

Abstract
The present work was aimed at the influence of ethanol on the complex formation of hydroxypropyl-β-cyclodextrin (HP-β-CD) with oleanolic acid (OA) and ursolic acid (UA), two insoluble isomeric triterpenic acids. Phase solubility studies were carried out to evaluate the solubilizing power of HP-β-CD, in association with ethanol, toward OA and UA. A mathematical model was applied to explain and predict the solubility of OA and UA influenced by HP-β-CD and ethanol. The solid complexes were prepared by evaporating the filtrate of samples which was prepared in different complexing media. The solubility of OA is much higher than that of UA in all the tested aqueous solutions. The solubility of OA and UA can be increased over 900 and 200 times, respectively, by forming complex with HP-β-CD. Ethanol (0.5%, v/v) can help the formation of OA-HP-β-CD complex, but is harmful to the formation of UA-HP-β-CD complex. Increasing solubility in water can be achieved by adding ethanol into the complexing media, but the concentration of ethanol should be optimized. The ring E of the chemical compounds has a great influence on the complexing process.
Key words: ethanol, hydroxypropyl-β-cyclodextrin, oleanolic acid, solubilization, ursolic acid
INTRODUCTION
General
Oleanolic acid (OA) and ursolic acid (UA) are pentacyclic triterpenoid isomers (see Fig. 1). The difference between the two isomers is only the location of one methyl group on ring E, but they show a great difference in physicochemical properties. Both OA and UA have numerous pharmacological effects such as hepatoprotection and chemotherapy, but their clinical use is limited by their poor aqueous solubility (1).
Fig. 1.

Chemical structures of OA and UA
It has been estimated that approximately 40% of the new chemical entities failed development because of poor bioavailability which is often associated with aqueous insolubility (2), so the solubilization of water-insoluble compounds is becoming more and more important in pharmaceutical preparations. Solubilization can be achieved by pH adjustment, particle size reduction, salt formation, solid dispersion, complexation, cosolvency, micellization, or a combination effect of any of the above (3).
Cyclodextrins (CDs) are capable of forming inclusion complexes with many drugs. This can result in improved chemical stability, an increase in the apparent aqueous solubility, and higher bioavailability without changing their pharmacokinetic properties (4–8). CDs have mainly been used as complexing agents in the pharmaceutical industry to increase aqueous solubility of poorly water-soluble drugs (9,10). However, the efficiency of complexation is often not very high, and therefore, relatively large amounts of CDs must be used to obtain the desired effect (11). On the other hand, for a series of reasons (e.g., including relatively high cost, possible toxicity, problems of bulk formulation, etc.), pharmaceutical dosage forms should contain as small amounts of CDs as possible (12). It is therefore important to develop methods which can be applied in order to enhance the efficiency of drug–CD complexation.
Cosolvency, the addition of water-miscible solvents to an aqueous system, is a powerful and popular method of increasing the equilibrium solubility of non-polar drugs in aqueous vehicles (13). It is reported that addition of cosolvents into the complexation media can enhance the complexation efficacy and solubility in water (14). Some researchers have established a mathematical equation to describe the combined effect of cosolvent and CD (15). In this article, the influence of ethanol on the complexation of HP-β-CD with OA and UA was evaluated according to phase solubility method, the data were fitted to the established mathematical model, and the solid complexes were also prepared.
The Solubilization and Complexation of CD
CD can form inclusion complexes with many chemical compound molecules by taking up the molecule or some lipophilic moiety of the molecule into their central cavity in aqueous solutions. No covalent bonds are formed or broken during the complexing process, and drug molecules in the complex are equilibrium with free molecules rapidly in the aqueous solution (16).
Higuchi and Connors (17) have classified complexes based on their effect on substrate solubility as indicated by phase solubility profiles. A-type phase solubility profiles are obtained when the solubility of the substrate (drug) increases with increasing ligand (CD) concentration. When the complex is first-order with respect to ligand and first- or higher order with respect to substrate, then AL-type phase solubility profiles are obtained. If the complex is first-order with respect to the substrate but second- or higher order with respect to the ligand, then Ap-type phase solubility profiles are obtained. AN-type phase solubility profiles can be difficult to interpret. B-type phase solubility profiles indicate formation of complexes with limited solubility in the aqueous complexation medium. In general, the water-soluble CD derivatives form A-type phase solubility profiles, and the most common type of complex is formed by one CD molecule and one chemical compound molecule (AL-type). In the AL-type phase solubility profiles, the total apparent drug solubility increases with the increasing concentration of cyclodextrion, and the linear dependence of the solubility of drugs on CD is described in Eq. 1:
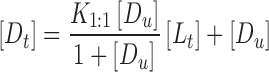 |
1 |
where [Dt] is the total apparent drug solubility, [Du] is the intrinsic drug solubility, K1:1 is the equilibrium constant for the binary complex, and [Lt] is the concentration of cyclodextrion.
According to Eq. 1, the intrinsic solubility ([Du]) should be equal to the intercept of the phase solubility profile (Sint), but there are numerous exemptions of this (Fig. 2). A profile with a positive deviation at low CD concentrations will cause 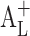 -type phase solubility profile, and a profile with a negative deviation at low CD concentrations will cause
-type phase solubility profile, and a profile with a negative deviation at low CD concentrations will cause 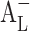 -type phase solubility profile (18).
-type phase solubility profile (18).
Fig. 2.

Linear phase solubility profiles. A normal profile (A
L); a profile with a positive deviation at low CD concentrations (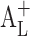 ); and a profile with a negative deviation at low CD concentrations (
); and a profile with a negative deviation at low CD concentrations (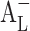 )
)
The value of the stability constant (K1:1) is used to compare the affinity of drugs for different CD or CD derivatives, and the stability constant (K1:1) of the complex can be calculated from the slope of phase solubility profile and the intrinsic solubility ([Du]) of the drug in the aqueous complexation media according to Eq. 2:
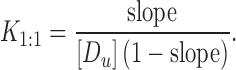 |
2 |
For poorly soluble drugs (aqueous solubility <0.1 mmol/L), the intrinsic solubility ([Du]) is in general much larger than the intercept of the phase solubility profile (Sint), resulting in nonlinearity of otherwise linear (AL-type) phase solubility profile. This can lead to an erroneous K1:1 value. A more accurate method for the determination of the solubilizing efficiency of CD is to determine their complexation efficiency (CE), i.e., the concentration ratio between CD in a complex and free CD. It is independent of both [Du] and Sint and more reliable. CE is calculated from the slope of the phase solubility profiles according Eq. 3:
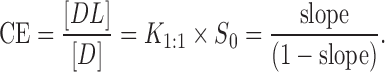 |
3 |
There is no correlation between the slope of the phase solubility profile and the deviation of Sint from [Du]. It is not clear why the intercept of the phase solubility profile is below [Du]; it could be due to the non-ideality of water as a solvent.
The Solubilization of Cosolvent
Cosolvent can raise the solubility of lipophilic compounds by destroying the hydrogen bond or reducing the polarity of aqueous solution (19). The exponential dependence of the solubility of non-polar drugs on cosolvent concentration in a cosolvent–water mixture is described in log-linear form as Eq. 4:
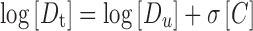 |
4 |
where σ is the cosolvent solubilization power for the particular cosolvent–solute system and [C] is the volume fraction of the cosolvent in the aqueous mixture.
The Combined Effect of Cosolvent and CD
He et al. (15) established a mathematical model which can describe and explain the combination effect of cosolvent and HP-β-CD on the solubilization of Fluasterone. The total concentration of Fluasterone in aqueous solution consisted of free Fluasterone, Fluasterone–CD binary complex, and Fluasterone–cosolvent–CD ternary complex. The total drug solubility as a function of both the ligand and cosolvent concentrations is described in Eq. 5:
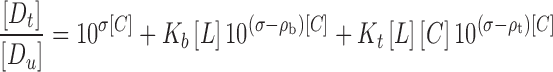 |
5 |
where Kb and Kt are the formation constants for the binary and ternary complexes, respectively; ρb and ρt are the cosolvent destabilizing powers for the binary complex and ternary complexes, respectively, i.e., the effect of the cosolvent on Kb and Kt as described by Li et al. (20).
MATERIALS AND METHODS
Materials
OA (QDGSXC-060218, purity >98%) and UA (XGSBS-060330, purity >98%) were obtained from Shanxi Huisheng Medicament Technology Corporation Limited. Hydroxypropyl-β-cyclodextrin (HP-β-CD, KLEPTOSE® HP, MS = 0.85) with an average molecular weight of 1,481 was kindly provided by Roquette Cyclodextrin Technologies Development Incorporation. High-performance liquid chromatography (HPLC) grade methanol was purchased from Shanghai Ludu Reagent Company. Ethanol (AR grade) was purchased from Sinopharm Chemical Reagent Co., Ltd. Acetic acid (AR grade) was purchased from Shanghai Lingfeng Chemical Reagent Co., Ltd. All water used was deionized water. HPLC system consisted of a pump (Shimadzu LC-10AT), a detector (SPD-10A), and a reverse-phase column (Dikma Kromasil, 100 A C18 5 μm 250 × 4.6 mm). The weight measurements were performed with an AY-220 electronic analytic weighting scale (Shimadzu, Japan). Constant Temperature Shaker (Hua Li Da HZ-9310K Refrigerator Shakers, Tai Cang Shi Ke Jiao Qi Cai Chang, China) was used in this experiment.
Methods
The HPLC Assay Method of OA and UA
The concentration of OA and UA was determined by HPLC method with UV detection at 210 nm (21). Dikma Kromasil (100 A C18 5 μm 250 × 4.6 mm) was used as the stationary phase. The column temperature was controlled at 30°C, the flow rate was 1.0 mL/min, and the sample injection volume was 20 μL.
Phase Solubility Studies
Phase solubility studies were carried out according to the method previously reported by Higuchi and Connors (17). A series of mixed solvents containing ethanol (0–80%, v/v) and HP-β-CD (0–60 mmol/L) were obtained by adding ethanol and HP-β-CD into distilled water. The phase solubility samples were prepared by adding excess OA or UA into 10 mL mixed solvent in a test tube. The samples were filtered with a 0.45-μm membrane after keeping shaking at 25°C for 48 h in Constant Temperature Shaker. The filtrates were diluted for proper times and then analyzed by HPLC.
Data Processing
When the concentration of CD ([L]) is 0, Eq. 5 can be rearranged to 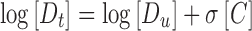 , and σ can be calculated according to the slope and intercept of the phase solubility profile. When the concentration of ethanol ([C]) is 0, Eq. 5 can be rearranged to
, and σ can be calculated according to the slope and intercept of the phase solubility profile. When the concentration of ethanol ([C]) is 0, Eq. 5 can be rearranged to 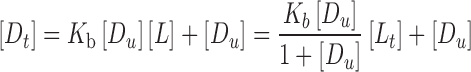 , and Kb can be calculated according to the slope and intercept of the phase solubility profile. When the concentration of ethanol is very low ([C] = 0.5%), the existence of ethanol does not influence the complexation process, and ρb and ρt can be supposed to be 0, so Eq. 5 can be rearranged to
, and Kb can be calculated according to the slope and intercept of the phase solubility profile. When the concentration of ethanol is very low ([C] = 0.5%), the existence of ethanol does not influence the complexation process, and ρb and ρt can be supposed to be 0, so Eq. 5 can be rearranged to 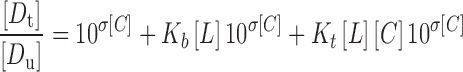 , and Kt can be calculated according to this equation. Then, ρb and ρt are calculated by a nonlinear regression process. Nonlinear regression results are obtained from DUD method using SAS software.
, and Kt can be calculated according to this equation. Then, ρb and ρt are calculated by a nonlinear regression process. Nonlinear regression results are obtained from DUD method using SAS software.
Preparation of Solid Samples
The isomers and HP-β-CD with 1:1 molar ratio were accurately weighed, then the mixtures were added into a series of complexing media containing ethanol (0%, 0.5%, 30%, 80%, v/v). The samples were stirred thoroughly for 6 h at 40°C. Filtration (0.45-μm membrane filter) was performed to remove undissolved drugs. The resulting solutions were evaporated under vacuum in a rotary evaporator until dryness. Then, the drug loading dosage and solubility of solid samples were detected. Drug loading dosage detection of solid complexes was carried out by extracting the drugs from 0.05 g solid complexes using 5 mL methanol, and then the concentration of the drugs were detected using the established HPLC method. Solubility detection of solid complexes was carried out by detecting the filtrate of complexes in water.
RESULTS
The HPLC Assay Method of OA and UA
Linear relationships between the peak areas and the concentrations of the two isomers were obtained: y = 8797.3x − 1300.7 (R2 = 0.9999, OA) and y = 7596.3x + 1724.1 (R2 = 0.9999, UA), where x is the concentration (μg/mL) and y is the peak area (mV s); R2 is the assessment coefficient. The concentrations of OA and UA from 1 to 40 μg/mL could be calculated by the established HPLC assay method.
Phase Solubility Studies
The solubility of OA and UA in different solvent systems is listed in Table I. Some measured values are missing because of the low apparent solubility ([Dt] <1 μg/mL). According to Table I, the solubility of OA is much higher than the solubility of UA in the same medium. The solubility of OA and UA in ethanol solutions with or without HP-β-CD is shown in Fig. 3a, b, respectively. According to Fig. 3a, the solubility of OA increases slightly at a low concentration of ethanol (0.5%, v/v) except for in the solvent containing 5 mmol/L HP-β-CD; the solubility of OA in the solvent containing 5 mmol/L HP-β-CD and 0.5% ethanol (v/v) is a little lower than the solubility of OA in 5 mmol/L HP-β-CD aqueous solution. After that, the solubility of OA decreases and then increases with the increasing concentration of ethanol. According to Fig. 3b, UA is a little different from OA; the slight increase of solubility is not shown when the concentration of ethanol is 0.5% (v/v). At a certain HP-β-CD level, the lowest solubility of both isomers is obtained in the complexing media containing 10% or 20% ethanol (v/v); the concentration of both isomers decreases and then approximating log-linear increases with the increasing concentration of ethanol.
Table I.
Solubility of OA and UA in Different Solvent Systems
| [C] (%, v/v) | [L] (mmol/L) | [D t] (mmol/L) | |
|---|---|---|---|
| OA | UA | ||
| 0 | 5 | 0.0880 | 0.0178 |
| 0 | 10 | 0.2099 | 0.0336 |
| 0 | 20 | 0.5120 | 0.0863 |
| 0 | 40 | 1.2645 | 0.2026 |
| 0 | 60 | 2.1518 | 0.3845 |
| 0.5 | 5 | 0.0840 | – |
| 0.5 | 10 | 0.2101 | 0.0113 |
| 0.5 | 20 | 0.5200 | 0.0596 |
| 0.5 | 40 | 1.2942 | 0.1990 |
| 0.5 | 60 | 2.3051 | 0.3801 |
| 5 | 5 | 0.0178 | – |
| 5 | 10 | 0.0437 | – |
| 5 | 20 | 0.1416 | 0.0192 |
| 5 | 40 | 0.6146 | 0.0512 |
| 5 | 60 | 1.0351 | 0.0760 |
| 10 | 5 | 0.0122 | – |
| 10 | 10 | 0.0284 | – |
| 10 | 20 | 0.0841 | – |
| 10 | 40 | 0.2760 | 0.0183 |
| 10 | 60 | 0.4400 | 0.0368 |
| 20 | 5 | 0.0152 | – |
| 20 | 10 | 0.0362 | – |
| 20 | 20 | 0.0882 | – |
| 20 | 40 | 0.2499 | 0.0172 |
| 20 | 60 | 0.4076 | 0.0315 |
| 40 | 0 | 0.0310 | 0.0085 |
| 40 | 5 | 0.0847 | 0.0151 |
| 40 | 10 | 0.1278 | 0.0162 |
| 40 | 20 | 0.2243 | 0.0211 |
| 40 | 40 | 0.4265 | 0.0319 |
| 40 | 60 | 0.7526 | 0.0477 |
| 60 | 0 | 0.4548 | 0.3051 |
| 60 | 5 | 0.4935 | 0.3532 |
| 60 | 10 | 0.5307 | 0.3561 |
| 60 | 20 | 0.6235 | 0.3979 |
| 60 | 40 | 0.8922 | 0.4754 |
| 60 | 60 | 1.1267 | 0.5686 |
| 80 | 0 | 3.4537 | 2.4564 |
| 80 | 5 | 3.6232 | 2.9685 |
| 80 | 10 | 3.8212 | 3.0515 |
| 80 | 20 | 4.1001 | 3.3876 |
| 80 | 40 | 4.9727 | 3.7864 |
| 80 | 60 | 5.7494 | 4.1173 |
[C] concentration of ethanol, [L] concentration of HP-β-CD, [D t] total drug in the solution, – represents [D t] < 1 μg/mL
Fig. 3.

a The solubility of OA in ethanol solutions with or without HP-β-CD. b The solubility of UA in ethanol solutions with or without HP-β-CD
The phase solubility profiles of OA and UA in HP-β-CD solutions with increasing concentration of ethanol are shown in Fig. 4a, b, respectively. All the phase solubility profiles can be considered to be AL-type (some UA phase solubility profiles only have two to three points). At a certain concentration of ethanol, the solubility of the isomers linear increases with the increasing concentration of HP-β-CD, and some of the phase solubility profiles show negative intercept deviation. CE of the two isomers calculated according to Fig. 4a, b is listed in Table II. The CE of both isomers is higher in 0.5% ethanol than in water. After that, the CE decreases and then increases with the increasing concentration of ethanol. The CE change trend of UA is similar to that of OA, but OA and UA show quiet different CE in the same condition; the CE of OA is much higher than that of UA in the same complexing media.
Fig. 4.

a The phase solubility profiles of OA in HP-β-CD solutions with increasing concentration of ethanol. b The phase solubility profiles of UA in HP-β-CD solutions with increasing concentration of ethanol
Table II.
Assessment Coefficients of Phase Solubility Profiles and Calculated CE
| [C] (%, v/v) | OA | UA | ||
|---|---|---|---|---|
| R 2 | CE | R 2 | CE | |
| 0 | 0.9936 | 0.0392 | 0.9812 | 0.0066 |
| 0.5 | 0.9896 | 0.0420 | 0.9890 | 0.0075 |
| 5 | 0.9812 | 0.0196 | 0.9947 | 0.0014 |
| 10 | 0.9907 | 0.0082 | 1.0000 | 0.0009 |
| 20 | 0.9933 | 0.0074 | 1.0000 | 0.0007 |
| 40 | 0.9839 | 0.0120 | 0.9833 | 0.0006 |
| 60 | 0.9959 | 0.0119 | 0.9922 | 0.0040 |
| 80 | 0.9973 | 0.0405 | 0.9863 | 0.0217 |
[C] concentration of ethanol, R 2 assessment coefficients of phase solubility profiles
Data Processing of Phase Solubility Studies
All the measured values were processed by SAS software. The parameters of the model are listed in Table III, and the fitting 3D graphs of combined effects of ethanol and HP-β-CD on solubilization of the isomers are shown in Fig. 5a, b, respectively. The assessment coefficients (R2) of OA and UA are 0.8670 and 0.7659, respectively. The change trend of the fitting 3D graphs is similar to that of Fig. 3a, b. According to the mathematical model, the increase of the solubility at low ethanol concentration is caused by the forming of drug–CD–cosolvent ternary complex, and the following decrease is due to the dissociation of drug–CD binary complex; the solubility of drugs increases as log-linear model because of the solubilization effect of cosolvent. According to Table III, σ of OA is lower than that of UA, which means that the interaction between UA and ethanol is stronger than the interaction between OA and ethanol. The higher ρb and ρt values of UA than OA represent that ethanol affects more the UA and HP-β-CD complexing process than the OA and HP-β-CD complexing process. The solubility of OA is much higher than the solubility of UA in the same aqueous solution, but Kb and Kt of OA are lower than Kb and Kt of UA. That is because the intrinsic complexation constants (Kb and Kt) are inversely proportional to the initial solubility, and the calculated initial solubility of OA is much higher than that of UA. We can find an increasing solubilization effect of HP-β-CD at a low concentration of ethanol in this model, which indicates that the solubilization effect can be increased by adding a little cosolvent into aqueous solution.
Table III.
Parameters of the Model
| Parameter and symbol | Calculated parameters | |
|---|---|---|
| OA | UA | |
Intrinsic binary complexation constant (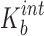 , L/mol) , L/mol) |
125.57 | 174.07 |
Intrinsic ternary complexation constant (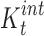 , L/mol) , L/mol) |
1740.17 | 4218.35 |
| Cosolvent solubilizing power (σ) | 0.0512 | 0.0615 |
| Cosolvent destabilizing power on binary complex (ρ b) | 9.3773 | 27.3151 |
| Cosolvent destabilizing power on ternary complex (ρ t) | 0.8839 | 0.9339 |
| Assessment coefficient (R 2) | 0.8670 | 0.7659 |
Fig. 5.

a Combined effects of ethanol and HP-β-CD on solubilization of OA. b Combined effects of ethanol and HP-β-CD on solubilization of UA
Preparation of Solid Samples
Fine powder was collected by evaporating the filtrate of samples, which is prepared in different complexing media, under vacuum in a rotary evaporator. The drug loading dosage and solubility of complexes prepared in different complexing media are listed in Table IV. The lowest drug loading dosage and solubility are obtained in the complexing media containing 30% (v/v) ethanol. A remarkable increase in the drug loading dosage of complexes is obtained in the complexing media containing 80% (v/v) ethanol, but the aqueous solubility decreases significantly at the same time. Adding 0.5% (v/v) ethanol into the complexing media increases both the drug loading dosage and aqueous solubility of OA-HP-β-CD complex; the solubility of OA can be increased more than 900 times. However, the addition of 0.5% ethanol (v/v) into the complexing media decreases both drug loading dosage and aqueous solubility of UA-HP-β-CD complex.
Table IV.
Drug Loading Dosage and Solubility of Complexes Prepared in Different Complexing Media
| Complexing media | OA | UA |
|---|---|---|
| Water | 1.35% (754.02 μg/mL) | 0.41% (220.88 μg/mL) |
| 0.5% Ethanol (v/v) | 1.37% (920.65 μg/mL) | 0.18% (200.68 μg/mL) |
| 30% Ethanol (v/v) | 0.43% (<1 μg/mL) | 0.04% (<1 μg/mL) |
| 80% Ethanol (v/v) | 6.30% (359.11 μg/mL) | 4.82% (32.75 μg/mL) |
Drug loading dosage of complexes (w/w) is listed outside the parentheses, and solubility of the complexes is listed inside the parentheses
DISCUSSION
The solubility of OA and UA in distilled water without the addition of CD and ethanol were too low to be detected ([Du] <1 μg/mL), so [Du] of the two isomers were calculated from Eq. 4. Poorly soluble drugs frequently show negative intercept deviation, i.e., Sint < [Du], resulting in 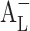 -type. For example, cinnarizine has a sharp
-type. For example, cinnarizine has a sharp 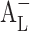 -type phase solubility profile that can be mistaken for Ap-type profile of cinnarizine/CD 1:2 complex (22). However, cinnarizine is a very lipophilic [log Poctanol/water 5.8 (23)] and water-insoluble compound ([Du] = 1 μg/mL at pH 11.9) with large aromatic planar regions or physicochemical characteristics that promote self-association in aqueous solutions (24). Self-association of the drug molecules and the drug–CD complexes, as well as non-inclusion complexation, can also lead to
-type phase solubility profile that can be mistaken for Ap-type profile of cinnarizine/CD 1:2 complex (22). However, cinnarizine is a very lipophilic [log Poctanol/water 5.8 (23)] and water-insoluble compound ([Du] = 1 μg/mL at pH 11.9) with large aromatic planar regions or physicochemical characteristics that promote self-association in aqueous solutions (24). Self-association of the drug molecules and the drug–CD complexes, as well as non-inclusion complexation, can also lead to 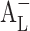 -type phase solubility profiles (25). CE is calculated from the slope of the phase solubility profiles; it is independent of both S0 and Sint and more reliable when the influences of different pharmaceutical excipients on the solubilization are being investigated, so the CE was used to evaluate the solubilizing efficiency of CD instead of stability constant.
-type phase solubility profiles (25). CE is calculated from the slope of the phase solubility profiles; it is independent of both S0 and Sint and more reliable when the influences of different pharmaceutical excipients on the solubilization are being investigated, so the CE was used to evaluate the solubilizing efficiency of CD instead of stability constant.
OA showed a slight increase of solubility at a low ethanol concentration (0.5%, v/v) in the phase solubility study, but UA showed a little decrease. The fitting 3D graph of OA is consistent with the phase solubility profiles, but the fitting 3D graph of UA is different from the phase solubility profiles at 0.5% ethanol (v/v). The different change trend of solubility of OA and UA at 0.5% ethanol (v/v) may be caused by the different binding affinity of ethanol to the isomers. According to Fig. 3, b, when the concentration of ethanol increased from 0.5% to 20% (v/v), the concentration of both isomers is approximating a log-linear decrease (R2 > 0.9). The decreased solubility of both isomers may be caused by the dissociation of the drug–CD binary complex or the competitive binding of ethanol to HP-β-CD molecules (26,27). And the log-linear increased solubility of the two isomers (R2 > 0.9) after that is caused by the solubilization effect of ethanol (19).
The decreased drug loading dosage and solubility of solid complexes prepared in 30% ethanol (v/v) is obtained by the competitive binding of ethanol to HP-β-CD molecules. The increased drug loading dosage of complexes prepared in 80% ethanol (v/v) is obtained by increasing the free drug moleculars in the samples, but the interaction between the isomers and HP-β-CD is weakened as the medium becomes more apolar, so the solubility of the isomers decreases. The samples of OA-HP-β-CD complex prepared in 0.5% ethanol (v/v) increase both the drug loading dosage (from 1.35% to 1.37%) and aqueous solubility (from 754.02 to 920.65 μg/mL), but the samples of UA-HP-β-CD complex prepared in 0.5% ethanol (v/v) are quite different. The drug loading dosage of UA-HP-β-CD complex, prepared in 0.5% ethanol, decreases from 0.41% to 0.18%, and the aqueous solubility decreases from 220.88 to 200.68 μg/mL. The different phenomena indicate that 0.5% ethanol (v/v) existing in the complexing media can help the formation of OA-HP-β-CD complex, but is harmful to the formation of UA-HP-β-CD complex. The increasing of drug loading dosage and solubility of solid complex samples prepared in 0.5% ethanol may be attributed to the forming of drug–CD–cosolvent ternary complex (15) and increasing aqueous solubility of OA in the complexing media, (19) which may be helpful to the formation of complex. The interaction between UA and ethanol is stronger than the interaction between OA and ethanol, so the existence of ethanol, even 0.5% in the complexing media, will lead to the dissociation of UA-HP-β-CD binary complex rather than formation of drug–CD–cosolvent ternary complex.
CONCLUSION
The CE of the two isomers can be increased by adding a little ethanol into complexing medium, but the addition amount should be optimized. The mathematical model can find application in describing and explaining the process of complexing or predicting the solubilization of OA and UA. The solubility of OA and UA can be increased over 900 and 200 times, respectively, by forming complex with HP-β-CD. Adding 0.5% (v/v) ethanol into complexing media can help the formation of OA-HP-β-CD complex, but is harmful to the formation of UA-HP-β-CD complex. Although the difference between the two compounds is only the location of one methyl group, quite different phenomena have been shown in this study, which indicates that the complex process is affected greatly by the structure of the ring E.
Acknowledgments
This work was supported by Nanjing Medical University (project no. 2005NYDZD12) and Jiangsu provincial Institute of Traditional Chinese Medicine (project no. KF2005004). Natural Science Fund Project of Colleges in Jiangsu Province (project no. 07KJD350152), Jiangsu provincial Bureau of Traditional Chinese Medicine (project no. HL07068). The authors are grateful to Lu Wang from School of Public Health Nanjing Medical University for her help on data processing. Thanks to the Department of Biotechnology School of Basic Medical Science Nanjing Medical University for their help during the experimental work.
Footnotes
Rui Li and Peng Quan have contributed equally to the work.
References
- 1.Liu J. Oleanolic acid and ursolic acid: research perspectives. J Ethnopharmacol. 2005;100:92–94. doi: 10.1016/j.jep.2005.05.024. [DOI] [PubMed] [Google Scholar]
- 2.Prentis RA, Lis Y, Walker SR. Pharmaceutical innovation by the seven UK-owned pharmaceutical companies (1964–1985) Br J Clin Pharmacol. 1988;25:387–396. doi: 10.1111/j.1365-2125.1988.tb03318.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Loftsson T, Jarho P, Másson M, Järvinen T. Cyclodextrins in drug delivery. Expert Opin Drug Deliv. 2005;2:335–351. doi: 10.1517/17425247.2.1.335. [DOI] [PubMed] [Google Scholar]
- 4.Ozkan Y, Atay T, Dikmen N, Isimer A, Aboul-Enein HY. Improvement of water solubility and in vitro dissolution rate of gliclazide by complexation with β-cyclodextrin. Pharm Acta Helv. 2000;74:365–370. doi: 10.1016/S0031-6865(99)00063-1. [DOI] [PubMed] [Google Scholar]
- 5.Tommasini S, Raneri D, Ficarra R, Calabro ML, Stancanelli R, Ficarra P. Improvement in solubility and dissolution rate of flavonoids by complexation β-cyclodextrin. J Pharm Biomed Anal. 2004;35:379–387. doi: 10.1016/S0731-7085(03)00647-2. [DOI] [PubMed] [Google Scholar]
- 6.Hirayama F, Uekama K. Cyclodextrin-based controlled drug release system. Adv Drug Deliv Rev. 1999;36:125–141. doi: 10.1016/S0169-409X(98)00058-1. [DOI] [PubMed] [Google Scholar]
- 7.Hara T, Arima H, Hirayama F, Uekama K. Enhanced bioavailability of a new thiazolidine derivative FPFS-410, an antidiabetic and lipid-lowering drug, after oral administration of its hydroxypropyl-β-cyclodextrin complex to bile duct-cannulated rats. J Pharm Sci. 2006;95:1771–1782. doi: 10.1002/jps.20655. [DOI] [PubMed] [Google Scholar]
- 8.Wang L, Jiang X, Xu W, Li C. Complexation of tanshinone IIA with 2-hydroxypropyl-b-cyclodextrin: effect on aqueous solubility, dissolution rate, and intestinal absorption behavior in rats. Int J Pharm. 2007;341:58–67. doi: 10.1016/j.ijpharm.2007.03.046. [DOI] [PubMed] [Google Scholar]
- 9.Loftsson T, Brewster ME. Pharmaceutical applications of cyclodextrins. 1. Drug solubilization and stabilization. J Pharm Sci. 1996;85:1017–1025. doi: 10.1021/js950534b. [DOI] [PubMed] [Google Scholar]
- 10.Yang CQ, Ding LY, Lin YL, Wu HY, Wang C. The phase-solubility study of the enhanced solubility of oleanolic acid by inclusion with HP-β-CD. Chinese Traditional Patent Medicine. 2005;27:888–889. [Google Scholar]
- 11.Loftsson T, Másson M. The effects of water-soluble polymers on cyclodextrins and cyclodextrin solubilization of drugs. J Drug Deliv Sci Technol. 2004;14:35–43. [Google Scholar]
- 12.Loftsson T, Másson M, Sigurjónsdóttir JF. Methods to enhance the complexation efficiency of cyclodextrins. ST P Pharma Sci. 1999;9:237–242. [Google Scholar]
- 13.Rytting E, Lentz KA, Chen XQ, Qian F, Venkatesh S. A quantitative structure–property relationship for predicting drug solubility in PEG 400/water cosolvent systems. Pharm Res. 2004;21:237–244. doi: 10.1023/B:PHAM.0000016237.06815.7a. [DOI] [PubMed] [Google Scholar]
- 14.Bodor N, Drustrup J, Wu W. Effect of cyclodextrins on the solubility and stability of a novel soft corticosteroid, loteprednol etabonate. Pharmazie. 2000;55:206–209. [PubMed] [Google Scholar]
- 15.He Y, Li P, Yalkowsky SH. Solubilization of fluasterone in cosolvent/cyclodextrin combinations. Int J Pharm. 2003;264:25–34. doi: 10.1016/S0378-5173(03)00389-2. [DOI] [PubMed] [Google Scholar]
- 16.Brewster ME, Loftsson T. Cyclodextrins as pharmaceutical solubilizers. Adv Drug Deliv Rev. 2007;59:645–666. doi: 10.1016/j.addr.2007.05.012. [DOI] [PubMed] [Google Scholar]
- 17.Higuchi T, Connors KA. Phase-solubility techniques. Adv Anal Chem Instrum. 1965;4:117–212. [Google Scholar]
- 18.Loftsson T, Hreinsdóttir D, Másson M. Evaluation of cyclodextrin solubilization of drugs. Int J Pharm. 2005;302:18–28. doi: 10.1016/j.ijpharm.2005.05.042. [DOI] [PubMed] [Google Scholar]
- 19.Millard JW, Alvarez-nunez FA, Yalkowsky SH. Solubilization by cosolvents: establishing useful constants for the log-linear model. Int J Pharm. 2002;245:153–166. doi: 10.1016/S0378-5173(02)00334-4. [DOI] [PubMed] [Google Scholar]
- 20.Li P, Zhao L, Yalkowsky SH. Combined effect of cosolvent and cyclodextrin on solubilization of nonpolar drugs. J Pharm Sci. 1999;88:1107–1111. doi: 10.1021/js990159d. [DOI] [PubMed] [Google Scholar]
- 21.Quan P, Liu DF, Li R, Zhang Q, Qian Y, Xu QW. The effects of water-soluble polymers on hydroxypropyl-β-cyclodextrin solubilization of oleanolic acid and ursolic acid. J Incl Phenom Macro. 2009;63:181–188. doi: 10.1007/s10847-008-9505-6. [DOI] [Google Scholar]
- 22.Okimoto K, Rajewski RA, Uekama K, Jona JA, Stella VJ. The interaction of charged and uncharged drugs with neutral (HP-β-CD) and anionically charged (SBE7-β-CD) β-cyclodextrins. Pharm Res. 1996;13:256–264. doi: 10.1023/A:1016047215907. [DOI] [PubMed] [Google Scholar]
- 23.Moffat AC, Osselton MD, Widdop B, editors. Clarke’s analysis of drugs and poisons. London: Pharmaceutical Press; 2004.
- 24.Yalkowsky SH. Solubility and solubilization in aqueous media. Washington: American Chemical Society; 1999. [Google Scholar]
- 25.Loftsson T, Másson M, Brewster ME. Self-association of cyclodextrins and cyclodextrin complexes. J Pharm Sci. 2004;93:1091–1099. doi: 10.1002/jps.20047. [DOI] [PubMed] [Google Scholar]
- 26.García-Río L, Hervés P, Leis JR, Mejuto JC, Pérez-Juste J, Rodríguez-Dafonte P. Evidence for complexes of different stoichiometries between organic solvents and cyclodextrins. Org Biomol Chem. 2006;4:1038–1048. doi: 10.1039/b513214b. [DOI] [PubMed] [Google Scholar]
- 27.Martin C, Sanchez F, Jimenez R, Prado R, Perez-Tejeda P, Lopez-Cornejo P. Salt and solvent effects on the kinetics and thermodynamics of the inclusion of the ruthenium complex [Ru(NH3)5(4,4′-bpy)]2+ in β-cyclodextrin. J Phys Chem B. 2006;110:12959–12963. doi: 10.1021/jp060659c. [DOI] [PubMed] [Google Scholar]