. PLoS Biol 2 E9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthomé R, Thomasset M, Maene M, Bourgeois N, Froger N, Budar F (2008) pur4 mutations are lethal to the male, but not the female, gametophyte and affect sporophyte development in Arabidopsis. Plant Physiol 147 650–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DM, Zeef LA, Ellis J, Goodacre R, Turner SR (2005) Identification of novel genes in Arabidopsis involved in secondary cell wall formation using expression profiling and reverse genetics. Plant Cell 17 2281–2295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson MR, Zhang B, Fang Z, Mischel PS, Horvath S, Nelson SF (2006) Gene connectivity, function, and sequence conservation: predictions from modular yeast co-expression networks. BMC Genomics 7 40–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daub CO, Steuer R, Selbig J, Kloska S (2004) Estimating mutual information using B-spline functions: an improved similarity measure for analysing gene expression data. BMC Bioinformatics 5 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies DL, Bouldin DW (1979) A cluster separation measure. IEEE Trans Pattern Anal Mach Intell 1 224–227 [PubMed] [Google Scholar]
- DeRisi JL, Iyer VR, Brown PO (1997) Exploring the metabolic and genetic control of gene expression on a genomic scale. Science 278 680–686 [DOI] [PubMed] [Google Scholar]
- Enright AJ, Van Dongen S, Ouzounis CA (2002) An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res 30 1575–1584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores-Pérez U, Sauret-Güeto S, Gas E, Jarvis P, Rodríguez-Concepción M (2008) A mutant impaired in the production of plastome-encoded proteins uncovers a mechanism for the homeostasis of isoprenoid biosynthetic enzymes in Arabidopsis plastids. Plant Cell 20 1303–1315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman TC, Goldovsky L, Brosch M, van Dongen S, Maziere P, Grocock RJ, Freilich S, Thornton J, Enright AJ (2007) Construction, visualization, and clustering of transcription networks from microarray expression data. PLoS Comput Biol 3 2032–2042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi TK, Zhong J, Mathivanan S, Karthick L, Chandrika KN, Mohan SS, Sharma S, Pinkert S, Nagaraju S, Periaswamy B, et al (2006) Analysis of the human protein interactome and comparison with yeast, worm and fly interaction datasets. Nat Genet 38 285–293 [DOI] [PubMed] [Google Scholar]
- Gibbons FD, Roth FP (2002) Judging the quality of gene expression-based clustering methods using gene annotation. Genome Res 12 1574–1581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartigan JA, Wong MA (1979) A k-means clustering algorithm. Appl Stat 28 100–108 [Google Scholar]
- Hirai MY, Sugiyama K, Sawada Y, Tohge T, Obayashi T, Suzuki A, Araki R, Sakurai N, Suzuki H, Aoki K, et al (2007) Omics-based identification of Arabidopsis Myb transcription factors regulating aliphatic glucosinolate biosynthesis. Proc Natl Acad Sci USA 104 6478–6483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubert L, Arabie P (1985) Comparing partitions. J Classification 13 193–218 [Google Scholar]
- Huttenhower C, Flamholz AI, Landis JN, Sahi S, Myers CL, Olszewski KL, Hibbs MA, Siemers NO, Troyanskaya OG, Coller HA (2007) Nearest neighbor networks: clustering expression data based on gene neighborhoods. BMC Bioinformatics 8 250–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihmels J, Levy R, Barkai N (2004) Principles of transcriptional control in the metabolic network of Saccharomyces cerevisiae. Nat Biotechnol 22 86–92 [DOI] [PubMed] [Google Scholar]
- Jeong H, Mason SP, Barabasi AL, Oltvai ZN (2001) Lethality and centrality in protein networks. Nature 411 41–42 [DOI] [PubMed] [Google Scholar]
- Jupiter DC, VanBuren V (2008) A visual data mining tool that facilitates reconstruction of transcription regulatory networks. PLoS One 3 e1717–e1724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King AD, Przulj N, Jurisica I (2004) Protein complex prediction via cost-based clustering. Bioinformatics 20 3013–3020 [DOI] [PubMed] [Google Scholar]
- Kitano H (2002) Systems biology: a brief overview. Science 295 1662–1664 [DOI] [PubMed] [Google Scholar]
- Lamport DT, Kieliszewski MJ, Showalter AM (2006) Salt stress upregulates periplasmic arabinogalactan proteins: using salt stress to analyse AGP function. New Phytol 169 479–492 [DOI] [PubMed] [Google Scholar]
- Li S, Armstrong CM, Bertin N, Ge H, Milstein S, Boxem M, Vidalain PO, Han JD, Chesneau A, Hao T, et al (2004) A map of the interactome network of the metazoan C. elegans. Science 303 540–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma S, Gong Q, Bohnert HJ (2007) An Arabidopsis gene network based on the graphical Gaussian model. Genome Res 17 1614–1625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manfield IW, Jen CH, Pinney JW, Michalopoulos I, Bradford JR, Gilmartin PM, Westhead DR (2006) Arabidopsis Co-expression Tool (ACT): Web server tools for microarray-based gene expression analysis. Nucleic Acids Res 34 W504–W509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mentzen WI, Wurtele ES (2008) Regulon organization of Arabidopsis. BMC Plant Biol 8 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millar AA, Gubler F (2005) The Arabidopsis GAMYB-like genes, MYB33 and MYB65, are microRNA-regulated genes that redundantly facilitate anther development. Plant Cell 17 705–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutwil M, Obro J, Willats WG, Persson S (2008) GeneCAT: novel Webtools that combine BLAST and co-expression analyses. Nucleic Acids Res 36 W320–W326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutwil M, Ruprecht C, Giorgi FM, Bringmann M, Usadel B, Persson S (2009) Transcriptional wiring of cell wall-related genes in Arabidopsis. Mol Plant. 2 1015–1024 [DOI] [PubMed] [Google Scholar]
- Newman MEJ, Girvan M (2004) Finding and evaluating community structure in networks. Phys Rev E Stat Nonlin Soft Matter Phys 69 026113. [DOI] [PubMed] [Google Scholar]
- Obayashi T, Hayashi S, Saeki M, Ohta H, Kinoshita K (2009) ATTED-II provides coexpressed gene networks for Arabidopsis. Nucleic Acids Res 37 D987–D991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obayashi T, Kinoshita K (2009) Rank of correlation coefficient as a comparable measure for biological significance of gene co-expression. DNA Res 16 249–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson S, Wei H, Milne J, Page GP, Somerville CR (2005) Identification of genes required for cellulose synthesis by regression analysis of public microarray data sets. Proc Natl Acad Sci USA 102 8633–8638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prieto C, Risueño A, Fontanillo C, De las Rivas J (2008) Human gene coexpression landscape: confident network derived from tissue transcriptomic profiles. PLoS One 3 e3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramamoorthy V, Cahoon EB, Thokala M, Kaur J, Li J, Shah DM (2009) Sphingolipid C-9 methyltransferases are important for growth and virulence but not for sensitivity to antifungal plant defensins in Fusarium graminearum. Eukaryot Cell 8 217–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schena M, Shalon D, Davis RW, Brown PO (1995) Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 270 467–470 [DOI] [PubMed] [Google Scholar]
- Schmid M, Davison TS, Henz SR, Pape UJ, Demar M, Vingron M, Schölkopf B, Weigel D, Lohmann JU (2005) A gene expression map of Arabidopsis thaliana development. Nat Genet 37 501–506 [DOI] [PubMed] [Google Scholar]
- Srinivasasainagendra V, Page GP, Mehta T, Coulibaly I, Loraine AE (2008) CressExpress: a tool for large-scale mining of expression data from Arabidopsis. Plant Physiol 147 1004–1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinhauser D, Usadel B, Luedemann A, Thimm O, Kopka J (2004) CSB.DB: a comprehensive systems-biology database. Bioinformatics 20 3647–3651 [DOI] [PubMed] [Google Scholar]
- Steuer R, Humburg P, Selbig J (2006) Validation and functional annotation of expression-based clusters based on gene ontology. BMC Bioinformatics 7 380–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart JM, Segal E, Koller D, Kim SK (2003) A gene-coexpression network for global discovery of conserved genetic modules. Science 302 249–255 [DOI] [PubMed] [Google Scholar]
- Thevissen K, Cammue BP, Lemaire K, Winderickx J, Dickson RC, Lester RL, Ferket KK, Van Even F, Parret AH, Broekaert WF (2000) A gene encoding a sphingolipid biosynthesis enzyme determines the sensitivity of Saccharomyces cerevisiae to an antifungal plant defensin from dahlia (Dahlia merckii). Proc Natl Acad Sci USA 97 9531–9536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thevissen K, Idkowiak-Baldys J, Im YJ, Takemoto J, François IE, Ferket KK, Aerts AM, Meert EM, Winderickx J, Roosen J, et al (2005) SKN1, a novel plant defensin-sensitivity gene in Saccharomyces cerevisiae, is implicated in sphingolipid biosynthesis. FEBS Lett 579 1973–1977 [DOI] [PubMed] [Google Scholar]
- Toufighi K, Brady SM, Austin R, Ly E, Provart NJ (2005) The Botany Array Resource: e-northerns, expression angling, and promoter analyses. Plant J 43 153–163 [DOI] [PubMed] [Google Scholar]
- Usadel B, Nagel A, Steinhauser D, Gibon Y, Bläsing OE, Redestig H, Sreenivasulu N, Krall L, Hannah MA, Poree F, et al (2006) PageMan: an interactive ontology tool to generate, display, and annotate overview graphs for profiling experiments. BMC Bioinformatics 18 535–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usadel B, Obayashi T, Mutwil M, Giorgi FM, Bassel GW, Tanimoto M, Chow A, Steinhauser D, Persson S, Provart NJ (2009) Co-expression tools for plant biology: opportunities for hypothesis generation and caveats. Plant Cell Environ 32 1633–1651 [DOI] [PubMed] [Google Scholar]
- Vandepoele K, Quimbaya M, Casneuf T, De Veylder L, Van de Peer Y (2009) Unraveling transcriptional control in Arabidopsis using cis-regulatory elements and coexpression networks. Plant Physiol 150 535–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dongen S (2000) Graph clustering by flow simulation. PhD thesis. University of Utrecht, Utrecht, The Netherlands
- van Noort V, Snel B, Huynen MA (2004) The yeast coexpression network has a small-world, scale-free architecture and can be explained by a simple model. EMBO Rep 5 280–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasserman S, Faust K (1994) Social Network Analysis. Cambridge University Press, Cambridge, UK
- Wei H, Persson S, Mehta T, Srinivasasainagendra V, Chen L, Page GP, Somerville C, Loraine A (2006) Transcriptional coordination of the metabolic network in Arabidopsis. Plant Physiol 142 762–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong R, Ye ZH (2007) Regulation of cell wall biosynthesis. Curr Opin Plant Biol 10 564–572 [DOI] [PubMed] [Google Scholar]
- Zimmermann P, Hirsch-Hoffmann M, Hennig L, Gruissem W (2004) GENEVESTIGATOR: Arabidopsis microarray database and analysis toolbox. Plant Physiol 136 2621–2632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zotenko E, Mestre J, O'Leary DP, Przytycka TM (2008) Why do hubs in the yeast protein interaction network tend to be essential: reexamining the connection between the network topology and essentiality. PLoS Comput Biol 4 e1000140. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.