

Abstract
The aim of the present study was to enhance the dissolution rate of meloxicam (MLX), a practically water-insoluble drug by preparation of solid dispersion using a hydrophilic polymer, poloxamer 188 (PXM). The kneading technique was used to prepare solid dispersions. A 32 full factorial design approach was used for optimization wherein the drug, polymer ratio (X1), and the kneading time (X2) were selected as independent variables and the dissolution efficiency at 60 min (%DE60) and yield percent were selected as the dependent variable. Multiple linear regression analysis revealed that for obtaining higher dissolution of MLX from PXM solid dispersions, a high level of X1 and a high level of X2 were suitable. The use of a factorial design approach helped in optimization of the preparation and formulation of solid dispersion. The optimized formula was characterized by solubility studies, angle of repose, and contact angle; Fourier transform infrared spectroscopy, differential scanning calorimetry, x-ray diffraction studies, and scanning electron microscopy demonstrated that enhanced dissolution of MLX from solid dispersion might be due to a decrease in the crystallinity of MLX and PXM. Analysis of dissolution data of optimized formula indicated the best fitting with Korsemeyer–Peppas model and the drug release kinetics as Fickian diffusion. In conclusion, dissolution enhancement of MLX was obtained by preparing its solid dispersion with PXM using kneading technique.
Key words: factorial design, kneading, poloxamer, poorly water-soluble drug, solid dispersion
INTRODUCTION
Meloxicam (MLX) is an oxicam derivative and a non-steroidal anti-inflammatory drug (NSAID) with anti-inflammatory, antipyretic, and analgesic activities. Unlike traditional nonselective NSAIDs, MLX preferentially inhibits the activity of cyclooxygenase II, resulting in a decreased conversion of arachidonic acid into prostaglandin precursors. The resulting decrease in prostaglandin synthesis is responsible for the therapeutic effects of MLX (1). The chemical structure of MLX is 4-hydroxy-2-methyl-N-(5-methyl-2-thiazolyl)-2H-1, 2-benzothiazine-3-carboxamide-1, 1-dioxide (Fig. 1) (2). The usual oral dosage of MLX in clinical treatment is 7.5–30 mg/day; the elimination half-life period of MLX in plasma is approximately 20 h after oral administration of 15 mg MLX to 24 healthy male volunteers (3). It is very efficient for the treatment of rheumatoid arthritis, osteoarthritis, and other joint diseases (4). Its therapeutic benefits combined with a good gastrointestinal tolerability are well documented in comparison with other NSAIDs; however, like many NSAIDs, MLX is practically insoluble in water (12 µg/ml). The low solubility of MLX and consequently the dissolution result in variations in bioavailability. So, enhancement of dissolution of MLX is useful for acceptable bioavailability.
Fig. 1.

The structure of meloxicam
Many studies have been done to enhance the solubility of MLX by using co-solvents (5) as well as by using other techniques (6). Solid dispersions of many poorly water-soluble drugs by incorporating them into a water-soluble polymer matrix have been considered as an effective method for improving drug dissolution rate and their saturation solubility in the gastrointestinal fluids (7).
Poloxamers are polyoxyethylene-polypropylene block copolymer nonionic surfactants that have been widely used as wetting and solubilizing agents and surface adsorption excipients (8).
They have been employed to enhance the solubility, dissolution, and bioavailability of many poorly water-soluble drugs using various techniques including melting and melting agglomeration (9). For some drugs, the improvement in solubility using poloxamers is higher compared to the other meltable polymers such as polyethylene glycol or complex-forming agents such as cyclodextrins (10). Poloxamer 188 (PXM) is empirically selected to prepare solid dispersions because of its low melting point (about 56–57°C), surfactant properties, and oral safety.
Kneading method is used to prepare valdecoxib-polyvinyl pyrrolidone binary systems, and results show that the dissolution rate of valdecoxib can be enhanced to a great extent by solid dispersion technique using an industrially feasible kneading method (11).
The use of factorial design experiment is an efficient method of indicating the relative significance of a number of variables in the formulation.
In addition, it offers the advantage to provide a way of analyzing the results to decide on most significant variables. Analysis of variance (ANOVA) is generally used, but with factorial design, a maximum outcome can be drawn out of these models with the use of a small number of experiments. In addition, they allow a means of assessing interactions which exist between different variables over the response (12).
MATERIALS AND METHODS
Materials
The MLX B.P. was obtained from Changzhou Longcheng Pharmaceuticals Co., Ltd. PXM was purchased from Sigma-Aldrich. Fine chemicals and all other chemicals/solvents used were of analytical grade and supplied by the Pharmacy Department, Faculty of Medicine, of the University of Malaya.
Method
Preparation of Binary System
Physical mixtures were prepared by mixing accurate weight of MLX with PXM in drug: polymer ratio of 1:2, 1:5, and 1:8 for 5 min using glass mortar and pestle. The physical mixture was triturated using a small volume of ethanol-water (1:1) solution to give a thick paste, which was kneaded at three kneading times, 10, 20, and 30 min, and then dried at 45°C in an oven. The dried mass was pulverized, passed through 30 mesh sieve size, stored in a vacuum desiccator (48 h), and passed through 60 mesh sieve size, then weighed, transferred to amber colored, airtight container, stored at 30 ± 1°C, and the yield was determined using the following formula:
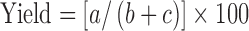 |
1 |
where, a is the weight of the solid dispersion sifted through a number 60 sieve, b is the weight of MLX taken for solid dispersion preparation, and c is the weight of PXM taken for solid dispersion preparation.
When ethanol alone was used for kneading, the thick paste got dried immediately. To avoid drying of the solvent during kneading, ethanol was previously mixed with water (1:1) and then used for the kneading process.
Drug Content
Solid dispersions equivalent to 10 mg of MLX were weighed accurately and dissolved in suitable quantity of methanol. The drug content was analyzed at 362 nm by UV spectrophotometer (Shimadzu, Japan). Each sample was analyzed in triplicate.
Phase-solubility Studies
Solubility measurements were performed in triplicate using the method reported by Higuchi and Connors (13).
An excess amount of MLX of about (20 mg) was added to 20 ml distilled water containing increasing concentrations of the PXM (i.e., 0.1%, 0.25%, 0.5%, 0.75%, and 1% w/v). The flasks were sealed and shaken at room temperature (28°C) for 48 h on a shaker, and the samples were filtered through a 0.22-µm cellulose nitrate membrane filter. The filtrate was suitably diluted and analyzed spectrophotometrically (Shimadzu, Japan) at 362 nm, a wavelength at which PXM does not interfere.
Experimental Design
A 32 full factorial design was used to systematically study the influence of the individual and combined effect of independent variables X1 and X2 on the dependent variables percent dissolution efficiency at 60 min (%DE60) and yield percent.
In this design, two factors are evaluated, each at three levels, and experimental trials are performed at all nine possible combinations (14).
Statistical model incorporating interactive and polynomial terms is used to evaluate the response.
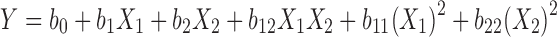 |
2 |
where, Y is the dependent variable, b0 is the arithmetic mean response of the nine runs, and bi is the estimated coefficient for the factor Xi. The main effects (X1 and X2) represent the average result of changing one factor at a time from its low to high value. The interaction terms (X1X2) show how the response changes when two factors are simultaneously changed. The polynomial terms [(X1)2 and (X2)2] are included to investigate nonlinearity. The composition of the factorial design batches C1 to C9 is shown in Table I.
Table I.
Composition of Factorial Design Batches
| Batch code | Variables levels in coded | %DE60 ± SDa | %Yield ± SD | |
|---|---|---|---|---|
| X 1 | X 2 | |||
| C1 | −1 | −1 | 32.83 ± 0.61 | 69.7 ± 0.80 |
| C2 | −1 | 0 | 34.75 ± 0.43 | 72.1 ± 1.05 |
| C3 | −1 | +1 | 36.98 ± 0.50 | 75.8 ± 1.21 |
| C4 | 0 | −1 | 39.63 ± 0.27 | 92.4 ± 0.85 |
| C5 | 0 | 0 | 40.98 ± 0.83 | 91.3 ± 1.05 |
| C6 | 0 | +1 | 42.41 ± 0.29 | 93.6 ± 0.75 |
| C7 | +1 | −1 | 42.91 ± 0.74 | 80.4 ± 0.60 |
| C8 | +1 | 0 | 44.00 ± 0.63 | 85.0 ± 0.40 |
| C9 | +1 | +1 | 44.86 ± 0.44 | 89.0 ± 0.29 |
| Actual values | ||||
| Coded terms | ||||
| −1 | 1:2 | 10 | ||
| 0 | 1:5 | 20 | ||
| +1 | 1:8 | 30 | ||
X 1 drug to polymer ratio, X 2 kneading time, %DE 60 dissolution efficiency at 60 min
aValues represent the mean ±SD of three experiments
Determination of Solubility
MLX and solid dispersions equivalent to 15 mg of MLX were added to 20 ml distilled water in screw-capped test tubes, vortexed for 2 min, and shaken at 28°C room temperature for 24 h. Resultant samples containing undissolved solid dispersions suspended in the test medium were centrifuged at 10,000 rpm for 5 min, and the clear supernatants obtained were filtered (0.22 µm membrane filter), suitably diluted with distilled water, and analyzed by spectrophotometer (Shimadzu, Japan) at 362 nm.
In Vitro Dissolution Studies
The dissolution study was performed using two media, distilled water and British Pharmacopeia buffer media.
In distilled water, in vitro dissolution studies of MLX, physical mixture, and solid dispersion were carried out using USP paddle method by dispersed powder technique (15). Sample equivalent to 15 mg of MLX was added to 900 ml distilled water containing 0.25% w/v sodium lauryl sulfate at 37 ± 0.5°C and stirred at 50 rpm. An aliquot of 5 ml was withdrawn at different time intervals with a syringe filter (pore size, 0.22 mm). The withdrawn volume was replenished immediately with the same volume of the pre-warmed (37°C) dissolution medium in order to keep the total volume constant. The filtered samples were suitably diluted, if necessary, and assayed spectrophotometrically at 362 nm. The mean of at least three determinations was used to calculate the drug release.
-
2.
In (British Pharmacopeia) dissolution medium, the procedure complies with the dissolution test for tablets and capsules in British Pharmacopeia, using Apparatus II. A medium of 900 ml of a buffer prepared was used by dissolving 13.61 g of potassium dihydrogen orthophosphate in 800 ml of water, then the pH was adjusted to 7.5 with 0.5 M sodium hydroxide, then sufficient water was added to produce 1,000 ml, and the paddle was rotated at 50 revolutions per minute. A 5-ml sample of the medium was withdrawn and filtered. The absorbance of the filtrate was measured, diluted with the dissolution medium if necessary, and assayed spectrophotometrically at 362 nm using dissolution medium in the reference cell.
Drug release data were appropriately corrected for losses of drug and dissolution medium volume during sampling by replacement using the following equation (16):
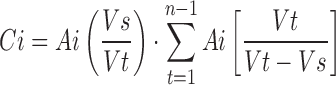 |
3 |
where, Ci is the corrected absorbance of ith observation, Ai is the observed specific absorbance, Vs is the sample volume, and Vt is the total volume of dissolution medium.
Percent dissolution efficiency (%DE) was also computed to compare the relative performance of various carriers in solid dispersion formulations (17). The magnitude of %DE at 60 min (%DE 60 min) for each formulation was computed as the percent ratio of area under the dissolution curve up to the time, t, to that of the area of the rectangle described by 100% dissolution at the same time
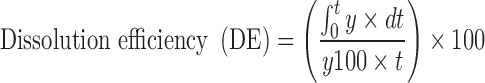 |
4 |
Similarity factor (f2) was calculated utilizing Moore and Flanner independent mathematical approach to compare the dissolution profile (18).
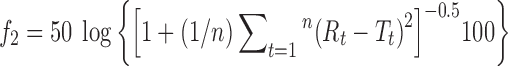 |
5 |
where Rt and Tt are the cumulative percentage dissolved at each of the selected n time points of the reference and test product, respectively.
Angle of Repose
To get an idea about flowability properties of the solid dispersions, angle of repose for optimized formula of experimental design was determined. If the angle exceeds 50°, the material will not flow satisfactorily, whereas materials having values near the minimum flow easily and well. The rougher and more irregular the surface of the particles, the higher is the angle of repose (19). The angle of repose was measured by passing solid dispersions through a sintered glass funnel of internal diameter 27 mm on the horizontal surface. The height (h) of the heap formed was measured with a cathetometer, and the radius (r) of the cone base was also determined. The angle of repose (Φ) was calculated from Eq. 6.
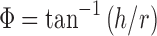 |
6 |
Contact Angle
Sample powder from pure drug or PXM or optimized formula of solid dispersion (300 mg) was compressed into a pellet by a hydraulic press at 5,000 kg/cm2 pressure (1 min). Water (20 µl) was placed from a microsyringe on the pellet surface, and the drop was photographed after 3 s for the determination of contact angle as described previously (20).
Differential Scanning Calorimetry
Thermal analyses were performed using differential scanning calorimeter (DSC-6-Perkin-Elmer). Under nitrogen flow of 20 ml/min, approximately 2 mg of MLX, PXM 188, physical mixture, or solid dispersion was placed in a sealed aluminum pan and heated at a scanning rate of 10°C/min from 30°C to 300°C. An empty aluminum pan was used as reference.
Fourier Transform Infrared Spectroscopy
Fourier transform infrared (FT-IR) spectroscopy was employed to characterize the possible interactions between the drug and the carrier in the solid state on an FT-IR spectrophotometer (Perkin-Elmer) by the conventional KBr pellet method. The spectra were scanned over a frequency range of 4,000–500 cm−1.
Scanning Electron Microscopy (SEM)
The scanning electron microscopy (SEM) analysis was carried out using scanning electron microscope (JSM 6100, Jeol, Japan). Samples of pure drug and the optimized formula of solid dispersion formulation were mounted onto the stubs using double-sided adhesive tape and then coated with a thin layer of gold palladium alloy (150–200A°). The scanning electron microscope was operated at an acceleration voltage of 20 KV, working distance (12–14 mm). The selected magnification was ×500.
Powder X-ray Diffraction Analysis (XRD)
Powder X-ray diffraction (XRD) patterns were recorded using D8 advance x-ray diffractometer Bruker AXS under the following conditions: target  monochromatized radiation, voltage 40 KV, and current 40 mA at ambient temperature. The data were collected in the continuous scan mode using a step size of 0.01° at 2θ/s. The scanned range was 5–50°.
monochromatized radiation, voltage 40 KV, and current 40 mA at ambient temperature. The data were collected in the continuous scan mode using a step size of 0.01° at 2θ/s. The scanned range was 5–50°.
Mechanism of Dissolution
In vitro drug release data were fitted to various release kinetic models (21) viz. zero-order, first-order, Higuchi, Hixson–Crowell cube root, and Korsemeyer–Peppas model employing the following set of Eqs. (7–11):
- Zero-order model
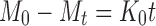
7 - First-order model
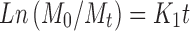
8 - Higuchi model
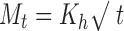
9 - Hixson–Crowell cube root model
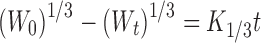
10 - Korsemeyer–Peppas model
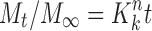
11
where Mo, Mt, and M1 correspond to the drug amount taken at time equal to zero, dissolved at a particular time, t, and at infinite time, respectively. The terms Wo and Wt refer to the weight of the drug taken initially and at time t, respectively.
Various other terms viz. ko, k1, Kh, k1/3, and Kk refer to the release kinetic constants obtained from the linear curves of zero-order, first-order, Higuchi, Hixson–Crowell cube root, and Korsemeyer–Peppas model, respectively.
RESULTS AND DISCUSSION
Drug Content
The drug content of the prepared solid dispersions was found to be in the range of 98.7–102.3% which is due to acceptable uniformity of content of the prepared solid dispersions.
Phase-solubility Studies
The solubility of MLX increase with the increase in polymer concentration (Fig. 2), which shows that a linear increase in drug solubility with increased carrier level, with R2 values of 0.9941, giving AL type solubility diagram (13). Similar results have been recorded about many drugs using hydrophilic polymers, due to formation of soluble complexes and/or cosolvent effect of carrier (22).
Fig. 2.

Effects of increasing concentration of poloxamer 188 on solubility of meloxicam. (R 2, 0.9941)
Results of Factorial Design
Preliminary experiments for preparation of solid dispersions indicated that factors X1 and X2 are effective variables on the in vitro dissolution and yield percent so were used for further systematic studies.
The %DE60 for the nine batches (C1 to C9) showed variation of 32.83% to 44.86% (Table I). The data clearly indicate that X1 and X2 strongly influence the %DE60 and yield percent. All the batches of factorial design exhibited yield greater than 69.7%.
The statistical evaluation of dependent variables was performed by using Design-Expert version 7.0.0 software. The regression analysis results (p value) of the variables on percentage dissolution efficiency at 60 min and percent of yield of solid dispersion are shown in Table II. The ANOVA results of solid dispersions are shown in Table III. According to p value, full model or reduced model can be selected, so in the present study, full model having both significant and non-significant p values was used in obtaining dependent variables because the fitness of full model to the system is better than reduced model. The coefficients for the equations representing the quantitative effect of the independent variables on percentage dissolution efficiency at 60 min and percent of yield of solid dispersion are shown in Table II. The equations for each polymer can be generated by putting values of coefficients in Eq. 2 in terms of coded factors.
Table II.
Results of Regression Analysis
| Response | Coefficients estimates | R 2 | |||||
|---|---|---|---|---|---|---|---|
| b 0 | b 1 | b 2 | b 12 | b 11 | b 22 | ||
| %DE60 | 40.99 | 4.54 | 1.48 | −0.55 | −1.62 | 0.031 | 0.9997 |
| p value | <0.0001 | <0.0001 | 0.0032 | 0.0004 | 0.7540 | ||
| %Yield | 91.98 | 6.13 | 2.65 | 0.63 | −13.77 | 0.68 | 0.9790 |
| p value | 0.0061 | 0.0572 | 0.6027 | 0.0029 | 0.6844 | ||
Table III.
The Results of Analysis of Variance
| Response | df (1,3) | SS | MS | F | R 2 | |
|---|---|---|---|---|---|---|
| %DE60 | Regression | 5 | 143.07 | 28.61 | 1,805.99 | 0.9997 |
| Error | 3 | 0.048 | 0.016 | |||
| %Yield | Regression | 5 | 649.38 | 27.95 | 27.95 | 0.9790 |
| Error | 3 | 13.94 | 4.65 |
Coefficients with one factor indicate the effect of that particular factor, while the coefficients with more than one factor and those with second-order terms represent the interaction between those factors and the quadratic nature of the phenomena, respectively. Positive sign of the term indicates positive (additive) effect, while negative sign indicates negative (antagonistic) effect of the factor on the response. It can be concluded from the equations that x1 (drug: polymer ratio) shows the larger positive effect than term x2 (kneading time) on percentage dissolution efficiency at 60 min and yield percent.
The quadratic terms of x1 and x2 also had effect on percentage dissolution efficiency at 60 min and yield percent.
Figures 3 and 4 show the contour plots and response surface plots for percentage dissolution efficiency at 60 min and yield percent, respectively. The contour lines indicated that the higher the polymer and longer kneading time, the more significant is the dissolution enhancement. However, for yield percent, decrease at higher polymer ratio was observed, which may be attributed to difficulty of sieving when higher polymer ratio was used.
Fig. 3.

Contour plots showing a percentage dissolution efficiency at 60 min; b percentage yield
Fig. 4.

Response surface plots showing a percentage dissolution efficiency at 60 min; b percentage yield
The reliability of the equations that describes the influence of factors on percentage dissolution efficiency at 60 min and yield percent was assessed by preparing two additional check points solid dispersions (batch C10 and batch C11) in triplicate using the amount of x1 and x2 −0.5 and +0.33 level (23). The experimental values and predicted values of each response are shown in Table IV. Equation 12 was used to calculate the percentage relative error between predicted values and experimental values of each response.
Table IV.
Validation of Model Obtained Using Experimental and Predicted Results of Checkpoint Batches
| Batch code | Variables | %DE60 predicted | %DE60 ± SD experimental | %Relative error | %Yield predicted | %Yield ± SD experimental | %Relative error | |
|---|---|---|---|---|---|---|---|---|
| X 1 | X 2 | |||||||
| C10 | −0.5 | +0.33 | 38.89 | 40.54 ± 0.65 | 0.042 | 86.31 | 83.54 ± 1.45 | 0.032 |
| C11 | +0.5 | −0.33 | 42.46 | 39.78 ± 0.78 | 0.063 | 90.68 | 94.34 ± 1.32 | 0.040 |
The percentage relative error obtained from checkpoint batches was in the range of 0.032 to 0.063. Low values of the relative error show that for both factors, there is a reasonable agreement of predicted values and experimental values. This proves the validity of model and confirms the effects of drug: polymer ratio and the kneading time on percentage dissolution efficiency at 60 min and yield percent.
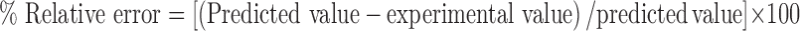 |
12 |
The optimized formula suggested by the factorial design was of drug: polymer ratio of 1:7.03 and 30min the time of kneading. Hence, this formula was prepared and characterized further.
Characterization
In Vitro Dissolution Studies
The dissolution profiles of optimized formulation (1:7.03 drug to polymer ratio), physical mixture (1:7.03 ratio), and pure drug are shown in Fig. 5. It is clearly evident from the figure that the dissolution efficiency of pure drug (19.81) and physical mixture (25.95) are very low as compared with the optimized formulation (44.45). The yield percent of the optimized formula was 93.6.
Fig. 5.

Dissolution profiles of optimized formula, physical mixture, and pure drug (n = 3, SD for all points in range of ±0.03–0.076)
The release of drug was analyzed also according to method B (using buffer solution). Similarity factor was calculated to compare both dissolution profiles of optimized formula, and the value was 79.85, which means that there was no significant variation between dissolution profile in water and buffer media.
Determination of Solubility
Solubility of MLX from optimized solid dispersion was increased from its 12.45 µg/ml aqueous solubility to 214.14 µg/ml.
Angle of Repose
The angle of repose for optimized formula of solid dispersion was found to be of mean 39.4 ± 0.26 which indicates acceptable free flowability.
Contact Angle
Wettability of solid dispersion (43°) was significantly improved compared with pure drug (78°).
Differential Scanning Calorimetry
Figure 6 shows the DSC curve of MLX, PXM, and optimized formula of solid dispersion. The MLX, PXM, optimized formula of solid dispersion at time of preparation and optimized formula of solid dispersion after 3 months show endothermic peak at 257.279°C, 56.123°C, 55.501°C, and 55.952°C, respectively. The endothermic peak corresponding to melting point of MLX is absent in the DSC thermogram of optimized formula of solid dispersion. It might be due the presence of the amorphous form of MLX in the solid dispersion. Moreover, this amorphous form was stable after 3 months as shown in the last thermogram.
Fig. 6.

Differential scanning calorimetry thermograms of meloxicam, poloxamer, optimized formula of solid dispersion at time of preparation, and optimized formula of solid dispersion after 3 months
Fourier Transform Infrared Spectroscopy
Figure 7 shows the spectrum of MLX, PXM, physical mixture, and optimized formula of solid dispersion. The spectrum of MLX shows characteristic peaks at 3,285.68 cm−1 (N-H stretching vibrations), 1,611.29 cm−1 (C = N stretching vibrations), and 1,168.49 cm−1 (S = O stretching vibrations), respectively. The PXM exhibits characteristic peaks at 3,465.32, 2,887.94, and 1,103.25 cm−1 due to stretching of O-H, C-H, and C-O groups. The spectrum of physical mixture was equivalent to the addition of spectrum of the drug and polymer indicating no interaction occurring in physical mixing. The spectrum of solid dispersion exhibited significant decrease in intensity of N-H stretching vibrations and C = N stretching vibrations which may be due to intermolecular hydrogen bonding. The spectra peaks of drug are almost unchanged in the optimized formula of solid dispersion which indicates that the overall symmetry of molecule is not significantly affected (24).
Fig. 7.

Fourier transform infrared images of solid system: a meloxicam, b poloxamer, c physical mixture, and d SD-optimized formula
Scanning Electron Microscopy (SEM)
The SEM images for pure drug and solid dispersion are shown in Fig. 8. Pure drug image shows crystalline rectangular shapes, whereas an image of solid dispersion of drug does not show any crystalline material.
Fig. 8.

Scanning electron microscopy images of meloxicam and optimized formula of solid dispersion
Powder X-ray Diffraction Analysis (XRD)
XRD patterns of MLX, PXM, and solid dispersion are shown in Fig. 9. In the x-ray diffractograms of MLX, sharp peaks at a diffraction angle (2θ) of 13°, 15°, 18.5°, and 26° indicate the presence of crystalline drug, while solid dispersion shows sharp peaks at 19° and 23.5°. These data reveal that the typical drug crystalline peaks were still detectable (with reduced intensity and less number) in the solid dispersion. This finding confirms the presence of little amount of crystalline drug in the solid dispersion despite the complete disappearance of its melting peak in the corresponding DSC curves; however, the sharp drug peaks corresponding to drug are absent in the solid dispersion. The XRD of solid dispersion exhibits peaks less than the sum of the number of peaks of MLX and PXM in their pure forms. This suggests that crystallinity of both drug and polymer is reduced in the solid dispersion.
Fig. 9.

Powder x-ray diffraction spectra of meloxicam, polymer (poloxamer), and optimized formula of solid dispersion
Decrease in crystallinity of the drug and polymer may contribute to enhancement of dissolution of the drug (25).
Mechanism of Dissolution
Table V lists the regression parameters obtained after fitting various release kinetic models to the in vitro dissolution data.
Table V.
Fitting of Drug Release from Optimized Solid Dispersion to Various Release Kinetic Models
| Mechanism | Zero-order | First-order | Higuchi | Hixson–Crowel | Korsemeyer | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Slope | r 2 | Slope | r 2 | Slope | r 2 | Slope | r 2 | Slope | r 2 | |
| Optimized formula | 0.3724 | 0.6559 | −0.003 | 0.8169 | 0.0878 | 0.9197 | 0.0064 | 0.8761 | 0.2091 | 0.9463 |
The goodness of fit for various models investigated for binary systems ranks in the order of Korsemeyer–Peppas > Higuchi > Hixson–Crowell cube root law > first-order > zero-order. The Korsemeyer–Peppas model describes drug release kinetics in the most befitting manner. The values of diffusional exponent “n” was obtained from the slopes of the fitted Korsemeyer–Peppas model. The solid dispersion tended to exhibit Fickian diffusional characteristics, as the corresponding values of n were lower than the standard value for declaring Fickian release behavior, the results point out the prevalence of diffusional mechanistic phenomena.
Conclusions
The results of the experimental study confirm that the factors X1 and X2 significantly influence the dependent variables %DE60 and yield percent. Characterization studies reveal that solid dispersion of MLX-PXM shows enhancement of MLX dissolution due to the conversion of MLX into a less crystalline and/or amorphous form. The application of experimental design techniques to the optimization of formulation helps in reaching the optimum point in the shortest time with minimum effort.
Acknowledgment
This research was supported by Department of Pharmacy, Faculty of medicine, University of Malaya by providing the facilities and requirements of research. I wish to thank all people that help during this project especially Prof. Kadhum A.H., Asst. Prof. Javary H.A., and Asst. Prof. Abdulla M.A.
References
- 1.Pairet M, van Ryn J, Schierok H, Mauz A, Trummlitz G, Engelhardt G. Differential inhibition of cyclooxygenases-1 and -2 by meloxicam and its 40-isomer. Inflamm Res. 1998;47(6):270–6. doi: 10.1007/s000110050329. [DOI] [PubMed] [Google Scholar]
- 2.Lugar P, Daneck K, Engel W, Trummlitz G, Wagner K. Structure and physicochemical properties of meloxicam, a new NSAID. Eur J Pharm Sci. 1996;4:175–87. doi: 10.1016/0928-0987(95)00046-1. [DOI] [Google Scholar]
- 3.Lipscomb GR, Wallis N, Armstrong G, Rees WD. Gastrointestinal tolerability of meloxicam and piroxicam: a double-blind placebo-controlled study. Br J Clin Pharmacol. 1998;46(2):133–7. doi: 10.1046/j.1365-2125.1998.00761.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Türck D, Roth W, Busch U. A review of the clinical pharmacokinetics of meloxicam. Br J Rheumatol. 1996;35(Suppl 1):13–6. doi: 10.1093/rheumatology/35.suppl_1.13. [DOI] [PubMed] [Google Scholar]
- 5.Naidu NB, Chowdary KP, Murthy KV, Satyanarayana V, Hayman AR, Becket G. Physicochemical characterization and dissolution properties of meloxicam-cyclodextrin binary systems. J Pharm Biomed Anal. 2004;35(1):75–86. doi: 10.1016/j.jpba.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 6.Vijaya Kumar SG, Mishra DN. Preparation and evaluation of solid dispersion of meloxicam with skimmed milk. Yakugaku Zasshi. 2006;126(2):93–7. doi: 10.1248/yakushi.126.93. [DOI] [PubMed] [Google Scholar]
- 7.Vijaya Kumar SG, Mishra DN. Preparation, characterization and in vitro dissolution of solid dispersion of meloxicam with PEG6000. Yakugaku Zasshi. 2006;126(8):657–64. doi: 10.1248/yakushi.126.657. [DOI] [PubMed] [Google Scholar]
- 8.Collett JH, Popli H, Kibbe AH, editors. Poloxamer. In: Handbook of Pharmaceutical Excipients, 3rd ed. London, UK: Pharmaceutical Press; 2000. p. 386–8.
- 9.Hang Y, Myung-Kwan C, Hoo-Kyun C. Preparation and characterization of piroxicam/poloxamer solid dispersion prepared by melting method and solvent method. J Korean Pharm Sci. 2007;37:1–5. doi: 10.4333/KPS.2007.37.1.001. [DOI] [Google Scholar]
- 10.Chutimaworapan S, Ritthidej GC, Yonemochi E, Oguchi T, Yamamoto K. Effect of water-soluble carriers on dissolution characteristics of nifedipine solid dispersions. Drug Dev Ind Pharm. 2000;26(11):1141–50. doi: 10.1081/DDC-100100985. [DOI] [PubMed] [Google Scholar]
- 11.Modi A, Tayade P. Enhancement of dissolution profile by solid dispersion (kneading) technique. AAPS PharmSciTech. 2006;7(3):68. doi: 10.1208/pt070368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elkheshen AS, Abdel-Gawad NA, Badawi AA. Factorial design and computer optimisation of a reconstitutable suspension of erythromycin ethyl succinate. Die Pharm Ind. 1997;59(5):439–43. [Google Scholar]
- 13.Higuchi T, Connors KA. “Phase-solubility techniques” in advances in analytical chemistry and instrumentation. New York: Reilly CN; 1965. pp. 117–212. [Google Scholar]
- 14.Bolton S, Charles S. Pharmaceutical statistics. New York, NY: Marcel Dekker Inc.; 2004. [Google Scholar]
- 15.Chiou WL, Riegelman S. Pharmaceutical applications of solid dispersion systems. J Pharm Sci. 1971;60:1281–302. doi: 10.1002/jps.2600600902. [DOI] [PubMed] [Google Scholar]
- 16.Singh B, Kaur T, Singh S. Correction of raw dissolution data for loss of drug and volume during sampling. Indian J Pharm Sci. 1997;59:196–9. [Google Scholar]
- 17.Khan KA. The concept of dissolution efficiency. J Pharm Pharmacol. 1975;27:48–9. doi: 10.1111/j.2042-7158.1975.tb09378.x. [DOI] [PubMed] [Google Scholar]
- 18.Gohel MC, Panchal MK. Novel use of similarity factor f2 and Sd for the development of diltiazem HCl modified-release tablets using a 32 factorial design. Drug Dev Ind Pharm. 2002;28:77–87. doi: 10.1081/DDC-120001488. [DOI] [PubMed] [Google Scholar]
- 19.McKenna A, McCafferty DF. Effect of particle size on the compaction mechanism and tensile strength of tablets. J Pharm Pharmacol. 1982;34(6):347–51. doi: 10.1111/j.2042-7158.1982.tb04727.x. [DOI] [PubMed] [Google Scholar]
- 20.Imai T, Nishiyama T, Ueno M, Otagiri M. Enhancement of the dissolution rates of poorly water-soluble drugs by water-soluble gelatin. Chem Pharm Bull. 1989;37(8):2251–2. doi: 10.1248/cpb.37.2251. [DOI] [PubMed] [Google Scholar]
- 21.Merchant HA, Shoaib HM, Tazeen J, Yousuf RI. Once-daily tablet formulation and in vitro release evaluation of cefpodoxime using hydroxypropyl methylcellulose: a technical note. AAPS PharmSciTech. 2006;7(3):78. doi: 10.1208/pt070378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cirri M, Mura P, Rabasco AM, et al. Characterization of ibuproxam binary and ternary dispersions with hydrophilic carriers. Drug Dev Ind Pharm. 2004;30(1):65–74. doi: 10.1081/DDC-120027513. [DOI] [PubMed] [Google Scholar]
- 23.Mashru RC, Sutariya VB, Sankalia MG, Sankalia JM, Parikh PP. Development and evaluation of fast dissolving film of salbutamol sulphate. Drug Dev Ind Pharm. 2005;31:25–34. doi: 10.1081/ddc-43947. [DOI] [PubMed] [Google Scholar]
- 24.Ambike AA, Mahadik KR, Paradkar A. Stability study of amorphous valdecoxib. Int J Pharm. 2004;282:151–62. doi: 10.1016/j.ijpharm.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 25.Heo MY, Piao ZZ, Kim TW, Cao QR, Kim A, Lee BJ. Effect of solubilizing and microemulsifying excipients in polyethylene glycol 6000 solid dispersion on enhanced dissolution and bioavailability of ketoconazole. Arch Pharm Res. 2005;28(5):604–11. doi: 10.1007/BF02977766. [DOI] [PubMed] [Google Scholar]