

Abstract
The aim of this study was to investigate the ability of liquid loadable tablets (LLT) to be loaded with a self-microemulsifying drug delivery system (SMEDDS) containing cyclosporine (CyA). LLT were prepared by direct compression of the porous carrier magnesium aluminometasilicate and subsequently loaded with SMEDDS by a simple absorption method. SMEDDS was evaluated regarding visual appearance and droplet size distribution after dispersion in aqueous media. The developed SMEDDS was found to be similar to Neoral®. LLT were characterized before and after loading regarding weight variation, tablet hardness, disintegration time, and in vitro drug release. It was found that LLT with high porosities suitable for liquid loading and further processing could be prepared. Adding a tablet disintegrant was found to improve in vitro drug release. Additionally, the volume-based loading capacity of LLT was evaluated and found to be comparable to soft gelatin and hard two-piece capsules. Furthermore, the pharmacokinetic performance of CyA from loaded LLT was tested in two PK-studies in dogs. Absorption of CyA from SMEDDS loaded into LLT was found in the first study to be significantly lower than the absorption of CyA from SMEDDS filled into a capsule. However, addition of a superdisintegrant improved the absorption markedly. The bioavailability of CyA from SMEDDS loaded into disintegrating LLT was found in the second study to be at the same level as from capsule formulation. In conclusion, the LLT technology is therefore seen as a promising alternative way of achieving a solid dosage form from liquid drug delivery systems.
Key words: cyclosporine, drug carrier, liquid loadable tablets, SMEDDS
INTRODUCTION
Transforming liquid active ingredients and liquid pharmaceutical preparations into solid dosage forms is a challenging task. Soft gelatin capsules have been used as unit dose containers for liquids for many years. As new equipment has become available that allows sealing of hard capsules, it has also become possible to prepare liquid formulations in two-piece hard capsules (1). However, the capsule technologies are associated with a range of problems such as leaking, leaching of components through the capsule shell, incompatibilities with capsule material, and slower manufacturing rate compared with tablet production. The soft capsules furthermore require specialized manufacturing equipment, and the manufacturing cost is higher compared with tablet production (2).
Solid formulation of liquid drug delivery systems, in general, has not been thoroughly investigated. Most attempts have focused on solid formulation of lipid systems and in particular formulation of liquid and semi-solid self-emulsifying drug delivery systems (SEDDS) (3). Lipid-based formulation systems have become increasingly important in the formulation of lipophilic drug substances as they can facilitate the dispersion of a drug in the gastrointestinal tract resulting in enhanced oral bioavailability (4). Especially self-microemulsifying drug delivery systems (SMEDDS) have gained increasing interest over the past years as a beneficial way of formulating drugs with low aqueous solubility and high membrane permeability, also known as biopharmaceutical classification system class II drugs (5).
The transformation of liquid lipid systems into a solid oral dosage form has been attempted by several methods such as capsule filling, spray drying (6), adsorption onto solid carriers (7–9), and melt granulation as well as other techniques (10). It is possible to obtain free-flowing powders from liquid formulations by adsorption onto solid carrier particles such as calcium silicate, magnesium aluminometasilicate, silicon dioxide, or carbon nanotube (10). These carriers can subsequently be filled directly into capsules or mixed with other excipients to allow for tablet compression. However, at least two concerns are associated with this approach. First, a large amount of carrier is required to solidify the liquid which subsequently implicates a large dosage volume (11). Second, when a liquid-loaded powder is compressed, problems with sticking, and lack of tablet hardness may occur at higher liquid load due to the liquid being squeezed out of the tablet during compression.
An alternative, as demonstrated in this study, is first to prepare a tablet by direct compression of solid carrier particles and subsequently load the liquid into the tablet. By this method, a higher liquid loading can be obtained without losing integrity of the tablet. The idea has previously been explored for tablets prepared from magnesium aluminometasilicate (MAMS) as carriers of chemical reagents and catalysts (12). A selection of chemical reagents were loaded into pre-compressed MAMS tablets (liquid-loadable tablets (LLT)) and used for accurate dosing in organic synthesis reactions. In this study, the concept is applied to a liquid lipid formulation instead of chemical reagents. Neusilin US2® (Fuji chemicals, JP) is a synthetic, amorphous form of MAMS prepared by spray drying resulting in a fine, ultra light granule. MAMS is practically insoluble in water and alcohol and partly soluble in acid and alkaline media. Neusilin® meets the requirements of the current US Pharmacopoeia/National Formulary and Japanese Pharmacopoeia. Based on the usage as an excipient in various formulations in Japan, up to 1.05 g Neusilin® can be used in oral formulations per day. Neusilin US2® is a neutral grade but alkaline grades are approved as antacid active ingredients with a maximum dosage of 4 g/day.
The work presented in this paper focuses on the ability of LLT prepared from MAMS to be loaded with a SMEDDS system containing the immunosuppressive agent cyclosporine (CyA) (13). The porous carrier, MAMS, is compressed into LLT and subsequently loaded with a SMEDDS system containing CyA (CyA-SMEDDS) corresponding to the Neoral® formulation. The tablet technology is evaluated regarding compactibility and liquid loading ability. Finally, the pharmacokinetic (PK) performance of CyA from loaded LLT is tested in a PK-study in beagle dogs.
MATERIALS AND METHODS
Materials
MAMS (Neusilin US2, Fuji Chemical, Japan) was used to manufacture LLT using magnesium stearate as lubricant (Peter Greven BV, The Netherlands) and crospovidone as disintegrant (Polyplasdone XL, ISP Technologies, KY). For the preparation of SMEDDS, propylene glycol (Lyondell, The Netherlands), ethanol anhydrous, linoleoyl macrogolglycerides (Labrafil M 2125 CS, Gattefossé, France), polyoxyl 40 hydrogenated castor oil (Cremophore RH40, BASF, Germany), and CyA (Euroasia and Sicor s.r.l.) were used. Acetonitrile, methanol, and phosphoric acid used for HPLC analysis were all of analytical grade. Neoral® capsules are commercialized by Novartis Healthcare A/S.
Methods
Preparation and Evaluation of SMEDDS
Preparation of CyA-SMEDDS
The CyA-SMEDDS was composed of polyoxyl 40 hydrogenated castor oil: linoleoyl macrogolglycerides/ethanol/CyA/propylene glycol (41:34:10:10:5 w/w%). All ingredients, except CyA, were mixed by magnetic stirring. CyA was added slowly using magnetic stirring and dissolved by heating up to 40°C. The composition of CyA-SMEDDS imitates the composition of Neoral®
Evaluation of SMEDDS
The prepared SMEDDS was evaluated visually, and the ability to form a microemulsion was tested by the method proposed by Khoo et al. (14). A 1-mL CyA-SMEDDS was added to 250 mL of purified water at 37°C and dispersed by magnetic stirring. After 10 min, the visual appearance was observed and the droplet size of the dispersion measured by photon correlation spectroscopy using a Malvern HPPS (HPP5001, Malvern Instruments Ltd., UK).
Preparation of CyA Formulations
Preparation of LLT
LLT were prepared with and without superdisintegrant (denoted LLT-2 and LLT-1, respectively). The composition of LLT-1 was MAMS/crospovidone/magnesium stearate (99:0:1 w/w%), whereas the composition of LLT-2 where MAMS/crospovidone/magnesium stearate (89:10:1 w/w%). Excipients for the preparation of the LLT shown were mixed in a cylindrical blender (Turbula Mixer, Bachenhofen Machinenfabrik, Switzerland) and subsequently compressed into tablets using an instrumented single punch excentric tablet press (Diaf TM20, Denmark). Tablets of approximately 250 mg were compressed using a round 12 mm flat-faced punch. The compression force applied measured in N was converted to pressure (MPa) by division with the cross section area of the tablet. Tablet hardness was determined using a hardness tester (Dr. Schleuniger Pharmatron, Switzerland). Tablet disintegration was performed in 0.1 N HCl using an automatic disintegration tester (Dr. Schleuniger Pharmatron, Switzerland).
Loading procedures
Tablets were placed in excess of CyA-SMEDDS and allowed to absorb the liquid until a constant tablet weight was reached. Before weighing, excess of liquid on the surface of the tablets was removed by tissue paper. After few hours of storage, the tablet surface is dry, comparable to the non-loaded tablet. The tablet surface was not further characterized in this study, but it might be necessary to coat the tablet to improve the cosmetic appearance of the final product.
The tablet porosity and the corresponding maximum loading capacity (milliliter) were determined before loading. In this study, the tablet is defined as being 100% loaded when the theoretical loading capacity has been reached and the tablet voids are completely saturated.
Preparation of capsule formulations
CyA-SMEDDS corresponding to 25 mg or 30 mg of CyA, respectively, was filled into two-piece hard gelatin capsules (size #2, Capsugel, Pfizer Inc.). CyA-SMEDDS was also loaded onto MAMS powder using a mortar and pestle. An amount of powder corresponding to 25 or 30 mg CyA was subsequently filled into two-piece hard gelatin capsules (size 000, Capsugel, Pfizer Inc.).
Tablet porosity
The tablet porosity (ε) is defined according to:
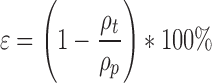 |
where ρp is the pycnometric tablet density and ρt the tablet density.
Pycnometric densities of LLT-1 and LLT-2 were 2.10 g/cm3 and 1.94 g/cm3, respectively (AccuPyc 1330, Micromeritics Instrument Corporation, GA). The tablet density was derived from the tablet weight divided by the tablet volume calculated from TabletCAD (Saga Software, Iceland).
In vitro drug release
Formulations of CyA were tested in vitro in 950 mL purified water at 37°C using USP type I (basket) at a rotation speed of 100 rpm. Samples were withdrawn at predetermined time intervals, filtered, and diluted 1:1 with acetonitrile before being analyzed by HPLC-UV.
Assay determination
An HPLC system (Shimadzu LC-10AD, SHIMADZU Deutchland GmbH, Germany) equipped with a 210 nm UV-detector and a 4.6 mm × 25 cm column (Synergi-Polar RP 80 Å, Phenomenex) was maintained at constant temperature of 60°C. A sample volume of 20 µL CyA solution was injected. The gradient system consisted of two mobile phases with the compositions/mobile phase A, acetonitrile/water/methanol/phosphoric acid (1000:920:72:0.8) and mobile phase B, acetonitrile/water/methanol/phosphoric acid (600:120:27:0.3). The proportion of mobile phase B was increased from 0% to 100% from 2 to 15 min. After 15 min, the proportion was immediately reduced to 0%. The flow rate was constantly kept at 2.0 mL per minute. Overall, an analytical process time of 25 min per sample was optimal, including 10 min after sample elution to stabilize the base line.
Bioanalysis
Bioanalysis of whole blood samples was performed at Aarhus Sygehus (Aarhus hospital, Denmark) using an analytical method routinely used in the monitoring of patients subjected to CyA treatment. An LC-MS/MS method with online extraction was used to quantify CyA from whole blood samples based on a column-switching approach. A 250 µl whole blood sample was mixed with 25 µl internal standard solution (cyclosporine D). A 500 µl precipitation reagent (0.1 M zinc sulphate/methanol 20/80) was added and the solution was mixed carefully and centrifuged at 10,900 rpm for 5 min. Subsequently, 5 µl of the supernatant was injected on the trap column (Phenomenex Luna C8(2) 10 × 2 mm, 5 µm) and washed with a mobile phase composed of 0.1% formic acid and 2 mM ammonium acetate/methanol (60/40) at a flow rate of 1 mL/min. After washing, the analytes were back-flushed from the trapping column onto the analytical column (Phenomenex Luna C18(2) 20 × 2 mm, 3 µm) where they were eluted isocratically with a mobile phase composed of 0.1% formic acid and 2 mM ammonium acetate/methanol (20/80) at a flow rate of 0.4 mL/min. Quantification was based on monitoring the transitions m/z 1219.8 → 1202.8 (CyA) and m/z 1233.8 → 1216.8 (CyD).
In Vivo Dog Study
The in vivo dog studies were performed at LAB Research (Lille Skensved, Denmark) according to all local, national, ethical, and regulatory principles and local licensing regulations.
Subjects
The pharmacokinetic performance of cyclosporine was tested in male beagle dogs (Harian Winkelmann GmbH, Germany). Before dosing, the dogs were fasted overnight. Food was given minimum 2 h after dosing. A 3 mL blood samples were drawn from the jugular vein before dosing and at the following times: 0.5, 1.0, 1.5, 2, 3, 6, 9, 24, 36, and 48 h after dosing. The blood samples were collected in tubes containing EDTA as anticoagulant. The blood samples were carefully mixed with the anticoagulant by gently turning the tube and subsequently stored at −20°C.
Study design
Formulations tested in vivo are described in Table I. In study no. 1, LLT loaded with CyA-SMEDDS (F1) were compared with CyA-SMEDDS loaded into capsules (F2) in six beagle dogs in a cross-over design with a wash-out period of 7 days between each treatment. In study no. 2, four formulations (F3–F6) were tested in a Latin Square study design in four beagle dogs, again using a wash-out period of 7 days between treatments. Pharmacokinetic calculations for both studies were performed using PC-based software WinNonlin version 4.1 (Pharsight Corporation, Mountain View, CA). Individual whole blood concentration profiles were subjected to non-compartmental pharmacokinetic analysis. From these profiles the maximum whole blood concentration, Cmax, and the time when it occurred, Tmax, were estimated. Area under the whole blood concentration curve until the last detectable time point, AUClast, and the area extrapolated to infinity, AUCinf, were determined by the log/linear trapezoidal method.
Table I.
Test Formulations Prepared for In Vivo Testing
| Formulation ID | Dosage form | Dosage strength (mg) | Capsule/tablet size (mm) | Formulation type |
|---|---|---|---|---|
| F1 | Capsule | 30 | 18 × 6.4 (#2) | CyA-SMEDDS filled capsule |
| F2 | Tablet | 30 | Ø 10 | LLT-1 tablet loaded with CyA-SMEDDS. Dose weight: 424 mg |
| F3 | Capsule | 25 | 13 × 7.5 | Neoral® soft gelatin capsule |
| F4 | Capsule | 25 | 18 × 6.4 (#2) | CyA-SMEDDS filled capsule |
| F5 | Capsule | 25 | 26 × 9.9 (#000) | MAMS powder loaded with CyA-SMEDDS |
| F6 | Tablet | 25 | Ø 12 | LLT-2 tablet loaded with CyA-SMEDDS. Dose weight, 514 mg |
Statistical calculations
Differences in the PK-parameter AUCinf were analyzed using single-factor analysis of variance ANOVA as described by Montgomery (15). Comparison of means was performed using Student's t test assuming equal variances (MS Office Excel version 2007, Microsoft Inc., WA).
RESULTS AND DISCUSSION
In Vitro Evaluation of SMEDDS
Visually, both CyA-SMEDDS and the content of Neoral® capsules were clear yellow homogenous liquid mixtures. When dispersed in 250 mL water, they formed opaque and slightly blue microemulsions. The droplet size of CyA-SMEDDS was determined as 29.9 nm ± 0.14 (mean ± SD, n = 3; Polydispersity Index, 0.053) and comparable to the droplet size of Neoral® of 26.2 nm ± 0.10 (mean ± SD, n = 3; Polydispersity Index, 0.026). The CyA-SMEDDS developed is therefore considered to be similar to Neoral® regarding the ability to disperse and form microemulsions.
Preparation of LLT
Neusilin US2 is a neutral grade of MAMS suitable for direct compression due to the spherical shape of the spray dried material, which contributes to an increased flowability. LLT prepared from MAMS with 10% disintegrant (Polyplasdone XL; corresponding to LLT-2) have a maximum porosity of approximately 80%. As shown in Fig. 1, the tablet porosity depends on the compression force applied. Even the use of a low compression force, corresponding to high tablet porosity, yielded tablets with a hardness that was satisfactory for tablet loading and further processing.
Fig. 1.

Correlation between tablet compression pressure and tablet porosity and tablet hardness. Composition of the tablets corresponds to LLT-2
Loading of LLT
Since LLT can absorb an amount of oil corresponding to the tablet porosity, a high porosity allows the tablet to be loaded with large amounts of, e.g., oil or other liquid systems. As shown in Table II, the weight variation of LLT after loading was below 3% reflecting a low variability of content of uniformity. After loading for ca. 1.5 h LLT of high porosity (corresponding to low compression force) had absorbed the maximum amount liquid possible, as shown in Fig. 2. The loading time, identified as the time to achieve 100% loading of LLT, was found to be affected by the compression force applied during tablet compression. As shown, application of higher compression force increased the loading time up to approximately 4 h and at the same time reduced the maximum theoretical loading amount due to the resulting lower tablet porosity.
Table II.
Tablet Characteristics of LLT
| LLT-1 | LLT-2 | |||||
|---|---|---|---|---|---|---|
| Weight (mg) | Hardness (N) | Disintegration time (h) | Weight (mg) | Hardness (N) | Disintegration time (min) | |
| Non-loaded LLT | 256.5 (±0.44, n = 20) | 74 (±23, n = 10) | >2 (n = 6) | 253.4 (±0.20, n = 10) | 61 (±113, n = 10) | <0.2 (n = 6) |
| Loaded with CyA-SMEDDS | 714.8 (±2.56, n = 6) | 62 (±42, n = 10) | >2 (n = 6) | 503.9 (±2.62,n = 6) | 53 (±74, n = 10) | 61 (±3.5, n = 6) |
Relative standard deviations (%) are stated in brackets
LLT liquid loadable tablets
Fig. 2.

Correlation between liquid loading and loading time. Tablets with a composition corresponding to LLT-2 prepared with increasing compression pressure were loaded with CyA-SMEDDS. Dashed lines indicate the maximum theoretical loading amount estimated as 553 mg (11 MPa), 374 mg (32 MPa), and 300 mg (46 MPa), respectively, derived from tablet porosity
The loading process can be seen as the replacement of air in the tablet pores by liquid. This means that the theory of capillary flow in porous media can be applied. The velocity by which the liquid is absorbed into the tablet can be described by the Washburn equation (16,17):
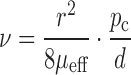 |
where v is the velocity of the interface between air and liquid, r is the radius and d the length of a pore, pc the capillary pressure and µeff is the effective viscosity. The equation applies to liquid and air because the two systems are not mixable. Therefore, the capillary flow in the loading process is controlled mainly by capillary action. According to the equation, decreasing the pore radius results in a decreased velocity and thereby an increased loading time. The pore size distribution of LLT with high porosity and low porosity was compared using mercury porosimetry (AutoPore IV 9500, Micromeritics®; data not shown). It was found that lower porosity, corresponding to higher compression pressure applied, would cause the formation of smaller pores. By decreasing the tablet porosity from approximately 80% down to 70%, the median pore diameter (volume) decreased from 107 to 23 nm. This explains why the loading time increases at higher compression pressures as shown in Fig. 2. It is also possible that closed cavities are formed inside the tablet at higher compression pressures. This would explain why the tablets subjected to high compression pressure did not absorb the maximum theoretical amount of liquid.
Liquid Loading Capacity of LLT Compared with Capsules
According to the product specification MAMS can absorb approximately 3 mL/g of an oil. After compression into LLT, between 0.92 and 2.21 mL/g could be absorbed by tablets with porosities ranging from 64–81%. This is comparable to the amounts of liquid contained in capsules. A limitation of the capacity volume of soft gelatin capsules is the thick capsule shell. In fact, up to about 33% of the capsule volume can be occupied by the shell. This number is based on the capacity stated by the manufacturer (SGcaps®, Capsugel) compared with the total outer capsule volume estimated by pycnometric determination (AccuPyc 1330, Micromeritics Instrument Corporation, GA). Two-piece hard gelatin capsules have thinner shells, but they can only be filled approximately 90% of their body volume in order to reduce the risk of spillage during machine movement, which leaves some space unused in the capsule cap. According to the specifications of two-piece hard capsules (Posilok®, Qualicaps Inc.), between 72% and 77% (v/v) of the total internal capsule volume can be occupied with liquid.
In comparison, up to approximately 80% (v/v) of the total outer volume of the LLT can be utilized for liquid filling corresponding to the tablet porosity. LLT do not require a gelatin shell but they may be coated with thin coatings for protection of the loaded drug formulation. Therefore, LLT provide an alternative to liquid filled capsules based on liquid loading capacity.
Dissolution Studies of loaded LLT
Capsules containing liquid CyA-SMEDDS (F4) immediately disintegrated and released their drug content comparable to Neoral® capsules (F3), as shown in Fig. 3. Loaded LLT without disintegrant (F2) showed a drug release of approximately 25% after 6 h when tested in vitro. Addition of 10% disintegrant (F6) improved the in vitro drug release up to approximately 50%. From loaded MAMS powder (F5), almost 80% was released. This illustrates the importance of obtaining complete tablet disintegration in order to obtain a high level of drug release.
Fig. 3.

Comparison of dissolution profile of loaded LLT without disintegrant (F2) and with disintegrant (F6). Profiles of Neoral® capsules (F3) and CyA-SMEDDS filled into capsules (F4) are also shown together with profiles of loaded Neusilin powder filled into capsules (F5). Dissolution performed in 950 mL purified water at 37°C using USP basket at 100 rpm
The in vitro release tests were performed in 950 mL purified water. The low aqueous solubility of CyA may give rise to the question of whether or not sink conditions were obtained. In purified water, however, it was possible to obtain immediate release of 100 mg Neoral® capsules and to detect 100% release of the dose after 45 min (data not shown). Therefore, the lower doses of 25 and 30 mg CyA, respectively, could be tested in vitro under sink conditions using purified water as the dissolution media.
When loaded LLT are introduced into an aqueous media or administered into the gastrointestinal environment the loaded liquid is being released from the tablet. Washburn’s equation may in some cases describe the release process if the media is not miscible with the loaded liquid. However, this is not the case with CyA-SMEDDS. Being an amphiphilic system, it is mixed with the aqueous phase and the release is controlled mainly by diffusive and convective mechanisms. To reduce the length of the diffusion path a disintegrant was incorporated into the tablet.
As shown in Table II, it was possible to obtain immediate disintegration of LLT by adding 10% disintegrant (crospovidone) to the formulation (LLT-2). Crospovidone facilitates tablet disintegration by a combination of swelling and wicking mechanisms (18). The addition of disintegrant also had an effect for loaded LLT, although the tablet disintegration time was significantly increased compared to non-loaded LLT.
The in vitro dissolution setup may not always reflect in vivo conditions. The positive effects of bile salts, gastric motility, and enzyme activity in the gastrointestinal tract were believed to be able to facilitate the release of CyA from loaded tablets. It was therefore decided to further evaluate loaded LLT without disintegrant in an in vivo dog study.
In Vivo Performance of Loaded LLT Versus Capsule Formulation
The goal of the first dog study was to compare the absorption profile of CyA-SMEDDS filled into a hard gelatin capsule (F1) with that of CyA-SMEDDS loaded into LLT (F2). The LLT tested in this study did not contain any disintegrant and therefore the absorption of CyA depended on the ability of CyA-SMEDDS to migrate through the tablet pores and subsequently be released from the formulation. A clear distinction in the pharmacokinetic profiles between F1 and F2 was found (Fig. 4, Table III). Based on the AUC of these whole blood profiles the relative bioavailability of CyA from the tablet formulation was found to be only 38% of that of the capsule formulation. A t test confirmed this to be a statistically significant difference (p < 0.01). This confirmed the need found in vitro of adding a tablet disintegrant in order to facilitate drug release.
Fig. 4.

Mean whole blood concentrations of CyA in six dogs following oral doses of 30 mg in the form of CyA-SMEDDS filled into capsules (F1) and CyA-SMEDDS loaded into LLT without disintegrant (F2). Error bars indicate standard deviations. The difference in AUC between F1 and F2 is statistically significant (p < 0.01)
Table III.
Mean Pharmacokinetic Parameters After Single-Dose Administration of CyA in Dogs
| Dog study no. 1 | Dog study no. 2 | |||||
|---|---|---|---|---|---|---|
| F1 (capsule 30 mg) | F2 (tablet 30 mg) | F3 (Neoral® 25 mg) | F4 (capsule 25 mg) | F5 (capsule 25 mg) | F6 (tablet 25 mg) | |
| Cmax (µg/L) | 337 (±124) | 81 (±29.7) | 331 (±57.4) | 231 (±119) | 370 (±121) | 227 (±76.9) |
| Tmax (h) | 1.25 (±0.49) | 2.17 (±0.68) | 0.88 (±0.25) | 1.25 (±0.65) | 1 (±0) | 1.88 (±0.86) |
| AUCinf (haµg/L) | 1,282 (±245) | 664 (±499)b | 1,269 (±256) | 883 (±382) | 1,267 (±265) | 1,040 (±272) |
| Relative bioavailabilitya | – | – | 1.00 | 0.70 | 1.00 | 0.82 |
All data are mean values ± SD, n = 4
a Relative bioavailability is compared with Neoral® (F3)
b Statistically significant difference to F1 (p < 0.01)
The second dog study included four formulations of cyclosporine. The commercial capsule formulation of Neoral® (F3) was compared with CyA-SMEDDS filled into hard gelatin capsules (F4), CyA-SMEDDS loaded onto MAMS powder (F5), and CyA-SMEDDS loaded into LLT containing 10% disintegrant (F6) as shown in Table I. CyA-SMEDDS filled into capsules (F4) was included in the study in order to be able to compare the performance of the SMEDDS developed in this study with the Neoral® SMEDDS. In the first dog study, disintegration of the LLT was found to be of importance. Therefore, a disintegrant was included in the LLT corresponding to the LLT-2 composition (F6). CyA-SMEDDS loaded onto MAMS powder (F5) was included in the study to imitate a fully disintegrated tablet.
Figure 5 shows whole blood profiles of CyA after oral administration of F3–F6. The bioavailability of CyA from each of the four formulations, estimated as the area under the whole blood curve, was compared using ANOVA. The ANOVA did not show any statistically significant differences between the four formulations (p = 0.2427). Based on this analysis and on the whole blood profiles we conclude that the bioavailability of CyA was at the same level from all four formulations tested. The pharmacokinetic parameters for F3–F6 are listed in Table III. There is a tendency, however, of a lower Cmax and higher Tmax from the loaded LLT (F6). This may be explained by the LLT possessing a controlled release mechanism resulting in a slower drug release initially. Thereby, the high peak concentration of CyA is avoided. As long as the total bioavailability of CyA is not impaired this may be considered an advantage as high peak concentrations may be associated with a higher incidence of adverse effects. One study with cyclosporine administration reported that adverse events mainly occurred 1–2 h after dosing corresponding to the time where maximum blood concentration was achieved (19). Also, the initial plasma peak has been associated with potential nephrotoxicity (20).
Fig. 5.

Mean whole blood concentrations of CyA in four dogs following oral doses of 25 mg CyA in study no. 2. Error bars indicate absolute standard deviations (SD). a Neoral® capsule (F3) and CyA-SMEDDS loaded onto MAMS powder (F5). b CyA-SMEDDS filled into capsule (F4) and LLT loaded with CyA-SMEDDS (F6). No statistically significant difference between AUC of formulations F3–F6 was found using a one-way ANOVA
The fact that the absorption of CyA is considered to be equivalent from the four tested formulations, leads to the conclusion that the developed CyA-SMEDDS was comparable to Neoral® with respect to being dispersed, released and absorbed in vivo. The loaded LLT were able to contain CyA-SMEDDS and release the complete dose in vivo, as evident from the fact that the absorption of CyA from loaded LLT (F6) was comparable to the liquid filled capsule formulations (F3 and F4).
Overall, the two dog studies show that LLT are able to release the drug content in vivo. Loaded LLT and the capsule formulations of CyA performed equally well.
Comparing Approaches to Solid Formulation of Liquids
The approach of absorbing a liquid into a preformed and non-collapsible carrier was used by Patil and Paradkar (21) to load porous polystyrene beads (PPB) with a SEDDS. The geometrical features such as bead size and pore architecture of PPB were found to influence the loading efficiency and subsequent in vitro drug release. They loaded 1 g PPB with 1.7 mL SEDDS. Compared with these results, the LLT tested in this study were able to absorb larger amounts of liquid. LLT were found in this study to be able to absorb 2.2 mL/g. A high loading degree is beneficial in terms of preparing solid dosage forms with a small size in combination with a high liquid content. The technology is thereby seen as a possible alternative to capsules and other solid liquid systems such as liquid-loaded powders, liquid-loaded beads, or solid SMEDDS. The manufacturing process of LLT stands out as being a simple procedure that can easily be scaled up since it is based on conventional tableting. The simple loading method allows tablets to be produced with a consistent content of active drug substance. Due to capillary forces the liquid is trapped inside the tablet pores and leaking of the liquid content has not been observed. By entrapping the liquid formulation inside a solid matrix, the chemical stability may also be improved.
CONCLUSION
LLT are seen as a potential alternative to soft gelatin capsules or hard two-piece capsules, with a high loading potential due to very high tablet porosity. Tablet disintegration of loaded LLT was found to influence the release of the loaded CyA-SMEDDS in vitro. Human in vivo studies are to be carried out to confirm the results obtained in dog in vivo studies.
Acknowledgement
The authors thank Carsten Schriver Højskov, Aarhus kommunehospital, for LC-MS analysis of cyclosporine in blood samples. For discussions regarding capillary absorption flow, we thank Jesper Qvist Thomassen. For reviewing this publication, we thank Ann Fullerton.
Abbreviations
- SEDDS
Self-emulsifying drug delivery system
- SMEDDS
Self-microemulsifying drug delivery system
- SNEDDS
Self-nanoemulsifying drug delivery system
- CyA
Cyclosporine A
- MAMS
Magnesium aluminometasilicate
- LLT
Liquid loadable tablets
- CyA-SMEDDS
SMEDDS containing CyA
- BCS
Biopharmaceutics classification system
References
- 1.Cole ET, Cade D, Benameur H. Challenges and opportunities in the encapsulation of liquid and semi-solid formulations into capsules for oral administration. Adv Drug Deliv Rev. 2008;60:747–756. doi: 10.1016/j.addr.2007.09.009. [DOI] [PubMed] [Google Scholar]
- 2.Bergstrom DH, Waranis RP, Rahman MS. Capsules, soft. In: Swarbrick J, Boylan JC, editors. Encyclopedia of pharmaceutical technology. 2. New York: Marcel Dekker; 2002. pp. 317–327. [Google Scholar]
- 3.Marchaud D, Hughes S. Solid dosage forms from self-emulsifying lipidic formulations. Pharmaceutical Technology Europe 2008;46–9
- 4.Porter CJ, Trevaskis NL, Charman WN. Lipids and lipid-based formulations: optimizing the oral delivery of lipophilic drugs. Nat Rev Drug Discov. 2007;6(3):231–248. doi: 10.1038/nrd2197. [DOI] [PubMed] [Google Scholar]
- 5.Pouton CW. Formulation of poorly water-soluble drugs for oral administration: physicochemical and physiological issues and the lipid formulation classification system. Eur J Pharm Sci. 2006;29(3–4):278–287. doi: 10.1016/j.ejps.2006.04.016. [DOI] [PubMed] [Google Scholar]
- 6.Yi T, Wan J, Xu H, Yang X. A new solid self-microemulsifying formulation prepared by spray-drying to improve the oral bioavailability of poorly water soluble drugs. Eur J Pharm Biopharm. 2008;70(2):439–444. doi: 10.1016/j.ejpb.2008.05.001. [DOI] [PubMed] [Google Scholar]
- 7.Patil P, Joshi P, Paradkar A. Effect of formulation variables on preparation and evaluation of gelled self-emulsifying drug delivery system (SEDDS) of ketoprofen. AAPS PharmSciTech. 2004;5(3):e42. doi: 10.1208/pt050342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dixit RP, Nagarsenker MS. Self-nanoemulsifying granules of ezetimibe: design, optimization and evaluation. Eur J Pharm Sci. 2008;35(3):183–192. doi: 10.1016/j.ejps.2008.06.013. [DOI] [PubMed] [Google Scholar]
- 9.Nazzal S, Nutan M, Palamakula A, Shah R, Zaghloul AA, Khan MA. Optimization of a self-nanoemulsified tablet dosage form of Ubiquinone using response surface methodology: effect of formulation ingredients. Int J Pharm. 2002;240(1–2):103–114. doi: 10.1016/S0378-5173(02)00130-8. [DOI] [PubMed] [Google Scholar]
- 10.Jannin V, Musakhanian J, Marchaud D. Approaches for the development of solid and semi-solid lipid-based formulations. Adv Drug Deliv Rev. 2008;60(6):734–746. doi: 10.1016/j.addr.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 11.Bansal T, Mustafa G, Khan ZI, Ahmad FJ, Khar RK, Talegaonkar S. Solid self-nanoemulsifying delivery systems as a platform technology for formulation of poorly soluble drugs. Crit Rev Ther Drug Carrier Syst. 2008;25(1):63–116. doi: 10.1615/critrevtherdrugcarriersyst.v25.i1.20. [DOI] [PubMed] [Google Scholar]
- 12.Ruhland T, Nielsen SD, Holm P, Christensen CH. Nanoporous magnesium aluminometasilicate tablets for precise, controlled, and continuous dosing of chemical reagents and catalysts: applications in parallel solution-phase synthesis. J Comb Chem. 2007;9(2):301–305. doi: 10.1021/cc060089q. [DOI] [PubMed] [Google Scholar]
- 13.Italia JL, Bhardwaj V, Kumar MN. Disease, destination, dose and delivery aspects of ciclosporin: the state of the art. Drug Discov Today. 2006;11(17–18):846–854. doi: 10.1016/j.drudis.2006.07.015. [DOI] [PubMed] [Google Scholar]
- 14.Khoo S, Humberstone AJ, Porter CJH, Edvards GA, Charman WN. Formulation design and bioavailability assessment of lipidic self-emulsifying formulations of halofantrine. Int J Pharm. 1998;167:155–164. doi: 10.1016/S0378-5173(98)00054-4. [DOI] [Google Scholar]
- 15.Montgomery DC. Design and analysis of experiments. 6. Hoboken: Wiley; 2005. [Google Scholar]
- 16.Washburn E. The dynamics of capillary flow. Phys Rev. 1921;17:273–283. doi: 10.1103/PhysRev.17.273. [DOI] [Google Scholar]
- 17.Aker E. A simulation model for two-phase flow in porous media, University of Oslo, Department of Physics; 1996
- 18.Rowe RC, Sheskey PJ, Owen SC. crospovidone. In: Rowe RC, Sheskey PJ, Owen SC, editors. Handbook of Pharmaceutical Excipients. 5. London: Pharmaceutical Press and APhA; 2006. pp. 214–216. [Google Scholar]
- 19.Odeberg JM, Kaufmann P, Kroon KG, Hoglund P. Lipid drug delivery and rational formulation design for lipophilic drugs with low oral bioavailability, applied to cyclosporine. Eur J Pharm Sci. 2003;20(4–5):375–382. doi: 10.1016/j.ejps.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 20.Muller RH, Runge SA, Ravelli V, Thunemann AF, Mehnert W, Souto EB. Cyclosporine-loaded solid lipid nanoparticles (SLN((R))): drug-lipid physicochemical interactions and characterization of drug incorporation. Eur J Pharm Biopharm. 2007. doi:10.1016/j.ejpb.2007.07.006 [DOI] [PubMed]
- 21.Patil P, Paradkar A. Porous polystyrene beads as carriers for self-emulsifying system containing loratadine. AAPS PharmSciTech. 2006;7(1):E28. doi: 10.1208/pt070128. [DOI] [PMC free article] [PubMed] [Google Scholar]