

Abstract
Electron transfers in photosynthesis and respiration commonly occur between metal-containing cofactors that are separated by large molecular distances. Employing laser flash-quench triggering methods, we have shown that 20-Å, coupling-limited FeII to RuIII and CuI to RuIII electron tunneling in Ru-modified cytochromes and blue copper proteins can occur on the microsecond timescale both in solutions and crystals. Redox equivalents can be transferred even longer distances by multistep tunneling, often called hopping, through intervening amino acid side chains. Our work has established that 20-Å hole hopping through an intervening tryptophan is two orders of magnitude faster than single-step electron tunneling in a Re-modified blue copper protein.
1. Introduction
Aerobic respiration and photosynthesis work in concert: dioxygen liberated by photosynthetic organisms sustains life in aerobic microbes and animals; and, in turn, the products of aerobic respiratory metabolism, carbon dioxide and water, nourish photosynthetic organisms. Electron flow through proteins and protein assemblies in the photosynthetic and respiratory machinery commonly occurs between metal centers or other redox cofactors that are separated by relatively large molecular distances, often in the 10 to 20 Å range.
In the mitochondrial respiratory chain (1–12), two equivalents from the NADH reduction of Complex I move ~140 Å down a sequence of 7–8 iron-sulfur clusters, entering a quinone with comcomitant translocation of four protons across the inner membrane (4,5). Quinol moves on to the bc1 complex (respiratory Complex III) where its two reducing equivalents follow a bifurcated pathway (9–11,13). The first electron moves along a high-potential pathway from a [2Fe-2S] cluster to cytochrome c1 in a step that likely involves domain movement, then finally to cytochrome c in the intermembrane space. The second quinol electron enters a low-potential chain of b-type cytochromes and migrates toward the mitochondrial matrix where it participates in the reduction of quinone to quinol, providing energy for proton pumping. Reduced cytochrome c diffuses to respiratory Complex IV (cytochrome c oxidase) where it delivers electrons to the binuclear CuA site (3,6,7). Electrons from CuA move through cytochrome a on their way to the cytochrome a3/CuB site of O2 reduction. Four additional protons are pumped across the inner mitochrondrial membrane by Complex IV as O2 is reduced to H2O. The proton gradient generated by proton pumping in Complexes I, III, and IV drives ATP synthesis by Complex V (ATP synthase) (3,12).
Understanding the underlying physics and chemistry of these and other biological oxidation-reduction (redox) processes is the goal of much of the work in our laboratory.
2. Historical background
In 1941, Albert Szent-Gyorgyi proposed that electrons travel between redox enzymes immobilized in membranes by using energy bands analogous to those found in semiconductors (14). Eight years later, Evans and Gergely pointed out that the very large band gaps in polypeptides would rule out thermal semiconductivity as the mechanism of electron flow (15). Various dynamical models were proposed whereby proteins would shuttle electrons through conformational motions. In 1956, for example, Chance suggested that partners along an electron-transport chain would alternately rotate to bring distant redox centers into contact (16). A common feature of the dynamical models was a strong temperature coefficient; the rates of these processes would slow dramatically at reduced temperatures.
The landscape changed dramatically in 1966 when DeVault and Chance demonstrated that a cytochrome in the photosynthetic bacterium Chromatium vinosum was oxidized with a half-time of 2μs following excitation with a pulsed ruby laser (17). More remarkable, however, was the observation that the time constant for this electron transfer reached a limiting value of 2 milliseconds at 100 K and remained constant down to a temperature of 4.5 K (17,18). Only quantum mechanical electron tunneling could account for the kinetics at low temperature. Chance proposed that a 1-eV tunneling barrier would produce an exponential distance dependence of the electron transfer rate, with a decay constant of 1 Å−1. He further argued that, depending on the actual barrier height, biological electron tunneling distances of 30–70 Å might be possible. Starting in 1974, we undertook experimental tests of these distance predictions, culminating in work on Ru-modified cytochrome c demonstrating that 15–20 Å electron tunneling can occur on biologically relevant timescales (19).
Theoretical descriptions of long-range electron transfer were being developed as well. The early nonadiabatic theories of Levich and Dogonadze (20) were refined and cast into square-barrier tunneling models to describe Chance’s Chromatium experiments (21,22). Drawing on ideas about superexchange interactions (23,24), subsequent tunneling models described how the chemical bonds of the intervening medium would couple distant electron transfer partners (25,26).
3. Single-step electron tunneling
3.1. The flash-quench method
Single electron transfer reactions can be found at the core of most biological redox processes. No fewer than 15 ET reactions, for example, are required to take reducing equivalents from NADH, deposit them in O2, and generate the electrochemical proton gradient that drives ATP synthesis (3–12). Most of these reactions involve quantum-mechanical electron tunneling between redox cofactors embedded in protein matrices separated by large molecular distances (>10 Å) (3–12). The semiclassical theory of ET (Eq. 1) provides a basic
| (1) |
framework for understanding the specific rates of reaction between an electron donor (D) and acceptor (A) held at fixed distance and orientation (kET) (27–29). These rates depend on three critical parameters: (1) the driving force for electron transfer (−ΔG°); (2) the extent of nuclear reorientation in D, A, and solvent that accompanies formation of D+ and A−(λ); and (3) the electronic coupling between reactants [D,A] and products [D+,A−] at the transition state (HAB). The first two parameters depend largely on the chemical composition and environments of the redox centers, whereas the third is a function the D-A distance and the structure of the intervening medium (27–33).
Our research program has been designed to elucidate the factors that determine the rates and efficiencies of biological electron flow (28,29). A key element of this effort has been the development of inter- and intramolecular laser flash-quench methods to trigger ET reactions (Figure 1) (28,29,34,35). We have used these methods to study the distance and medium dependences of long-range electron tunneling reactions (28,29), to trigger redox enzyme catalysis (36), and initiate multistep tunneling processes (37). In a typical reaction sequence, a laser-excited M-diimine sensitizer (*Ms) directly donates (accepts) an electron to (from) a redox partner (*ET), generating the oxidized (reduced) diimine complex and the reduced (oxidized) partner (Fig. 1, 1→2→3 (6→4→5)) (35). The intermediate formed in this excited-state ET reaction decays in a subsequent charge recombination to regenerate the original D-A complex (3→1 (5→6)). This scheme is viable only when intramolecular ET competes effectively with excited-state deactivation (typically 100 ns – 1μs). If this were the only technique available, the short lifetimes of the excited M-diimine complexes would limit measurements of ET rates to systems in which the M-diimine and the protein active site were well coupled.
Figure 1.
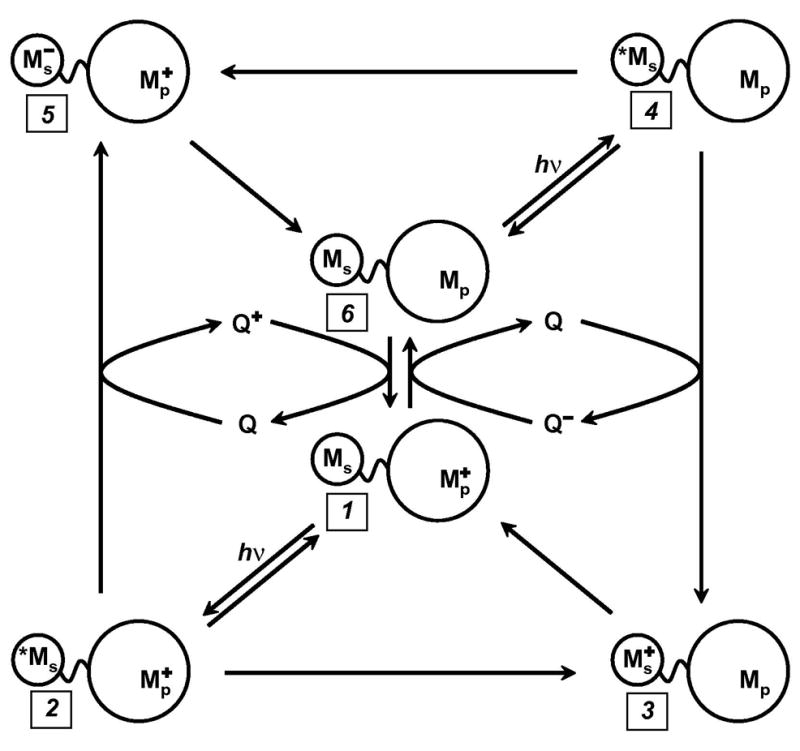
Flash-quench scheme for measuring intraprotein ET rates, and generating oxidized and reduced metal centers in proteins. Ms is a metal-diimine photosensitizer; Mp is the protein metal center.
We have developed intermolecular flash-quench methods specifically to study systems in which charge separation does not compete with excited-state decay, or when the lifetime of the charge-separated state must be protracted (34,35). The method takes advantage of bimolecular quenching and scavenging reactions to separate photogenerated holes and electrons onto different molecules that diffuse away from one another. Once the charges are separated onto different molecules, their timescale for recombination moves into the millisecond range – a thousand-fold improvement over intramolecular charge separation. In the flash-quench procedure (Fig. 1: oxidative, 6→4→3→1; reductive, 1→2→5→6), a quencher (Q) is added to the solution to react with *M in a bimolecular ET reaction. This quenching process generates the same intermediates as the intramolecular quenching reaction (3 or 5), but with greater efficiency. Once generated, the intermediate will react via intramolecular ET (3→1, 5→6). Then, on a much longer timescale (~ms), the reduced (oxidized) quencher will react with the oxidized (reduced) protein to regenerate the original complex (6 or 1). Even longer time windows can be examined (seconds) if irreversible redox quenchers are employed.
3.2. Ru-azurins
Work on Ru-modified P. aeruginosa azurin has provided a reference point for electron tunneling through folded polypeptide structures (38,39). The copper center in azurin is situated at one end of an eight-stranded β-barrel, ligated in a trigonal plane by two imidazoles (His46, His117) and a thiolate (Cys112); in addition, there are weak axial interactions (Met121 thioether sulfur, Gly45 carbonyl oxygen) (40,41). The P. aeruginosa azurin has two additional histidine residues, one of which (His83) reacts readily with Ru-labeling reagents.
Measurements of high-driving-force intraprotein ET kinetics were made possible by Ru-diimine labeling protocols and flash-quench triggering (34). Our early investigations of blue copper proteins involved Ru(bpy)2(im)(His83)2+-CuI-azurin (Ru(His83)2+-CuI-Az). Pulsed laser irradiation of the metal-to-ligand charge transfer (MLCT) absorption band on the Ru chromophore, followed by bimolecular quenching with an external oxidant (Ru(NH3)63+), produced the kinetic intermediate, Ru(His83)3+-CuI-Az. This transient species relaxes via long-range electron transfer to generate Ru(His83)2+-CuII-Az, which subsequently returns to the pre-flash state in a bimolecular ET reaction with Ru(NH3)62+. The specific rate of CuI to Ru(His83)3+ ET at ambient temperature (~295 K) is 1.0(1) × 106 s−1 (39). Subsequent investigations demonstrated that CuI to RuIII or OsIII ET rates in His-83 labeled azurin crystals are nearly identical with solution values (42).
3.3. Theory
Semiclassical theory (eq 1) predicts that the rates of intramolecular ET reactions depend on the driving force (ΔG°), the reorganization energy (λ), and the reactant-product electronic coupling (HAB) (27). Studies of the temperature and driving force dependences of CuI→Ru(His83)3+ rates are consistent with λ = 0.7(1) eV and HAB = 0.070(5) cm−1 (43,44). Indeed, the activationless character the of CuI→Ru(His83)3+ ET is manifested by the absence of any rate variation in the 308–220 K temperature range. Remarkably, the specific rate increases somewhat with a further reduction in temperature to 170 K, possible owing to compaction of the protein structure (43). The temperature independence arises from the near equality of the reaction driving force and reorganization energy, combined with the small entropy change for CuI→RuIII ET. Reorganization energies less than 0.6 eV and greater than 0.8 eV would lead to a significant decrease in ET rate at 170 K.
3.4. Electronic coupling
One focus of our ET work has been the elucidation of distant electronic couplings between redox sites in Ru-proteins (28,29,45). To explore the distance dependence of CuI→RuIII ET in azurin, we prepared a H83Q base mutant and five different mutant proteins with single His residues at different locations on β-strands extending from the Cys112 and Met121 ligands (K122H, T124H, T126H, Q107H, M109H) (38). Tunneling distances (Ru-Cu) in these five Ru(bpy)2(im)(HisX)2+- and Ru(bpy)2(im)(His83)2+-azurins span ~10 Å (15.9 – 26.0 Å). Measurements of CuI → Ru(HisX)3+ ET (−ΔG° = 0.7 eV) in the set of Ru-azurins firmly established the distance dependence of electron transfer along β-strands (38). The driving-force-optimized azurin tunneling timetable reveals a nearly perfect exponential distance dependence, with a decay constant (β) of 1.1 Å−1, and an intercept at close contact (ro = 3 Å) of 1013 s−1 (Figure 2). This decay constant is remarkably close to that suggested 40 years ago by Chance and is quite similar to that found for superexchange-mediated tunneling across saturated alkane bridges (β ≈ 1.0 A−1) (46,47), strongly indicating that a similar coupling mechanism is operative in folded polypeptides.
Figure 2.
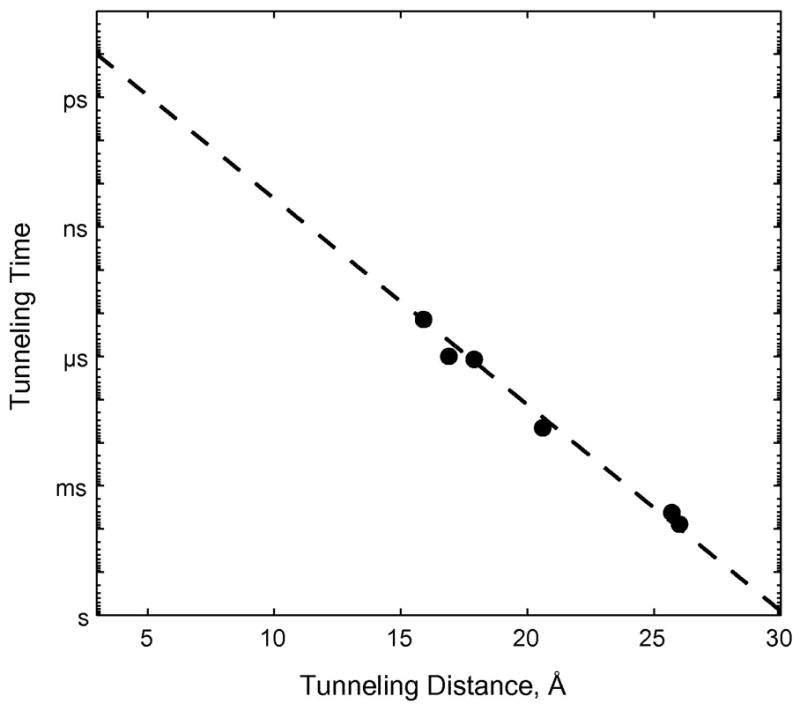
Distance dependence of driving-force-optimized ET rates in Ru-labeled P. aeruginosa azurin. The dashed line is the best fit to the data (β= 1.1 Å −1).
The intercept of 1013 s−1 at ro = 3 Å is the specific rate expected for activationless ET between two redox centers in van der Waals contact. It is of interest to compare this value to results from measurements of ultrafast ET in model systems. Direct comparisons between our extrapolated value and ultrafast data are complicated by the effects of medium dynamics: as donor-acceptor electronic coupling increases, a regime is reached where rates are no longer limited by the strength of this coupling, but by the dynamics of solvent reorientation. This so-called solvent controlled adiabatic limit will typically be below the nonadiabatic limiting rate constant (k°NA) determined by extrapolation of rate-distance data to a close-contact intercept.
With the foregoing caveats in mind, we consider two model systems that provide insights into limiting ET rates at close contact. Barbara and coworkers examined thermal ET following metal-to-metal charge transfer (MMCT) excitation in (NH3)5RuIIINCMII(CN)5− (M = Fe, Ru) (48,49). These mixed-valence complexes fall in the Robin-Day Class II category (50); the MMCT extinction coefficients are ~3 × 103 M−1cm−1 and the estimated electronic coupling parameters are 1900 (M = Ru) and 1500 cm−1 (M = Fe) (48). In solvents with relatively fast dielectric relaxation (e.g., H2O, formamide, N-methyl-formamide), observed FeII→RuIII and RuII→RuIII ET rate constants are ~1013 s−1. Solvent relaxation dynamics typically are divided into two distinct timescales: fast components corresponding to inertial motions of the solvent occur in tens of femtoseconds; and slow processes (>0.5 ps) corresponding to overdamped motions. Because ET in (NH3)5RuIIINCMII(CN)5− is faster than slow solvent relaxation components, Barbara concluded that overdamped solvent motions play only a minor role in the ultrafast reaction (48,49). Hence, in these binuclear metal complexes, specific ET rates are ~1013 s−1 at a metal-metal separation of ~5 Å (51). The path between the two metals is fully covalent, so the coupling should be greater than that for two centers in van der Waals contact.
Photoinitiated ET in charge transfer (CT) complexes provides a second comparison to our extrapolated maximum rate constant. Spears and coworkers examined viologen (methyl viologen, MV; heptyl viologen, HV) complexes with 4,4 -biphenol (BP) (52). The noncovalent interaction between the two redox groups is weaker than in the bimetallic complexes; the CT extinction coefficients are ~4 × 101 M−1cm−1 and the estimated electronic coupling parameters are 361 (MV) and 381 cm−1 (HV). Ab initio calculations of the optimum ground-state geometry in the MV-BP complex suggest a 4 Å separation between planes of the aromatic rings. The specific thermal ET rates following ultrafast excitation into the CT absorption band in fast-relaxing solvents (e.g., methanol, acetonitrile) are ~2.5 × 1012 s−1. Each of these ET reactions likely falls in the solvent controlled adiabatic limit; the estimated specific rate in the absence of solvent control is 2.5 × 1013 s−1 (52).
Results of the foregoing ultrafast ET experiments are consistent with the kET value (1013 s−1) extrapolated to contact between redox sites in Ru-azurins. The separation distance at which this limit is achieved depends on whether the two sites are covalently coupled or are in van der Waals contact. In the case of intraprotein ET between two metal complexes (i.e., Ru(bpy)2(im)(HisX)3+ and Cu(His46)(His117)(Cys112)(Met121)+), we believe that van der Waals contact between the two metal centers, not the metal-ligand complexes, is more consistent with an ET rate of 1013 s−1. Hence, in our analyses of protein ET rates, we consider metal-to-metal distances with a contact separation of 3 Å.
3.5. Tunneling or Hopping?
The energy gap between the donor-acceptor redox levels and those of oxidized or reduced intermediate states is the primary criterion in determining when hole or electron hopping becomes important (53,54). In Ru-azurin, photogenerated Ru(bpy)2(im)(His)3+ (E° = 1.0 V vs. NHE) (34) potentially could oxidize Trp or Tyr residues (28). If the CuI center is replaced by redox-inert ZnII in the protein, however, we find that photogenerated holes in Ru(bpy)2(im)(HisX)3+ complexes remain localized on the metal center. The energy gap between the Ru(HisX)3+ hole and oxidized bridge states must therefore be greater than 75 meV (3kBT at 295 K). Our finding that the CuI →Ru(HisX)3+ ET rate in Ru(bpy)2(im)(HisX)-azurin does not decrease in going from 300 to 240 K and actually increases slightly at 170 K demonstrates that hopping does not occur in this case: a reaction with an endergonic step would be highly disfavored at low temperature. We conclude that the Ru-azurin timetable (Figure 2) provides a benchmark for superexchange-mediated electron tunneling through proteins.
3.6. Tunneling timetables
The rates of high-driving-force ET reactions have been measured for more than 30 Ru(diimine)-labeled metalloproteins (7, 29, 30, 38). Driving-force-optimized values are scattered around the Ru-azurin 1.1 Å−1 exponential distance decay. Rates at a single distance can differ by as much as a factor of 103 and D/A distances that differ by as much as 5 Å can produce virtually identical rates (Figure 3). Owing to the structural heterogeneity of polypeptide matrices in folded proteins, long-range ET rate constants do not exhibit a uniform dependence on donor-acceptor distance. In seminal work, Beratan and Onuchic developed a generalization of the McConnell superexchange coupling model that accounts for rate scatter attributable to protein structural complexity (26,55,56). In this tunneling-pathway model, the medium between D and A is decomposed into smaller subunits linked by covalent bonds, hydrogen bonds, and through-space jumps. More elaborate computational protocols also have shed light on the factors that determine distant couplings in proteins (33,57–70).
Figure 3.
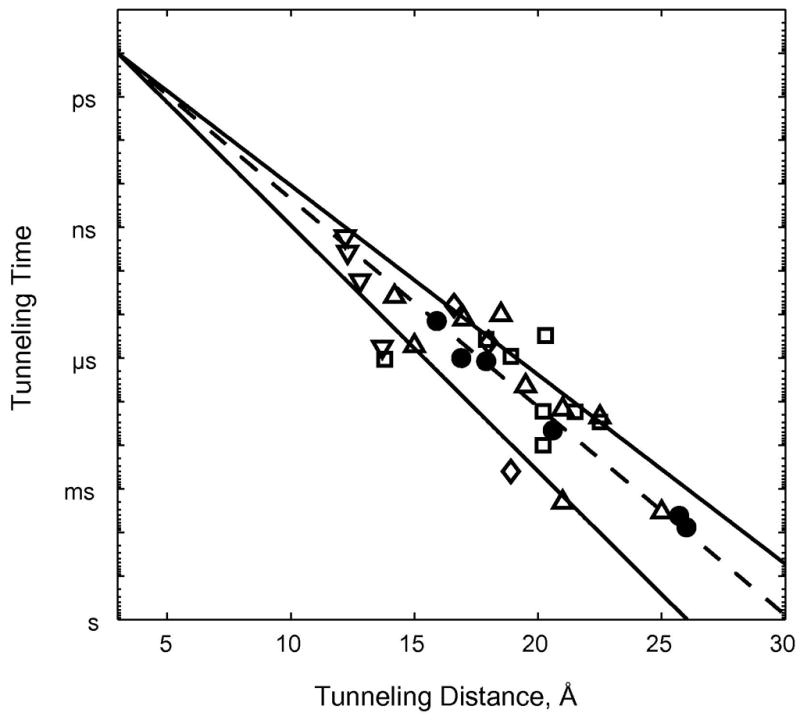
Tunneling timetable for intraprotein ET in Ru-modified azurin (●), cytochrome c (□), myoglobin (◇), cytochromes b562 (Δ), HiPIP (▽). The solid lines illustrate distance decay factors of 1.3 (lower) and 1.0 Å−1 (upper); the dashed line illustrates a 1.1 Å−1 decay.
Empirical data from Ru-modified proteins demonstrate conclusively that long-range electron tunneling rates depend critically on the composition and structure of the medium between two redox centers. The polypeptide matrices of folded proteins are not homogeneous media and consequently electron tunneling rates in proteins do not exhibit uniform exponential distance dependences. It is convenient to characterize the coupling efficiency of a particular polypeptide matrix by an effective exponential decay constant (β′). Taking the data in Figure 3, we can define β′ values for each Ru-modified protein, assuming an intercept at close contact (ro = 3 Å) of 1013 s−1. The histogram of β′ values from Ru-modified proteins (Figure 4) reveals an asymmetric distribution peaking near 1.1 Å−1, with extremes of 0.85 and 1.5 Å−1. This range of β′ values is entirely consistent with studies of ET in synthetic donor-bridge-acceptor complexes (46,47).
Figure 4.

Histogram of the distribution of distance decay factors extracted from Ru-protein ET data. The solid curve is an approximate continuous representation of the distribution constructed from an asymmetric Gaussian function.
Heterogeneous polypeptide matrices bear some similarities to frozen solvent glasses as electron tunneling media. We have found that decay constants for tunneling through aqueous and tetrahydrofuran glasses (1.6 Å−1) are substantially greater than those found for most proteins (Figure 4). Tunneling across oligoxylene bridges, an all-covalent analogue of glassy toluene (1.2 Å−1), is far more efficient (0.75 Å−1). It is interesting to note that values of frozen-solvent tunneling rate constants extrapolated to close contact (0.5 – 5 ×1013 s−1) are in good agreement with that found for Ru-modified azurin. In terms of long-range coupling efficiency, however, frozen solvent glasses are greatly inferior to polypeptide matrices as ET media; the larger β values in solvents are likely a consequence of the predominance of van der Waals contacts over covalent contacts between donors and acceptors.
4. Multistep electron tunneling (hopping)
4.1 Kinetics modeling
Electron tunneling times must be in the millisecond to microsecond range for biological redox machines to function properly. As a result, the maximum center-to-center distance for single-step tunneling through proteins can be no greater than ~20 Å (Figure 5). The structures of several redox enzyme assemblies, however, suggest that charge transport may occur over distances that far exceed this single-step limit (28,71,72). How can charge transport in proteins cover distances well over 20 Å? One possibility is by hopping, as it can be shown that coupled tunneling reactions, particularly with endergonic steps, can in favorable cases deliver electrons or holes rapidly to very distant sites (28,73,74). Requirements for functional hopping include optimal positioning of redox centers and fine tuning of reaction driving forces.
Figure 5.
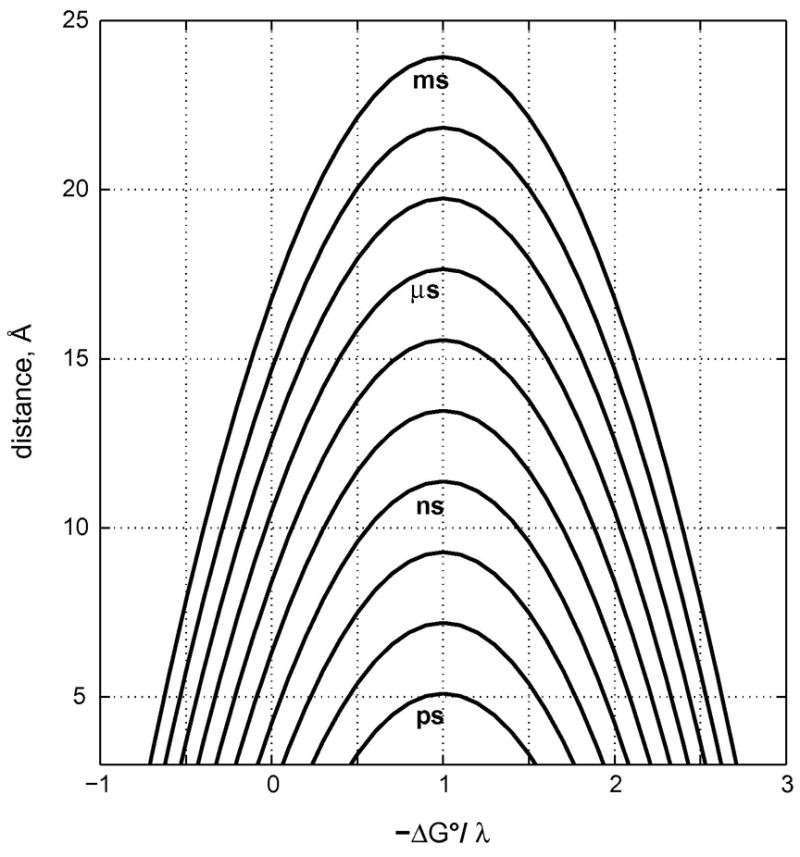
Tunneling-time (1/kobs) contours as functions of donor-acceptor distance (β = 1.1 Å−1) and driving force [in units of λ; kBT/λ= kB(295 K)/(0.8 eV) = 0.318].
Modeling the kinetics of electron hopping is a straightforward problem that can be solved analytically without employing simplifying approximations (28). Using the well defined properties of ET reactions (eq. 2), and the average distance dependence defined by Ru-protein tunneling timetables, it is possible to predict hopping rates for any set of driving-force, temperature, and distance parameters. Consider the two-step tunneling reaction defined in (2) (reactants, R
| (2) |
= D-I-A; redox intermediate, H = D+-I−-A or D-I+-A−; products, P = D+-I-A−). The general solution to the rate law for this process calls for biexponential production of P, although under some circumstances the appearance of P can be approximated by a single exponential function. Taking a value of λ = 0.8 eV for both tunneling reactions (i.e., R→H and H→P) and a distance decay constant of 1.1 Å−1, we can calculate the time dependence of the populations of all three reacting species for various values of ΔG°RH, ΔG°HP, rRH, and rHP (28). Results for the particular case in which ΔG°RH = −ΔG°HP and rRH = rHP are illustrated in Figure 6. This model approximates biological electron transport (ΔG°RP = 0) with a single endergonic step. Transport across 20 Å is 104 times faster than a single tunneling step at this distance and submillisecond transfers can be realized. An important conclusion is that hopping can facilitate electron flow over distances greater than 20 Å in cases where the free-energy changes for endergonic intermediate steps are no greater than 0.2 eV.
Figure 6.
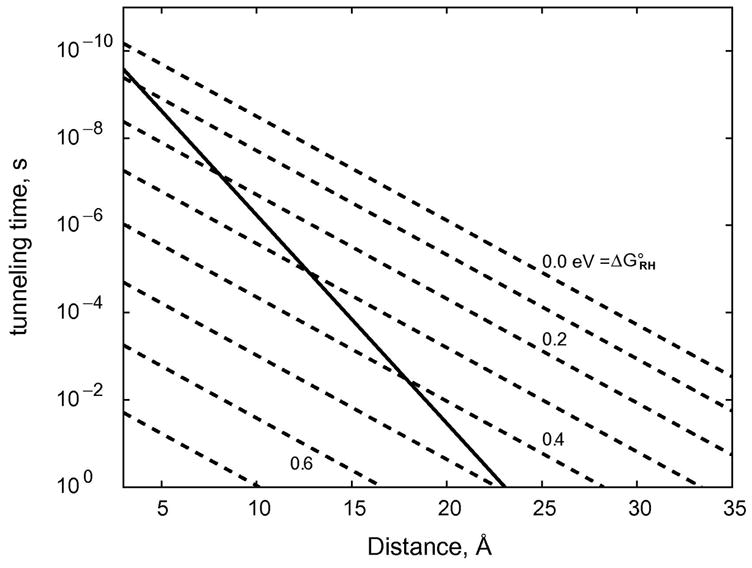
Distance dependences of the rates of single-step and two-step electron tunneling reactions. Solid line indicates theoretical distance dependence for a single-step, ergoneutral (ΔG°RP = 0) tunneling process (β = 1.1 Å−1). Dashed lines indicate distance dependence calculated for two-step ergoneutral tunneling (R←H←P) with the indicated standard free-energy changes for the R←H step.
4.2. Re-azurin
Kinetics modeling suggests that charge transfer rates can be greatly enhanced by multistep electron tunneling in which redox-active amino acid side chains act as intermediate donors or acceptors. Our work on Ru-azurins has shown that single-step, coupling-limited (activationless) CuI to RuIII electron tunneling rates decrease exponentially with a decay constant of 1.1 Å−1 over a wide distance range (Figure 2) (28,38,42,43). Electron transport over distances greater than 25 Å likely involves multistep tunneling (71,72,74–79) in which redox-active amino acid side chains act as intermediate donors or acceptors rather than tunneling bridges (28,29,80). Calculations of hole hopping based on decay constants between 1.0 and 1.2 Å−1 and reorganization energies in the 0.7 to 0.8 eV range indicate that electrons could be transported 30 Å or more in hundreds of nanoseconds if an intervening redox center (Int) with a reduction potential (E(Int+/0)) well above that of the donor (E(D+/0) but not more than 200 mV above that of the acceptor (E(A0/−) is placed between D and A (28,29).
We expressed three mutant azurins to test the proposition that an intervening tryptophan or tyrosine can facilitate electron transfer between distant metal redox centers. In these mutants, a histidine ligand is at position 124 on the β strand extending from Met121, and either tryptophan, tyrosine, or phenylalanine is at position 122. The construction was completed by attaching the sensitizer ReI(CO)3(4,7-dimethyl-1,10-phenanthroline) (ReI(CO)3(dmp)) to His124. ReI(CO)3(dmp)(His124) is a powerful oxidant in its 3MLCT excited state: E°[*ReII(CO)3(dmp•−)(His124)/ReI(CO)3(dmp•−)(His124)] = 1.4 V vs. NHE (81). The crystal structure of the Re-labeled Trp122 variant (ReI(CO)3(dmp)(His124)|(Trp122)|AzCuII) revealed that dmp and the Trp122 indole group are near van der Waals contact (~4 Å), and the Cu-Re distance is 19.4 Å (Figure 7) (37).
Figure 7.
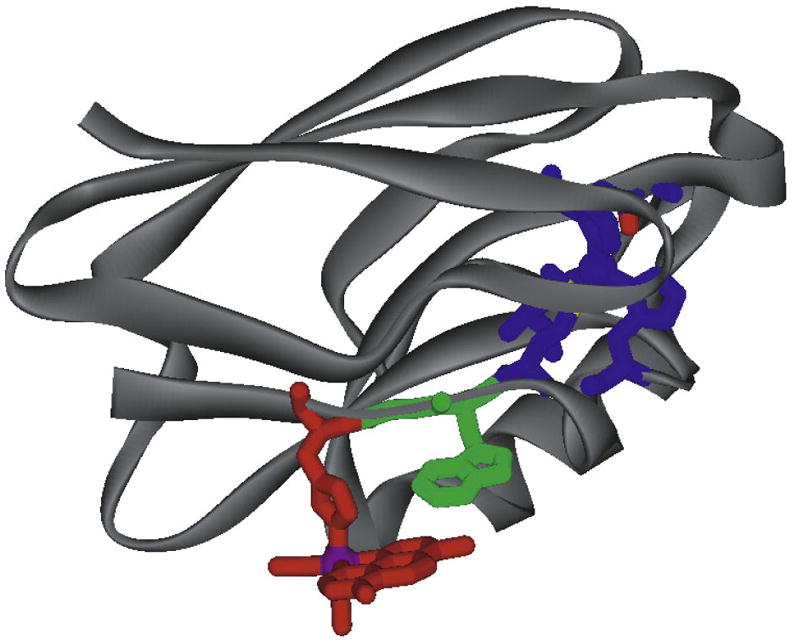
Model of the Cu-W-Re electron-tunneling architecture from the 1.5 Å resolution x-ray crystal structure of ReI(CO)3(dmp)(His124)|(Trp122)|AzCuII. The aromatic rings of dmp (red) and Trp122 (green) slightly overlap, with one dmp methyl group projecting over the indole ring and the plane of the respective -systems making a 20.9° angle. The average separation of atoms on the overlapped six-membered rings is 3.82 Å, whereas 4.1 Å separates the edge of the Trp122 indole and the His124 imidazole. Distances between redox centers: Cu (blue) to Trp122 aromatic centroid, 11.1 Å; Trp122 aromatic centroid to Re (purple), 8.9 Å; Cu to Re, 19.4 Å.
Luminescence from MLCT-excited ReI(CO)3(dmp)(His124)|(Trp122)|AzCuI is substantially quenched relative to that of the Phe122 analogue. Transient absorption measurements reveal rapid (<50 ns) formation of CuII following pulsed laser excitation of ReI(CO)3(dmp)(His124)|(Trp122)|AzCuI (Figure 8); data at 500 nm are consistent with concomitant formation of ReI(CO)3(dmp•−)(His124); charge recombination to regenerate ReI(CO)3(dmp)(His124)|(Trp122)|AzCuI proceeds with a time constant of ~3μs. No evidence for AzCuII formation was found following excitation of ReI(CO)3(dmp)(His124)|(Phe122)|AzCuI or ReI(CO)3(dmp)(His124)|(Tyr122)|AzCuI.
Figure 8.

Transient kinetics of ReI(CO)3(dmp)(His124)|(Trp122)|AzCuI. (A) Time-resolved luminescence (grey: λobs > 450 nm; λex = 355 nm, 10 ps pulsewidth; pH 7.2), instrument response function (grey), and fit to a three-exponential kinetics model [black: τ1 = 35 ps (growth); τ2 = 363 ps (decay); τ3 = 25 ns (decay)]. (B) Visible transient absorption [λobs = 632.8, 500 nm; λex = 355 nm, 1.5 mJ, 8 ns pulsewidth; pH 7.2]. Black lines are fits to a biexponential kinetics model [τ1 = 25 ns (growth); τ2 = 3.1 ms (decay)].
Time-resolved infrared absorption (TRIR) spectroscopy was employed to probe both the electronic ground and MLCT excited states of ReI(CO)3(dmp)(His124) as well as the ET intermediate ReI(CO)3(dmp•−)(His124). We found that Trp122 is required for rapid formation of ReI(CO)3(dmp•−)(His124) and that dmp reduction occurs following excitation of ReI(CO)3(dmp)(His124)|(Trp122)|AzCuII and ReI(CO)3(dmp)(His124)|(Trp122)|AzZnII. Importantly, ET is not observed upon excitation of ReI(CO)3(dmp)(His124)|(Tyr122)|AzZnII.
We interpreted the data in terms of a two-step electron tunneling model (Figure 9). Optical excitation of ReI(CO)3(dmp)(His124) creates a 1MLCT excited state, which undergoes ~150 fs intersystem crossing (82) to a vibrationally excited triplet (*3MLCT). Subpicosecond generation of ReI(CO)3(dmp•−)(His124) is attributable to ET from Trp122 to the ReI(CO)3(dmp)(His124) excited singlet (1MLCT). In prior work, we found that equilibration of *3MLCT in ReI(CO)3(phen)(HisX)|AzCuII (X = 83, 109) involving both vibrational and surrounding-medium relaxation proceeds on the 10 to 100 ps timescale (83). Hence, the ~35 ps formation of ReI(CO)3(dmp•−)(His124) in ReI(CO)3(dmp)(His124)|(Trp122)|AzCuI likely involves parallel relaxation and reduction of the *3MLCT state. The source of reducing equivalents in these fast ET reactions is the indole side chain of Trp122; no evidence for ReI(CO)3(dmp•−)(His124) formation was observed in either the Phe122 or Tyr122 protein.
Figure 9.
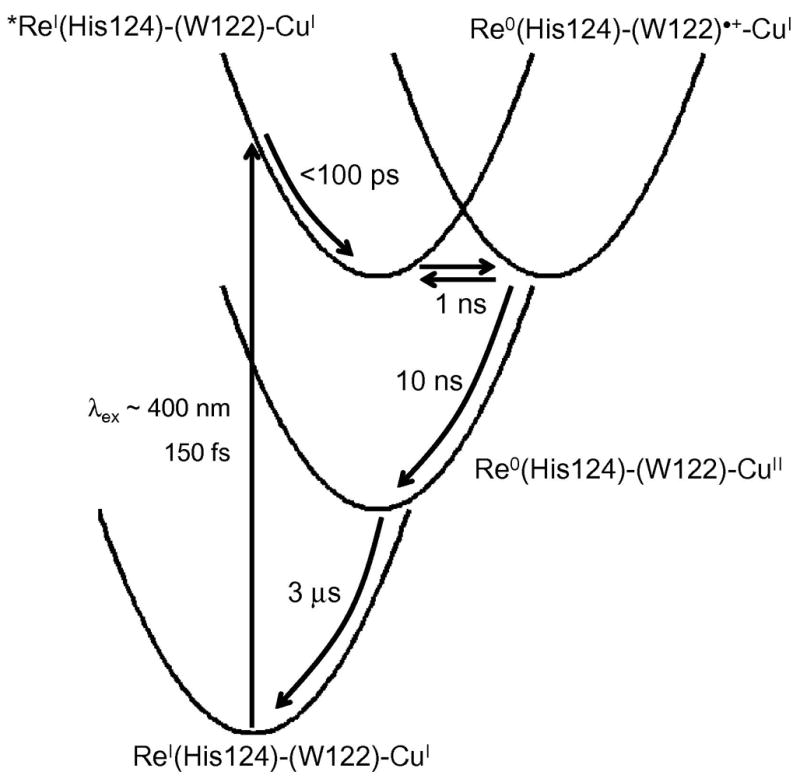
Kinetics model of photoinduced electron transfer in ReI(CO)3(dmp)(His124)|(Trp122)|AzCuI. Light absorption produces electron and hole separation in the MLCT-excited ReI complex. Vibrational relaxation of *Re occurs in ~100 ps, followed by migration of the hole to CuI via (Trp122)•+ in less than 50 ns. Charge recombination proceeds on the microsecond timescale. Elementary rate constants were extracted from fits to time-resolved luminescence, visible absorption, and infrared spectroscopic data.
We proposed that a 350-ps kinetics phase is due to equilibration between the 3MLCT state and ReI(CO)3(dmp•−)(His124)|(Trp122)•+|AzCuI, and a subsequent 30-ns process to reduction of (Trp122)•+ by AzCuI to generate ReI(CO)3(dmp•−)(His124)|(Trp122)|AzCuII. Ground-state repopulation proceeds in 3μs via single-step long-range ET from ReI(CO)3(dmp•−)(His124) to AzCuII. Rate constants corresponding to elementary reaction steps were extracted using a numerical procedure to fit all of the time-resolved data to a two-step tunneling kinetics model. Transient spectra calculated using this model and the resulting elementary rate constants were found to be in excellent agreement with the experimental data.
The key finding is that CuI oxidation in ReI(CO)3(dmp)(His124)|(Trp122)|AzCuI is more than two orders of magnitude faster than expected for electron tunneling over 19 Å. Analysis of the reaction kinetics revealed that the reduction potential of *ReII(CO)3(dmp•−)(His124) is just 28 mV greater than that of (Trp122)•+/0, but this is sufficient for very rapid (~ns) ET between adjacent dmp and Trp122 aromatic rings. Replacement of Trp122 by Tyr or Phe inhibits the initial ET event, presumably because the (Tyr122)•+/0 and (Phe122)•+/0 potentials are more than 200 mV above E°(*ReII(CO)3(dmp•−)(His124)/ReI(CO)3(dmp•−)(His124)). Concerted oxidation and deprotonation of Tyr122 by *ReII(CO)3(dmp•−)(His124) could be thermodynamically favorable, but likely would be accompanied by a significant activation barrier. The Trp radical cation is a relatively weak acid (pKa = 4.5(2)) (84,85); its deprotonation, which is energetically favorable at pH 7, likely would proceed on a microsecond timescale (76). Hence, (Trp122)•+ can rapidly oxidize CuI in the azurin active site as it remains protonated in the hopping intermediate.
4.2. Hopping maps
Two-step electron tunneling maps can be created by solving the rate law corresponding to the elementary reaction steps given in expr. (2). The
| (3) |
elementary rate constants kXY were defined in terms of reaction driving force (ΔG°), reorganization energy (λ), and donor-acceptor distance (rXY) according to semiclassical electron transfer theory (3) (28):
where R is the gas constant, T is absolute temperature, and β = 1.1 Å−1 is the empirical distance decay constant for electron tunneling through proteins (28). The constant α is eliminated in analyses of the rate advantage of two-step vs. single-step tunneling.
The differential equations corresponding to single- and two-step tunneling can be solved analytically. Two-step tunneling is strictly a biexponential process and one-step tunneling follows single-exponential kinetics. In order to compare the two, we defined an average electron transport time (τ) according to (4). The
| (4) |
relative advantage of two-step over single-step tunneling is defined as τET/τ, where τET is the time constant for single-step tunneling.
The specific rate of each elementary ET reaction depends on three parameters: driving force, reorganization energy, and electronic coupling (the overall advantage of two-step over single-step tunneling depends on seven independent parameters). The hopping map of driving-force effects on two-step (CuI→Int→*ML) and single-step (CuI→*ML) tunneling rates for a molecular framework analogous to ReI(CO)3(dmp)(His124)|(Trp122)|AzCuI is shown in Figure 10. The bounded region in the map corresponds to driving-force regimes in which two-step hopping is faster than single-step tunneling. Our analysis indicates that the overall charge separation rate is more sensitive to the free-energy change for the first of the two tunneling steps. Indeed, the rate advantage of the multistep process is lost if the first tunneling step is too endergonic (ΔG° (Int→*ML) > 200 meV) (28,29). The map predicts a ~100-ns time constant for CuI oxidation, in good agreement with the experimental value of 31 ns. Strikingly, two-step hopping is over 300 times faster than single-step CuI to *ReII(CO)3(dmp•−)(His124) tunneling.
Figure 10.

Two-step hopping map for electron tunneling through ReI-modified azurin. Colored contours reflect electron-transport timescales as functions of the driving force for the first tunneling step (ordinate, Int→*ML) and the overall electron-transfer process (abscissa, CuI →*ML). The heavy black lines enclose the region in which two-step hopping is faster than single-step tunneling. The dashed black line indicates the driving force for *ReII(CO)3(dmp•−)(His124)|(Trp122)|AzCuI → ReI(CO)3(dmp•−)(His124)|(Trp122)•+|AzCuI ET; the black dot corresponds to *ReII(CO)3(dmp•−)(His124)|(Trp122)|AzCuI → ReI(CO)3(dmp•−)(His124)|(Trp122)•+|AzCuI → ReI(CO)3(dmp•−)(His124)|(Trp122)|AzCuII hopping.
5. Concluding remarks
Biological redox machines often require multistep electron tunneling architectures that can move charges rapidly over long distances with only a small loss of free energy. A hole originating on the Tyr122 radical in E. coli ribonucleotide reductase is transferred some 35 Å to the active site, retaining sufficient oxidizing power to generate the Cys439 radical that initiates conversion of nucleotides to deoxynucleotides (71,72,75,79); and the photochemically generated hole in the P680 pigment (E°(P680+•/0) ~ 1.3 V) of the photosynthetic oxygen evolving center is transferred via YZ to the Mn-cluster active site where H2O is oxidized to O2 (E°= 1.23 V) (77,86). Our modeling demonstrates that the ReI(CO)3(dmp)(His124)|(Trp122)|AzCuI architecture could provide a suitable framework for an artificial solar energy storage device. The weak dependence of rates on −ΔG° (CuI→*ML) confirms that more than 2 eV can be stored in a photochemical charge separation process without a substantial sacrifice in rate. We anticipate no change in charge separation rate constant if, through site-directed mutagenesis, the reduction potential of the blue copper active site in ReII(CO)3(dmp•−)(His124)|(Trp122)|AzCuI were raised by 0.6 V. If the potential of the copper site were high enough to oxidize water, charge separation would proceed with a 1.4 μs time constant, more than a 600-fold improvement over a direct CuI→ReII single-step process.
Acknowledgments
We thank NIH, NSF, GCEP (Stanford), CCSER (Gordon and Betty Moore Foundation), and the Arnold and Mabel Beckman Foundation for support of our research program.
2. Glossary
- Distance decay constant (β)
the exponential decay constant characterizing the decrease of ET rates with increasing donor-acceptor separation
- Electronic coupling
the interaction between the electronic states of reactants and products at the transition state configuration for ET; characterized by the matrix element HAB
- Electron tunneling
nonadiabatic electron transfer in which the electron goes ‘through’ rather than over the intervening barrier
- ET
electron transfer
- Distance decay constant (β)
the exponential decay constant characterizing the decrease of ET rates with increasing donor-acceptor separation
- Inverted region
the regime in which rates decrease with increasing reaction driving force
- Reorganization energy
the extent of nuclear rearrangement that accompanies charge movement is characterized by the reorganization energy parameter (λ)
Biographies
H. B. Gray
Harry B. Gray studied inorganic chemistry at Northwestern University (1957–60) and the University of Copenhagen (1960–61) before joining the chemistry faculty of Columbia University. In 1966, he moved to the California Institute of Technology, where he is the Arnold O. Beckman Professor of Chemistry and the Founding Director of the Beckman Institute. His main research interests are in inorganic spectroscopy, solar photochemistry, and biological inorganic chemistry.
J. R. Winkler
Jay R. Winkler studied inorganic chemistry at the California Institute of Technology (1978–83) before joining the chemistry department at Brookhaven National Laboratory. In 1990, he returned to Caltech, where he is a Member of the Beckman Institute, Director of the Beckman Institute Laser Center, and a Faculty Associate in Chemistry. His research interests are time-resolved spectroscopy and its application to solar photochemistry and biological inorganic chemistry.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Harry B. Gray, Email: hbgray@caltech.edu.
Jay R. Winkler, Email: winklerj@caltech.edu.
References
- 1.DiMauro S, Schon EA. Mechanisms of disease: Mitochondrial respiratory-chain diseases. New England Journal of Medicine. 2003;348:2656–68. doi: 10.1056/NEJMra022567. [DOI] [PubMed] [Google Scholar]
- 2.Lenaz G, Genova ML. Kinetics of integrated electron transfer in the mitochondrial respiratory chain: random collisions vs. solid state electron channeling. American Journal of Physiology-Cell Physiology. 2007;292:C1221–C39. doi: 10.1152/ajpcell.00263.2006. [DOI] [PubMed] [Google Scholar]
- 3.Saraste M. Oxidative phosphorylation at the fin de siecle. Science. 1999;283:1488–93. doi: 10.1126/science.283.5407.1488. [DOI] [PubMed] [Google Scholar]
- 4.Hinchliffe P, Sazanov LA. Organization of iron-sulfur clusters in respiratory complex I. Science. 2005;309:771–74. doi: 10.1126/science.1113988. [DOI] [PubMed] [Google Scholar]
- 5.Sazanov LA, Hinchliffe P. Structure of the hydrophilic domain of respiratory complex I from Thermus thermophilus. Science. 2006;311:1430–36. doi: 10.1126/science.1123809. [DOI] [PubMed] [Google Scholar]
- 6.Tsukihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, Shinzawa-Itoh K, Nakashima R, Yaono R, Yoshikawa S. Structures of Metal Sites of Oxidized Bovine Heart Cytochrome c Oxidase at 2.8 Å. Science. 1995;269:1071–74. doi: 10.1126/science.7652554. [DOI] [PubMed] [Google Scholar]
- 7.Iwata S, Ostermeier C, Ludwig B, Michel H. Structure at 2.8 Å Resolution of Cytochrome c Oxidase from Paracoccus denitrificans. Nature. 1995;376:660–69. doi: 10.1038/376660a0. [DOI] [PubMed] [Google Scholar]
- 8.Sun F, Huo X, Zhai YJ, Wang AJ, Xu JX, Su D, Bartlam M, Rao ZH. Crystal structure of mitochondrial respiratory membrane protein complex II. Cell. 2005;121:1043–57. doi: 10.1016/j.cell.2005.05.025. [DOI] [PubMed] [Google Scholar]
- 9.Zhang ZL, Huang LS, Shulmeister VM, Chi YI, Kim KK, Hung LW, Crofts AR, Berry EA, Kim SH. Electron transfer by domain movement in stockbroker bc(1) Nature. 1998;392:677–84. doi: 10.1038/33612. [DOI] [PubMed] [Google Scholar]
- 10.Xia D, Yu CA, Kim H, Xian JZ, Kachurin AM, Zhang L, Yu L, Deisenhofer J. Crystal structure of the cytochrome bc(1) complex from bovine heart mitochondria. Science. 1997;277:60–66. doi: 10.1126/science.277.5322.60. [DOI] [PubMed] [Google Scholar]
- 11.Iwata S, Lee JW, Okada K, Lee JK, Iwata M, Rasmussen B, Link TA, Ramaswamy S, Jap BK. Complete structure of the 11-subunit bovine mitochondrial cytochrome bc(1) complex. Science. 1998;281:64–71. doi: 10.1126/science.281.5373.64. [DOI] [PubMed] [Google Scholar]
- 12.Abrahams JP, Leslie AGW, Lutter R, Walker JE. Structure at 2.8-Angstrom Resolution of F1-Atpase from Bovine Heart-Mitochondria. Nature. 1994;370:621–28. doi: 10.1038/370621a0. [DOI] [PubMed] [Google Scholar]
- 13.Xia D, Esser L, Yu L, Yu CA. Structural basis for the mechanism of electron bifurcation at the quinol oxidation site of the cytochrome bc1 complex. Photosynthesis Research. 2007;92:17–34. doi: 10.1007/s11120-007-9155-3. [DOI] [PubMed] [Google Scholar]
- 14.Szent-Györgyi A. Towards a New Biochemistry? Science. 1941;93:609–11. doi: 10.1126/science.93.2426.609. [DOI] [PubMed] [Google Scholar]
- 15.Evans MG, Gergely J. A Discussion of the Possibility of Bands of Energy Levels in Proteins. Electronic Interactions in Non Bonded Systems. Biochimica et Biophysica Acta. 1949;3:188–97. [Google Scholar]
- 16.Chance B, Williams GR. The Respiratory Chain and Oxidative Phosphorylation. Advances in Enzymology. 1956;17:65–134. doi: 10.1002/9780470122624.ch2. [DOI] [PubMed] [Google Scholar]
- 17.De Vault D, Chance B. Studies of Photosynthesis Using a Pulsed Laser. I. Temperature Dependence of Cytochrome Oxidaton Rate in Chromatium. Evidence for Tunneling. Biophysical Journal. 1966;6:825–46. doi: 10.1016/s0006-3495(66)86698-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.De Vault D, Parkes JH, Chance B. Electron Tunnelling in Cytochromes. Nature. 1967;215:642–44. doi: 10.1038/215642a0. [DOI] [PubMed] [Google Scholar]
- 19.Winkler JR, Nocera DG, Yocom KM, Bordignon E, Gray HB. Electron-Transfer Kinetics of Pentaammineruthenium(III)(histidine-33)-Ferricytochrome c. Measurement of the Rate of Intramolecular Electron Transfer between Redox Centers Separated by 15 Å in a Protein. Journal of the American Chemical Society. 1982;104:5798–800. [Google Scholar]
- 20.Levich VG, Dogonadze RR. Theory of Non-Radiation Electron Transitions from Ion to Ion in Solutions. Doklady Akademii Nauk SSSR. 1959;124:123–26. [Google Scholar]
- 21.Hopfield JJ. Electron Transfer between Biological Molecules by Thermally Activated Tunneling. Proceedings of the National Academy of Sciences, USA. 1974;71:3640–44. doi: 10.1073/pnas.71.9.3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jortner J. Temperature Dependent Activation Energy for Electron Transfer Between Biological Molecules. Journal of Chemical Physics. 1976;64:4860–67. [Google Scholar]
- 23.Halpern J, Orgel LE. The Theory of Electron Transfer between Metal Ions in Bridged Systems. Discussions of the Faraday Society. 1960:32–41. [Google Scholar]
- 24.McConnell HM. Intramolecular Charge Transfer in Aromatic Free Radicals. Journal of Chemical Physics. 1961;35:508–15. [Google Scholar]
- 25.Larsson S. Electron-Transfer in Chemical and Biological-Systems - Orbital Rules for Non-Adiabatic Transfer. Journal of the American Chemical Society. 1981;103:4034–40. [Google Scholar]
- 26.Beratan DN, Betts JN, Onuchic JN. Protein Electron Transfer Rates Set by the Bridging Secondary and Tertiary Structure. Science. 1991;252:1285–88. doi: 10.1126/science.1656523. [DOI] [PubMed] [Google Scholar]
- 27.Marcus RA, Sutin N. Electron Transfers in Chemistry and Biology. Biochimica et Biophysica Acta. 1985;811:265–322. [Google Scholar]
- 28.Gray HB, Winkler JR. Electron Tunneling through Proteins. Quarterly Review of Biophysics. 2003;36:341–72. doi: 10.1017/s0033583503003913. [DOI] [PubMed] [Google Scholar]
- 29.Gray HB, Winkler JR. Long-range electron transfer. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:3534–39. doi: 10.1073/pnas.0408029102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Beratan DN, Betts JN, Onuchic JN. Protein Electron Transfer Rates Set by the Bridging Secondary and Tertiary Structure. Science. 1991;252:1285–88. doi: 10.1126/science.1656523. [DOI] [PubMed] [Google Scholar]
- 31.Skourtis SS, Beratan DN. Theories of Structure-Function Relationships for Bridge-Mediated Electron Transfer Reactions. Advances in Chemical Physics. 1999;106:377–452. [Google Scholar]
- 32.Beratan DN, Skourtis SS. Electron Transfer Mechanisms. Current Opinion in Chemical Biology. 1998;2:235–43. doi: 10.1016/s1367-5931(98)80065-3. [DOI] [PubMed] [Google Scholar]
- 33.Prytkova TR, Kurnikov IV, Beratan DN. Coupling coherence distinguishes structure sensitivity in protein electron transfer. Science. 2007;315:622–25. doi: 10.1126/science.1134862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chang I-J, Gray HB, Winkler JR. High-Driving-Force Electron Transfer in Metalloproteins: Intramolecular Oxidation of Ferrocytochrome c by Ru(2,2′-bipyridine)2(imidazole)(histidine-33)3+ Journal of the American Chemical Society. 1991;113:7056–57. [Google Scholar]
- 35.Bjerrum MJ, Casimiro DR, Chang I-J, Di Bilio AJ, Gray HB, Hill MG, Langen R, Mines GA, Skov LK, Winkler JR, Wuttke DS. Electron Transfer in Ruthenium-Modified Proteins. Journal of Bioenergetics and Biomembranes. 1995;27:295–302. doi: 10.1007/BF02110099. [DOI] [PubMed] [Google Scholar]
- 36.Berglund J, Pascher T, Winkler JR, Gray HB. Photoinduced Oxidation of Horseradish Peroxidase. Journal Of the American Chemical Society. 1997;119:2464–69. [Google Scholar]
- 37.Shih C, Museth AK, Abrahamsson M, Blanco-Rodriguez AM, Di Bilio AJ, Sudhamsu J, Crane BR, Ronayne KL, Towrie M, Vlcek A, Richards JH, Winkler JR, Gray HB. Tryptophan-accelerated electron flow through proteins. Science. 2008;320:1760–62. doi: 10.1126/science.1158241. [DOI] [PubMed] [Google Scholar]
- 38.Langen R, Chang I-J, Germanas JP, Richards JH, Winkler JR, Gray HB. Electron Tunneling in Proteins: Coupling through a β-Strand. Science. 1995;268:1733–35. doi: 10.1126/science.7792598. [DOI] [PubMed] [Google Scholar]
- 39.Regan JJ, Di Bilio AJ, Langen R, Skov LK, Winkler JR, Gray HB, Onuchic JN. Electron Tunneling in Azurin: Coupling across a β Sheet. Chemistry and Biology. 1995;2:489–96. doi: 10.1016/1074-5521(95)90266-x. [DOI] [PubMed] [Google Scholar]
- 40.Adman ET, Jensen LH. Structural Features of Azurin at 2.7 Å Resolution. Israel Journal of Chemistry. 1981;21:8–12. [Google Scholar]
- 41.Nar H, Messerschmidt A, Huber R, van de Kamp M, Canters GW. Crystal-Structure Analysis of Oxidized Pseudomonas aeruginosa Azurin at pH 5.5 and pH 9.0 - a pH Induced Conformational Transition Involves a Peptide-Bond Flip. Journal of Molecular Biology. 1991;221:765–72. doi: 10.1016/0022-2836(91)80173-r. [DOI] [PubMed] [Google Scholar]
- 42.Crane BR, Di Bilio AJ, Winkler JR, Gray HB. Electron Tunneling in Single Crystals of Pseudomonas aeruginosa Azurins. Journal of the American Chemical Society. 2001;123:11623–31. doi: 10.1021/ja0115870. [DOI] [PubMed] [Google Scholar]
- 43.Skov LK, Pascher T, Winkler JR, Gray HB. Rates of Intramolecular Electron Transfer in Ru(bpy)2(im)(His83)-Modified Azurin Increase below 220 K. Journal of the American Chemical Society. 1998;120:1102–03. [Google Scholar]
- 44.Di Bilio AJ, Hill MG, Bonander N, Karlsson BG, Villahermosa RM, Malmström BG, Winkler JR, Gray HB. Reorganization Energy of Blue Copper: Effects of Temperature and Driving Force on the Rates of Electron Transfer in Ruthenium- and Osmium-Modified Azurins. Journal of the American Chemical Society. 1997;119:9921–22. [Google Scholar]
- 45.Gray HB, Winkler JR. Electron Transfer in Proteins. Annual Review of Biochemistry. 1996;65:537–61. doi: 10.1146/annurev.bi.65.070196.002541. [DOI] [PubMed] [Google Scholar]
- 46.Smalley JF, Feldberg SW, Chidsey CED, Linford MR, Newton MD, Liu Y-P. The Kinetics of Electron Transfer through Ferrocene-Terminated Alkanethiol Monolayers on Gold. Journal of Physical Chemistry. 1995;99:13141–49. [Google Scholar]
- 47.Smalley JF, Finklea HO, Chidsey CED, Linford MR, Creager SE, Ferraris JP, Chalfant K, Zawodzinsk T, Feldberg SW, Newton MD. Heterogeneous Electron-Transfer Kinetics for Ruthenium and Ferrocene Redox Moieties through Alkanethiol Monolayers on Gold. Journal of the American Chemical Society. 2003;125:2004–13. doi: 10.1021/ja028458j. [DOI] [PubMed] [Google Scholar]
- 48.Tominaga K, Kliner DAV, Johnson AE, Levinger NE, Barbara PF. Femtosecond Experiments and Absolute Rate Calculations on Intervalence Electron Transfer of Mixed-Valence Compounds. Journal of Chemical Physics. 1993;98:1228–43. [Google Scholar]
- 49.Son DH, Kambhampati P, Kee TW, Barbara PF. Femtosecond multicolor pump-probe study of ultrafast electron transfer of [(NH3)(5)(RuNCRuII)-N-III(CN)(5)](−) in aqueous solution. Journal of Physical Chemistry A. 2002;106:4591–97. [Google Scholar]
- 50.Brunschwig BS, Creutz C, Sutin N. Optical transitions of symmetrical mixed-valence systems in the Class II-III transition regime. Chemical Society Reviews. 2002;31:168–84. doi: 10.1039/b008034i. [DOI] [PubMed] [Google Scholar]
- 51.Vance FW, Slone RV, Stern CL, Hupp JT. Comparative absorption, electroabsorption and electrochemical studies of intervalence electron transfer and electronic coupling in cyanide-bridged bimetallic systems: ancillary ligand effects. Chemical Physics. 2000;253:313–22. [Google Scholar]
- 52.Ponnu A, Sung JH, Spears KG. Ultrafast electron-transfer and solvent adiabaticity effects in viologen charge-transfer complexes. Journal of Physical Chemistry A. 2006;110:12372–84. doi: 10.1021/jp0617322. [DOI] [PubMed] [Google Scholar]
- 53.Sheu S-Y, Yang D-Y, Selzle HL, Schlag EW. Charge Transport in a Polypeptide Chain. European Physical Journal D. 2002;20:557–63. [Google Scholar]
- 54.Schlag EW, Sheu SY, Yang DY, Selzle HL, Lin SH. Distal charge transport in peptides. Angewandte Chemie-International Edition. 2007;46:3196–210. doi: 10.1002/anie.200601623. [DOI] [PubMed] [Google Scholar]
- 55.Beratan DN, Onuchic JN. Electron Tunneling Pathways in Proteins: Influences on the Transfer Rate. Photosynthesis Research. 1989;22:173–86. doi: 10.1007/BF00048296. [DOI] [PubMed] [Google Scholar]
- 56.Onuchic JN, Beratan DN, Winkler JR, Gray HB. Pathway Analysis of Protein Electron Transfer Reactions. Annual Review of Biophysics and Biomolecular Structure. 1992;21:349–77. doi: 10.1146/annurev.bb.21.060192.002025. [DOI] [PubMed] [Google Scholar]
- 57.Balabin IA, Onuchic JN. A New Framework for Electron-Transfer Calculations - Beyond the Pathways-like Models. Journal of Physical Chemistry B. 1998;102:7497–505. [Google Scholar]
- 58.Kumar K, Kurnikov IV, Beratan DN, Waldeck DH, Zimmt MB. Use of Modern Electron Transfer Theories to Determine the Electronic Coupling Matrix Elements in Intramolecular Systems. Journal of Physical Chemistry A. 1998;102:5529–41. [Google Scholar]
- 59.Skourtis SS, Balabin I, Kawatsu T, Beratan DN. Protein dynamics and electron transfer: Electronic decoherence and non-Condon effects. Proceedings of the National Academy of Sciences, USA. 2005;102:3552–57. doi: 10.1073/pnas.0409047102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lin JP, Balabin IA, Beratan DN. The Nature of Aqueous Tunneling Pathways between Electron-Transfer Proteins. Science. 2005;310:1311–13. doi: 10.1126/science.1118316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kawatsu T, Beratan DN, Kakitani T. Conformationally averaged score functions for electronic propagation in proteins. Journal of Physical Chemistry B. 2006;110:5747–57. doi: 10.1021/jp052194g. [DOI] [PubMed] [Google Scholar]
- 62.Balabin IA, Beratan DN, Skourtis SS. Persistence of Structure Over Fluctuations in Biological Electron-Transfer Reactions. Physical Review Letters. 2008;101 doi: 10.1103/PhysRevLett.101.158102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Regan JJ, Onuchic JN. Electron-Transfer Tubes. Advances in Chemical Physics. 1999;107:497–553. [Google Scholar]
- 64.Tan ML, Balabin I, Onuchic JN. Dynamics of electron transfer pathways in cytochrome c oxidase. Biophysical Journal. 2004;86:1813–19. doi: 10.1016/S0006-3495(04)74248-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Onuchic JN, Kobayashi C, Baldridge KK. Quantum tunneling in biological reactions: The interplay between theory and experiments. Journal of the Brazilian Chemical Society. 2008;19:206–10. [Google Scholar]
- 66.Skourtis SS, Beratan DN. Electron Transfer Contact Maps. Journal of Physical Chemistry B. 1997;101:1215–34. [Google Scholar]
- 67.Stuchebrukhov AA. Tunneling Currents in Electron-Transfer Reactions in Proteins. 2. Calculation of Electronic Superexchange Matrix Element and Tunneling Currents using Nonorthogonal Basis Sets. Journal of Chemical Physics. 1996;105:10819–29. [Google Scholar]
- 68.Stuchebrukhov AA. Long-distance electron tunneling in proteins. Theoretical Chemistry Accounts. 2003;110:291–306. [Google Scholar]
- 69.Zheng XH, Georgievskii Y, Stuchebrukhov AA. On the electron tunneling in molecules: A generalized orthogonalization procedure for finding tunneling orbitals. Journal of Chemical Physics. 2004;121:8680–86. doi: 10.1063/1.1796751. [DOI] [PubMed] [Google Scholar]
- 70.Zheng XH, Medvedev DM, Stuchebrukhov AA. Does internal water influence electron Tunneling in proteins? Example of cytochrome c oxidase. International Journal of Quantum Chemistry. 2005;102:473–79. [Google Scholar]
- 71.Sjöberg BM. Ribonucleotide Reductases - A Group of Enzymes with Different Metallosites and a Similar Mechanism. Structure and Bonding. 1997;88:139–73. [Google Scholar]
- 72.Stubbe J, Nocera DG, Yee CS, Chang MCY. Radical Initiation in the Class I Ribonucleotide Reductase: Long-Range Proton-Coupled Electron Transfer? Chemical Reviews. 2003;103:2167–201. doi: 10.1021/cr020421u. [DOI] [PubMed] [Google Scholar]
- 73.Winkler JR, Di Bilio A, Farrow NA, Richards JH, Gray HB. Electron Tunneling in Biological Molecules. Pure and Applied Chemistry. 1999;71:1753–64. [Google Scholar]
- 74.Page CC, Moser CC, Chen X, Dutton PL. Natural Engineering Principles of Electron Tunnelling in Biological Oxidation-Reduction. Nature. 1999;402:47–52. doi: 10.1038/46972. [DOI] [PubMed] [Google Scholar]
- 75.Chang MCY, Yee CS, Nocera DG, Stubbe J. Site-Specific Replacement of a Conserved Tyrosine in Ribonucleotide Reductase with an Aniline Amino Acid: A Mechanistic Probe for a Redox-Active Tyrosine. Journal of the American Chemical Society. 2004;126:16702–03. doi: 10.1021/ja044124d. [DOI] [PubMed] [Google Scholar]
- 76.Aubert C, Vos MH, Mathis P, Eker APM, Brettel K. Intraprotein Radical Transfer during Photoactivation of DNA Photolyase. Nature. 2000;405:586–90. doi: 10.1038/35014644. [DOI] [PubMed] [Google Scholar]
- 77.Tommos C, Babcock GT. Proton and Hydrogen Currents in Photosynthetic Water Oxidation. Biochimica Et Biophysica Acta-Bioenergetics. 2000;1458:199–219. doi: 10.1016/s0005-2728(00)00069-4. [DOI] [PubMed] [Google Scholar]
- 78.Frey PA. Importance of Organic Radicals in Enzymatic Cleavage of Unactivated C-H Bonds. Chemical Reviews. 1990;90:1343–57. [Google Scholar]
- 79.Stubbe J, van der Donk WA. Protein Radicals in Enzyme Catalysis. Chemical Reviews. 1998;98:705–62. doi: 10.1021/cr9400875. [DOI] [PubMed] [Google Scholar]
- 80.Berlin YA, Hutchison GR, Rempala P, Ratner MA, Michl J. Charge hopping in molecular wires as a sequence of electron-transfer reactions. Journal of Physical Chemistry A. 2003;107:3970–80. [Google Scholar]
- 81.Connick WB, Di Bilio AJ, Hill MG, Winkler JR, Gray HB. Tricarbonyl(1,10-phenanthroline)(imidazole)rhenium(I): A Powerful Photooxidant for Investigations of Electron Tunneling in Proteins. Inorganica Chimica Acta. 1995;240:169–73. [Google Scholar]
- 82.Cannizzo A, Blanco-Rodríguez AM, Nahhas A, Šebera J, Záliš S, Vlček A, Jr, Chergui M. Femtosecond Fluorescence and Intersystem Crossing in Rhenium(I) Carbonyl–Bipyridine Complexes. Journal of the American Chemical Society. 2008;130:8967–74. doi: 10.1021/ja710763w. [DOI] [PubMed] [Google Scholar]
- 83.Blanco-Rodriguez AM, Busby M, Gradinaru C, Crane BR, Di Bilio AJ, Matousek P, Towrie M, Leigh BS, Richards JH, Vlcek A, Gray HB. Excited-State Dynamics of Structurally Characterized [ReI(CO)3(phen)(HisX)]+ (X=83,109) Pseudomonas aeruginosa Azurins in Aqueous Solution. Journal of the American Chemical Society. 2006;128:4365–70. doi: 10.1021/ja057451+. [DOI] [PubMed] [Google Scholar]
- 84.Harriman A. Further Comments on the Redox Potentials of Tryptophan and Tyrosine. Journal of Physical Chemistry. 1987;91:6102–04. [Google Scholar]
- 85.Solar S, Getoff N, Surdhar PS, Armstrong DA, Singh A. Oxidation of Tryptophan and N-Methylindole by N3•, Br2−, and (SCN)2− Radicals in Light-Water and Heavy-Water Solutions - a Pulse-Radiolysis Study. Journal of Physical Chemistry. 1991;95:3639–43. [Google Scholar]
- 86.Ferreira KN, Iverson TM, Maghlaoui K, Barber J, Iwata S. Architecture of the photosynthetic oxygen-evolving center. Science. 2004;303:1831–38. doi: 10.1126/science.1093087. [DOI] [PubMed] [Google Scholar]