

Abstract
Traumatic brain injury (TBI) is one of the most disabling injuries in the population, with 1.5 million Americans new cases each year and 5.3 million Americans overall requiring long-term daily care as a result of their injuries. One critical aspect in developing effective treatments for TBI is determining if new, specific receptor populations emerge in the early phase after injury that can subsequently be targeted to reduce neuronal death after injury. One specific glutamate receptor subtype, the calcium-permeable AMPA receptor (CP-AMPAR), is becoming increasingly recognized for its role in physiological and pathophysiological processes. Although present in relatively low levels in the mature brain, recent studies show that CP-AMPARs can appear following ischemic brain injury or status epilepticus, and the mechanisms that regulate the appearance of these receptors include alterations in transcription, RNA editing, and receptor trafficking. In this report, we use an in vitro model of TBI to show a gradual appearance of CP-AMPARs four hours following injury to cortical neurons. Moreover, the appearance of these receptors is mediated by the phosphorylation of CaMKIIα following injury. Selectively blocking CP-AMPARs after mechanical injury leads to a significant reduction in the cell death that occurs 24 h following injury in untreated controls, and is similar in protection offered by broad-spectrum NMDA and AMPA receptor antagonists. These data point to a potentially new and more targeted therapeutic approach for treating TBI.
Key words: calcium-permeable AMPA receptors, CaMKII, mechanical injury, trafficking, traumatic brain injury
Introduction
Despite its high incidence and morbidity, there are very limited treatment options for improving outcome after traumatic brain injury (TBI). One critical feature that can yield new insight into treatments for TBI is defining how the mechanical injury leads to progressive changes in neuronal receptors that can strongly influence cell fate or function. A major issue with targeting most glutamate receptors for treating TBI is that these same receptors participate in normal neuronal signaling. We aim to identify an injury induced target which would have little to no effect on physiologic function, but would still inhibit the neuronal cell death associated with TBI.
Although AMPA receptors (AMPAR) are often not associated with significant calcium influx, there are two specific subunits of the AMPAR (GluR1 and GluR2) that can lead to forming a calcium-permeable AMPAR (CP-AMPAR) under certain conditions. The GluR1 and GluR2 subunits are distributed in high levels throughout the hippocampus and cortex (Ozawa et al., 1998). One form of CP-AMPAR occurs when the GluR2 mRNA is not edited, leading to a GluR2 containing AMPAR with significantly higher calcium permeability than the edited GluR2 subunit. However, the majority of AMPARs in the mature brain contain the edited GluR2 subunit, and are therefore calcium-impermeable (Hollman et al., 1991). A second form of CP-AMPAR appears when the AMPAR is formed entirely from GluR1 subunits, termed GluR1 homomers. Under basal conditions, though, only 8% of AMPARs in CA1/CA2 neurons are GluR1 homomers and therefore calcium-permeable (Wenthold et al., 1996).
Despite this relatively low level of expression, calcium-permeable AMPARs are emerging as an AMPA receptor subtype that can significantly contribute to both physiological and pathophysiological processes in the nervous system. For example, CP-AMPARs modulate forms of long-term potentiation (LTP), can activate specific downstream signaling cascades (Barria et al., 1997; Kakegawa et al., 2002; Okada et al., 2003; Tian and Feig, 2006), and is one important alteration that occurs during activity-dependent changes at the synapse (Liu and Cull-Candy, 2000; Thiagarajan et al., 2005). More recent evidence points to the possible role that CP-AMPARs play in governing neuronal fate after disease or injury. For example, ischemic brain injury leads to changes in the RNA editing efficiency of ADAR2, an enzyme responsible for conferring the calcium-impermeable property to GluR2 containing AMPA receptors (Sommer et al., 1991; Peng et al., 2006). Alternatively, in both ischemia and following status epilepticus, the gene-silencing factor REST serves to inhibit GluR2 at the transcriptional level, leading to a higher balance of calcium-permeable AMPA receptors over days in the CA1 region of the hippocampus (Huang et al., 2002; Calderone et al., 2003). Alternatively, many rapid changes in calcium-permeable AMPA receptors are linked to the kinases that control AMPA receptor trafficking, with studies showing that protein kinase A and protein kinase C, as well as CaMKIIα, contribute to early AMPA mediated changes in models of pain, ischemia, and mild trauma (Semyanov and Godukhin, 2001; Jones and Sorkin, 2005; Bell et al., 2007). Across most in vivo pathological conditions, the targeted inhibition of CP-AMPARs leads to protection against neuronal death or pain (Sorkin et al., 1999; Jones and Sorkin, 2004; Noh et al., 2005), suggesting a potential new direction for treatments for several neurological disorders.
We report the appearance of CP-AMPARs in cortical neurons following in vitro mechanical injury, and show that these CP-AMPARs contribute to delayed enhanced cytosolic calcium transients elicited by AMPA stimulation after injury. We show that the appearance of CP-AMPARs following injury occurs despite only a brief period of increased cytosolic calcium, and is mediated by the phosphorylation of CaMKIIα. Importantly, our data show that blockade of CP-AMPARs after mechanical injury in vitro offers significant protection against neuronal death. Collectively, these data suggest that CP-AMPARs may be an especially encouraging new therapeutic target for TBI. Future studies can reveal how controlling the expression and activation of calcium-permeable AMPA receptors can be optimized for therapies that may eventually translate into improving the outcome of traumatically brain injured patients.
Methods
Primary cell culture
Animal procedures were performed in accordance with the Institutional Animal Care and Use Committee at the University of Pennsylvania. Primary cortical neurons were isolated from embryonic day 18 (E18; Charles River, Wilmington, MA) rats, and 250,000 cells were plated on a 1.227-in2 area of poly-L-lysine (PLL; Sigma, St. Louis, MO) coated silicone membrane. The culture surface (membrane) was attached to a stainless steel well. The silicone substrates used (Sylgard 184 + 186 mix; Dow Corning, Midland, MI) were transparent, flexible, and elastic, which allowed us to mimic the mechanical forces that occur during TBI (Meaney et al., 1995). Neurons were cultured in Neurobasal media (Invitrogen) with B27 in a humidified incubator at 37°C, 5% CO2 for 18–21 days in vitro (DIV). The length of time for culturing was used to allow for the full expression of glutamate receptors (Lin et al., 2002; Hall et al., 2007), and to avoid the spontaneous calcium oscillations that can appear in DIV11–14 cultures. These spontaneous calcium oscillations are considered a characteristic of immature neurons and will render the neurons insensitive to mechanical injury (Geddes-Klein et al., 2006b). Moreover, published reports that show the expression profile of the AMPA subunits begins to plateau after 16DIV (Kumar, 2002; Pellegrini-Giampietro, 1991), suggesting cells attain sufficient stable AMPAR profiles at DIV18–21.
Drug treatments
All compounds were solubilized in buffered saline solution (NBS; in mM: 51.3 NaCl, 5.4 KCl, 2 MgCl2, 1.8 CaCl2, 26 NaHCO3, 0.9 NaH2PO4, 10 HEPES, 0.001 TTX, pH 7.3), added to the cells 5 min prior to injury, and remained on the cultures for the duration of the experiment unless otherwise noted. Cultures were pre-incubated with compounds to inhibit either all AMPARs (NBQX; 20 μM), all NMDA receptors (APV, 5 μM), or CP-AMPARs (Joro Spider Toxin; Jstx; 1 μM; Biomol International, Plymouth Meeting, PA). A calcium response dose curve for AMPA stimulation was created for our cultures using Fura-2AM (Invitrogen; data not shown). A submaximal dose was chosen (10 μM AMPA; Sigma) to keep all measurements within the effective working range of the Fura-2 indicator.
Injury
We used an in vitro model of TBI that reproduces the mechanical forces that occur during injury on a cultured monolayer of neurons (Lusardi et al., 2004a). Cultures at DIV18–21 on flexible membranes were placed on a stainless steel plate and covered with a top plate to form a sealed chamber. Increasing the chamber pressure to its peak level in 15 msec caused the compliant silicone membrane to stretch, in turn applying a stretch to the cultured neurons. This pressurization phase was immediately followed by a release of the pressure, typically within 25 msec of achieving the peak pressure. Although cells were cultured over a circular area (23 mm diameter), we designed the supporting stainless steel plate to expose only cells in a defined region (a 6 mm × 18 mm rectangular region; approximately one third of the culture surface area) to a stretch, designing the system to provide a uniform membrane stretch over 95% of the surface within that region (Lusardi et al., 2004a). The membrane in the injured region was stretched uniaxially, where the width was extended 80% beyond its initial width before returning to its original dimension. We did not observe any obvious detachment of the cells from the membrane following stretch injury, nor did we observe any gross morphological changes in the cultures. Naïve unstretched neurons served as reference conditions for statistical comparison.
Calcium imaging
A group of cultures were mechanically injured and then assessed for either (a) the level of cytosolic calcium at different times postinjury, or (b) the response in cytosolic calcium to an application of AMPA (10 μM) to the media. Within each experimental condition, a minimum of four separate wells was tested. Only one timepoint was measured for each culture well. In some cultures designated for calcium measurements at increasing time points post-injury (30 min, 60 min, 2 h, 4 h, and 6 h), the calcium response to AMPA stimulation was also measured. None of the cultures used for calcium imaging were also used for electrophysiology and viability measurements. Forty-five minutes prior to a scheduled observation time, neurons were loaded with the calcium-sensitive fluorescent dye, Fura-2AM (Molecular Probes, Eugene, OR; 5 μM), at 37°C. Immediately prior to imaging, neuronal cultures were rinsed in buffered saline solution and placed on a Nikon TE300 microscope (Optical Apparatus, Ardmore, PA) for calcium fluorescence imaging. Cells were alternately excited at 340 and 380 nm using an excitation shutter filter wheel (Sutter Instruments, Novato, CA), and the corresponding emission images (510 nm) were collected using a 14-bit Hamamatsu camera (Optical Apparatus, Ardmore, PA) at a rate of approximately every 3 sec. The fluorescence ratio from excitation at the two wavelengths (F340, F380) was used to calculate the Fura ratio:
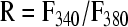 |
(1) |
To determine the response of neurons to AMPA stimulation, a baseline set of images was acquired for 30 sec prior to the addition of 10 μM AMPA, a concentration selected from pilot studies (1–100 μM) to avoid saturating the receptor response (Glaum, 1990) and maintain fluorescence measurements within the linear range of the Fura-2 indicator. Immediately following AMPA treatment, the response was captured for 2.5 min in the presence of AMPA. In each cell, we normalized the response to AMPA stimulation to its baseline fluorescence ratio. Cells with abnormally high baseline ratios, defined as a fluorescence ratio greater than 1 or less than 0.3, were excluded from analysis. The excluded cells were typically less than 5% of observed cells. All cells in the field of view, averaging approximately 100 cells per well, were imaged across three to four wells for each experimental condition studied. A nested analysis of variance, with the culture well for each cell as the nesting variable, was used to detect significant differences across experimental conditions. Tukey posthoc procedures were used for planned multiple comparisons following the analysis of variance (ANOVA). For all statistical conditions, p-values of less than 0.05 were considered significant.
Cell death
Cultures used to evaluate cell death were pretreated for 5 min with antagonist or control NBS, and then injured. The drugs remained on the culture for the 24 h until imaging. None of these cultures were used for either the calcium imaging or the electrophysiological measurements. Neurons were incubated with Hoechst (5 μg/mL) and ethidium homodimer (2 μM) for 30 min and then imaged using the fluorescence imaging system described above. Three images were taken from randomly chosen fields of view (approximately 0.55 mm2 each) from either the injured or uninjured regions of cultures. Typically, 300 cells per well were captured, and at least four culture wells from each treatment condition were used to obtain sufficient statistical power in our analysis. Cell viability was reported as the number of ethidium positive cells divided by the number of Hoechst positive cells, expressed as a percent. Cell death data was analyzed using an analysis of variance, with three independent images from each culture well serving as a nesting variable. As no significant differences were observed between drug treatments and control saline media in uninjured cultures, we used a one-way analysis with treatment as the independent variable. Tukey posthoc testing (p < 0.05) was used to detect differences between individual drug treatments and the untreated, injured condition.
Electrophysiology
Whole-cell patch clamp recordings were performed on 10 neurons each from the uninjured and injured groups. Neurons selected had a pyramidal like morphology, and recordings were performed using an Axoclamp-2B amplifier (Molecular Devices, Sunnyvale, CA) at room temperature. Only one cell per culture well was measured, and none of the wells used for electrophysiology were used for either the viability or calcium imaging measurements. Patch pipettes were fabricated from borosilicate glass capillaries (1.5 mm OD; World Precision Instruments, Inc., Sarasota, FL) and were drawn to a resistance between 4 and 6 MΩ using a pipette puller (Sutter Instrument, Novato, CA). Neurons were bathed in solution that contained (in mM): 130 NaCl, 4 KCl, 3 CaCl2, 10 HEPES, 11 glucose, 0.001 TTX, 0.010 nimodipine, 0.010 bicuculline, 0.025 APV, 0.001 CdCl2. Intracellular pipette solution contained (in mM): 120 CsCl, 5KCl, 2NaCl, 0.2 CaCl2, 10 EGTA, 10 HEPES, 2 MgATP, and 0.6 GTP.
Four hours after injury or NBS application, neurons were voltage clamped between −65 to −70 mV and cells with resting leak currents less than −100 pA were discarded. A ramp between −80 mV and 40 mV was applied, and currents were recorded. IAMPA was recorded approximately 2 min after perfusion with 10 μM AMPA. Based on a perfusion chamber volume of 0.5 mL and an estimated average flow rate of 3–4 mL per minute, we concluded that 3 min of perfusion was sufficient to exchange the initial solution with the AMPA solution. Baseline currents were subtracted from IAMPA and normalized to the baseline current at −80 mV. A nonlinear I-V relationship is characteristic of CP-AMPARs and is a result of the internal polyamine block at higher membrane potential (Bowie, 1995). Past studies use the rectification index (RI) as an indicator of the AMPAR calcium permeability in each cell and was calculated according to equation 2, where I40 is the recorded current at +40 mV, I−60 is the current at −60 mV, and Erev is approximated as 0 mV (Iino et al., 1996):
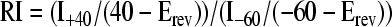 |
(2) |
Inward rectification was determined to occur for cells exhibiting an RI of <0.5, which is within the range of thresholds used in previous studies (Jia et al., 2002; Van Damme et al., 2002). A Chi-squared test was used to determine if injury increased the frequency of cells showing inward rectification.
Western blot
For injured samples, the marked injured region of membrane was excised from the rest of the culture. Therefore, only protein from the injured region was used for immunoblotting. Neurons were lysed in RIPA buffer (in mM 10 HEPES, 200 NaCl, 30 EDTA, 0.5% TritonX-100, pH 7.4 with Complete mini protease inhibitor [Roche Molecular Biochemicals], 0.5 NaF, 0.1 K4P2O7, 1 NaVO4). Cell lysates were cleared by centrifugation at 16,000g for 30 min at +4°C. 10 μg of protein per lysate was resolved on 4–12% Bis-Tris NuPage gels (Invitrogen) at 150 V for 1 h 30 min then transferred to a polyvinylidene difluoride (PVDF) membrane (Invitrogen) at 10 V overnight in transfer buffer (Invitrogen) containing 20% methanol. The membranes were blocked in TBST (20 mM Tris-HCL, 1.5 M NaCl, 0.1% Tween-20, pH 7.4) with 5% dry milk for 1 h at room temperature. The blots were then incubated with primary antibodies for phospho-CaMKIIα (1:1000; AB3827; Chemicon) in TBST with 5% dry milk overnight at 4°C. The blots were then rinsed in TBST and incubated with horseradish peroxidase-conjugated anti-rabbit secondary antibody (1:4000; Amersham Biosciences, Piscataway, NJ) for 2 h at room temperature. Blots were visualized using ECL detection (Pierce Biotechnology, Rockford, IL). The antibodies were stripped from the blots using Restore Stripping Buffer (Pierce; 15 min, 37°C) and reprobed for total CaMKIIα (0.1 μg/mL; MAB 3119, Chemicon-1:5000 HRP conjugated anti-mouse secondary, Jackson Immuno-Research) followed by actin (1:10000; Chemicon-1:20000 HRP conjugated anti-mouse secondary). Protein bands were quantified with a computer assisted densitometric scanning (Kodak 1D Image Analysis Software; Eastman Kodak Company, Rochester, NY). After subtracting for background intensity, the average area intensity for the phospho-CaMKIIα band was divided by the average area intensity for the total CaMKIIα band. Averages of the intensity ratios were calculated for injured and uninjured groups, and compared with Student's t-testing (p < 0.05).
Results
Mechanical injury leads to neuronal death at 24 hours, but only a transient increase in cytosolic calcium
One hallmark event in TBI is the immediate change in cytosolic calcium due to a combination of glutamate release and alteration in synaptic receptor properties following injury (Faden et al., 1989; Zhang et al., 1996). Previous use of the in vitro model in this report examined a stretch level (50% peak) that does not cause cortical neuronal death 24 h following injury (Geddes-Klein et al., 2006b). We increased the level of mechanical injury in 10% increments, and found the mechanical threshold for cortical neuronal death was 80% peak stretch (data not shown). We tested if this higher level of mechanical injury would lead to a persisting increase in cytosolic calcium, suggesting either long-term membrane damage or continued receptor activation such as continuously open NMDARs or Na+/Ca2+ exchangers. However, we found that the increase in cytosolic calcium did not last longer than 10 min following 80% peak stretch injury (Fig. 1A). We also tested if there was a possible secondary elevation in cytosolic calcium, owing to potential changes in ionic or mitochondrial dysregulation which can occur over several hours following mechanical injury in vitro (Tavalin et al., 1997; Weber et al., 1999, 2001; Ahmed et al., 2002). However, we did not observe elevations in baseline fluorescence ratios either 4 or 6 h following injury (Fig. 1A).
FIG. 1.

Mechanical injury to cultured cortical neurons leads to only a transient increase in cytosolic calcium, but a gradual and enhanced calcium influx from AMPA stimulation. (A) At levels of mechanical injury (80% peak stretch) that cause neuronal death 24 h after injury, the Fura-2AM fluorescence ratio was significantly increased only five minutes following stretch injury. Beyond this timepoint, the cytosolic calcium ratio was not different from pre-stretch control cultures. (B) At increasing time points following 80% stretch injury, calcium influx in response to AMPA (10 μM) application were measured and compared to the response to AMPA of uninjured control cultures. Sample Fura ratio traces are shown following the application of AMPA in control, unstretched cultures. and mechanically injured cultures (80% stretch) at 4 h after injury. (C) The mean response of all injured cells to AMPA stimulation (10 μM), when normalized to time matched uninjured controls, was enhanced at 4 and 6 h following injury (*p < 0.05).
Mechanical injury causes a gradual enhancement in AMPA-mediated increase in cytosolic calcium
A limited number of reports show the composition or function of synaptic glutamate receptors changes after mechanical injury and subsequently affect post-injury calcium signaling (Arundine et al., 2003, 2004). We specifically considered if changes to the AMPA receptor population in mechanically injured neurons would change over the first 6 hours following injury, leading to a potential change in cytosolic calcium signaling over this time period. At early time-points following injury (30 min, 60 min), we found that a modest 10 μM AMPA stimulation would elicit cytosolic calcium increases that were lower in magnitude than uninjured controls (Fig. 1C). Although we detected no difference between uninjured and injured samples 2 h after injury, we observed a progressive and enhanced response to this modest AMPA stimulation at 4 and 6 h after injury (Fig. 1B,C). CP-AMPARs appear after mechanical injury and contribute to an enhanced, AMPA mediated increase in cytosolic calcium.
A number of synaptic changes could explain the enhanced cytosolic calcium response to AMPA stimulation following mechanical injury. Past studies show that AMPA receptors lose their normal desensitization property following traumatic injury, and that this change persists for at least 24 h following mechanical injury (Goforth et al., 1999, 2004). However, this would likely lead to an enhanced response across all time points following injury, rather than the delayed enhancement of the calcium response we observed 4 h after stretch injury.
We tested if the potentiation of the cytosolic calcium response to AMPA was due to the appearance of calcium-permeable AMPA receptors. We used whole cell patch clamp measurements of the AMPA I-V relationship in mechanically injured neurons 4 h following injury, the same time point where we first noticed an enhanced response to AMPA stimulation. Of 10 control neurons, we noticed inward rectification behavior in only one neuron (Fig. 2A). In comparison, five of the 10 mechanically injured neurons showed a rectification index of less than 0.5, which was the threshold we developed in control cells to discern neurons with significant inward rectification from those neurons showing little to no rectification (Fig. 2A). As a group, mechanically injured neurons with low rectification index showed a clear inward rectification at positive membrane potentials. The relative frequency of neurons showing inward rectification behavior in the injured group was significantly different from the control, uninjured group (Fig. 2B). In our injury condition, there was no change in current density between control cells and cells 4 h post-injury (data not shown).
FIG. 2.

The increased calcium influx from AMPA stimulation is partly due to the appearance of calcium-permeable AMPA receptors. (A) Cells were injured, returned to the incubator, and whole cell voltage clamp recordings 4 h following injury were used to measure changes in the AMPA I-V relationship following injury. In nearly all (9/10) uninjured control cells (left), an outward rectification and linear I-V relationship was observed. In half (5/10) of the injured neurons (right), this linear I-V relationship was maintained. However, in the remaining half of injured neurons, there was a marked inward rectification (filled circles, right). Data shown as mean + SEM. (B) The difference in the proportion of neurons showing inward rectification between uninjured controls and mechanically injured neurons was significant using a Chi-squared test (p < 0.05). (C) Consistent with the voltage clamp recordings, the enhancement in cytosolic calcium caused by modest AMPA stimulation was significantly reduced by incubating cultures with an inhibitor to calcium-permeable AMPA receptors (Joro spider toxin, 1 μM). No significant difference was observed between uninjured and Jstx treated, injured neurons (p < 0.05).
These electrophysiological data showed that a higher fraction of mechanically injured neurons exhibited the delayed appearance of CP-AMPARs, a possible reason for the enhanced calcium response to AMPA stimulation. We tested if a blocker of CP-AMPA receptors (Joro spider toxin) would influence the response of injured neurons to AMPA stimulation. Consistent with the appearance of CP-AMPARs in a fraction of the population, we found that blocking calcium-permeable AMPARs 4 h following injury led to a significant reduction in the cytosolic calcium increase from AMPA stimulation, bringing the response back to control, uninjured levels (Fig. 2C). The enhanced response to AMPA stimulation in mechanically injured neurons is mediated by CaMKIIα phosphorylation.
Several past studies point to possible mechanisms that regulate the appearance of CP-AMPARs. Due to the relatively rapid appearance of the receptors, we considered if kinases involved in AMPAR trafficking were activated following injury, and if this activation was important for the subsequent enhancement of the calcium response to AMPA stimulation. Of the potential kinases, we investigated the role of CaMKIIα in more detail, given its role in AMPA receptor insertion at the synapse (Hayashi et al., 2000). Recent data show that CaMKIIα is activated rapidly following in vivo TBI (Atkins et al., 2006; Schwarzbach et al., 2006) and that CaMKIIα activation is a key step in the loss of AMPA receptor desensitization following mechanical injury in vitro (Goforth et al., 2004). Immunoblot data showed that the autophosphorylated form of CaMKIIα was significantly increased 4 h following mechanical injury (Fig. 3A). Although it seems that there might be a decrease in total CaMKIIα expression post-injury, this change was not statistically significant. In addition, we found that inhibiting the CaMKIIα phosphorylation using KN-93 (2 μM) led to a reduction in the enhanced calcium response to AMPA stimulation 4 h following injury (Fig. 3B).
FIG. 3.

Mechanical injury of cortical neurons leads to CaMKIIα phosphorylation, which mediates the appearance of CP-AMPARs. (A) In mechanically injured cultures, a noticeable increase in the autophosphorylated form of CaMKIIα appeared at 4 h after injury (left), leading to a significant difference in the ratio of the autophosphorylated CaMKIIα/total CaMKII between injured (n = 5) and uninjured (n = 4) cultures. (B) Normalized increases in the calcium influx following AMPA stimulation was significantly reduced with KN-93 treatment (2 μM; n = 4; p < 0.05), an inhibitor of CaMKIIα phosphorylation, when compared to uninjured cultures 4 h following injury.
Blockade of CP-AMPARS leads to protection against neuronal death following mechanical injury
Given the appearance of CP-AMPARs following mechanical injury, we next tested if these receptors would influence neuronal fate 24 h after injury. Incubating cultures with an inhibitor of CP-AMPARs (Jstx) led to a significant reduction in ethidium homodimer staining 24 h after mechanical injury (Fig. 4A,B). Similar results were observed when all AMPA receptors were inhibited during injury using NBQX (20 μM) or with a broad spectrum NMDA receptor antagonist (APV, 5 μM), which has been shown to be effective in reducing neuronal death in several past in vitro studies (Cater et al., 2007; DeRidder et al., 2006).
FIG. 4.

Significant neuronal protection occurs with inhibition of calcium-permeable AMPARs after mechanical injury. (A) Cells were pretreated with saline, Joro spider toxin (Jstx; 1 μM), NBQX (20 μM), and APV (5 μM), and cell death was measured 24 h post-injury using ethidium homodimer (2 μM) and hoechst (5 μg/mL). The ratio of double labeled cells to those only hoechst positive were compared between control and injured cultures (scale bar = 20 μm). (B) Injury-induced cell death was equally prevented with a global AMPAR blockade or CP-AMPAR block before injury (n = 4; p < 0.05). In comparison, pretreatment with an NMDAR antagonist (APV) also led to significant neuronal protection.
Discussion
In this report, we show the appearance of calcium-permeable AMPA receptors in mechanically injured cortical neurons. At levels of mechanical injury sufficient to cause neuronal death at 24 h, we first identified changes in AMPA mediated increases in cytosolic calcium four hours following injury. We used whole cell voltage clamp to measure the AMPA I-V relationship in mechanically injured neurons four hours after injury, and observed inward rectification of AMPA currents in half of the injured cells. We found the AMPA mediated increase in cytosolic calcium 4 h following injury was significantly reduced if CP-AMPARs were pharmacologically blocked, and that the enhanced response to AMPA was not present if cultures are pretreated with an inhibitor for CaMKIIα phosphorylation. Perhaps most importantly, incubating cultures with inhibitors for calcium-permeable AMPARs led to significant protection against cell death 24h following injury. Together, these data show that the appearance of calcium-permeable AMPARs in cortical neurons following lethal mechanical injury is mediated by CaMKIIα phosphorylation and CP-AMPARs are an effective target for inhibiting the effects of mechanical injury on cortical neurons in vitro.
This current work continues to build upon past work showing dramatic changes in the properties of AMPA receptors after mechanical injury. In a similar in vitro model using mixed neuronal/glial cultures, past studies showed that mechanical injury can cause a loss of AMPA receptor desensitization within 15–20 min following mechanical injury, a subsequent enhancement of AMPA currents, and a persistence in these changes that last at least 24 h following injury (Goforth et al., 1999, 2004). These changes in the AMPAR properties were linked to the initial activation of the NMDA receptor following mechanical injury, an activation that could be due to the change in the properties of the NMDA receptor or, alternatively, the release of glutamate from the mechanical stimulation in vitro (LaPlaca and Thibault, 1998).
These data also parallel a recent report using cerebellar Purkinje neurons showing that calcium-permeable AMPARs appear following mild mechanical injury combined with a modest AMPA stimulation (Bell et al., 2007). Focusing on identifying the role of CP-AMPARs in a combined mechanical and excitotoxic insult that would model mild traumatic brain injuries, Bell et al. (2007) showed a rapid appearance of calcium-permeable AMPA receptors 15 min following the combined injury. Moreover, the cell death that ensued 20 h following the combined mechanical and chemical insult could be prevented if cultures were treated with a CP-AMPAR antagonist. Our study examined a more severe stretch level that would lead to neuronal death at 24 h in the absence of an additional excitotoxic insult, using cortical neurons instead of cerebellar neurons. The different stretch magnitudes may account for the difference in the relative timing for calcium-permeable AMPA receptors to appear following injury, but we cannot discount other experimental factors that include the maturation age in culture (Tian and Feig, 2006), the relative difference in the level of NMDA receptors between cortical and cerebellar neurons (Williams et al., 1993; Okada et al., 2003), or the influence of the different strain fields applied to the cultured neurons between the two studies (Geddes-Klein et al., 2006a). Nevertheless, the consistent stretch-induced appearance of CP-AMPARs across two different neuronal types and injury severities suggest this phenomenon is not unique to one culturing system or region of the brain, but has broader implications on the neuronal response to mechanical trauma in several brain regions.
The mechanisms that govern the level of calcium-permeable AMPA receptors in neurons are complex, influenced by the activation of PKC and/or CaMKIIα, the relative amount of synaptic activity, and the influence of other molecules that regulate the insertion and removal of synaptic AMPA receptors (Cull-Candy et al., 2006). The role of CaMKIIα is intriguing, as the autophosphorylated form of CaMKIIα can persist for periods much longer than the calcium transient required for its initial activation. Indeed, our data show that CaMKIIα is phosphorylated 4 h after injury, despite increases in cytosolic calcium that subside within 10 min after injury. Past studies point to the role of activated CaMKIIα in phosphorylating specific serine residues on the GluR1 subunit of the AMPA receptor, its role in inserting AMPARs at the synapse following activation, and its central role in NMDA receptor mediated LTP (Barria et al., 1997; Hayashi et al., 2000; Lisman and Zhabotinsky, 2001). Despite this past evidence, the presence and extent of CaMKIIα activation following TBI remains relatively unexplored. Our data show that CaMKIIα is centrally involved in explaining the appearance of CP-AMPARs in approximately half of the mechanically injured cortical neurons, as the inhibition of CaMKIIα phosphorylation led to the inhibition of an AMPA mediated increase in cytosolic calcium we normally observed four hours following injury. Although calcium sensitive PKC isoforms may also contribute to the subsequent appearance of CP-AMPARs (Boehm et al., 2006), we found the inhibition of PKC in naïve cultures led to an enhanced cytosolic calcium response to AMPA treatment (data not shown), making further comparisons to injured cultures difficult. We are careful not to assume the role of CaMKIIα is equally important for the persisting expression of CP-AMPARs, as changes in RNA editing may also lead to the expression of unedited GluR2 subunits that will confer calcium-permeable to GluR2-containing AMPARs that appear over several days following injury (Noh et al., 2005; Peng et al., 2006). Determining how the expression of CP-AMPARs may persist, and the possible difference in mechanisms that regulate the long-term expression of these CP-AMPARs, would lead to a more complete approach for regulating the appearance of CP-AMPARs for days following injury.
One of the potentially most important observations from our data is that the blockade of calcium-permeable AMPA receptors led to protection against neuronal death 24 h following mechanical injury similar to that of global AMPAR and NMDAR blockade. These data suggest that the initial activation of AMPARs and NMDARs, may lead to the delayed insertion of CP-AMPARs. We do not expect inhibiting voltage gated sodium channels (VGSC) would offer significant neuroprotection in this model of injury, as the VGSC contributes little to the initial calcium response to mechanical injury (Geddes-Klein et al., 2006a). Our data also show that prolonged periods of cytosolic calcium dysregulation are not needed for the appearance of CP-AMPARs, as the changes in cytosolic calcium following these lethal mechanical injury levels returned to baseline values within ten minutes. From one perspective, this observation highlights the possible importance of early transient changes in acute calcium signaling on long-term outcome. Certainly, there may be additional secondary excitotoxic insults appearing following traumatic injury that can lead to additional periods of calcium transients. One key feature, though, in our data is the relatively long 4-h time period between the initial injury and the resulting enhanced cytosolic calcium response to AMPA stimulation. This long time interval suggests there may be a correspondingly wide therapeutic time window to deliver CP-AMPAR antagonists and still maintain their effectiveness.
Despite the encouraging evidence for using CP-AMPAR antagonists to reduce neuronal death after mechanical injury, one must also be prudent about the potential side effects of CP-AMPARs, a problem which has limited the practical use of other glutamate receptor antagonists. It is encouraging to note that inhibiting CP-AMPARs in dissociated neurons does not significantly change network activity, the frequency of miniature excitatory postsynaptic currents (mEPSCs) (Liu et al., 2006), and only slightly changes the magnitude of mEPSCs (Kato et al., 2007). Moreover, current in vivo work has reported no motor function deficits associated with the treatment of animals with CP-AMPAR antagonists (Jones and Sorkin, 2004). However, these data are also balanced by recent studies showing the uncertain role of CP-AMPARs in long-term potentiation. Some research suggests that CP-AMPAR antagonists may interfere with this cellular process associated with memory (Plant et al., 2006) while others discount any role of CP-AMPARs in LTP (Adesnik and Nicoll, 2007). Nevertheless, combined with the recent demonstration that CP-AMPAR antagonists also provide neuroprotection against a combined mild mechanical injury and excitotoxic insult (Bell et al., 2007), these data offer a promising new and more focused AMPAR-based therapeutic direction for treating TBI in vivo. Several past studies using broad spectrum AMPA receptor antagonists indicate the promise that these compounds have for reducing lesion size or improving neurobehavioral outcome following injury (Ikonomidou and Turski, 1996; Furukawa et al., 2003). However, the blockade of all AMPARs may lead to long-term changes in synaptic AMPAR expression (Liu and Cull-Candy, 2000; Esteban et al., 2003; Lee et al., 2004; Thiagarajan et al., 2005), and will also interfere with the molecular mechanisms that can regulate the process of synaptic strengthening during the acute recovery period. Due to the relatively low level of CP-AMPAR expression throughout most of the mature brain (Wenthold et al., 1996), a CP-AMPAR specific approach offers a potentially more targeted approach than a complete AMPAR blockade. If the expression of CP-AMPARs continued for days following injury, this targeted approach may also be effective as a delayed therapy and represent a significant step forward for treating TBI. Indeed, recent work using models of ischemic brain injury demonstrate the clear neuroprotective benefits offered by CP-AMPAR antagonists, and also indicate the therapy can be delayed with only a modest reduction in efficacy (Noh et al., 2005). Although the role of CP-AMPARs in physiological processes such as long-term potentiation and depression are actively being studied and may need to be considered when designing in vivo approaches for CP-AMPAR-based treatments, there is now evidence building that CP-AMPARs may be a reasonable and effective in vivo target for treating TBI.
Acknowledgments
Funding was provided by the NIH (grants RO1 HD41699 and NS 35712).
Author Disclosure Statement
The authors declare that they have no competing financial interests.
References
- Adesnik H. Nicoll R.A. Conservation of glutamate receptor 2-containing AMPA receptors during long-term potentiation. J. Neurosci. 2007;27:4598–4602. doi: 10.1523/JNEUROSCI.0325-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed S.M. Weber J.T. Liang S. Willoughby K.A. Sitterding H.A. Rzigalinski B.A. Ellis E.F. NMDA receptor activation contributes to a portion of the decreased mitochondrial membrane potential and elevated intracellular free calcium in strain-injured neurons. J. Neurotrauma. 2002;19:1619–1629. doi: 10.1089/089771502762300274. [DOI] [PubMed] [Google Scholar]
- Arundine M. Aarts M. Lau A. Tymianski M. Vulnerability of central neurons to secondary insults after in vitro mechanical stretch. J. Neurosci. 2004;24:8106–8123. doi: 10.1523/JNEUROSCI.1362-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arundine M. Chopra G.K. Wrong A. Lei S. Aarts M.M. Macdonald J.F. Tymianski M. Enhanced vulnerability to NMDA toxicity in sublethal traumatic neuronal injury in vitro. J. Neurotrauma. 2003;20:1377–1395. doi: 10.1089/089771503322686166. [DOI] [PubMed] [Google Scholar]
- Atkins C.M. Chen S. Alonso O.F. Dietrich W.D. Hu B.R. Activation of calcium/calmodulin-dependent protein kinases after traumatic brain injury. J. Cereb. Blood Flow Metab. 2006;26:1507–1518. doi: 10.1038/sj.jcbfm.9600301. [DOI] [PubMed] [Google Scholar]
- Barria A. Muller D. Derkach V. Griffith L.C. Soderling T.R. Regulatory phosphorylation of AMPA-type glutamate receptors by CaM-KII during long-term potentiation. Science. 1997;276:2042–2045. doi: 10.1126/science.276.5321.2042. [DOI] [PubMed] [Google Scholar]
- Bell J.D. Ai J. Chen Y. Baker A.J. Mild in vitro trauma induces rapid Glur2 endocytosis, robustly augments calcium permeability and enhances susceptibility to secondary excitotoxic insult in cultured Purkinje cells. Brain. 2007;130:2528–2542. doi: 10.1093/brain/awm164. [DOI] [PubMed] [Google Scholar]
- Boehm J. Kang M. Johnson R. Esteban J. Huganir R. Malinow R. Synaptic Incorporation of AMPA Receptors during LTP is controlled by a PKC phosphorylation site on GluR1. Neuron. 2006;51:213–225. doi: 10.1016/j.neuron.2006.06.013. [DOI] [PubMed] [Google Scholar]
- Bowie D. Mayer M. Inward rectification of both AMPA and kainate subtype glutamate receptors generated by polyamine-mediated ion channel block. Neuron. 1995;15:453–462. doi: 10.1016/0896-6273(95)90049-7. [DOI] [PubMed] [Google Scholar]
- Calderone A. Jover T. Noh K.M. Tanaka H. Yokota H. Lin Y. Grooms S.Y. Regis R. Bennett M.V. Zukin R.S. Ischemic insults derepress the gene silencer REST in neurons destined to die. J. Neurosci. 2003;23:2112–2121. doi: 10.1523/JNEUROSCI.23-06-02112.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cater H.L. Gitterman D. Davis S.M. Benham C.D. Morrison B., III Sundstrom L.E. Stretch-induced injury in organotypic hippocampal slice cultures reproduces in vivo post-traumatic neurodegeneration: role of glutamate receptors and voltage-dependent calcium channels. J. Neurochem. 2007;101:434–447. doi: 10.1111/j.1471-4159.2006.04379.x. [DOI] [PubMed] [Google Scholar]
- Cull-Candy S. Kelly L. Farrant M. Regulation of Ca2+-permeable AMPA receptors: synaptic plasticity and beyond. Curr. Opin. Neurobiol. 2006;16:288–297. doi: 10.1016/j.conb.2006.05.012. [DOI] [PubMed] [Google Scholar]
- Deridder M. Simon M.J. Siman R. Auberson Y.P. Raghupathi R. Meaney D.F. Traumatic mechanical injury to the hippocampus in vitro causes regional caspase-3 and calpain activation that is influenced by NMDA receptor subunit composition. Neurobiol. Dis. 2006;22:165–176. doi: 10.1016/j.nbd.2005.10.011. [DOI] [PubMed] [Google Scholar]
- Esteban J.A. Shi S.H. Wilson C. Nuriya M. Huganir R.L. Malinow R. PKA phosphorylation of AMPA receptor subunits controls synaptic trafficking underlying plasticity. Nat. Neurosci. 2003;6:136–143. doi: 10.1038/nn997. [DOI] [PubMed] [Google Scholar]
- Faden A.I. Demediuk P. Panter S.S. Vink R. The role of excitatory amino acids and NMDA receptors in traumatic brain injury. Science. 1989;244:798–800. doi: 10.1126/science.2567056. [DOI] [PubMed] [Google Scholar]
- Furukawa T. Hoshino S. Kobayaski S. Asakura T. Takahashi M. Atsumi T. Teramoto A. The glutamate AMPA receptor antagonist, YM872, attenuates cortical tissue loss, regional cerebral edema, and neurological motor deficits after experimental brain injury in rats. J. Neurotrauma. 2003;20:269–278. doi: 10.1089/089771503321532851. [DOI] [PubMed] [Google Scholar]
- Geddes-Klein D.M. Schiffman K.B. Meaney D.F. Mechanisms and consequences of neuronal stretch injury in vitro differ with the model of trauma. J. Neurotrauma. 2006a;23:193–204. doi: 10.1089/neu.2006.23.193. [DOI] [PubMed] [Google Scholar]
- Geddes-Klein D.M. Serbest G. Mesfin M.N. Cohen A.S. Meaney D.F. Pharmacologically induced calcium oscillations protect neurons from increases in cytosolic calcium after trauma. J. Neurochem. 2006b;97:462–474. doi: 10.1111/j.1471-4159.2006.03761.x. [DOI] [PubMed] [Google Scholar]
- Goforth P.B. Ellis E.F. Satin L.S. Enhancement of AMPA-mediated current after traumatic injury in cortical neurons. J. Neurosci. 1999;19:7367–7374. doi: 10.1523/JNEUROSCI.19-17-07367.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goforth P.B. Ellis E.F. Satin L.S. Mechanical injury modulates AMPA receptor kinetics via an NMDA receptor-dependent pathway. J. Neurotrauma. 2004;21:719–732. doi: 10.1089/0897715041269704. [DOI] [PubMed] [Google Scholar]
- Hall J.H. Ripley B. Ghosh A. NR2B signaling regulates the development of synaptic AMPA receptor current. J. Neurosci. 2007;27:13446–13456. doi: 10.1523/JNEUROSCI.3793-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi Y. Shi S.H. Esteban J.A. Piccini A. Poncer J.C. Malinow R. Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science. 2000;287:2262–2267. doi: 10.1126/science.287.5461.2262. [DOI] [PubMed] [Google Scholar]
- Henninger N. Dützmann S. Sicard K.M. Kollmar R. Bardutzky J. Schwab S. Impaired spatial learning in a novel rat model of mild cerebral concussion injury. Exp. Neurol. 2005;195:447–457. doi: 10.1016/j.expneurol.2005.06.013. [DOI] [PubMed] [Google Scholar]
- Hollmann M. Hartley M. Heinemann S. Ca2+ channels of KA-AMPA-gated glulamate receptor channels depends on subunit composition. Science. 1991;252:851–853. doi: 10.1126/science.1709304. [DOI] [PubMed] [Google Scholar]
- Huang Y. Doherty J.J. Dingledine R. Altered histone acetylation at glutamate receptor 2 and brain-derived neurotrophic factor genes is an early event triggered by status epilepticus. J. Neurosci. 2002;22:8422–8428. doi: 10.1523/JNEUROSCI.22-19-08422.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iino M. Koike M. Isa T. Ozawa S. Voltage-dependent blockage of Ca2+-permeable AMPA receptors by joro spider toxin in cultured rat hippocampal neurones. J. Physiol. 1996;496(2):431–437. doi: 10.1113/jphysiol.1996.sp021696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonomidou C. Turski L. Prevention of trauma-induced neurodegeneration in infant and adult rat brain: glutamate antagonists. Metab. Brain Dis. 1996;11:125–141. doi: 10.1007/BF02069500. [DOI] [PubMed] [Google Scholar]
- Jia Y. Jeng J.-M. Sensi S.L. Weiss J.H. Zn2+ currents are mediated by calcium-permeable AMPA/kainate channels in cultured murine hippocampal neurons. J. Physiol. 2002;543:35–48. doi: 10.1113/jphysiol.2002.020172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones T.L. Sorkin L.S. Calcium-permeable alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid/kainate receptors mediate development, but not maintenance, of secondary allodynia evoked by first-degree burn in the rat. J. Pharmacol. Exp. Ther. 2004;310:223–229. doi: 10.1124/jpet.103.064741. [DOI] [PubMed] [Google Scholar]
- Jones T.L. Sorkin L.S. Activated PKA and PKC, but not CaMKIIα, are required for AMPA/kainate-mediated pain behavior in the thermal stimulus model. Pain. 2005;117:259–270. doi: 10.1016/j.pain.2005.06.003. [DOI] [PubMed] [Google Scholar]
- Kakegawa W. Yamada N. Iino M. Kameyama K. Umeda T. Tsuzuki K. Ozawa S. Postsynaptic expression of a new calcium pathway in hippocampal CA3 neurons and its influence on mossy fiber long-term potentiation. J. Neurosci. 2002;22:4312–4320. doi: 10.1523/JNEUROSCI.22-11-04312.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato K. Sekino Y. Takahashi H. Yasuda H. Shirao T. Increase in AMPA receptor-mediated miniature EPSC amplitude after chronic NMDA receptor blockade in cultured hippocampal neurons. Neurosci. Lett. 2007;418:4–8. doi: 10.1016/j.neulet.2007.02.058. [DOI] [PubMed] [Google Scholar]
- Laplaca M.C. Thibault L.E. Dynamic mechanical deformation of neurons triggers an acute calcium response and cell injury involving the N-methyl-d-aspartate glutamate receptor. J. Neurosci. Res. 1998;52:220–229. doi: 10.1002/(SICI)1097-4547(19980415)52:2<220::AID-JNR10>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Lee S.H. Simonetta A. Sheng M. Subunit rules governing the sorting of internalized AMPA receptors in hippocampal neurons. Neuron. 2004;43:221–236. doi: 10.1016/j.neuron.2004.06.015. [DOI] [PubMed] [Google Scholar]
- Lin Y.-C. Huang Z.-H. Jan I.-S. Yeh C.-C. Wu H.-J. Chou Y.-C. Chang Y.-C. Development of excitatory synapses in cultured neurons dissociated from the cortices of rat embryos and rat pups at birth. J. Neurosci. Res. 2002;67:484–493. doi: 10.1002/jnr.10077. [DOI] [PubMed] [Google Scholar]
- Lisman J.E. Zhabotinsky A.M. A model of synaptic memory: a CaMKII/PP1 switch that potentiates transmission by organizing an AMPA receptor anchoring assembly. Neuron. 2001;31:191–201. doi: 10.1016/s0896-6273(01)00364-6. [DOI] [PubMed] [Google Scholar]
- Liu B. Liao M. Mielke J.G. Ning K. Chen Y. Lei Li L. El-Hayek Y.H. Gomez E. Zukin R.S. Fehlings M.G. Wan Q. Ischemic insults direct glutamate receptor subunit 2-lacking AMPA receptors to synaptic sites. J. Neurosci. 2006;26:5309–5319. doi: 10.1523/JNEUROSCI.0567-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S.J. Zukin R.S. Ca2+-permeable AMPA receptors in synaptic plasticity and neuronal death. Trends Neurosci. 2007;30:126–134. doi: 10.1016/j.tins.2007.01.006. [DOI] [PubMed] [Google Scholar]
- Liu S.Q. Cull-Candy S.G. Synaptic activity at calcium-permeable AMPA receptors induces a switch in receptor subtype. Nature. 2000;405:454–458. doi: 10.1038/35013064. [DOI] [PubMed] [Google Scholar]
- Lusardi T.A. Rangan J. Sun D. Smith D.H. Meaney D.F. A device to study the initiation and propagation of calcium transients in cultured neurons after mechanical stretch. Ann. Biomed. Eng. 2004a;32:1546–1558. doi: 10.1114/b:abme.0000049038.75368.75. [DOI] [PubMed] [Google Scholar]
- Lusardi T.A. Wolf J.A. Putt M.E. Smith D.H. Meaney D.F. Effect of acute calcium influx after mechanical stretch injury in vitro on the viability of hippocampal neurons. J. Neurotrauma. 2004b;21:61–72. doi: 10.1089/089771504772695959. [DOI] [PubMed] [Google Scholar]
- Meaney D.F. Smith D.H. Shreiber D.I. Bain A.C. Miller R.T. Ross D.T. Gennarelli T.A. Biomechanical analysis of experimental diffuse axonal injury. J. Neurotrauma. 1995;12:689–694. doi: 10.1089/neu.1995.12.689. [DOI] [PubMed] [Google Scholar]
- Noh K.M. Yokota H. Mashiko T. Castillo P.E. Zukin R.S. Bennett M.V. Blockade of calcium-permeable AMPA receptors protects hippocampal neurons against global ischemia-induced death. Proc. Natl. Acad. Sci. USA. 2005;102:12230–12235. doi: 10.1073/pnas.0505408102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada T. Yamada N. Tsuzuki K. Horikawa H.P. Tanaka K. Ozawa S. Long-term potentiation in the hippocampal CA1 area and dentate gyrus plays different roles in spatial learning. Eur. J. Neurosci. 2003;17:341–349. doi: 10.1046/j.1460-9568.2003.02458.x. [DOI] [PubMed] [Google Scholar]
- Peng P.L. Zhong X. Tu W. Soundarapandian M.M. Molner P. Zhu D. Lau L. Liu S. Liu F. Lu Y. ADAR2-dependent RNA editing of AMPA receptor subunit GluR2 determines vulnerability of neurons in forebrain ischemia. Neuron. 2006;49:719–733. doi: 10.1016/j.neuron.2006.01.025. [DOI] [PubMed] [Google Scholar]
- Plant K. Pelkey K.A. Bortolotto Z.A. Morita D. Terashima A. McBain C.J. Collingridge G.L. Isaac J.T.R. Transient incorporation of native GluR2-lacking AMPA receptors during hippocampal long-term potentiation. Nat. Neurosci. 2006;9:602–604. doi: 10.1038/nn1678. [DOI] [PubMed] [Google Scholar]
- Schwarzbach E. Bonislawski D.P. Xiong G. Cohen A.S. Mechanisms underlying the inability to induce area CA1 LTP in the mouse after traumatic brain injury. Hippocampus. 2006;16:541–550. doi: 10.1002/hipo.20183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semyanov A. Godukhin O. Epileptiform activity and EPSP-spike potentiation induced in rat hippocampal CA1 slices by repeated high-K+: involvement of ionotropic glutamate receptors and Ca2+/calmodulin-dependent protein kinase II. Neuropharmacology. 2001;40:203–211. doi: 10.1016/s0028-3908(00)00147-7. [DOI] [PubMed] [Google Scholar]
- Sommer B. Kohler M. Sprengel R. Seeburg P.H. RNA editing in brain controls a determinant of ion flow in glutamate-gated channels. Cell. 1991;67:11–19. doi: 10.1016/0092-8674(91)90568-j. [DOI] [PubMed] [Google Scholar]
- Sorkin L.S. Yaksk T.L. Doom C.M. Mechanical allodynia in rats is blocked by a Ca2+-permeable AMPA receptor antagonist. Neuroreport. 1999;10:3523–3526. doi: 10.1097/00001756-199911260-00011. [DOI] [PubMed] [Google Scholar]
- Tavalin S.J. Ellis E.F. Satin L.S. Inhibition of the electrogenic Na pump underlies delayed depolarization of cortical neurons after mechanical injury or glutamate. J. Neurophysiol. 1997;77:632–638. doi: 10.1152/jn.1997.77.2.632. [DOI] [PubMed] [Google Scholar]
- Thiagarajan T.C. Lindskog M. Tsien R.W. Adaptation to synaptic inactivity in hippocampal neurons. Neuron. 2005;47:725–737. doi: 10.1016/j.neuron.2005.06.037. [DOI] [PubMed] [Google Scholar]
- Thurman D. Alverson C. Dunn K. Guerrero J. Sniezek J. Traumatic brain injury in the United States: a public health perspective. J. Head Trauma Rehabil. 1999;14:602–615. doi: 10.1097/00001199-199912000-00009. [DOI] [PubMed] [Google Scholar]
- Tian X. Feig L.A. Age-dependent participation of Ras-GRF proteins in coupling calcium-permeable AMPA glutamate receptors to Ras/Erk signaling in cortical neurons. J. Biol. Chem. 2006;281:7578–7582. doi: 10.1074/jbc.M512060200. [DOI] [PubMed] [Google Scholar]
- Van Damme P. Van Den Bosch L. Van Houtte E. Callewaert G. Robberecht W. GluR2-dependent properties of AMPA receptors determine the selective vulnerability of motor neurons to excitotoxicity. J. Neurophysiol. 2002;88:1279–1287. doi: 10.1152/jn.2002.88.3.1279. [DOI] [PubMed] [Google Scholar]
- Weber J.T. Rzigalinski B.A. Ellis E.F. Traumatic injury of cortical neurons causes changes in intracellular calcium stores and capacitative calcium influx. J. Biol. Chem. 2001;276:1800–1807. doi: 10.1074/jbc.M009209200. [DOI] [PubMed] [Google Scholar]
- Weber J.T. Rzigalinski B.A. Willoughby K.A. Moore S.F. Ellis E.F. Alterations in calcium-mediated signal transduction after traumatic injury of cortical neurons. Cell Calcium. 1999;26:289–299. doi: 10.1054/ceca.1999.0082. [DOI] [PubMed] [Google Scholar]
- Wenthold R.J. Petralia R.S. Blahos J., II Niedzielski A.S. Evidence for multiple AMPA receptor complexes in hippocampal CA1/CA2 neurons. J. Neurosci. 1996;16:1982–1989. doi: 10.1523/JNEUROSCI.16-06-01982.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams K. Russell S.L. Shen Y.M. Molinoff P.B. Developmental switch in the expression of NMDA receptors occurs in vivo and in vitro. Neuron. 1993;10:267–278. doi: 10.1016/0896-6273(93)90317-k. [DOI] [PubMed] [Google Scholar]
- Zhang L. Rzigalinski B.A. Ellis E.F. Satin L.S. Reduction of voltage-dependent Mg2+ blockade of NMDA current in mechanically injured neurons. Science. 1996;274:1921–1923. doi: 10.1126/science.274.5294.1921. [DOI] [PubMed] [Google Scholar]