

Abstract
The aim of the present study was to explore the cogrinding technique as a tool to slow down the drug release from capsule formulations. To this end, the physical mixtures of theophylline–magnesium stearate were prepared and subjected to different milling times (1, 15, 30, 120 min). In order to investigate the effect of magnesium stearate concentration on drug release, various concentrations of magnesium stearate (1%, 3%, 5%, and 10%, w/w) were used. The dissolution rate of the drug from coground samples and physical mixtures were determined at pH 6.5 according to USP. The results showed that all coground formulations showed slower release rates than their physical mixture counterparts. The effect of cogrinding time on the drug release was complex. Cogrinding time had no significant effect on drug release when the amount of magnesium stearate was 1% (w/w). When the amount of magnesium stearate was increased from 1% to 3% and cogrinding time increased from 1 to 5 min, there was a significant reduction in drug release. Beyond 5-min cogrinding, the drug release increased again. For coground samples containing 5% or 10% (w/w) magnesium stearate, generally, the highest drug release was obtained at higher cogrinding time. This was due to a significant increase in surface area of particles available for dissolution as proven by scanning electron microscopy results. Fourier transform infrared and differential scanning calorimetry results ruled out any significant interaction between theophylline and magnesium stearate in solid state.
Key words: cogrinding, dissolution, grinding time, magnesium stearate, ratio of drug to carrier, solid-state characterization
INTRODUCTION
Cogrinding is widely performed for reducing the particle size of powdered poorly water-soluble drugs with the aim of enhancing their dissolution rates and, consequently, their bioavailability (1,2). Cogrinding is a process where an active substance and a hydrophilic or hydrophobic carrier in powder form are dry-coground in a mill. At the end of the cogrinding process, the binary (sometimes ternary if there is more than one carrier) composition obtained is unloaded from the mill and optionally sieved to eliminate possible aggregates. This process works by the two components that are the active ingredient and the carrier closely being absorbed into one another at the nano or microcrystalline level. Several papers reported the favorable effect of cogrinding with polymers such as microcrystalline cellulose, lactose, and chitosan on the dissolution properties and bioavailability of crystalline drugs (1–7). The increase in bioavailability after micronization of drugs, which is most often performed by jet-milling or ball mill, is well known, and the technique has been applied to a variety of poorly water-soluble drugs (2,6,8–11). For example, cogrinding of indomethacin in the presence of hydrophilic carriers (PEG) caused a considerable increase in the dissolution rate of indomethacin (8). In another study, cogrinding used as an effective technique in increasing the dissolution of carbamazepine due to reduced crystallinity, as seen in X-ray pattern, enhanced wettability, and decreased particle size reported that the cogrinding with some excipients is a powerful tool to speed up the dissolution of poorly water-soluble drugs without converting the drug to the amorphous form or changing the particle size (2). Yang et al. (12) showed a slower dissolution rate of prednisone compared to its crystalline form. An improvement in the dissolution rate and bioavailability of indomethacine by interaction with silica was reported by Watanabe et al. (13) Most of the previous studies on cogrinding reported a dramatic increase in the dissolution rate of drug amorphized by cogrinding technique with carrier (14,15). The product obtained from the cogrinding process can be packaged into sachets as powder form with pharmaceutically acceptable excipients. In addition, it can be incorporated into other formulation such as capsules, suppositories, and transdermal plasters.
To the best of our knowledge, there is little information in literature showing that cogrinding technique can be employed as a tool to produce efficient sustained release formulations (16). Therefore, in the present study, an attempt was made to explore the use of ball mill (grinding technique) as a tool to sustain the drug release from binary mixtures of theophylline–magnesium stearate. The effect of milling time and concentration of magnesium stearate ranging 0–10% (w/w) on drug release from capsule formulations were investigated. The solid state of binary mixture of drug–magnesium stearate before and after milling was also studied.
MATERIALS AND METHODS
Materials
Theophylline powder (anhydrous form) with melting point of 271–273°C and magnesium stearate were purchased from Sigma Pharmaceuticals, USA and Acrose, UK, respectively. Empty gelatine capsules (Capsugel, USA) were used to encapsulate the different prepared powder mixtures and coground powders.
Preparation of Coground Formulations
Pure theophylline powder and magnesium stearate with various concentrations (1%, 3%, 5%, 10%, w/w) were charged into the chamber (made of stainless steel) of the vibration ball mill (Fritsch, Germany) with maximum capacity of 250 ml such that all core formulations contained 10 g theophylline. Ten stainless steel balls (size, mass, and density of each ball were 2 cm, 32.3 g, and 7.72 g/cm3, respectively) were added in the ball mill chamber so that the total volume of powder mixture and balls equalled one third the volume of the ball mill chamber. The powder mixture was then ground at 400 rpm for 1, 5, 15, 30, and 120 min. The samples were collected, labelled, and stored in glass vial before use.
Preparation of Physical Mixtures
For comparison purposes, the physical mixtures of drug/magnesium stearate were prepared by simply mixing the drug powder and magnesium stearate powder with the same percentages that have been used for coground formulations in the turbular blender (Copley, UK). The mixture was subjected to different mixing times, 1, 3, 5, 15, 30, and 120 min. The samples were collected, labelled, and stored in glass vial before use.
Preparation of Capsules
The mass of selected samples for dissolution study was 100 mg for the pure theophylline powder and 101, 103, 105, and 110 mg for the coground and physical mixtures formulations with 1%, 3%, 5%, and 10% magnesium stearate, respectively, such that each sample contained 100 mg theophylline. The accurately weighed sample was poured into the empty gelatine capsules size 0, the cap replaced tightly, and then stored in a corresponding labelled vial before use.
In Vitro Release Study
The dissolution of theophylline from coground samples, physical mixtures, and untreated pure drug powders were measured at least in triplicate using a USP 27 standard dissolution apparatus I (Erweka DT-6R, Germany), basket method, rotating at 100 rpm maintained at 37°C in 900 ml phosphate buffer solution (pH 6.5). Capsules equivalent to 100 mg of theophylline were placed into 900 ml of dissolution media. From the dissolution flask, samples were withdrawn at selected time intervals (10, 20, 30, 40, 50, 60, 70, 80, 90, 100, 110, 120, 135, 150, 165, 180, 195, 210, 225, 240, 270, 300, 330, 360, 390, 420, 450, 480, 510, 540, 570, and 600 min) using a peristaltic pump, and the concentrations of theophylline in the samples were determined by UV spectrophotometer at 271 nm.
DSC Studies
A differential scanning calorimeter (DSC7, Mettler Toledo, Switzerland) was used to measure enthalpy and melting point of different formulations. The equipment was calibrated using indium and zinc. Samples were heated ranging 25–350°C at a scanning rate of 10°C/min in aluminum pans under nitrogen gas. The onsets of the melting points and enthalpies of fusion were calculated by the software (Mettler).
FT-IR Studies
Fourier transform infrared (FT-IR) spectra were used to investigate any interaction between theophylline and magnesium stearate during the grinding process. These spectra were taken by FT-IR (Perkin Elmer, USA) equipment for pure theophylline powder, pure magnesium stearate powder, coground, and physical mixtures. The scanning range was 450–4,000 cm−1, and the resolution was 1 cm−1.
SEM Studies
Electron micrographs of coground and physical mixtures of drug and magnesium stearate were obtained using a scanning electron microscope (Philips XL 20, Eindhoven, Netherlands) operating at 15 kV. The specimens were mounted on a metal stub with double-sided adhesive tape and coated under vacuum with gold in an argon atmosphere prior to observation.
Dissolution Parameters
The dissolution efficiency (DE) of a pharmaceutical dosage form is defined as the area under the dissolution curve up to the time, t, expressed as the percentage of the area of the rectangle (17).
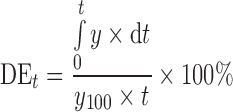 |
1 |
where y is the percent of drug dissolved at time t.
The mean dissolution rate (MDR) and mean dissolution time (MDT) were calculated according to the following equations:
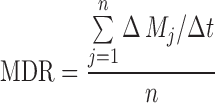 |
2 |
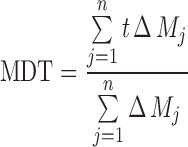 |
3 |
where j is the sample number, n is the number of dissolution sample times, Δt is the time at midpoint between t and t − 1 (easily calculated with (t + t − 1)/2), and ΔMj is the additional amount of drug dissolved between t and t − 1.
RESULTS AND DISCUSSION
It can be anticipated that two key parameters might control the drug release from coground samples: amount of hydrophobic excipient (carrier) and cogrinding (milling) time. In this study, magnesium stearate was chosen as the retarding excipient because of its high hydrophobicity and low melting point (about 110°C). The results of the effect of magnesium stearate concentration on drug release from the samples subjected to different cogrinding times are shown in Fig. 1a–d. It is clear that as concentration of magnesium stearate increases, the amount of drug release from the samples decreases. This could be due to the presence of more hydrophobic particles, magnesium stearate, around the drug particles at high concentrations. For example, samples containing 1% magnesium stearate coground for 1 min released 100% of the drug within 1 h (Fig. 1a), whereas the sample containing 10% (w/w) magnesium stearate released only 12% of the drug within 1 h (Fig. 1a). Similar results were obtained for the samples coground for 5, 15, and 30 min, which are apparent from Fig. 1. It has already been shown that in order to prepare theophylline sustained release tablets with approximately 90% drug release within 10 h, at least 30% HPMC K4M should be incorporated into tablet formulations (18). To prepare theophylline sustained release powders/granules embedded in hard gelatine capsules, even more manufacturing processes are needed. The present results showed that the presence of 5% magnesium stearate coground with theophylline powder could be enough to produce similar release pattern as sustained release tablets when the coground powders are embedded in hard gelatine capsules. Since magnesium stearate is highly hydrophobic excipient, it has been reported that a simple mixture of drugs with magnesium stearate for long periods of time could reduce the dissolution of drug (19). Therefore, it is important to compare release rates of physical mixtures with coground samples to explore advantages of coground samples over simple physical mixtures. The physical mixtures of theophylline with various concentrations of magnesium stearate (1%, 3%, 5%, and 10%, w/w) were prepared at different mixing times (1, 5, 15, and 30 min), and the results of drug release from these formulations were presented in Fig. 2. It can be seen from Fig. 2 that similar conclusion to coground samples can be drawn for the physical mixtures of theophylline–magnesium stearate samples. In other words, the results showed that like coground samples, simple mixing of theophylline with magnesium stearate in turbular blender can produce slow release pattern as concentration of magnesium stearate increases in the formulation. However, comparing the drug release profile of physical mixtures with coground counterparts revealed that cogrinding process has synergistic effect on retardation of drug release from binary mixtures. In order to have better comparison between physical mixtures and coground samples, independent dissolution parameters such as DE, MDT, and MDR were calculated, and the results are listed in Table I. It can be seen from Table I that in most cases, coground samples showed lower DE and MDR and higher MDT in comparison with physical mixture counterparts particularly at high concentration of magnesium stearate. This indicates that cogrinding process is a more efficient technique than simple physical mixing in sustaining the drug release from theophylline capsules containing magnesium stearate. This might be due to an increase in the temperature of milling balls around magnesium particles as a result of friction between particles and balls/wall of the jar. The generation of heat, in turn, can melt some magnesium stearate particles (melting point of magnesium stearate is around 110°C), and the molten hydrophobic particles can coat the drug particle surfaces and increase the hydrophobicity of the drug particle surfaces. This could be the main reason for the reduction in drug release in coground samples in comparison with simple physical binary mixtures of drug–magnesium stearate.
Fig. 1.

Effect of magnesium stearate and cogrounding time on release profiles of theophylline capsules (samples coground for a 1 min; b 5 min; c 15 min; and d 30 min)
Fig. 2.

Effect of magnesium stearate and mixing time on release profiles of theophylline capsules (samples mixed for a 1 min; b 5 min; c 15 min; and d 30 min)
Table I.
Dissolution Parameters Obtained for Different Formulations
| Mg stearate (%) | Time (min) | DE (%) | MDT (min) | MDR (%/min) | |||
|---|---|---|---|---|---|---|---|
| Physical mixture | Coground | Physical mixture | Coground | Physical mixture | Coground | ||
| 1 | 1 | 93.41 | 94.65 | 19.90 | 30.48 | 0.13 | 0.26 |
| 5 | 91.96 | 98.78 | 22.17 | 23.37 | 0.18 | 0.24 | |
| 15 | 100.25 | 97.64 | 18.63 | 21.02 | 0.14 | 0.21 | |
| 30 | 96.89 | 98.01 | 21.67 | 21.88 | 0.17 | 0.23 | |
| 3 | 1 | 92.66 | 84.72 | 66.19 | 89.18 | 0.29 | 0.29 |
| 5 | 90.21 | 53.38 | 64.13 | 213.87 | 0.31 | 0.18 | |
| 15 | 90.21 | 91.24 | 64.13 | 52.66 | 0.31 | 0.24 | |
| 30 | 92.50 | 99.46 | 57.06 | 55.13 | 0.31 | 0.29 | |
| 5 | 1 | 91.62 | 52.21 | 65.42 | 194.24 | 0.30 | 0.17 |
| 5 | 91.23 | 50.05 | 73.66 | 205.88 | 0.32 | 0.17 | |
| 15 | 87.15 | 45.27 | 74.45 | 206.85 | 0.29 | 0.15 | |
| 30 | 86.60 | 97.21 | 64.00 | 40.22 | 0.30 | 0.29 | |
| 10 | 1 | 89.95 | 33.98 | 87.10 | 216.58 | 0.31 | 0.12 |
| 5 | 87.16 | 31.43 | 78.50 | 236.83 | 0.30 | 0.10 | |
| 15 | 73.56 | 31.41 | 155.81 | 230.47 | 0.25 | 0.11 | |
| 30 | 70.73 | 56.34 | 164.46 | 177.41 | 0.24 | 0.17 | |
DE dissolution efficiency, MDT mean dissolution time, MDR mean dissolution rate
As described previously, milling time is one of the key parameters to modify the drug release pattern. To examine the effect of milling time on drug release from formulation, mixtures of theophylline–magnesium stearate samples were subjected to different milling times ranging from 1 to 30 min. It was interesting to note that generally, the effect of milling time on dissolution rate of theophylline from samples is not predictable. Table I showed that in the case of formulations containing 1% magnesium stearate, an increase in milling time had no significant effect on DE, MDT, and MDR values (p > 0.05). The release results for samples containing 3% magnesium stearate showed that the effect of milling time on the drug release was remarkably different. As milling time increases from 1 to 5 min, there is a considerable reduction in drug release, whereas an increase in milling time from 5 to 15 or 30 min increased the release rate, which is apparent from the data reported in Table I. Considering release profiles (Fig. 1) and data reported in Table I for coground samples containing 5% and 10% magnesium stearate showed that milling time had no significant effect on drug release profiles as the time of milling was increased from 1 to 15 min. Milling time beyond 15 min caused a significant increase in the dissolution rate of theophylline. On the basis of this information, it can be concluded that at high concentration of magnesium stearate, optimum milling time is required to obtain slow release profile for coground samples. A further milling beyond the optimum milling time would deteriorate the retardation action of magnesium stearate. The investigation was extended to see if similar results can be obtained for physical mixtures of theophylline and magnesium stearate with different mixing times. In contrast to coground formulations, physical mixtures containing 1%, 3%, and 5% showed a slight decrease in drug release in early time of dissolution (1–20 min), and after 20 min, no significant difference (p > 0.05) was observed. When a high concentration of magnesium stearate (10%, w/w) was mixed for different periods of time, a reduction in DE was more profound than physical mixtures containing low concentration of magnesium stearate. For example, DE was reduced from 89.95% to 70.73% for samples containing 10% magnesium stearate as mixing time was increased from 1 to 30 min, whereas this value for physical mixtures containing 5% magnesium stearate was reduced from 91.62% to 86.6%. It is noted that at a reduced milling time of 1 min, as the concentration of magnesium stearate increases, a remarkable reduction in the release rate (Table I) was observed (DE was reduced from 94.65% to 33.98% at 1-min milling time). This indicates that 1-min milling of theophylline–magnesium stearate blend is enough to induce slow release pattern.
So far, it can be concluded that the coground formulations generally exhibited a substantially lower dissolution rate than the physical mixtures when the same percentage of magnesium stearate and blending/milling time were used. However, it should be kept in mind that a high milling time can enhance the dissolution rate particularly at high concentrations of magnesium stearate. In order to find an evidence for this behavior, cogrinding time and mixing time were extended to 120 min for the samples containing low concentrations of magnesium stearate (1%, w/w) and high concentrations of magnesium stearate (10%, w/w). An interesting result was obtained as described below.
Scanning electron microscopy (SEM) of theophylline samples mixed for 1 and 120 min containing 1% and 10% magnesium stearate is shown in Fig. 3a–d. It is clear from SEM that there is no reduction in the particle size of the drug after 120 min of simple blending (Fig. 3a, b). However, longer mixing time increases the tendency of magnesium stearate to adhere to the drug surfaces and hence reduces the dissolution rate. This is more evident in physical mixtures containing high concentrations of magnesium stearate (Fig. 3c, d). This indicates that mixing time should have considerable impact on the release of theophylline from samples when the number (concentration) of magnesium stearate particles is high. Covering the drug particle surfaces by more magnesium stearate particles at longer mixing time could be the main reason for reduction in drug release in comparison with shorter mixing times.
Fig. 3.

SEM of physical mixtures of theophylline with different concentrations of magnesium stearate: a, b 1% (w/w) magnesium stearate; c, d 10% (w/w) magnesium stearate (all samples were mixed for 1 and 120 min)
When the physical mixtures were subjected to various milling times, different results were obtained as shown in Fig. 4a–f. The micrographs show that cogrinding of the mixtures caused a reduction in drug particle size and also modified the shape of the particles (compare SEM in Fig. 3 with Fig. 4). For example, when the milling time was increased from 1 to 120 min, the mean particle size was decreased from 18.1 ± 5.2 to 6.8 ± 2.9 µm for the samples containing 10% magnesium stearate. Similar results were obtained for the samples containing 1% magnesium stearate. A remarkable reduction in particle size occurred within the first minute of cogrinding. The mean particle size of physical mixtures of theopylline–magnesium stearate (concentration of magnesium stearate was 1%) was reduced from 120 ± 35 to 21 ± 5 µm as milling time was increased from 0 to 1 min. As milling time increased from 1 to 120 min for the samples containing 1% magnesium stearate, small thread-like particles appeared on drug particle surfaces (Fig. 4c). These particles could be recrystallized magnesium stearate or theophylline particles. The dissolution results showed that these changes caused by longer milling time had no significant effect on dissolution behavior (Fig. 5) of samples containing 1% magnesium stearate. When the amount of magnesium stearate in coground samples was increased from 1% to 10%, at shorter milling time, round-shaped drug particles coated by melted magnesium stearate particles were observed (Fig. 4d). When the milling time was increased from 1 to 120 min, very fine drug particles with new surfaces without magnesium stearate were generated as a result of fragmentation of theophylline particles (Fig. 4f). Therefore, this sample should show faster dissolution than coground samples subjected to shorter cogrinding time. Indeed, the drug release profiles showed that samples ground for 120 min had faster dissolution rate than samples subjected to 1-min cogrinding (Fig. 5).
Fig. 4.

SEM of mixtures of theophylline with different concentrations of magnesium stearate: a–c 1% (w/w) magnesium stearate; c, d, f 10% (w/w) magnesium stearate (all samples were coground for 1, 30, and 120 min)
Fig. 5.

The effect of milling time on drug release from binary mixtures of theophylline–magnesium stearate
In order to examine the possible interaction between theophylline and magnesium stearate, two milling or mixing times, 1 and 120 min, for the mixtures containing 10% magnesium stearate and 90% theophylline were selected. All selected samples were subjected to FT-IR and differential scanning calorimetry (DSC) analyses. Figure 6 shows the thermograms of theophylline, magnesium stearate, and their binary mixtures prepared by cogrinding technique and simple physical mixing for 1 and 120 min. Obviously, there was no change for the original raw materials of theophylline and for magnesium stearate before and after milling since the DSC curve for each pair of samples was almost similar. By magnifying DSC thermograms of coground sample between 80°C and 120°C, it is clear that the main peaks of magnesium stearate are still detectable (Fig. 6). The results showed that (Table II and Fig. 6) samples containing theophylline showed a sharp endothermic peak around 272°C, indicating the fusion of theophylline at this temperature. The endothermic peak at 116.8°C for magnesium stearate corresponds to the fusion of the lubricant. The results showed that there is no difference in the melting points of theophylline present at different samples (Table II). Although the enthalpy of fusion of theophylline is slightly higher for coground and physical mixtures (Table II), the statistical analysis showed that this increase in the enthalpy of theophylline is not significant (p > 0.05). This indicates that the cogrinding process (i.e., preparation method of binary mixtures of theophylline and magnesium stearate) has no significant effect on interaction between the drug and lubricant.
Fig. 6.

a Pure magnesium stearate; b pure theophylline; c theophylline–magnesium stearate (90:10%) mixed for 1 min; d theophylline–magnesium stearate (90:10%) mixed for 120 min; e theophylline–magnesium stearate (90:10%) milled for 1 min; f theophylline–magnesium stearate (90:10%) milled for 120 min
Table II.
Thermogram Data of Theophylline, Magnesium Stearate, and their Binary Mixtures Prepared Under Different Conditions (n = 3)
| Formulation | Theophylline | Magnesium stearate | ||
|---|---|---|---|---|
| Melting point (°C) | Enthalpy (J/g) | Melting point (°C) | Water evaporation (°C) | |
| Theophylline alone | 272.7 ± 1.0 | 160.8 ± 3.2 | – | – |
| Magnesium stearate alone | – | – | 116.8 ± 3.5 | 93.5 ± 2.5 |
| Theo-MagS (90:10) milled for 1 min | 272.5 ± 1.5 | 161.8 ± 2.9a | 116.0 ± 2.8 | 105.0 ± 2.1 |
| Theo-MagS (90:10) milled for 30 min | 272.5 ± 1.2 | 164.6 ± 3.0a | 116.0 ± 3.2 | 103.0 ± 2.4 |
| Theo-MagS (90:10) mixed for 1 min | 272.0 ± 1.5 | 164.3 ± 3.1a | 117.9 ± 2.7 | 102.1 ± 1.9 |
| Theo-MagS (90:10) mixed for 120 min | 272.1 ± 1.4 | 164.4 ± 2.8a | 117.5 ± 3.5 | 104 ± 2.0 |
aThese data are normalized
Magnesium stearate has been shown to be incompatible with an important number of drugs, such as glibenclamide or aspirin (20,21). It is clear from thermograms that magnesium stearate presents two molecules of water hydration. The dehydration process occurs in several steps at over 70°C. Additionally, it presents a fusion melting peak at 110–120°C, which sometimes appears at lower temperatures or even superimposes the dehydration events as a consequence of the product being a mixture of magnesium palmitate and stearate (22,23). The result obtained in the present study was in agreement with the previous studies showing wide endothermic peak with two events at 93°C and 116°C. The first peak is the indication of water evaporation, and the second peak can be attributed to the fusion of magnesium stearate.
FT-IR spectra of physical mixtures and coground samples were shown in Fig. 7. The FT-IR absorption bands at 1,705 and 1,661 cm−1 for theophylline were assigned to the C=O stretching vibration for carbonyl groups, and the band at 1,566 cm−1 was attributed to the stretching vibration of the imine group of theophylline. Comparing binary mixtures of theophylline samples with pure theophylline showed that still those bands are apparent in the binary mixtures and there is no modification in the peak positions and trends, indicating no interaction between theophylline and magnesium stearate.
Fig. 7.

FTIR spectra of a pure theophylline; b theophylline–magnesium stearate milled for 1 min; c theophylline–magnesium stearate milled for 30 min; d theophylline–magnesium stearate mixed for 1 min; e theophylline–magnesium stearate mixed for 30 min; f magnesium stearate
Bands at 1,464, 1,574, 2,850, and 2,917 cm−1 are distinctive for magnesium stearate. Comparing all FT-IR spectra showed that in physical mixtures, the presence of the last two bands are apparent, but in coground samples, the intensity of these two bands is very low, although all binary mixtures have the same concentration of magnesium stearate. It is clear from FT-IR of coground samples as milling time increases the intensity of bands at 2,850 and 2,917 cm−1 decrease. This could be due to the evaporation of water from the samples as a result of generation of heat during milling. It has been reported that magnesium stearate has around 5% moisture (24).
CONCLUSION
Cogrinding time and magnesium stearate concentration had significant effect on drug release from theophylline capsule formulations. It seems that sustained release action is amplified with coground approach. It was shown that cogrinding time is of crucial importance as additional cogrinding time could deteriorate the retardation effect of magnesium stearate. FT-IR and DSC results ruled out any significant interaction between theophylline and magnesium stearate in solid state.
Reference
- 1.Vogt M, Kunath K, Dressman JB. Cogrinding enhances the oral bioavailability of EMD 57033, a poorly water soluble drug in dogs. Eur J Pharm Biopharm. 2008;68:338–345. doi: 10.1016/j.ejpb.2007.06.011. [DOI] [PubMed] [Google Scholar]
- 2.Barzegar-Jalali M, Nayebi AM, Valizadeh H, Hanaee J, Barzegar-Jalali A, Adibkia K, et al. Evaluation of in vitro–in vivo correlation and anticonvulsive effect of carbamazepine after cogrinding with microcrystalline cellulose. J Pharm Pharmaceut Sci. 2006;9:307–316. [PubMed] [Google Scholar]
- 3.Nakai Y, Nakajima S, Yamamoto K, Terada K, Konno T. Effects of grinding on the physical and chemical properties of crystalline medicinals with microcrystalline cellulose. IV. Chem Pharm Bull. 1980;28:652. doi: 10.1248/cpb.28.652. [DOI] [PubMed] [Google Scholar]
- 4.Sawayanagi Y, Nambu N, Nagai T. Enhancement of dissolution properties of prednisolone from ground mixtures with chitin or chitosan. Chem Pharm Bull. 1983;31:2507–2509. doi: 10.1248/cpb.31.2064. [DOI] [PubMed] [Google Scholar]
- 5.Yamamoto K, Nakano M, Arita K, Takayama Y, Nakai Y. Dissolution behavior and bioavailability of phenytoin from a ground mixture with microcrystalline cellulose. J Pharm Sci. 1976;65:1484. doi: 10.1002/jps.2600651017. [DOI] [PubMed] [Google Scholar]
- 6.Vogt M, Kunath K, Dressman JB. Dissolution improvment of four poorly water soluble drugs by cogrinding with commonly used excipients. Eur J Pharm Biopharm. 2008;68:330–337. doi: 10.1016/j.ejpb.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 7.Vogt M, Kunath K, Dressman JB. Dissolution enhancement of fenofibrate by micronization, cogrinding and spray-drying: comparison with commercial preparations. Eur J Pharm Biopharm. 2008;68:283–288. doi: 10.1016/j.ejpb.2007.05.010. [DOI] [PubMed] [Google Scholar]
- 8.Shokri J, Hanaee J, Barzegar-Jalali M, Changizi R, Rahbar M, Nokhodchi A. Improvement of the dissolution rate of indomethacin by a cogrinding technique using polyethylene glycols of various molecular weights. J Drug Del Sci Tech. 2006;16:203–209. [Google Scholar]
- 9.Nokhodchi A, Javadzadeh Y, Siahi MR, Barzegar-Jalali M. The effect of type and concentration of vehicles on the dissolution rate of a poorly soluble drug (indomethacin) from liquisolid compacts. J Pharm Pharmaceut Sci. 2005;8:18–25. [PubMed] [Google Scholar]
- 10.Nokhodchi A, Talari R, Valizadeh H, Barzegar-Jalali M. An investigation on the solid dispersions of chordazepoxide. Int J Biomed Sci. 2007;3:211–217. [PMC free article] [PubMed] [Google Scholar]
- 11.Liversidge GG, Cundy KC. Particle size reduction for improvement of oral bioavailability of hydrophobic drugs: absolute oral bioavailability of nanocrystalline danazol in beagle dogs. Int J Pharm. 1995;125:91–97. doi: 10.1016/0378-5173(95)00122-Y. [DOI] [Google Scholar]
- 12.Yang KY, Glemza R, Jarowski CI. Effects of amorphous silicon dioxides on drug dissolution. J Pharm Sci. 1979;68:560–565. doi: 10.1002/jps.2600680511. [DOI] [PubMed] [Google Scholar]
- 13.Watanabe T, Ohno I, Wakiyama N, Kusai A, Senna M. Controlled dissolution properties of indomethacin by compounding with silica. STP Pharma Sciences. 2002;12:363–367. [Google Scholar]
- 14.Bahl D, Hudak J, Bogner R. Comparison of the ability of various pharmaceutical silicates to amorphize and enhance dissolution of indomethacin upon co-grinding. Pharm Dev Technol. 2008;13:255–269. doi: 10.1080/10837450802012869. [DOI] [PubMed] [Google Scholar]
- 15.Bahl D, Bogner RH. Amorphization alone does not account for the enhancement of solubility of drug co-ground with silicate: the case of indomethacin. AAPS Pharm Sci Tech. 2008;9:146–153. doi: 10.1208/s12249-007-9013-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Durig T, Fassihi R. Mechanistic evaluation of binary effects of magnesium stearate and talc as dissolution retardants at 85% drug loading in an experiment. J Pharm Sci. 1997;86:1092–1098. doi: 10.1021/js970052v. [DOI] [PubMed] [Google Scholar]
- 17.Khan KA. Concept of dissolution efficiency. J Pharm Pharmacol. 1975;27:48–49. doi: 10.1111/j.2042-7158.1975.tb09378.x. [DOI] [PubMed] [Google Scholar]
- 18.Nokhodchi A, Khaseh P, Ghafourian T, Siahi-Shadbad MR. The role of various surfactants and fillers in controlling the release rate of theophylline from HPMC matrices. STP Pharma Sciences. 1999;9:555–560. [Google Scholar]
- 19.Li-Hua W, Chowhan ZT. Drug–excipient interactions resulting from powder mixing. V. Role of sodium lauryl sulphate. Int J Pharm. 1990;60:61–78. doi: 10.1016/0378-5173(90)90190-F. [DOI] [Google Scholar]
- 20.Pyramides G, Robinson JW, Zito SW. The combined use of DSC and TGA for the thermal analysis of atenolol tablets. J Pharm Biomed Anal. 1995;13:103–110. doi: 10.1016/0731-7085(94)00112-F. [DOI] [PubMed] [Google Scholar]
- 21.Oliveira GG, Ferraz HG, Matos JSR. Thermoanalytical study of glibenclamide and excipients. J Therm Anal Calorimetry. 2005;79:267–270. doi: 10.1007/s10973-005-0047-5. [DOI] [Google Scholar]
- 22.Marini A, Berbenni V, Moioli S, Bruni G, Cofrancesco P, Margheritis C, et al. Drug–excipient compatibility studies by physico-chemical techniques—the case of indomethacin. J Therm Anal Calorimetry. 2003;73:529–545. doi: 10.1023/A:1025426012578. [DOI] [Google Scholar]
- 23.Verma RK, Garg S. Compatibility studies between isosorbide mononitrate and selected excipients used in the development of extended release formulations. J Pharm Biomed Anal. 2004;35:449–458. doi: 10.1016/j.jpba.2004.01.012. [DOI] [PubMed] [Google Scholar]
- 24.Cunha-Filho MSS, Martínez-Pacheco R, Landín M. Compatibility of the antitumoral β-lapachone with different solid dosage forms excipients. J Pharm Biomed Anal. 2007;45:590–598. doi: 10.1016/j.jpba.2007.08.016. [DOI] [PubMed] [Google Scholar]