

Abstract
Praziquantel (PZQ), the primary drug of choice in the treatment of schistosomiasis, is a highly lipophilic drug that possesses high permeability and low aqueous solubility and is, therefore, classified as a Class II drug according to the Biopharmaceutics Classification System (BCS). In this work, β-cyclodextrin (β-CD) and hydroxypropyl-β-cyclodextrin (HP-β-CD) were used in order to determine whether increasing the aqueous solubility of a drug by complexation with CDs, a BCS-Class II compound like PZQ could behave as BCS-Class I (highly soluble/highly permeable) drug. Phase solubility and the kneading and lyophilization techniques were used for inclusion complex preparation; solubility was determined by UV spectroscopy. The ability of the water soluble polymer polyvinylpyrolidone (PVP) to increase the complexation and solubilization efficiency of β-CD and HP-β-CD for PZQ was examined. Results showed significant improvement of PZQ solubility in the presence of both cyclodextrins but no additional effect in the presence of PVP. The solubility/dose ratios values of PZQ-cyclodextrin complexes calculated considering the low (150 mg) and the high dose (600 mg) of PZQ, used in practice, indicate that PZQ complexation with CDs may result in drug dosage forms that would behave as a BCS-Class I depending on the administered dose.
Key words: biopharmaceutics classification, cyclodextrins, inclusion complex formation, praziquantel, solubility/dose ratio, solubility enhancement
INTRODUCTION
The Biopharmaceutics Classification System (BCS) developed by Amidon et al. (1) in 1995 in conjunction to the relevant FDA guidance (2) permitted drug classification in one of four classes based on its aqueous solubility and intestinal permeability. Recently, Rinaki et al. (3) developed a quantitative version of BCS, termed QBCS, using the solubility/dose ratio as the key parameter for solubility classification since it is inextricably linked to the dynamic characteristics of the dissolution process (4). The QBCS utilizes a solubility/dose ratio-permeability plane with scientifically physiologically based cutoff values for compound classification. Additionally, Gu et al. (5) provided conclusive evidence that the solubility/dose ratio is the key parameter for the enhancement of the extent of absorption by food for Class II drugs. Accordingly, if the solubility of a BCS-Class II drug can be enhanced to produce solubility/dose ratio values greater than unity, which is the reported cutoff value (3), the drug would behave as a BCS-Class I compound.
Praziquantel has been characterized as the primary drug of choice in the treatment of schistosomiasis and is recognized as an essential drug by WHO (6,7). It is a highly lipophilic drug that possesses low aqueous solubility and erratic absorption from the gastrointestinal (GI) tract. It is classified as a Class II drug according to BCS (8,9). Praziquantel also exhibits high first pass effect, and consequently, high doses are required for effective per os administration. Improvement of the solubility of praziquantel in aqueous media would result not only in improved GI absorption but also in the IV administration of the drug which is prohibited currently by its low aqueous solubility.
The ability of cyclodextrins (CDs) to form inclusion complexes with various molecules is widely used in pharmaceutical industry in order to increase aqueous solubility and bioavailability of lipophilic drugs (10–17). Toxicity studies have shown that when administered orally, cyclodextrins are practically nontoxic due to the lack of absorption from the gastrointestinal tract (11,12). However, due to toxicological considerations, natural β-cyclodextrin cannot be used in parenteral formulations (11,12). On the contrary, its hydrophilic derivative, hydroxypropyl-β-cyclodextrin, has been proven nontoxic and can be found in marketed parenteral formulations (11–13). From the regulatory point of view, both β- and hydroxypropyl-β-cyclodextrin are described in the European and US Pharmacopoeias (Eur. Pharm., USP/NF) while the latter is also cited in the FDAs list of Inactive Pharmaceutical Ingredients.
The aim and novelty of this study was to determine whether increasing the aqueous solubility of a drug by complexation with β-cyclodextrin and hydroxypropyl-β-cyclodextrin, a BCS-Class II compound like PZQ could behave as a BCS-Class I drug. Although complexation of PZQ with β-CD has been reported in the literature (18–20), its contribution to the biopharmaceutical behavior of the drug has not been considered, while the interaction of PZQ with HP-β-CD is investigated for the first time. Moreover, solubility improvement of PZQ by HP-β-CD complexation would possibly enable intravenous administration of PZQ. Phase solubility and kneading and lyophilization techniques were used for inclusion complex (binary systems) preparation, while solubility was determined by UV spectroscopy. The effect of sample heating at 70°C for 1 h before phase solubility experiments was also studied, while the ability of the water soluble polymer polyvinylpyrolidone to increase the complexation efficiency of β-CD and HP-β-CD for PZQ was also examined.
MATERIALS AND METHODS
Apparatus
A Perkin Elmer Lambda 6 double-beam UV/VIS spectrometer (Illinois, USA) was used for spectrophotometric measurements. The operational conditions were: scan speed 100 nm/min, scan range 240–500 nm. Lyophilizator, Telstar, Model Kryodos–50, (Terrassa, Spain) was used for complex preparation in solid state. Thermostated shaking bath (Julabo SW-1, Julabo Labortechnik GmbH, Germany) with externally adapted thermostating unit (Edmund, Buhler, UK) was used for solubility experiments.
Reagents and Materials
All chemicals were of analytical purity grade. 2-Hydroxypropyl-β-cyclodextrin (HP-β-CD) of molar substitution 0.6  was purchased from Sigma–Aldrich (St. Louis, MO). β-cyclodextrin (β-CD)
was purchased from Sigma–Aldrich (St. Louis, MO). β-cyclodextrin (β-CD)  was purchased from Serva Electrophoresis GmbH (Heidelberg, Germany). Polyvinylpyrolidone (PVP) (KOLLIDON 30, BASF Aktiengesellschaft) and praziquantel (PZQ)
was purchased from Serva Electrophoresis GmbH (Heidelberg, Germany). Polyvinylpyrolidone (PVP) (KOLLIDON 30, BASF Aktiengesellschaft) and praziquantel (PZQ)  were kindly donated by the local companies Roumpoulakis S.A. and Veterin S.A., respectively. Water purified with Labconco water pro ps system (Kansas City, MO, USA) was used in all procedures. Nylon syringe filters Titan® of 5 μm pore size and regenerated cellulose syringe filters Titan® of 0.45 μm pore size, were purchased from Scientific Resources Inc. (Eatontown, NJ, USA).
were kindly donated by the local companies Roumpoulakis S.A. and Veterin S.A., respectively. Water purified with Labconco water pro ps system (Kansas City, MO, USA) was used in all procedures. Nylon syringe filters Titan® of 5 μm pore size and regenerated cellulose syringe filters Titan® of 0.45 μm pore size, were purchased from Scientific Resources Inc. (Eatontown, NJ, USA).
Procedures
Calibration Curves
Calibration curves for the spectrophotometric determination of PZQ were constructed using PZQ standard solutions, in a concentration range of 0.5–5.0 × 10−4 M, prepared on one hand in phosphate buffer 0.1 M at pH 7.4, in the absence and presence of PVP 0.1% w/v and on the other hand in purified water. The absorbance of all solutions was measured at λmax = 262 nm.
Solubility Studies—Phase Solubility Experiments
Determination of Equilibration Time
An excess amount of PZQ (40 mg) was added to 10 ml of 0.01 M cyclodextrin solution in 0.1 M phosphate buffer at pH 7.4, and the resulted suspensions were either equilibrated in a thermostated water bath at 25°C or heated at 70°C for 1 h and then equilibrated in a thermostated shaking bath at 25°C for up to 96 h. Aliquots were withdrawn at 0, 3, 6, 9, 12, 15, 24, 48, 72, and 96 h, filtered through regenerated cellulose syringe filters (pore size 0.45 μm) and analyzed spectrophotometrically for PZQ concentration.
Phase Solubility Experiments
An excess amount of PZQ (40 mg) was added to solutions containing from 0 to 0.01 M cyclodextrin, prepared in 0.1 M phosphate buffer at pH 7.4. Samples were either placed in a thermostated shaking bath and equilibrated at 25°C for 24 h or heated in a water bath at 70°C for 1 h and then placed in the thermostated at 25°C shaking bath and equilibrated for 24 h. After equilibration, suspensions were filtered through regenerated cellulose syringe filters of 0.45 μm pore size, and PZQ concentration was determined spectrometrically. Stability constants (K) of drug-cyclodextrin complexes were calculated according to the method of Higuchi and Connors (21), applying the following equation:
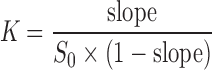 |
1 |
where slope is the slope of the corresponding phase-solubility diagrams, i.e., plots of measured PZQ solubility expressed as molar concentration versus cyclodextrin molar concentration, and S0 is the drug solubility in the absence of cyclodextrin.
Effect of PVP on Solubilization and Complexation Ability of β-CD and HP-β-CD
An excess amount of PZQ (40 mg) was added to solutions containing 0.01 M of β-CD or HP-β-CD and PVP in the concentration range of 0–1% w/v, prepared in 0.1 M phosphate buffer at pH 7.4. Samples were either equilibrated in a thermostated water bath at 25°C for 24 h or heated at 70°C, under sonication, for 1 h prior to equilibration at 25°C for 24 h. After equilibration, suspensions were filtered through regenerated cellulose syringe filters of 0.45 μm pore size, and PZQ concentration was determined spectrometrically after appropriate dilution with 0.1 M phosphate buffer at pH 7.4. PVP concentration in the measured samples was not greater than 0.1% w/v.
Preparation of PZQ–CD Solid State Binary Systems
Kneading and Lyophilization Procedure
For the preparation of PZQ–CD binary systems in the solid state, a procedure developed previously in our laboratory (22) was used and appropriately modified for the needs of the present study. More specifically, a quantity of 0.080 g of PZQ was mixed in a mortar with the appropriate amount of cyclodextrin (at 1:1, 1:2, and 1:4 molar ratios). The above mixture was ground for 20 min until a homogeneous powder was obtained. Then, 1 mL of water was added, and the mixture was kneaded for 5 min. Again, 1 mL of water was added, and kneading was continued for another 5 min in order to obtain a homogeneous paste. The paste was transferred into a beaker with 50 ml of water at 40°C. The suspension was stirred for 20 min at 40°C. Then, it was filtered, first through nylon membrane syringe filters (5.0 μm pore size) and then through regenerated cellulose syringe filters (0.45 μm pore size). An appropriate amount of the filtrate was used for quantification of PZQ, and the remaining amount was lyophilized to obtain solid state PZQ–CD binary systems.
PZQ Measurement in Lyophilized Products
Determination of PZQ content in the lyophilized products was performed spectrometrically according to the following procedure: An accurately weighed amount of lyophilized product was transferred into a 5.0-ml volumetric flask, dissolved in a small amount of purified water, and diluted to volume with the same solvent. The absorbance of this solution against purified water was measured at 262 nm. Concentration of PZQ was determined using the respective calibration curve, and the % w/w content of the lyophilized product with respect to PZQ was calculated taking into account the weighted amount of the lyophilized product.
RESULTS
Calibration Curves
Analytical parameters of PZQ calibration curves constructed in 0.1 M phosphate buffer at pH 7.4 in the absence and presence of PVP 0.1% w/v and in purified water are given in Table I. Statistical evaluation of the results using Student’s t test revealed no statistically significant differences between their slopes (P > 0.05); therefore, calibration curves for the quantification of PZQ were constructed in phosphate buffer of pH 7.4. The statistically equal values of slope and the zero intercept of the calibration curves constructed in the absence and presence of PVP reflect the specificity of the applied analytical method and the absence of interferences from the used polymer. Limits of detection and quantification of PZQ were also determined and found equal to 6.5 × 10−6 and 2.0 × 10−5 M, respectively. The within-day %RSD of the method was less than 2% while the between day value was less than 5% in the entire range of concentrations used for the calibration curves.
Table I.
Analytical Parameters of the Calibration Curves of PZQ in the Concentration Range of 0.5–5 × 10−4 M in (1) Phosphate Buffer pH 7.4, (2) the Same Phosphate Buffer Including PVP 0.1% w/v, and (3) Deionized Water
| Solvent | Slope (b ± s) | Intercept (a ± s) (×10−4) | r 2 (n) |
|---|---|---|---|
| Phosphate buffer pH 7.4 | 354.7 ± 5.2 | −3.5 ± 6.9 | 0.9999 (6) |
| PVP 0.1% w/v in phosphate buffer pH 7.4 | 349.7 ± 6.1 | −5.9 ± 6.8 | 0.9999 (6) |
| Deionized water | 355.4 ± 5.3 | −6.8 ± 7.0 | 0.9998 (6) |
Solubility Studies—Phase Solubility Experiments
Equilibration Time
Equilibration was reached within 12 h in the case of β-CD, while a 24-h period was needed for equilibration when HP-β-CD was used as complexing agent. Therefore, the 24-h time interval was decided to be the optimum equilibration time. In addition, the amount of PZQ added initially to the CDs solutions was proven adequate for the phase solubility experiments since in all cases, solid drug remained at the end of the experiment.
Equilibration time was also determined when samples were preheated at 70°C under sonication for 1 h prior to solubility experiments at 25°C. Preheating at 70°C under sonication for 1 h is a commonly used polymer activation procedure when water soluble polymers are used with cyclodextrins as solubility enhancement agents (11–13). However, the exact mechanism of activation is not known. According to our results, heating at 70°C under sonication for 1 h prior to equilibration at 25°C not only accelerated but also increased PZQ–CD binary system formation as reflected on the measured PZQ solubility values. In addition, the concentration of PZQ in the formed PZQ–CD binary system remained unchanged for more than 72 h in the thermostated at 25°C shaking bath, excluding thus the possibility of the formation of supersaturated solutions due to the polymer activation procedure. Based on these results, in conjunction with those obtained from samples that were equilibrated at 25°C without preheating at 70°C for 1 h, the 24-h time interval was chosen as optimum equilibration time for all experiments in order to obtain comparable data. Again, the amount of PZQ added initially to the CDs solutions was assured to be adequate for the phase solubility experiments.
Cyclodextrin and Preheating Effect on PZQ Solubility
Results from phase solubility experiments are presented in Fig. 1. The presence of β-CD and HP-β-CD was proven to increase the solubility of PZQ by a solubility enhancement factor, SCD/S0, of about 4.5, calculated as the ratio of PZQ solubility in 0.01 M CD solution (SCD) versus drug solubility value in the absence of CD (S0 = 0.54 × 10−3 M). These results are consistent with literature data (16–18) reporting a 4–5-fold increase in PZQ solubility in presence of β-CD. Preheating at 70°C for 1 h under sonication prior to equilibration resulted in further increase of drug solubility, and the calculated solubility enhancement factor was 5.5 and 6.0 for β-CD and HP-β-CD, respectively. Probably, the increase in aqueous solubility of pure PZQ caused by the elevated temperature during the preheating procedure resulted in enhancement of drug complexation with β-CD and HP-β-CD, since the dissolved amount of PZQ that was able to interact with β-CD and HP-β-CD to form soluble inclusion complexes was increased.
Fig. 1.

Phase solubility diagrams of PZQ with β-CD a and HP-β-CD b at 25°C and pH 7.4, without (filled circles) and with preheating at 70°C for 1 h under sonication (filled triangles)
Phase solubility diagrams shown in Fig. 1 are all linear, and complexation constants of PZQ with β-CD and HP-β-CD can be calculated from slopes and intercepts of the corresponding regression lines after linear regression data analysis. Such estimated complexation constant values are presented in Table II along with analytical parameters of the phase solubility diagrams and values of the solubility enhancement factor. PZQ complexation with β-CD was stronger than that with HP-β-CD as the calculated K values revealed (Table II), while the calculated K value for PZQ-β-CD interaction was in good agreement with that previously reported (18–20). It should be mentioned, however, that these previous studies deal only with the interaction of PZQ with either β-CD (19,20) or α-, β-, and γ-CD (18) while the interaction of PZQ with HP-β-CD is investigated for the first time in the present work. Results given in Table II also revealed that complexation affinity of CDs for PZQ was enhanced when preheating procedure was applied. In addition, enhancement was more profound for PZQ-HP-β-CD interaction, as reflected on the respective complexation constant, K, values. These results support the assumption that application of the preheating procedure lead to the increase of PZQ solubility through the enhancement of its complexation with β-CD and HP-β-CD.
Table II.
Analytical Parameters of Phase Solubility Diagrams and Estimated Values of Solubility Enhancement Factor, S CD/S 0, and Complexation Constant, K, of PZQ–β-CD and PZQ–HP-β-CD Binary Systems at 25°C, pH 7.4
| CD | Preheatinga | Slope (b ± SD) | Intercept (a ± SD) (×10−4 M) | r 2 (n)b | S CD/S 0 c | Complexation constant, K (±SD)d |
|---|---|---|---|---|---|---|
| β-CD | No | 0.180 ± 0.002 | 5.3 ± 0.1 | 0.9997 (6) | 4.3 | 412.2 ± 5.3 |
| Yes | 0.245 ± 0.005 | 6.1 ± 0.3 | 0.999 (6) | 5.3 | 531 ± 30 | |
| HP-β-CD | No | 0.164 ± 0.002 | 5.4 ± 0.1 | 0.9994 (6) | 4.0 | 365 ± 14 |
| Yes | 0.265 ± 0.002 | 5.6 ± 0.1 | 0.9998 (6) | 5.8 | 642 ± 13 |
aPreheating was performed for 1 h at 70°C under sonication, prior to equilibration at 25°C
bNumber of replications in parenthesis
cSolubility enhancement factor calculated as the ratio of PZQ solubility in 0.01 M CD solution (S CD) versus drug solubility value measured in the absence of CD (S 0 = 0.54 × 10−3 M)
dCalculated according to Eq. 1 using the slope and intercept (predicted S 0) of the respective phase solubility plots. The calculated K values become 407, 600, 365, and 667 M−1, respectively, when the experimental S 0 = 0.54 × 10−3 M is used in Eq. 1
Since all phase solubility plots were of AL type, and calculated slopes after linear regression analysis were found to be less than 1, it could be assumed, according to Higuchi and Connors (21), that stoichiometry of the formed binary systems was 1:1. This assumption is supported by a recent study (19) where molecular modeling and NMR analysis demonstrated that PZQ forms 1:1 inclusion complexes with β-CD and that the isoquinoline ring system of PZQ is embedded in the cavity of β-CD.
Effect of PVP on β-CD and HP-β-CD Solubilization Ability
Water soluble polymers such as hydroxypropyl methylcellulose, polyvinylpyrolidone, carboxymethyl cellulose, etc., have been previously reported (11,14,15) to increase complexation of various drug molecules with cyclodextrins. Choice of PVP was based on literature data (23) where this polymer was referred to increase both solubility and dissolution rate of PZQ by complex forming between drug and polymer. Effect of polymer activation by heating at 70°C for 1 h, under sonication, was also examined. Results are shown in Fig. 2. It is obvious that presence of PVP at concentrations of 0.1% and 1% w/v had no effect on the solubilization ability of both β-CD and HP-β-CD for PZQ, and only when the polymer was present at a concentration of 0.5% w/v, a slight but statistically not significant increase of β-CD and HP-β-CD solubilization ability was observed. In addition, in the absence of CDs, PVP had no effect on the aqueous solubility of PZQ (Fig. 2) irrespective of polymer activation by sample preheating. Furthermore, phase solubility experiments of PZQ with β-CD and HP-β-CD, performed in the presence of PVP 0.5% w/v and sample preheating (data not shown), resulted in linear plots and K estimates equal to 544 ± 34 and 627 ± 53 M−1, for PZQ-β-CD and PZQ-HP-β-CD complexes, respectively. These values are almost identical to those obtained in the absence of PVP under the same experimental conditions (Table II). Accordingly, presence of PVP affected neither the solubilization ability nor the complexation affinity of both β-CD and HP-β-CD for PZQ. This behavior is rather surprising since “bulky” polymers, such as PVP and HPMC, which are able to form hydrogen bonds, are reported to create non-inclusion complexes with cyclodextrins by hydrogen bond formation between the CD molecule and OH groups (or other groups) on the polymers (11,15). Therefore, enhancement of solubilization and complexation abilities of CDs is observed since ternary complexes between drug/CD/polymer can be formed. On the other hand, it was recently reported that drug/cyclodextrin complexes can self-associate through hydrogen bond formation to form water-soluble aggregates or micelles, which can further contribute to solubilize drugs through non-inclusion complexation (24). It could be assumed, therefore, that hydrogen bond formation between PZQ and CD molecules probably hinders any similar bond formation between PVP and CD molecules, inhibiting thus ternary PVP/drug/CD complex formation.
Fig. 2.

Effect of PVP concentration on the solubility of PZQ, at 25°C after 24 h equilibration, in the presence of β-CD a, HP-β-CD b, and in the absence of CDs c (all samples were preheated at 70°C for 1 h under sonication)
PZQ–CD Solid State Binary Systems Production
Effect of PZQ/CD Molar Ratio
The effect of molar ratio of PZQ/CD solid substances, used for the preparation of PZQ–CD binary systems, was studied. Results are shown in Fig. 3. For β-CD, a molar ratio of 1:2 (PZQ/CD) gave the highest PZQ concentration in the produced binary systems, while for HP-β-CD, the optimum molar ratio (PZQ/CD) was found to be 1:4. The different behavior of β-CD and HP-β-CD, regarding the effect of PZQ/CD molar ratio on the solubility of the produced PZQ/CD binary system, may be considered in parallel with the solubility order of the cyclodextrins employed [i.e., solubility of β-CD is much lower than that of HP-β-CD (10–17)] and the recent observation that hydrogen bond formation between drug/cyclodextrin complexes can further contribute to drug solubilization by cyclodextrins through non-inclusion complexation (24). It should be also taken into account that the low solubility of β-CD is attributed to the strong inter- and intramolecular hydrogen bond formation between the secondary hydroxyl groups of the CD molecules, while no such bonding is observed in the case of HP-β-CD due to partial substitution of the secondary hydroxyl groups of the CD molecule with hydroxypropyl groups. Thus, one can speculate that the contribution of non-inclusion complexation in the solubilization ability of HP-β-CD is more evident compared to β-CD, resulting in the almost linear increase of the solubilization ability of the former when its contribution to drug/CD molar ratio is increased from 1:1 to 1:4.
Fig. 3.

Effect of PZQ/β-CD and PZQ/HP-β-CD molar ratio of the solid substances used for the preparation of PZQ–CD binary systems on the concentration of PZQ in the produced binary system
Storage Stability of the PZQ–CD Solid State Binary Systems
Solid (lyophilized) PZQ–CD binary systems were produced using the optimum solid molar ratio (PZQ/CD) for each CD, i.e., 1:2 for β-CD and 1:4 for HP-β-CD, by lyophilization of the respective filtrates after the kneading procedure. Storage stability of the obtained solid products was studied over a time period of 1 month at room temperature in dark closed containers using UV spectrometry. No changes of the UV absorption spectra of PZQ were observed with respect either to the intensity or to the λmax value. Accordingly, solid binary systems were considered to be stable for the examined time period under the applied experimental conditions. Results from storage stability study are presented in Fig. 4.
Fig. 4.

A bar plot diagram showing the stability of the produced binary systems of PZQ with β-CD (filled bars) and HP-β-CD (gray bars) in solid state at room temperature using the kneading and lyophilization technique
Effect of CD Complexation on the Solubility/Dose Ratio of PZQ
Use of cyclodextrins to improve solubility is mainly related to BCS Class II drugs (16). These drugs are characterized by low aqueous solubility, where the solubility/dose ratio value is the limiting step for their GI absorption. Thus, cyclodextrins are particularly applied for production of drug dosage forms with dimensionless solubility/dose ratio values ≥1. These drug dosage forms would behave as BCS Class I drugs having higher bioavailability, since drugs of this class are completely dissolved and completely absorbed from GI tract.
As mentioned above, praziquantel is recognized as an essential drug by WHO (6,7). It is a highly lipophilic (clogP = 3.36) and was recently characterized (25) as “highly permeable” (Papp = 4.4 × 10−5 cm/s) drug. It also possesses low aqueous solubility (CS = 0.4 mg/mL) (25,26) and erratic absorption from the gastrointestinal tract. It is, therefore, classified as a BCS Class II drug (8,9). Regarding dosage forms, this drug is given per os as 150- and 600-mg tablets. Accordingly, dose-dependent biopharmaceutics classification may be observed. To this end, the effect of dose and PZQ complexation with β-CD and HP-β-CD on the calculated value of the solubility/dose ratio and the resulted biopharmaceutics classification of the drug were evaluated.
Solubility/Dose Ratio Calculation
The dimensionless solubility/dose ratio (1/q) of PZQ was calculated using the following formula (3,4):
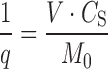 |
2 |
where M0 is the dose in milligrams, V is the intestinal volume content in milliliters, and CS is the saturation solubility in milligrams per milliliter. Solubility values of PZQ in the absence and presence of β-CD and HP-β-CD were used for calculation of the dimensionless solubility/dose ratio (1/q), in relation to the dose (150 or 600 mg) and the volume of the intestinal content (250 or 500 ml). More specifically, PZQ solubility values in the presence of β-CD and HP-β-CD were obtained from kneading experiment. Furthermore, consideration of two different volume values is based on the common practice to administer oral drugs with a glass of water (250 mL), while the value of 500 mL was also used to account for volume variability. In addition, the volume of 500 mL was recently proposed as the optimum volume for in vitro dissolution studies in the draft of the revised guideline on the investigation of bioequivalence of the European Medicines Agency (EMEA) (27) that was released in July 2008.
Biopharmaceutics Classification of PZQ Binary Systems
Results for PZQ biopharmaceutics classification according to the abovementioned assumptions are shown in Fig. 5a and b. Actually, these plots correspond to BCS-Classes I and II drugs since, as mentioned above, according to its Papp value, PZQ is characterized as highly permeable drug. Thus, β-CD/PZQ and HP-β-CD/PZQ molar ratio are used in the ordinate of Fig. 5a and b, respectively, in order to study the dose and cyclodextrin effect on the biopharmaceutics classification of this drug. In the absence of β-CD or HP-β-CD, PZQ behaves as a BCS-Class II drug irrespectively of the dose considered. It should be noted, however, that consideration of the low dose (150 mg) value resulted to an obvious right shift of the respective solubility/dose ratio value, reflecting the importance of this parameter to the biopharmaceutics classification of drug substances.
Fig. 5.

Biopharmaceutics classification of PZQ–CD binary systems produced by the kneading technique. Effect of PZQ/β-CD a and PZQ/HP-β-CD b molar ratio, dose of 150 mg (circles) or 600 mg (triangles) and volume of intestinal content 250 mL (black symbols) or 500 ml (gray symbols)
In the presence of β-CD and HP-β-CD, the calculated solubility/dose ratio, 1/q, values are shifted to the right, i.e., to higher values, as cyclodextrin concentration increases and dose is decreased (Fig. 5a and b). More specifically, when a low dose of 150 mg is considered, PZQ is classified as a BCS-Class I drug for all β-CD/PZQ or HP-β-CD/PZQ molar ratios used. On the contrary, when a high dose of 600 mg is examined, PZQ behaves as BCS-Class II drug unless the high volume of 500 mL is considered. In other words, not only cyclodextrin but also dose-dependent biopharmaceutics classification of PZQ is observed.
DISCUSSION
The phase solubility technique (21) and a kneading and lyophilization procedure were used to prepare binary systems of PZQ with β-CD and HP-β-CD. Improved aqueous solubility was observed compared to the parent drug that would probably result to enhanced oral bioavailability. Application of the kneading and lyophilization procedure resulted in binary systems with higher PZQ content in comparison to those obtained after application of the phase solubility technique, for both CDs examined (Table III), as reflected on the calculated solubility enhancement factor (SCD/S0) values. More specifically, when the kneading and lyophilization procedure was applied, the calculated solubility enhancement factor (SCD/S0), was twice greater than that calculated after application of the phase solubility technique. It should be mentioned, however, that application of the phase solubility technique was limited by the low solubility of β-CD; therefore, the drug/CD molar ratio was approximately 1:1 for the highest CD concentration used (0.01 M). On the other hand, when the kneading technique was applied, the optimum drug/CD molar ratio was used, i.e., 1:2 and 1:4 for β-CD and HP-β-CD, respectively. Overall, the solubility enhancement factor values calculated for β-CD and HP-β-CD are not significantly different when the same method of binary system preparation is considered (Table III). Therefore, taking into account the fact that the amount of HP-β-CD needed to produce the same PZQ solubility enhancement is twice as much of that needed from β-CD, due to the lower molecular weight of β-CD, and the limitations arising from the upper limit in size of conventional oral solid dosage forms (about 800 mg), it is concluded that β-CD should be preferred as solubilizing agent for PZQ when preparation of oral solid dosage forms is considered. On the other hand, the high value of the solubility enhancement factor calculated for HP-β-CD (Table III) in conjunction to its low toxicity (HP-β-CD is approved by FDA for intravenous administration) permit the assumption that PZQ-HP-β-CD binary systems produced by the kneading technique using 1:4 drug/CD molar ratio could be considered for intravenous administration. In addition, the lyophilized product prepared in the same molar ratio could be also considered for the same purpose. Furthermore, it should be mentioned that the complexation affinity of both CDs studied for PZQ, as reflected on the determined complexation constant, K, values (Table II), is in the range reported to be accepted for pharmaceutical application (10, 17). Finally, the observation that addition of PVP has no effect on the solubilization and complexation ability of CDs for PZQ should be correlated to the ability of the polymer to form hydrogen bonds with the PZQ–CD binary system. Recently, spectrophotometric and thermodynamic studies on the effect of PVP on the formation of naproxen–β-CD complex (28) have shown that PVP interacts with the drug–β-CD complex to form a ternary system in which PVP partially or totally coats the drug–β-CD complex, interacting with the CD and the drug through hydrogen bonds. However, the presence of different portions of PVP, in a range of 0–1% w/w, has not increased the ability of drug–CD complexation. This finding is in accordance to our results where neither the complexation nor the solubilization ability of CDs was found to be affected by the presence of PVP in the same concentration range.
Table III.
PZQ Content in the Produced Binary Systems with β-CD and HP-β-CD Prior and after Lyophilization
| Composition | Method of preparation | PZQ concentration prior to lyophilization (±SD)a (×10−3 M) | SCD/S0 a | PZQ content % (w/w) in the lyophilized product (±SD)b |
|---|---|---|---|---|
| PZQ/β-CD (1:2) | Kneading | 4.85 ± 0.1 | 9.0 | 11.5 ± 0.3 |
| PZQ, β-CD (0.01 M) | Phase solubility | 2.68 ± 0.04 | 4.9 | 7.4 ± 0.3 |
| PZQ/HP-β-CD (1:4) | Kneading | 5.05 ± 0.03 | 9.4 | 5.5 ± 0.2 |
| PZQ, HP-β-CD (0.01 M) | Phase solubility | 2.24 ± 0.03 | 4.0 | 4.8 ± 0.2 |
aSolubility enhancement factor calculated as the ratio of the concentration of PZQ in the solution of PZQ–CD binary systems prior to lyophilization, S CD, to the aqueous solubility of the drug, S 0
bDerived from more than three measurements
Regarding the Biopharmaceutics Classification of PZQ–CDs binary systems, the methodology used in this work could be seen as a useful tool for testing whether or not complexation with a cyclodextrin would result in enhanced biopharmaceutics behavior of a poor water soluble drug such as PZQ. In addition, despite the static nature of BCS according to which a static physicochemical drug property such as the aqueous solubility is considered to characterize a dynamic process such as in vivo dissolution, the use of all available doses in conjunction with the two volume levels applied, permit a rough estimate of the dissolution behavior of the produced PZQ–CDs binary systems.
CONCLUSIONS
The concept and the methodology suggested in this work would provide a useful tool for a rough prediction of the effect of CD complexation on the biopharmaceutics behavior of low solubility drugs according to the Biopharmaceutics Classification System. More specifically, Biopharmaceutics Classification of PZQ was found to be dose- and cyclodextrin-dependent since PZQ/CDs products moved from BCS-Class II to BCS-Class I when the low dose of 150 mg was considered irrespectively of the intestinal volume used. These findings were based on the results of the study on the effect of β-cyclodextrins on the aqueous solubility and solubility/dose ratio values of PZQ. In fact, PZQ solubility was increased by a factor of four in the presence of both β-CD and HP-β-CD, measured by the phase solubility technique, while presence of PVP did not affect either the solubilization ability or the complexation affinity of both cyclodextrins for PZQ. In addition, application of the kneading and lyophilization procedure resulted in binary systems with improved aqueous solubility compared not only to the parent drug but also to the binary systems prepared with the phase solubility technique.
Acknowledgments
This work was financially supported by the Special Account for Research Grants of the N&K University of Athens.
References
- 1.Amidon G, Lennarnas H, Shah V, Crison J. A theoretical basis for a biopharmaceutics drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res. 1995;12:413–429. doi: 10.1023/A:1016212804288. [DOI] [PubMed] [Google Scholar]
- 2.FDA/CDER, Guidance for industry, waiver of in vivo bioavailability and bioequivalence studies for immediate release solid oral dosage forms based on a biopharmaceutics classification system. Washington D.C., August 2000.
- 3.Rinaki E, Valsami G, Macheras P. Quantitative biopharmaceutics classification system: the central role of dose/solubility ratio. Pharm Res. 2003;20:1917–1925. doi: 10.1023/B:PHAM.0000008037.57884.11. [DOI] [PubMed] [Google Scholar]
- 4.Rinaki E, Dokoumetzidis A, Macheras P. The mean dissolution time depends on dose/solubility ratio. Pharm Res. 2003;20:406–408. doi: 10.1023/A:1022652004114. [DOI] [PubMed] [Google Scholar]
- 5.Gu C, Li H, Levons J, Lentz K, Gandhi R, Raghavan K, Smith R. Predicting effect of food on extent of drug absorption based on physicochemical properties. Pharm Res. 2007;24:1118–1130. doi: 10.1007/s11095-007-9236-1. [DOI] [PubMed] [Google Scholar]
- 6.WHO . Report of the WHO informal consultation on the use of Praziquantel during pregnancy/lactation and Albendazole/Mebendazole in children under 24 months. Geneva: WHO; 2002. [Google Scholar]
- 7.WHO . Prevent ion and control of schistosomiasis and soil-transmitted helminthiasis. Geneva: WHO; 2002. [Google Scholar]
- 8.Linderberg M, Kopp S, Dressman J. Classification of orally administered drugs on the World Health Organization Model list of Essential Medicines according to the biopharmaceutics classification system. Eur J Biopharm. 2004;58:265–278. doi: 10.1016/j.ejpb.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 9.Kasim N, Whitehouse M, Ramachandran C, Bermejo M, Lennernas H, Hussain A, et al. Molecular properties of WHO essential drugs and provisional biopharmaceutical classification. Molecular Pharmaceutics. 2004;1:85–96. doi: 10.1021/mp034006h. [DOI] [PubMed] [Google Scholar]
- 10.Duchêne D (ed) Cyclodextrins and their industrial uses. Editions de Santé, Paris, France; 1987.
- 11.Brewster M, Loftsson T. Cyclodextrins as pharmaceutical solubilizers. Adv Drug Del Rev. 2007;59:645–666. doi: 10.1016/j.addr.2007.05.012. [DOI] [PubMed] [Google Scholar]
- 12.Loftsson T, Duchene D. Cyclodextrins and their pharmaceutical applications. Int J Pharm. 2007;329:1–11. doi: 10.1016/j.ijpharm.2006.10.044. [DOI] [PubMed] [Google Scholar]
- 13.Gould S, Scott RC. 2-Hydroxypropyl-β-cyclodextrin (HP-β-CD): a toxicology review. Food Chem Toxicol. 2005;43:1451–1459. doi: 10.1016/j.fct.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 14.Loftsson T. Increasing the cyclodextrin complexion of drugs and drug bioavailability through addition of water-soluble polymers. Pharmazie. 1998;53:733–740. [PubMed] [Google Scholar]
- 15.Loftsson T, Masson M. The effects of water-soluble polymers on cyclodextrins and cyclodextrin solubilization of drugs. J Drug Del Sci Tech. 2004;14:35–43. [Google Scholar]
- 16.Loftsson T. Cyclodextrins and the biopharmaceutics classification system of drugs. J Inc Phen Macroc Chem. 2002;44:63–67. doi: 10.1023/A:1023088423667. [DOI] [Google Scholar]
- 17.Uekama K. Design and evaluation of cyclodextrin-based drug formulation. Chem Pharm Bull. 2004;52:900–915. doi: 10.1248/cpb.52.900. [DOI] [PubMed] [Google Scholar]
- 18.Becket G, Schep L, Tan M-Y. Improvement of the in vitro dissolution of praziquantel by complexation with α-, β- and γ-cyclodextrins. Int J Pharm. 1999;179:65–71. doi: 10.1016/S0378-5173(98)00382-2. [DOI] [PubMed] [Google Scholar]
- 19.De Jesus M, Pinto L, Fraceto L, Takahata Y, Lino A, Jaime C, De Paula E. Theoretical and experimental study of praziquantel and β-cyclodextrin inclusion complex using molecular mechanic calculations and 1H-nuclear magnetic resonance. J Pharm Biomed Anal. 2006;41:1428–1432. doi: 10.1016/j.jpba.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 20.El-Arini S, Leuenberger H. Dissolution properties of praziquantel-cyclodextrin systems. Pharm Dev Techn. 1996;1:307–315. doi: 10.3109/10837459609022600. [DOI] [PubMed] [Google Scholar]
- 21.Higuchi T, Connors K. Phase-solubility techniques. Adv Αnal Chem Instr. 1965;4:117–212. [Google Scholar]
- 22.Vertzoni M, Kartezini Th, Archontaki H, Reppas Ch, Valsami G. Solubilization and quantification of lycopene in aqueous media in the form of cyclodextrin binary systems. Int J Pharm. 2006;309:115–122. doi: 10.1016/j.ijpharm.2005.11.021. [DOI] [PubMed] [Google Scholar]
- 23.El-Arini S, Leuenberger H. Dissolution properties of praziquantel-PVP systems. Pharm Acta Helv. 1998;7:89–94. doi: 10.1016/S0031-6865(97)00051-4. [DOI] [PubMed] [Google Scholar]
- 24.Loftsson T, Masson M, Brewster M. Self-association of cyclodextrins and cyclodextrin complexes: minireview. J Pharm Sci. 2004;93:1091–1099. doi: 10.1002/jps.20047. [DOI] [PubMed] [Google Scholar]
- 25.Dinora G, Julio R, Nelly C, Lilian Y, Cook H. In vitro characterization of some biopharmaceutical properties of praziquantel. Int J Pharm. 2005;295:93–99. doi: 10.1016/j.ijpharm.2005.01.033. [DOI] [PubMed] [Google Scholar]
- 26.El-Arini S, Giron D, Leuenberger H. Solubility properties of racemic praziquantel and its enantiomers. Pharm Dev Technol. 1998;3:557–564. doi: 10.3109/10837459809028638. [DOI] [PubMed] [Google Scholar]
- 27.EMEA draft guideline on the investigation of bioequivalence, 24 July, 2008.
- 28.Valero M, Mura P. Effect of PVP-25 on the formation of the naproxen:β-cyclodextrin complex. Int J Pharm. 2003;253:97–110. doi: 10.1016/S0378-5173(02)00664-6. [DOI] [PubMed] [Google Scholar]