

Abstract
Alterations in the expression, molecular composition, and localization of voltage-gated sodium channels play major roles in a broad range of neurological disorders. Recent evidence identifies sodium channel proteolysis as a key early event after ischemia and traumatic brain injury, further expanding the role of the sodium channel in neurological diseases. In this study, we investigate the protease responsible for proteolytic cleavage of voltage-gated sodium channels (NaChs). NaCh proteolysis occurs after protease activation in rat brain homogenates, pharmacological disruption of ionic homeostasis in cortical cultures, and mechanical injury using an in vitro model of traumatic brain injury. Proteolysis requires Ca2+ and calpain activation but is not influenced by caspase-3 or cathepsin inhibition. Proteolysis results in loss of the full-length α-subunits, and the creation of fragments comprising all domains of the channel that retain interaction even after proteolysis. Cell surface biotinylation after mechanical injury indicates that proteolyzed NaChs remain in the membrane before noticeable evidence of neuronal death, providing a mechanism for altered action potential initiation, propagation, and downstream signaling events after Ca2+ elevation.
Introduction
Serving an integral role in neurotransmission, it is not surprising that voltage-gated sodium channels (NaChs) underlie or participate in a broad array of neurological disorders brought about by genetic mutations within channel subunits or alterations in subunit expression levels [for a recent review, see Waxman (2007)]. NaChs also play major pathophysiological roles in the early stages of ischemia, spinal cord injury, and traumatic brain injury where their inhibition preserves electrophysiological properties, leads to tissue sparing, and promotes functional recovery (Stys et al., 1992; Agrawal and Fehlings, 1996).
Despite this growing knowledge on NaChs and neurological disease, less well known is how an active intracellular event—NaCh proteolysis—contributes to either the incidence or progression of disease or injury in the nervous system. Overexpression of β-amyloid cleaving enzyme (BACE1) and δ-secretases leads to cleavage of the internal domain of the NaCh β2 subunit (Kim et al., 2005), while axonal stretch injury, ischemic injury to the isolated spinal cord, and pharmacological increases in membrane permeability lead to proteolysis of the pore forming α-subunit of the channel (Paillart et al., 1996; Iwata et al., 2004; Jette et al., 2006). However, the site of α-subunit proteolysis, the protease responsible for this cleavage, and the downstream consequences of proteolysis are not well defined. Here, we investigate NaCh proteolysis using models mimicking pathological elevations in cytosolic calcium or, alternatively, traumatic mechanical injury. We show that activated calpain causes NaCh proteolysis, suggesting that calpain-mediated NaCh cleavage may play an important pathophysiological role in recovery or demise after injury.
Materials and Methods
Calpain and caspase-3 activation in rat brain homogenates.
All reagents were purchased from Sigma-Aldrich unless noted otherwise. Brains from anesthetized adult female CDIGS rats (Charles River Laboratories) were removed and homogenized in 10 ml of homogenization buffer [10 mm HEPES, 5 mm EDTA, 0.035% β-mercaptoethanol, 0.5 mm phenylmethylsulphonyl fluoride (PMSF), and 1 μm pepstatin A, pH 7.4]. Homogenates were centrifuged (1000 × g, 10 min, 4°C) to remove the nuclear fraction. Protein concentrations were analyzed using the Bio-Rad Dc Protein Assay. Homogenates were pretreated with or without inhibitors to calpain (50 μm MDL28170, Calbiochem) or the caspases (30 μm z-VAD-FMK, Calbiochem) for 15 min on ice. Calcium (1, 5, 10, or 50 mm CaCl2) or active caspase 3 (20 U/ml, Calbiochem) was added to homogenates and samples were incubated either at 24°C for 30 min (calpain activation) or 37°C for 60 min (caspase 3 activation). Control samples were incubated on ice. The reaction was stopped after addition of electrophoresis sample buffer and 2.5% β-mercaptoethanol.
Calpain activation in rat brain membrane preparations.
Adult rat brains were removed from anesthetized adult female CDIGS rats (Charles River Laboratories) and homogenized in 10 ml of high sucrose buffer for membrane preparation (300 mm sucrose, 10 mm Tris, 2 mm EDTA, 0.5 mm PMSF, and 1 μm pepstatin A, pH 8.1) and centrifuged for 10 min (4000 × g, 4°C). The supernatant was subjected to an additional centrifugation step (100,000 × g, 1 h, 4°C), and the pellet was resuspended in Tris-acetate buffer (100 mm Tris-acetate, 0.1 mm EGTA, 0.5 mm PMSF, and 1 μm pepstatin A, pH 7.4) and centrifuged (16,000 × g, 10 min, 4°C). Supernatant (membrane preparation) protein concentrations were analyzed using the Bio-Rad Dc Protein Assay. Membrane preparations were pretreated with or without the calpain inhibitor (100 μm MDL28170, Calbiochem) for 15 min on ice. Calcium (1 mm CaCl2) and/or calpain 1 (5 and 50 U, Calbiochem) was added to membrane preparations and samples were incubated for 30 min at room temperature (24°C). The reaction was stopped after addition of electrophoresis sample buffer and 2.5% β-mercaptoethanol.
Cell culture.
Cortical neurons from embryonic day 18 rats were isolated and maintained as described previously (Geddes-Klein et al., 2006). For the in vitro stretch injury model, cultures were plated on poly-l-lysine-coated deformable silicone membranes (Sylgard 184 and 186, Dow Corning).
Ionomycin application and protease inhibition.
At days in vitro (DIV) 18–21, cultures were rinsed and incubated for 15 min in a saline solution (media composition, in mm: 100 NaCl, 5.4 KCl, 2 MgCl2, 1.8 mm CaCl2, 26 NaHCO3, 0.9 NaH2PO4, 10 HEPES, 25 glucose, pH 7.4). Where applicable, inhibitors of calpain (10 μm MDL28170), caspases (3 μm z-VAD-FMK), cathepsins (10 μm z-Phe-Gly-NHO-Bz, Calbiochem), the proteasome (1 μm lactacystin, Calbiochem), and the lysosome (100 μm chloroquine or 50 mm NH4Cl), as well as a 0.2% dimethyl sulfoxide (DMSO) control, remained in the media throughout the length of the experiment. In calcium-free media, CaCl2 was omitted and 1 mm EDTA was added. Cultures were incubated for various times (0–60 min) in an incubator with media containing ionomycin (1 μm, Invitrogen) and lysed (10 mm HEPES, 200 mm NaCl, 30 mm EDTA, 0.5% Triton-X, broad spectrum protease inhibitor cocktail [Complete Protease Inhibitor (Roche)], 50 mm NaF, 10 mm K4P2O7, 1 mm NaVO4). Lysates were sonicated and centrifuged (1000 × g, 10 min, 4°C) to remove the nuclear fraction and unlysed material.
Calcium imaging.
Increases in intracellular [Ca2+] were measured by using the ratiometric cell permeable, fluorescent Ca2+ indicator, Fura-2 A.M. (Invitrogen). Dissociated neurons were incubated with Fura-2 A.M. (5 μm in 0.1% Pluronic F-127, Invitrogen) for 40 min in saline solution at 37°C and 5% CO2. Using the same time course from the ionomycin studies (see above), cultures were excited at 340 and 380 nm every two seconds, and the images of the resulting emission (510 nm) intensities were captured with a Hamamatsu digital camera (C4742–98, Universal Imaging) and analyzed using MetaFlour (Molecular Devices). Cell bodies were selected as regions of interest and the average ratios of the intensities resulting from the two excitation wavelengths (340 nm/380 nm) over a 1 min period were collected. A Student's t test was used to compare treated and untreated groups at each time point.
Viral vector generation and transduction.
Recombinant adeno-associated viral (AAV) vectors were generated by the Vector Core at the University of Pennsylvania. The human calpastatin gene (clone ID 3878564 from Open Biosystems) was amplified by PCR using pCMV-SPORT6-hCAST as a template. The product of the PCR was cloned into ApaI and KpnI sites of the p1262 plasmid (designed by the University of Pennsylvania Vector Core). The vector coexpressed calpastatin and enhanced green fluorescent protein (EGFP) under the control of a cytomegalovirus (CMV) promoter by using a cis-acting hydrolase element (CHYSEL) sequence (AAV2/1.CMV.EGFP-T2A-hCAST). The CHYSEL sequence allowed bicistronic expression from a single transcript by using a self-cleaving peptide (Szymczak et al., 2004). Recombinant adeno-associated viral (AAV) vectors were generated by triple transfection of 293 cells, and purified by cesium chloride gradient sedimentation as described previously (Fisher et al., 1997). Vector genome titers were measured by real-time PCR using primer/probe set corresponding to the polyA region of the vectors. The AAV2/1 vector designed to express EGFP under the control of a CMV promoter was provided by the University of Pennsylvania Vector Core.
Cortical cultures at DIV 8 were transduced with 1 × 1010 genome copies of AAV 2/1 expressing EGFP, and human calpastatin or EGFP and transduction efficiency was assessed as described previously (Bevers et al., 2009). At DIV 19, 76 ± 3% and 76 ± 2% efficiency was achieved with human calpastatin/EGFP or EGFP, respectively.
Fragment isolation through immunoprecipitation.
Six 60 mm cortical culture dishes were treated with or without ionomycin and lysed in 600 μl of lysis buffer (in mm: 20 Tris-HCl, 200 NaCl, 1 mm EDTA, pH 7.4, inhibitor cocktail). Lysates were centrifuged (16,000 × g, 10 min, 4°C), the supernatants were precleared during a 1 h incubation with 40 μl of Protein G Plus Agarose slurry (Pierce) at 4°C, and then incubated with 2 μg of Nav1.2 C-terminal antibody or 2 μg of mouse IgG overnight at 4°C on a roller. Protein G slurry (40 μl) was added to the antibody/antigen complex and incubated for 1.5 h at 4°C, flow through was collected, and samples were eluted with 2× sample buffer plus 2% β-mercaptoethanol for 30 min at 37°C. Samples were analyzed via Western blot as stated below, except that a polyclonal pan (Alomone Labs, 1:1000) instead of a monoclonal pan (Sigma-Aldrich) antibody was used in this study to reduce IgG immunoreactivity.
In vitro stretch injury.
An in vitro injury model, as described previously, was used to stretch injure DIV 12–16 cortical neurons (Lusardi et al., 2004). In designated experimental groups, neurons were pretreated with protease inhibitors 15 min before injury. After stretch injury, cultures were returned to the incubator for 1 or 6 h (37°C, 5% CO2). All inhibitors remained in the culture media for the duration of the experiment. For Western blot analysis, the injured and uninjured regions were separated and lysed in the lysis buffer, sonicated, and centrifuged for 10 min (1000 × g, 4°C).
Cell death analysis.
For cell death assays, stretch injured cultures were incubated with the cell permeant nuclear marker Hoechst 33342 (5 μg/ml, Invitrogen) and cell impermeant marker ethidium homodimer-1 (2 μm, Invitrogen) for 30 min before the 6 h time point after injury. Dual images were captured using the appropriate excitation/emission filters (Hoechst: 71000A filter set, Chroma Technology; Ethidium homodimer: 41004 filter set, Chroma Technology) and analyzed using MetaMorph (Molecular Devices). Three separate fields of view were imaged and analyzed within each injured or uninjured culture. Hoechst-positive regions of interest (nuclear region) were identified and counted from the Hoechst emission image to attain overall cell numbers. The ethidium homodimer emission image was then overlaid with the Hoechst image, and dual positive cells, indicating loss of membrane integrity and cell death, were counted and normalized to the total number of cells (Hoechst positive) in an image. These ratios were averaged over the three images to give a final ratio per culture. Statistical significance for the data was determined by ANOVA (p < 0.05). If the data were found to be significant through ANOVA, post hoc analysis was conducted using Tukey–Kramer honestly significant difference (HSD) with p < 0.05 being considered significant.
Biotinylation.
Surface proteins from cultures were biotinylated 6 h after stretch injury using an established procedure (Wu et al., 2007). Cultures were placed on ice and incubated in a PBS solution containing 1 mg/ml biotin (Sulfo-NHS-Biotin, Pierce), rinsed, and the injured region was dissected out and lysed in RIPA lysis buffer. Biotinylated proteins were purified by adhesion to immobilized monomeric avadin (Pierce) while the unbound lysate was collected as the internal fraction. The biotinylated, surface proteins were eluted from the beads with electrophoresis sample buffer with 2% β-mercaptoethanol during heating to 37°C for 30 min. Initial lysate, internal, and biotinylated fractions were subjected to Western blot analysis as mentioned below.
Western blot.
Equal protein amounts (10 μg) from denatured samples were run on a 3–8% Tris-acetate gel (Invitrogen), transferred to PVDF membranes (Bio-Rad), and probed for the NaCh α-subunit III–IV loop (pan: Sigma Aldrich, 1:1000), the Nav1.2 C terminal [University of California (UC), Davis/National Institutes of Health (NIH) NeuroMab Facility, 1:1000], the Nav1.2 I–II loop (Alomone Labs, 1:1000), and the Nav1.1 C terminal (UC Davis/NIH NeuroMab Facility, 1:500) of the NaCh. Blots were stripped and reprobed for actin (Millipore Bioscience Research Reagents, 1:10,000), α-spectrin (MAB1622: Millipore Bioscience Research Reagents, 1:10,000) using an antibody that recognizes the full-length protein and calpain/caspase 3-specific breakdown products (Siman et al., 1984), or human calpastatin (Millipore Bioscience Research Reagents, 1:1000). Blots were incubated with corresponding HRP-conjugated antibodies (Jackson ImmunoResearch Laboratories). Blots were quantified using a computer assisted two-dimensional densitometric scanning program (Kodak 1D Image Analysis Software, Eastman Kodak Company). All proteins were normalized to corresponding negative/positive control groups. Statistical significance was determined by ANOVA (p < 0.05). Post hoc analysis was conducted using Tukey-Kramer HSD with a p < 0.05 being considered significant. Data were plotted as mean ± SEM.
Results
Calcium elevation leads to calpain-dependent proteolysis of the NaCh in rat brain homogenates
To investigate NaCh proteolysis in a simplified system, we incubated adult rat brain homogenates isolated in a 5 mm EDTA buffer for 30 min with 10 mm Ca2+ at room temperature (24°C). After Western blot, the α-spectrin antibody recognized 150 kDa bands, a known calpain/caspase 3-mediated breakdown product (supplemental Fig. S1A,B, available at www.jneurosci.org as supplemental material). Probing for NaChs using a pan antibody showed a concurrent accumulation of an ∼160 kDa and 110 kDa fragment and a loss of the full-length channel (supplemental Fig. S1A,B, available at www.jneurosci.org as supplemental material). Pretreatment with the calpain inhibitor MDL28170, but not the caspase inhibitor z-VAD-FMK, prevented both spectrin and pan NaCh fragment formation, suggesting that calcium addition leads to only calpain activation and subsequent proteolysis of both proteins (supplemental Fig. S1A,B, available at www.jneurosci.org as supplemental material). To further validate that NaCh proteolysis is calpain specific, active caspase 3 was added to rat brain homogenates at 37°C for 1 h. The 150 kDa spectrin breakdown product appeared and was prevented during pretreatment with the caspase inhibitor, while no proteolysis of the NaCh was observed (supplemental Fig. S1A,B, available at www.jneurosci.org as supplemental material). Therefore, NaCh proteolysis appears to be specific to calpain activation in this model.
Calpain cleaves multiple NaCh α-subunits in membrane preparations
The NaCh isoform targeted by calpain-mediated proteolysis was investigated further in isolated whole rat brain membranes. Calpain 1 and 1 mm Ca2+ were added to preparations and incubated for 30 min at room temperature. The addition of 1 mm Ca2+ alone was enough to induce fragment formation recognized by the NaCh pan, Nav1.2 (I–II loop), and spectrin antibodies. Adding calpain produced 160 and 110 kDa NaCh bands (pan, Nav1.2 C terminal) or 150, 100, and 80 kDa bands (Nav1.2 I–II loop) as well as the 150 kDa spectrin band (Fig. 1A–G). A significant loss of full-length protein immunoreactivity occurred for all proteins after 50 U calpain addition, while 5 U was enough to produce significant loss as recognized by the pan NaCh antibody. For Nav1.1 (C terminal), no increase in fragment accumulation occurred, even after a significant decrease in the full-length band, suggesting a loss in immunoreactivity after proteolysis. This indicates that the NaCh α-subunit is highly susceptible to calpain-mediated proteolysis, but that proteolysis may differ among α-subunits.
Figure 1.

Activation of calpain in rat whole-brain membrane preparations results in NaCh proteolysis. A–F. Representative Western blots displaying propensity or resistance to loss of full-length and appearance of lower-molecular-weight products recognized by the pan NaCh (A), Nav1.2 C terminal (B), Nav1.2 I–II loop (C), Nav1.1 C terminal (D), spectrin (E), and actin (F) antibodies after incubations with Ca2+ (1 mm), calpain, and/or MDL28170 (MDL) for 30 min at room temperature. Overexposed blots highlight NaCh breakdown fragments (A, C). G, Quantification of the representative blots seen in A. F, Data normalized to the full-length intensity of the corresponding room temperature control (n = 3–4, *p < 0.05 from the room temperature control).
Calpain-mediated proteolysis of the NaCh in dissociated cortical cultures occurs after elevating intracellular Ca2+
We next determined whether Ca2+/calpain-dependent proteolysis occurs in cortical neurons. Treating cortical neurons with a calcium ionophore (1 μm ionomycin) allowed us to gradually elevate the intracellular [Ca2+] over 1 h (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). Immunoblotting showed 150 kDa spectrin breakdown products, increasing in density with the time of ionomycin application, indicative of either calpain or caspase 3 activation (supplemental Fig. S3A,B, available at www.jneurosci.org as supplemental material). Over this time course, the ∼160 and 110 kDa proteolytic fragments appeared concurrently, as recognized by the pan NaCh antibody. Removing Ca2+ from the bathing solution and adding 1 mm EDTA (supplemental Fig. S3D,F, available at www.jneurosci.org as supplemental material) eliminated spectrin and NaCh breakdown.
Cortical neurons were then treated for 15 min with inhibitors targeting calpain I and II (MDL28170), caspases (z-VAD-FMK), cathepsins B, L, and S (z-Phe-Gly-NHO-Bz), the lysosome (chloroquine or NH4Cl), or the proteasome (lactacystin) before ionomycin application (supplemental Fig. S3C,E,F, available at www.jneurosci.org as supplemental material). Only the calpain inhibitor significantly reduced the density of the ionomycin-mediated NaCh and spectrin breakdown products to the level of the sham cultures. Lysosomal inhibition with chloroquine caused a small, yet significant, reduction in NaCh fragment formation. However, pretreatment with a second lysosome inhibitor (NH4Cl) or the cathepsin inhibitor had no significant effect on proteolysis (supplemental Fig. S3C,E,F, available at www.jneurosci.org as supplemental material), suggesting that the proteolytic protection provided by MDL28170 was primarily derived from its inhibition of calpain and not other proteases contained in the lysosome (Bevers and Neumar, 2008).
Thus far, the only highly specific inhibitor of calpain 1 and 2 is their endogenous inhibitor, calpastatin (Bevers and Neumar, 2008). To further validate that proteolysis was calpain specific, we overexpressed human calpastatin and EGFP in our rat neuronal cultures, transducing EGFP alone as a control. At DIV 19, 12 d after transduction, no significant loss of full-length NaChs or spectrin was observed in cultures overexpressing calpastatin after a 1 h ionomycin treatment, while significant loss was seen in all cultures only expressing EGFP (Fig. 2). This further supports the calpain specificity of NaCh proteolysis in cortical cultures after Ca2+ elevation.
Figure 2.

Overexpression of human calpastatin in rat cortical cultures prevents ionomycin-induced NaCh proteolysis. A, Representative Western blots showing the effect of calpastatin on NaCh proteolysis. At DIV 19, 10 d after transduction using an AAV with EGFP alone or EGFP and human calpastatin, cortical cultures were incubated with ionomycin (1 μm) for 1 h. Cultures transduced with calpastatin/EGFP displayed no significant ionomycin-induced NaCh α-subunit (pan, Nav1.2 C terminal) or spectrin proteolysis compared with those transduced with EGFP alone. B, Quantification of the representative blots seen in A. Data normalized to the corresponding untreated EGFP control (n = 4, *p < 0.05 from the EGFP control).
Nav1.2 fragment analysis suggests either 2 or 3 cleavage sites exist along the channel
As shown in the previous studies, both the pan NaCh antibody as well as the Nav1.2 (C-terminal) antibody recognize 160 and 110 kDa fragments, while the Nav1.2 (I–II loop) antibody recognizes 150, 100, and 80 kDa fragments. We hypothesized that the pan and Nav1.2 C-terminal antibodies were recognizing the same fragments, and therefore fragments immunoprecipitated by one antibody would be recognized by the other. Pulling down with the Nav1.2 (C-terminal) antibody and probing with the pan (III–IV loop) antibody displayed an enrichment in immunoreactivity for the full-length, 160, and 110 kDa bands, compared with the original lysate, indicating that the antibodies are indeed recognizing the same parts of the channel (Fig. 3A). The fragments recognized by the Nav1.2 I–II loop antibody are theoretically too small to contain the C terminal, and we expected to see a loss of fragment immunoreactivity during probing for Nav1.2 I–II after pulling down with the Nav1.2 C-terminal antibody. Indeed, while enrichment of immunoreactivity for the full-length Nav1.2 still occurred, immunoreactivity for all I–II loop containing fragments decreased, compared with the original lysate, suggesting a loss of direct binding to the Nav1.2 C-terminal antibody. The small amount of I–II loop containing fragments in the elution suggests that even after proteolysis, the domains of Nav1.2 retain their association in the membrane. Figure 3B summarizes these findings, displaying estimated cleavage sites within Nav1.2. The first predicted site occurs within the I–II loop, resulting in a 160 kDa fragment (containing the III–IV loop and C-terminal) and a 100 kDa fragment (containing the I–II loop). The second predicted site occurs within the II–III loop, resulting in a 110 kDa fragment (containing the III–IV loop and C terminal) and a 150 kDa fragment (containing the I–II loop). The origin of the final fragment (80 kDa, I–II loop) is less clear in this model. One possibility is that it indicates a third cleavage site within the N terminal or the I–II loop, creating a shortened version of the 100 kDa fragment containing domain I. However, nonspecific recognition of different NaCh isoform fragments cannot be ruled out, since a corresponding 180 kDa fragment recognized by the Nav1.2 C terminal antibody does not appear on these blots.
Figure 3.

Calpain-mediated NaCh proteolysis results in cleavage at two sites along Nav1.2. A, The Nav1.2 channel was immunoprecipitated from ionomycin-treated (Io; 1μm, 1 h) or control cultures using the Nav1.2 C-terminal antibody and probed for using a pan NaCh (III–IV loop) and Nav1.2 (I–II loop) antibody. In ionomycin-treated cultures, the pan antibody recognized the immunoprecipitated full-length channel and the 160 and 110 kDa fragments while the Nav1.2 (I–II loop) antibody displayed a loss of immunoreactivity for the 150, 100, and 80 kDa fragments. No immunoreactivity was seen in mouse IgG-treated lysates (n = 3). B, Cleavage sites and corresponding fragment creation within Nav1.2, as recognized by domain-specific antibodies and immunoprecipitation. Estimated cleavage sites are within the II–III loop and I–II loop. Sites of glycosylation (G), phosphorylation (P), ankyrin G binding (Ank G), and endocytosis (E) are indicated on the figure.
Proteolysis of the NaCh in an in vitro model of traumatic brain injury
Next, we investigated NaCh proteolysis in an in vitro model of traumatic brain injury that produces little immediate change in membrane permeability, but rather causes the activation of neuronal receptors and channels that include the NMDA receptor (Geddes-Klein et al., 2006). We used a level of stretch causing no significant increase in neuronal death 6 h after injury (supplemental Fig. S4, available at www.jneurosci.org as supplemental material), yet causing significant neuronal death 24 h after injury (Spaethling et al., 2008). Subsequently, we injured at these strain levels (60–80%) and collected lysates at 1 and 6 h time points. The pan NaCh antibody was chosen in these studies for robust recognition of fragment formation and channel loss. No significant accumulation of NaCh fragments appeared 1 h after injury (data not shown), yet a significant increase in both α-spectrin (150 kDa) and NaCh fragments (110 kDa) over uninjured controls was seen 6 h after mechanical injury (Fig. 4A,B). Consistent with previous data, mechanically injured cultures pretreated with the calpain inhibitor displayed a reduction in spectrin and NaCh α-subunit fragment accumulation (Fig. 4A,B).
Figure 4.
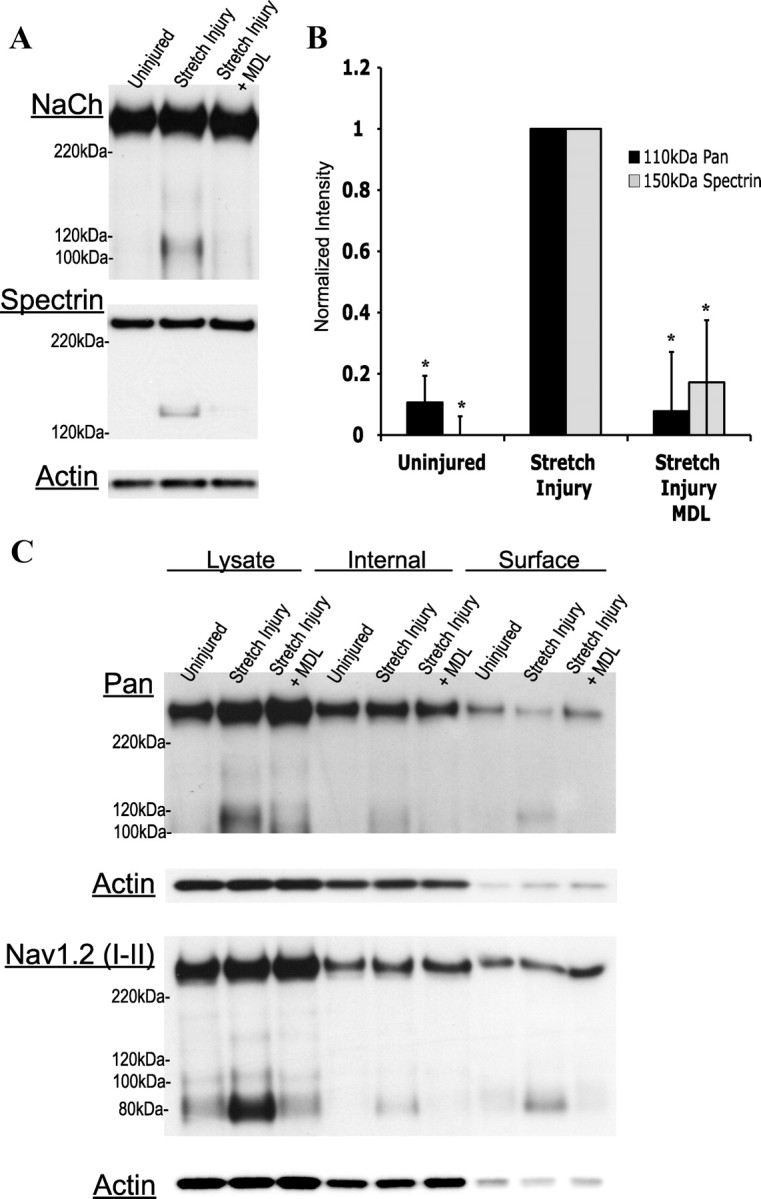
Calpain-dependent NaCh proteolysis occurs in an in vitro model of cortical stretch injury, targeting a specific internal loop of the channel. A, Representative Western blots displaying NaCh proteolysis after a 60–80% stretch injury, with or without pretreatment with the calpain inhibitor (MDL28170; MDL). B, Quantification of the representative blots seen in A. Data normalized to the corresponding stretch injured culture (n = 4, *p < 0.05 from the uninjured control). C, Representative Western blot showing the cellular distribution of proteolyzed NaChs after stretch injury. Six hours after injury, surface membrane biotinylation was performed on cultures with or without a 15 min MDL28170 pretreatment. The 110 and 80 kDa NaCh fragments appeared in the surface fraction, as recognized by the pan and Nav1.2 (I–II loop) antibody, respectively, suggesting that both a C- and an N-terminal portion of the channel remain in the membrane after proteolysis (n = 4).
A portion of cleaved NaChs remain at the membrane surface
To investigate cellular locations of proteolyzed sodium channels accumulating 6 h after stretch injury, biotinylated neuronal surface proteins were probed with the pan (III–IV loop) and Nav1.2 (I–II loop) antibodies, along with actin as a control for evaluating the purity of the fractions. Both 110 kDa III–IV loop containing fragments and 80 kDa I–II loop containing fragments were identified in the biotinylated, surface fraction, indicating that the majority of the channel remains at the surface after proteolysis (Fig. 4C). Fragments were also identified in the internal fraction, indicating that either proteolyzed channels eventually become internalized, or that calpain-mediated proteolysis is not specific to surface NaChs and also targets those located in the intracellular pool (Schmidt et al., 1985). Pretreatment of cultures with the calpain inhibitor resulted in a reduction of accumulation in the biotinylated fraction. These data indicate that calpain-mediated proteolysis of the channel does not lead to immediate internalization.
Discussion
In this study, we find that NaCh proteolysis occurs consistently after three experimental manipulations—activating calpain in rat brain preparations, after application of the Ca2+ ionophore ionomycin to elevate cytosolic Ca2+ in cortical neurons, and after mechanical stretch injury to cortical neurons. All conditions lead to robust calpain activation, as seen from the formation of calpain-specific spectrin breakdown products. Blocking calpain pharmacologically or through overexpression of its endogenous inhibitor calpastatin prevents both NaCh and spectrin proteolysis, while blocking other cysteine proteases including the caspases and cathepsin B, L, and S, as well as normal protein turnover mechanisms of the lysosome and proteasome, does not reduce proteolysis to control levels. At least two sites of proteolysis exist, within the I–II and II–III loops of Nav1.2, and some degree of association remains between fragments after proteolysis. In stretch injured cultures, surface biotinylation identifies fragments comprising both the C- and N-terminal parts of Nav1.2, indicating that proteolyzed channels remain in the membrane after cleavage.
Proteolysis of NaCh α-subunits appears after ischemic injury to the isolated spinal cord, after elevation of intracellular Na+ in cultured neurons, and after mechanical injury to cultured axons (Paillart et al., 1996; Iwata et al., 2004; Jette et al., 2006). Unlike the highly Na+-dependent removal of NaChs from the surface preceding degradation in the lysosome (Paillart et al., 1996), our data demonstrates a primarily calpain-mediated/Ca2+-dependent proteolysis. In addition, our proteolysis requires more time than the relatively rapid (<20 min) loss of the immunoreactivity to the NaCh inactivation loop observed in cultured axons after mechanical injury. However, calpain is one protease family targeted by the broad spectrum protease inhibition technique used by Iwata et al. (2004), pointing out the possibility that calpain may also play a role with channel proteolysis in cultured axons.
Based on fragment formation/migration using several α-subunit antibodies, our data indicates that proteolysis occurs within the I–II and II–III loop of Nav1.2, providing several possible roles in which the calpain-mediated process may affect neurological function. Proteolysis within the II–III linker could interfere with cytoskeletal anchoring through ankyrin-G and spectrin (Jenkins and Bennett 2001) or endocytosis by affecting the internalization sequence found within this loop (Fache et al., 2004). The I–II loop contains multiple phosphorylation sites, and proteolysis along this region could alter kinase- and phosphatase-mediated channel regulation (Cantrell and Catterall, 2001; Beacham et al., 2007). Within Nav1.1, proteolysis resulted in loss of immunoreactivity for the C terminal without fragment formation, indicating that sites of proteolysis may differ between α-subunits.
Perhaps the most intriguing is our finding that proteolysis occurs before the loss of membrane integrity, a hallmark end stage indicator of neuronal death. In combination with our finding that proteolyzed NaChs remain at the surface, this suggests that proteolyzed NaChs can contribute to the complex cascades triggered by cytosolic calcium elevations and subsequent calpain activation. Calpain activation occurs in axons and neuronal somata throughout the brain after traumatic brain and spinal cord injury, stroke, and within a large number of degenerative neurological diseases (Saatman et al., 1996; Siman et al., 1996; Büki et al., 1999; Ray et al., 2003; Vosler et al., 2008). Future work on the calpain-mediated proteolysis of NaChs will determine whether this proteolytic event is evidence of a clear commitment to degeneration or, alternatively, a step that could be used ultimately as a survival signal. Once the implications of NaCh proteolysis are better understood, one can examine the potential benefits of calpain-specific therapies already available and ready for preclinical testing.
Footnotes
This work was supported by National Institutes of Health (NIH) Grants NS 35712 and NS PPG PO1 NS 056202. The monoclonal antibodies Nav1.1 and Nav1.2 were developed by and/or obtained from the University of California, Davis/NIH NeuroMab Facility, supported by NIH Grant U24NS050606, and maintained by the Department of Neurobiology, Physiology and Behavior, College of Biological Sciences, University of California, Davis. Support for vector production was provided by the NHBLI Gene Therapy Resource Program and the University of Pennsylvania Preclinical Vector Core facility.
References
- Agrawal SK, Fehlings MG. Mechanisms of secondary injury to spinal cord axons in vitro: role of Na+, Na(+)-K(+)-ATPase, the Na(+)-H+ exchanger, and the Na(+)-Ca2+ exchanger. J Neurosci. 1996;16:545–552. doi: 10.1523/JNEUROSCI.16-02-00545.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beacham D, Ahn M, Catterall WA, Scheuer T. Sites and molecular mechanisms of modulation of Na(v)1.2 channels by Fyn tyrosine kinase. J Neurosci. 2007;27:11543–11551. doi: 10.1523/JNEUROSCI.1743-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevers MB, Neumar RW. Mechanistic role of calpains in postischemic neurodegeneration. J Cereb Blood Flow Metab. 2008;28:655–673. doi: 10.1038/sj.jcbfm.9600595. [DOI] [PubMed] [Google Scholar]
- Bevers MB, Lawrence E, Maronski M, Starr N, Amesquita M, Neumar RW. Knockdown of m-calpain increases survival of primary hippocampal neurons following NMDA excitotoxicity. J Neurochem. 2009;108:1237–1250. doi: 10.1111/j.1471-4159.2008.05860.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Büki A, Siman R, Trojanowski JQ, Povlishock JT. The role of calpain-mediated spectrin proteolysis in traumatically induced axonal injury. J Neuropathol Exp Neurol. 1999;58:365–375. doi: 10.1097/00005072-199904000-00007. [DOI] [PubMed] [Google Scholar]
- Cantrell AR, Catterall WA. Neuromodulation of Na+ channels: an unexpected form of cellular plasticity. Nat Rev Neurosci. 2001;2:397–407. doi: 10.1038/35077553. [DOI] [PubMed] [Google Scholar]
- Fache MP, Moussif A, Fernandes F, Giraud P, Garrido JJ, Dargent B. Endocytotic elimination and domain-selective tethering constitute a potential mechanism of protein segregation at the axonal initial segment. J Cell Biol. 2004;166:571–578. doi: 10.1083/jcb.200312155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher KJ, Jooss K, Alston J, Yang Y, Haecker SE, High K, Pathak R, Raper SE, Wilson JM. Recombinant adeno-associated virus for muscle directed gene therapy. Nat Med. 1997;3:306–312. doi: 10.1038/nm0397-306. [DOI] [PubMed] [Google Scholar]
- Geddes-Klein DM, Schiffman KB, Meaney DF. Mechanisms and consequences of neuronal stretch injury in vitro differ with the model of trauma. J Neurotrauma. 2006;23:193–204. doi: 10.1089/neu.2006.23.193. [DOI] [PubMed] [Google Scholar]
- Iwata A, Stys PK, Wolf JA, Chen XH, Taylor AG, Meaney DF, Smith DH. Traumatic axonal injury induces proteolytic cleavage of the voltage-gated sodium channels modulated by tetrodotoxin and protease inhibitors. J Neurosci. 2004;24:4605–4613. doi: 10.1523/JNEUROSCI.0515-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins SM, Bennett V. Ankyrin-G coordinates assembly of the spectrin-based membrane skeleton, voltage-gated sodium channels, and L1 CAMs at Purkinje neuron initial segments. J Cell Biol. 2001;155:739–746. doi: 10.1083/jcb.200109026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jette N, Coderre E, Nikolaeva MA, Enright PD, Iwata A, Smith DH, Jiang Q, Stys PK. Spatiotemporal distribution of spectrin breakdown products induced by anoxia in adult rat optic nerve in vitro. J Cereb Blood Flow Metab. 2006;26:777–786. doi: 10.1038/sj.jcbfm.9600226. [DOI] [PubMed] [Google Scholar]
- Kim DY, Ingano LA, Carey BW, Pettingell WH, Kovacs DM. Presenilin/gamma-secretase-mediated cleavage of the voltage-gated sodium channel beta2-subunit regulates cell adhesion and migration. J Biol Chem. 2005;280:23251–23261. doi: 10.1074/jbc.M412938200. [DOI] [PubMed] [Google Scholar]
- Lusardi TA, Rangan J, Sun D, Smith DH, Meaney DF. A device to study the initiation and propagation of calcium transients in cultured neurons after mechanical stretch. Ann Biomed Eng. 2004;32:1546–1558. doi: 10.1114/b:abme.0000049038.75368.75. [DOI] [PubMed] [Google Scholar]
- Paillart C, Boudier JL, Boudier JA, Rochat H, Couraud F, Dargent B. Activity-induced internalization and rapid degradation of sodium channels in cultured fetal neurons. J Cell Biol. 1996;134:499–509. doi: 10.1083/jcb.134.2.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray SK, Hogan EL, Banik NL. Calpain in the pathophysiology of spinal cord injury: neuroprotection with calpain inhibitors. Brain Res Brain Res Rev. 2003;42:169–185. doi: 10.1016/s0165-0173(03)00152-8. [DOI] [PubMed] [Google Scholar]
- Saatman KE, Bozyczko-Coyne D, Marcy V, Siman R, McIntosh TK. Prolonged calpain-mediated spectrin breakdown occurs regionally following experimental brain injury in the rat. J Neuropathol Exp Neurol. 1996;55:850–860. doi: 10.1097/00005072-199607000-00010. [DOI] [PubMed] [Google Scholar]
- Schmidt J, Rossie S, Catterall WA. A large intracellular pool of inactive Na channel alpha subunits in developing rat brain. Proc Natl Acad Sci U S A. 1985;82:4847–4851. doi: 10.1073/pnas.82.14.4847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siman R, Baudry M, Lynch G. Brain fodrin: substrate for calpain I, an endogenous calcium-activated protease. Proc Natl Acad Sci U S A. 1984;81:3572–3576. doi: 10.1073/pnas.81.11.3572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siman R, Bozyczko-Coyne D, Savage MJ, Roberts-Lewis JM. The calcium-activated protease calpain I and ischemia-induced neurodegeneration. Adv Neurol. 1996;71:167–174. discussion 174–165. [PubMed] [Google Scholar]
- Spaethling JM, Klein DM, Singh P, Meaney DF. Calcium-permeable AMPA receptors appear in cortical neurons after traumatic mechanical injury and contribute to neuronal fate. J Neurotrauma. 2008;25:1207–1216. doi: 10.1089/neu.2008.0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stys PK, Waxman SG, Ransom BR. Ionic mechanisms of anoxic injury in mammalian CNS white matter: role of Na+ channels and Na(+)-Ca2+ exchanger. J Neurosci. 1992;12:430–439. doi: 10.1523/JNEUROSCI.12-02-00430.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szymczak AL, Workman CJ, Wang Y, Vignali KM, Dilioglou S, Vanin EF, Vignali DA. Correction of a multi-gene deficiency in vivo using a single ‘self-cleaving’ 2A peptide-based retroviral vector. Nat Biotechnol. 2004;22:589–594. doi: 10.1038/nbt957. [DOI] [PubMed] [Google Scholar]
- Vosler PS, Brennan CS, Chen J. Calpain-mediated signaling mechanisms in neuronal injury and neurodegeneration. Mol Neurobiol. 2008;38:78–100. doi: 10.1007/s12035-008-8036-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman SG. Channel, neuronal and clinical function in sodium channelopathies: from genotype to phenotype. Nat Neurosci. 2007;10:405–409. doi: 10.1038/nn1857. [DOI] [PubMed] [Google Scholar]
- Wu HY, Hsu FC, Gleichman AJ, Baconguis I, Coulter DA, Lynch DR. Fyn-mediated phosphorylation of NR2B Tyr-1336 controls calpain-mediated NR2B cleavage in neurons and heterologous systems. J Biol Chem. 2007;282:20075–20087. doi: 10.1074/jbc.M700624200. [DOI] [PMC free article] [PubMed] [Google Scholar]