

Abstract
Objective
This aim of this study was to determine the impact of diabetes on oxidant balance and mitochondrial metabolism of carbohydrate- and lipid-based substrates in myocardium of type 2 diabetic patients.
Background
Heart failure represents a major cause of death among diabetics, and it has been proposed that derangements in cardiac metabolism and oxidative stress may underlie the progression of this co-morbidity, but scarce evidence exists in support of this mechanism in humans.
Methods
Mitochondrial O2 consumption and H2O2 emission were measured in permeabilized myofibers prepared from samples of right atrial appendage obtained from non-diabetic (n=13) and diabetic (n=11) patients undergoing non-emergent coronary artery bypass graft surgery.
Results
Mitochondria in atrial tissue of type 2 diabetic individuals display a sharply decreased capacity for glutamate and fatty acid-supported respiration, in addition to an increased content of myocardial triglycerides, as compared to non-diabetics. Furthermore, diabetics display an increased mitochondrial H2O2 emission during oxidation of carbohydrate- and lipid-based substrates, depleted glutathione, and evidence of persistent oxidative stress in their atrial tissue.
Conclusions
These findings are the first to directly investigate the effects of type 2 diabetes on a panoply of mitochondrial functions in the human myocardium using cellular and molecular approaches, and they demonstrate that mitochondria in diabetic human heart have specific impairments in maximal capacity to oxidize fatty acids and glutamate, yet increased mitochondrial H2O2 emission, providing insight into the role of mitochondrial dysfunction and oxidative stress in the pathogenesis of heart failure in diabetic patients.
Keywords: human heart, mitochondria, diabetes mellitus, lipids, oxidative stress
Introduction
The cascade of metabolic events that progressively leads to type 2 diabetes consists of early hyperlipidemia and hyperinsulinemia, followed eventually by β-cell demise and hyperglycemia, the latter defining the disease. Each of these metabolic ‘perturbations’ is thought to contribute individually, but also collectively, to altered cellular structure and electromechanical function in the diabetic myocardium, a condition known clinically as diabetic cardiomyopathy (1). In Western societies, the rapidly increasing number of type 2 diabetics, coupled with the obesity epidemic, illustrates the need for studies which specifically focus on addressing the cellular and molecular mechanisms driving the pathology of this co-morbidity.
In diabetes, the elevated levels of serum triglycerides and free fatty acids results in pronounced accumulation of myocardial triglycerides, a phenomenon which has been well-established in experimental models (2,3) and humans (4,5). This unbalanced lipid metabolism leads to cardiac steatosis, a condition proposed to play a causative role in the development of contractile dysfunction in the diabetic human myocardium. A decreased ratio of ATP produced per O2 consumed (measure of mitochondrial efficiency) is also evident in the diabetic myocardium, raising the possibility that mitochondrial dysfunction may be an underlying cause of the cardiomyopathy (6). Furthermore, increased mitochondrial reactive oxygen species (ROS) production has been shown to accompany this mitochondrial dysfunction (7).
To date, the lack of approaches to investigate metabolism of carbohydrate- and lipid-based substrates at the sub-cellular level (e.g. mitochondria) in human myocardium represents a significant obstacle to identifying the mechanisms responsible for myopathy in the diabetic heart. Recently, the use of permeabilized myofibers as an in vitro model of mitochondrial function has provided a number of mechanistic insights concerning the role of mitochondria in diseases affecting human skeletal (8,9) and cardiac muscle (10). In this study we have utilized this system in a biochemical approach to investigate the effects of type 2 diabetes on mitochondrial respiration and oxidant emission under a diverse range of substrate conditions, as well as the global redox environment in atrial appendage tissue from patients undergoing non-emergent coronary artery bypass graft (CABG) surgery.
Methods
Patient demographics and clinical characteristics
Approval for this study was granted by the Institutional Review Board of East Carolina University. Informed consent was obtained from patients at Pitt County Memorial Hospital undergoing CABG using cardiopulmonary bypass and hypothermic cardioplegic arrest. All demographic and clinical data pertaining to the patients who participated in this study are shown in Table 1. The patients were grouped either as non-diabetic or diabetic according to two major variables: 1) clinical diagnosis of diabetes; and 2) glycated hemoglobin (HbA1c) values of ≥ 6.1 extending up to approximately 1 year prior to surgery. The vast majority of diabetic patients were given intra-venous insulin for ≥ 48 hours prior to the procedure (standard of care). Patients with enlarged atria, history of arrhythmia, or left ventricular ejection fractions ≤ 30% were excluded from this study.
Table 1.
Patient demographics and clinical characteristics for all patients enrolled in this study.
| Non-Diabetics (n = 13) |
Diabetics (n = 11) |
p | |
|---|---|---|---|
| Age, mean ± SD, years | 52.9 ± 8.9 | 56.6 ± 7.2 | 0.26 |
| Sex (M/F) | 11/2 | 10/1 | 1 |
| Race (C/AA) | 12/1 | 10/1 | 1 |
| Clinical characteristics | |||
| LVEF (%) | 54.0 ± 2.4 | 57.1 ± 1.9 | 0.8 |
| Smoking, n (%) | 11 (83) | 8 (73) | 0.63 |
| Hypertension, n (%) | 11 (83) | 10 (91) | 1 |
| BMI | 32.0 ± 5.2 | 33.1 ± 5.3 | 0.9 |
| HbA1C | 5.78 ± 0.38 | 7.26 ± 2.49 | < 0.001* |
| HDL | 36.2 ± 10.9 | 34.5 ± 5.4 | 0.26 |
| LDL | 129.0 ± 41.4 | 66.42 ± 30.1 | < 0.001* |
| TG | 164.7 ± 72.7 | 232.6 ± 46.7 | 0.03 * |
| Preoperative Medications n(%) | |||
| Aspirin | 11 (85) | 10(91) | 0.26 |
| Plavix | 3 (23) | 4 (36) | 0.35 |
| Beta Blocker | 13 (100) | 11 (100) | 1 |
| Statin | 9 (69) | 9 (82) | 0.52 |
| Other lipid lowering agent | 0 | 3 (27) | 0.026 * |
| Nitrates | 7 (54) | 10 (91) | 0.08 |
| Calcium Channel Blocker | 2 (15) | 2 (18) | 0.31 |
| Amiodarone | 0 | 1 (9) | 0.34 |
| Digoxin | 0 | 1 (9) | 0.34 |
| ACE Inhibitor/ARB | 8 (62) | 8 (73) | 0.4 |
| Diuretic | 1 (8) | 2 (18) | 0.42 |
| Insulin | 0 | 10 (91) | < 0.001* |
| Oral Hypoglycemic Agent | 0 | 1 (9) | 0.78 |
LVEF – Left Ventricular Ejection Fraction
Values are mean ± S.D., and values in parentheses are percent of total n for each group.
P < 0.05
Human atrial appendage biopsy and tissue processing
After median sternotomy, and prior to institution of cardiopulmonary bypass, a purse-string suture was placed in the right atrial appendage to allow for placement of the venous cannula. A sample of the appendage directly superior to the purse-string was dissected and immediately rinsed in ice-cold Buffer X (11). This process results in a pristine, human myocardial specimen that has not been subjected to potentially confounding variables such as cardioplegic arrest, mechanical handling, and contact with tubing. The sample was then blotted on gauze to remove excess buffer, trimmed of the epicardial layer and pericardial fat, and a small portion immediately frozen in liquid N2 for protein and mRNA analysis.
Permeabilized muscle fiber preparation, mitochondrial O2 and H2O2 measurements
The technique of permeabilized fiber bundle preparation is adapted from previous methods (11,12). Mitochondrial O2 consumption measurements were made using the Oroboros O2K Oxygraph (Innsbruck, Austria), and mitochondrial H2O2 emission was measured using an Horiba Jobin Yvon spectrofluorometer according to methods described previously (9).
Atrial tissue glutathione (GSH/GSSG) and triglyceride measurements
Atrial muscle samples frozen in liquid N2 were pulverized, homogenized, and protein samples prepared for glutathione measurements as described previously (9). Total GSH and GSSG were measured using the reagents and calibration set provided by the GSH/GSSG assay (Oxis Research) according to the manufacturer's instructions, with some small modifications. Triglycerides were measured in atrial muscle homogenate using a colorimetric assay kit provided by BioVision, according to the manufacturer's instructions.
Immunoblotting and protein quantification
Samples of atrial muscle protein homogenate were separated using SDS-PAGE, transferred to PVDF membranes and subjected to immunoblot using antibodies for PPARα (Abcam), PGC1α (Cell Signaling), 3-Nitrotyrosine (Abcam), and HNE-adduct (Oxis Research). Densitometric analysis was performed using Image J software (http://rsb.info.nih.gov/ij/).
Statisical analysis
All statistical and graphical analysis was performed using GraphPad Prism 5.0 (GraphPad software, San Diego, CA). Categorical variables were compared by using Fisher's exact test, and all interval variables compared using Student's un-paired t-test. Data shown in Table 1 are expressed as means ± S.D. All experimental data are expressed as means ± S.E.M. Differences between Non-Diabetics and Diabetics were considered statistically significant for P < 0.05. Regression analysis was used to examine the correlation between HbA1c and myocardial triglycerides and maximal rate of fatty acid oxidation in both groups. For measurements of kinetics of mitochondrial O2 consumption and H2O2 emission in each group, best-fit curves were obtained using non-linear regression analysis, and statistically significant differences between groups were confirmed by comparison of the R2 values.
Results
Triglyceride levels and mitochondrial fatty acid-supported respiration
Atrial tissue obtained from diabetic patients undergoing CABG contained an ∼2-fold greater level of intramyocellular (IMCL) triglyceride content than non-diabetics (Figure 1A), which positively correlated with HbA1c in these patients (Figure 1B). To determine whether this increased IMCL triglyceride may be linked to reduced mitochondrial oxidation of fatty acids in the atrium of diabetic patients, we investigated the kinetics of O2 consumption supported by palmitoyl-L-carnitine (an activated fatty acid) in permeabilized atrial myofibers prepared from diabetic and non-diabetic patients. A glucose/hexokinase ADP-regenerative system was used to maintain a continuous ADP-stimulated respiratory state to insure substrate oxidation was not impeded by thermodynamic constraints. By titrating palmitoyl-L-carnitine during continuous maximal state 3 respiration, we were able to measure both the sensitivity of mitochondrial transporters and β-oxidation enzymes for palmitoyl-L-carnitine (K0.5) as well as its maximal oxidation rate (Vmax). While the K0.5 for palmitoyl-L-carnitine was unchanged, Vmax supported by palmitoyl-L-carnitine was significantly reduced in permeabilized atrial fibers from diabetic compared with non-diabetic patients (Figure 1D). This reduced maximal fatty acid oxidation capacity in atrial fibers was negatively correlated with levels of blood HbA1c (Figure 1E). Citrate synthase activity was similar in atrial tissue homogenates between groups (30.8 ± 3.3 vs. 29.8 ± 2.4 μmol · min -1 · mg -1 protein in non-diabetic vs. diabetic), providing evidence that the reduced fatty acid oxidative capacity in atrial tissue of diabetic patients is not due to an overall reduction in mitochondrial content.
Figure 1. High levels of IMCL triglycerides in atrium of type 2 diabetic patients is linked to reduced maximal capacity of mitochondrial fatty acid oxidation.
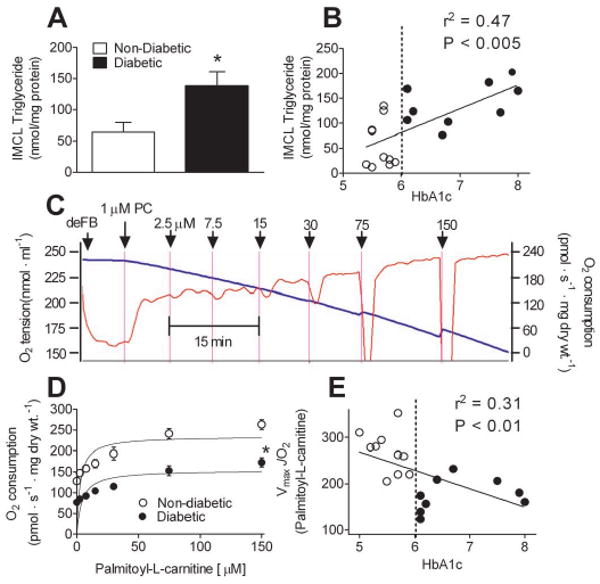
A, Levels of IMCL triglycerides in atrial tissue homogenate prepared from type 2 Diabetic and Non-Diabetic patients. B, Plot showing correlation between myocardial triglycerides and HbA1c in Diabetic (○) and Non-Diabetic (●) patients (B). Shown in C is a representative trace of ADP-stimulated O2 consumption in permeabilized human atrial myofibers in response to incrementally increasing concentrations of palmitoyl-L-carnitine. Permeabilized fibers in the absence of substrate (deFB) were added to respiratory medium in the presence of 3 mM ADP, 5 mM glucose, 1 U/ml hexokinase (to create permanent, maximally phosphorylating respiratory state), followed by incrementally increasing concentrations of palmitoyl-L-carnitine (PC) in the presence of 2 mM malate. The blue line is O2 concenctration in the medium (left Y-axis), the red line is rate of O2 consumption (right Y-axis). D, Kinetic plots of palmitoyl-L-carnitine supported respiration (i.e. fatty acid oxidation) in permeabilized atrial myofibers prepared from both groups of patients, and the correlation of Vmax with HbA1c (E). Quantified data are means ± S.E.M, N = 9-12 patients in each group. * P < 0.05 vs Non-Diabetic.
Expression of PPARα and PGC1α
As a nuclear receptor responsible for determining substrate preference in the heart, PPARα coordinates expression of most key regulators of fatty acid metabolism (13). Because palmitoyl-L-carnitine supported respiration was so strongly diminished in diabetic atrial tissue, we postulated that this may be due to altered expression of PPARα or its transcriptional co-activator PGC1α, a crucial transcriptional co-activator of numerous mitochondrial genes. Although mean PPARα protein levels were slightly lower, and PGC1α slightly higher in diabetic atrial tissue, the difference did not reach statistical significance in this cohort of patients (data not shown), providing evidence that reduced palmitoyl-carnitine respiration observed in diabetic atrium is not due to reduced expression of these key regulatory proteins.
Respiration supported by TCA-cycle substrates and kinetics of ADP-stimulated respiration
Next we examined whether respiratory control and/or capacity during respiration supported by carbohydrate-based substrates was altered in atrial tissue prepared from diabetic verses non-diabetic patients. Titration of ADP during respiration supported by maximal pyruvate (Figure 2A) or succinate (Figure 2B) revealed similar submaximal and maximal respiratory kinetics, indicating that activities of the pyruvate dehydrogenase, succinate dehydrogenase (complex II of the respiratory system), and the adenine nucleotide translocase (a key regulator of energy transfer in cardiomyocytes (14,15)) are not different in atrial muscle from diabetics as compared to non-diabetics.
Figure 2. Glutamate-specific impairment in maximal respiratory capacity in atrium of type 2 diabetic patients.
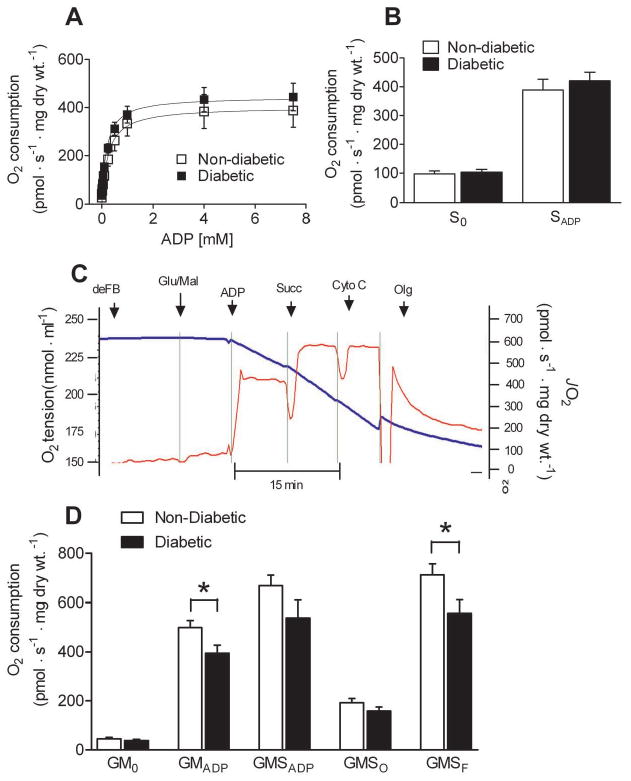
A, Kinetic plots of ADP-stimulated respiration supported by 10 mM pyruvate and 2 mM malate in permeabilized atrial myofibers prepared from Diabetic and Non-Diabetic patients. B, Minimal (S0) and maximal (SADP) respiration supported by succinate in both groups. C, representative trace of a typical O2 consumption experiment in permeabilized human atrial myofibers using sequential addition of oxidative substrates, nucleotides and inhibitors to assess contribution of multiple oxidative substrates to total respiratory flux. Permeabilized fibers in the absence of substrate were added to respiratory medium (deFB), followed by 5 mM glutamate/2 mM malate (GM0), 5 mM ADP (GMADP), 10 mM succinate (GMSADP), 20 μM cytochrome C (to test for intactness of outer mitochondrial membrane), 10 μg/ml oligomycin (GMSO), and 3 μM FCCP (GMSF). D, Quantified rates of glutamate-supported respiration in permeabilized atrial myofibers of Diabetic and Non-Diabetic patients. Quantified data are means ± S.E.M, N = 7-8 patients in each group. * P < 0.05 vs Non-Diabetic.
We then asked whether maximal respiration supported by glutamate was altered in atrial tissue of diabetics, as glutamate oxidation provides a significant source of fuel to the mammalian heart (16,17). Maximal glutamate oxidation was reduced in permeabilized atrial myofibers prepared from diabetic patients, as assessed by respiration in the presence of glutamate and malate. This was not due to disruption of the outer mitochondrial membrane and/or cytochrome C deficiency, as addition of exogenous cytrochrome C failed to restore respiration in diabetic atrial myofibers (Figure 2C and D).
Mitochondrial H2O2 emission and oxidative stress
As a previous report has demonstrated oxidative damage to mitochondria in diabetic mononuclear cells (18), we sought to determine whether the propensity for mitochondria to emit oxidants (i.e. mitochondrial oxidant emitting potential (9)) is elevated in atrial tissue from type 2 diabetics. First, we measured H2O2 emission in the presence of incrementally increasing concentrations of succinate (with oligomycin present to inhibit mitochondrial ATP-ase) in permeabilized atrial myofibers prepared from diabetic and non-diabetic patients. Mitochondria in diabetic atrial tissue displayed both a greater maximal rate of H2O2 emission supported by succinate, as well as a greater rate of H2O2 emission at low succinate concentrations (Figure 3A), indicating a greater mitochondrial oxidant emitting potential in the myocardium of these patients. This increased H2O2 emission with succinate was not due to increased flux through complex II (i.e. increased succinate dehydrogenase activity), as parallel experiments revealed similar rates of O2 consumption (Figure 3B).
Figure 3. Mitochondria in atrium of type 2 diabetic patients display high levels of mitochondrial H2O2 emission while oxidizing carbohydrate- and lipid-based substrates.
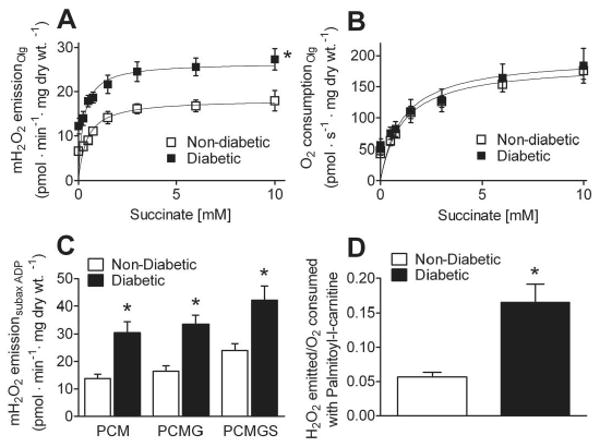
A, Kinetic plots of mitochondrial H2O2 emission and (B) O2 consumption in permeabilized atrial myofibers prepared from Diabetic and Non-Diabetic patients supported by incrementally increasing concentrations of succinate in the presence of 10 μg/ml of oligomycin (to inhibit ATPase and ensure basal respiratory state) + 5 mM glutamate, 2 mM malate. C, Quantified rates of mitochondrial H2O2 emission during respiration in the presence of 125 μM ADP, 5 mM glucose, 1 U/ml hexokinase (to create permanent, submaximally phosphorylating respiratory state), supported by 75μM palmitol-L-carnitine + 2 mM malate (PCM), 5 mM glutamate (PCMG) and 10 mM succinate (PCMGS). D, Ratio of moles of H2O2 emitted per mole of O2 consumed during respiration supported by palmitoyl-L-carnitine. Quantified data are means ± S.E.M, N = 7-9 patients in each group. * P < 0.05 vs Non-Diabetic.
To determine the mitochondrial oxidant emitting potential of atrial fibers under conditions more representative of the physiological state, we measured H2O2 emission during continuous submaximal state 3 respiration (i.e., using glucose/hexokinase ADP-regenerative system) supported by palmitoyl-L-carnitine alone, and following addition of glutamate, malate and succinate to simulate oxidation of substrates from multiple sources. Mitochondria in diabetic atrial tissue displayed greater rates of H2O2 emission than non-diabetics under all substrate conditions examined (Figure 3C). Surprisingly, a greater rate of mitochondrial H2O2 emission in diabetic atrial tissue with palmitoyl-L-carnitine was evident despite the decreased capacity for palmitoyl-L-carnitine supported respiration, which translates to a much greater ratio of moles of H2O2 emitted per mole of O2 consumed with this substrate (Figure 3D).
As a result of the presence of increased mitochondrial H2O2 emission in diabetic atrial tissue, we speculated that markers of oxidative stress would be present in this tissue. Atrial tissue from diabetics displayed both a greater concentration of oxidized glutathione (GSSG) and reduced concentration of total glutathione (GSHt) than non-diabetics (Figure 4A), which corresponded to a decreased GSH/GSSG ratio in the diabetics (Figure 4B). Higher steady-state levels of lipid peroxidation and nitrosative stress were also detected in diabetic atrial tissue, as assessed by immunoblot analysis of proteins from atrial homogenate using antibodies that react with hydroxynonenal (HNE)- and 3-nitrotyrosine modified proteins, respectively (Figure 4C-F).
Figure 4. Altered glutathione redox status and evidence of persistent lipo-peroxyl and nitrosative stress in atrial tissue of type 2 diabetic patients.
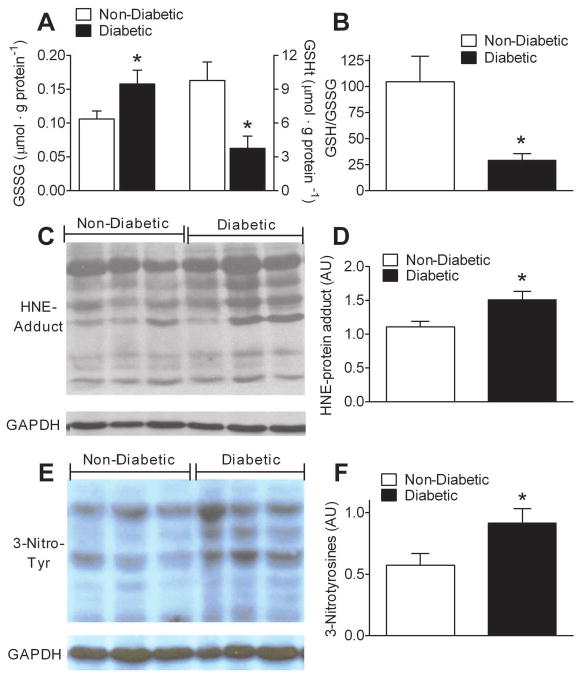
A, Quantified levels of oxidized glutathione (GSSG), and total glutathione (GSHt), in atrial tissue of Diabetic and Non-Diabetic patients. B, Redox environment of atrial tissue as determined by GSH/GSSG ratio. C, Representative immunoblot and D, densitometric quantification of hydroxynonenal (HNE)-modified proteins in atrial tissue from both groups of patients. E, Representative immunoblot and F, densitometric quantification of 3-nitrotyrosine-modified proteins in atrial tissue from both groups of patients. Quantified data are means ± S.E.M, N = 8-11 patients in each group. * P < 0.05 vs Non-Diabetic.
Discussion
By using a permeabilized fiber approach on atrial tissue obtained during coronary bypass surgery, the present study is the first to comprehensively address the effect of type 2 diabetes on mitochondrial metabolism of lipid and carbohydrate-based substrates, and ROS emission in human myocardium, providing evidence that 1) maximal capacity for mitochondrial oxidation of palmitoyl-carnitine is decreased in atrium of type 2 diabetic human myocardium; 2) mitochondrial content and expression of PPARα and PGC1α are unchanged in atrium of diabetics; 3) glutamate, but not pyruvate or succinate oxidation, is decreased in atrium of diabetics; and 4) mitochondrial H2O2 emission supported by both carbohydrate- and lipid-based substrates is greater in diabetic atrium, corresponding to increased oxidative stress in this tissue. These findings demonstrate that mitochondria in diabetic human heart have specific impairments that limit the maximal capacity to oxidize palmitoyl-carnitine and glutamate. Furthermore, they suggest that despite these impairments, the propensity for the electron transport system to emit oxidants is elevated in the diabetic mitochondria, likely contributing to increased oxidative stress and the marked decline in cardiac electromechanical function which is known to occur in diabetic myocardium over time, ultimately leading to heart failure.
Cardiac steatosis in diabetes
Hyperlipidemia, a prominent characteristic of type 2 diabetes mellitus, has been shown to cause a decrease in glucose and lactate utilization and an increase in fatty acid uptake and oxidation by the myocardium (1,19,20). However, in diabetes, the increased levels of serum free fatty acids and triglycerides often exceed their demand in the working heart, leading to a buildup of IMCL triglycerides (i.e. steatosis) in the tissue (2,3). In the present study, IMCL triglyceride content was significantly higher in diabetic human atrial tissue, likely a consequence of the elevated serum triglycerides, supporting the concept of a chronic mismatch between substrate supply and metabolic demand. In addition, the maximal capacity for mitochondrial fatty acid oxidation was markedly lower in diabetic atrial fibers, implying a maladaptive response or impairment at the level of β-oxidation and/or the respiratory system. Whether a reduced capacity to oxidize fatty acids is present throughout the heart, particularly in left ventricle (LV) of these patients, remains to be determined, although an exquisite series of recent studies using non-invasive spectroscopic techniques have shown the presence of elevated IMCL triglyceride in LV of obese and type 2 diabetic patients (4,5). Importantly, the increased IMCL triglyceride content was found to strongly correlate with diastolic dysfunction in type 2 diabetic patients, independent of BMI, age and hypertension (5). These authors hypothesized that as a result of LV steatosis, diastolic dysfunction manifests early on in the disease, progressively leading to global contractile dysfunction (i.e. systolic dysfunction) in the LV with time. The role of steatosis in contractile dysfunction is controversial, however. Recently, intriguing studies have suggested that accumulation of triglycerides in both skeletal (21) and cardiac muscle (22) as a result of diet-induced obesity is not at all pathogenic but even protective against obesity-associated maladies such as insulin resistance, implying that alternative explanations for the association between cardiac steatosis and diabetic cardiomyopathy need to be explored. We and others have shown that activated fatty acids generate a substantial amount of mitochondrial ROS (11), representing another possible route by which adverse effects are generated in the diabetic myocardium.
Mitochondrial fatty acid oxidation in diabetic myocardium
In the present study, the finding that maximal fatty-acid supported respiration is decreased in atrial tissue from diabetic patients is somewhat surprising and unexpected, in light of a recent study by Herrero et al which found that myocardial fatty acid oxidation was elevated in diabetic patients in vivo (23). However, that study was conducted on young (25-35 yrs) patients in a precisely controlled post-prandial state, whereas in the present study, all patients were much older (45-65 yrs) and fasted for a prolonged period of time (∼12-18 hours) prior to surgery (i.e. when the biopsy was taken). It is conceivable that diabetic myocardium preferentially oxidizes lipids to a greater extent than non-diabetics in the post-prandial state, but then is unable to appropriately ‘ramp up’ capacity for lipid oxidation during the transition to the fasted state. This is similar to the observation by Young et al which showed that the myocardium in the obese, insulin-resistant Zucker rat was unable to appropriately up-regulate fatty acid oxidation to levels comparable to lean rats in response to fasting (24). In addition, myocardial insulin-resistance is a plausible explanation for the impaired palmitoyl-carnitine and glutamate oxidation evident in the diabetic atrial tissue, as a recent study by Boudina et al showed that cardiac mitochondria in mice with a cardiac-specific deletion of the insulin receptor showed a strikingly similar phenotype (25).
Mitochondrial ROS and oxidative stress in diabetic myocardium
In the present study, mitochondria in diabetic human atrial tissue displayed much greater rates of H2O2 emission than non-diabetics, regardless of the substrate used or respiratory state. These findings suggest that alterations in electron transport system and/or antioxidant capacity are present in the diabetic human myocardium, and provide evidence that mitochondria may be a significant source of the ROS in this tissue.
Moreover, persistent oxidative stress is evident in diabetic human atrial tissue as demonstrated by the increased levels of hydroxynonenal (HNE)- and 3-nitrotyrosine-modified proteins, which can only occur as a result of constitutively increased ROS production (or decreased removal) in the tissue. Interestingly, both HNE and tyrosine nitrosylation have been shown to alter specific proteins involved in metabolism in the mitochondrial inner membrane and matrix in a manner that alters their activity (26).
Clinical relevance of present study
Collectively, the findings of the present study demonstrate that mitochondria in diabetic human heart have specific impairments in maximal capacity to oxidize fatty acids and glutamate, in addition to an increased propensity for mitochondrial H2O2 emission and evidence of persistent oxidative stress, thus providing important insight on the effects of type 2 diabetes in the human myocardium. As this study was performed on atrial tissue, an important follow-on study would be to examine whether the same events are occurring in ventricular tissue, given the well-established anatomical and physiological differences between these two myocardial compartments. Since ventricular tissue is extremely difficult to obtain from viable human heart, studies in large animal models of diabetes which more closely mimic the human condition (e.g. use of drugs for glycemic and lipidemic control, aging models) should be considered to more precisely investigate the effects of diabetes on myocardial metabolism in a translational manner. Ultimately, this could lead to better therapeutic approaches and proper evaluation of diabetic patients in the future.
Acknowledgments
The authors would like to thank Robert Lust for helpful discussions, and T. Bruce Ferguson and W. Randolph Chitwood for their kind support on this project.
Funding Sources: This work was supported by grant DK073488 and DK074825 from the NIH.
Abbreviations
- CABG
coronary artery bypass graft
- ROS
reactive oxygen species
- GSH
reduced glutathione
- GSSG
oxidized glutathione
- IMCL
intramyocellular lipid
- HNE
hydroxynonenal
- PPAR
peroxisome proliferator-activated receptor
- LV
left ventricle
Footnotes
Disclosures: The authors have no conflicts of interest with industry to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Boudina S, Abel ED. Diabetic cardiomyopathy revisited. Circulation. 2007;115:3213–23. doi: 10.1161/CIRCULATIONAHA.106.679597. [DOI] [PubMed] [Google Scholar]
- 2.Zhou YT, Grayburn P, Karim A, et al. Lipotoxic heart disease in obese rats: implications for human obesity. Proc Natl Acad Sci U S A. 2000;97:1784–9. doi: 10.1073/pnas.97.4.1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Saddik M, Lopaschuk GD. Triacylglycerol turnover in isolated working hearts of acutely diabetic rats. Can J Physiol Pharmacol. 1994;72:1110–9. doi: 10.1139/y94-157. [DOI] [PubMed] [Google Scholar]
- 4.McGavock JM, Lingvay I, Zib I, et al. Cardiac steatosis in diabetes mellitus: a 1H-magnetic resonance spectroscopy study. Circulation. 2007;116:1170–5. doi: 10.1161/CIRCULATIONAHA.106.645614. [DOI] [PubMed] [Google Scholar]
- 5.Rijzewijk LJ, van der Meer RW, Smit JW, et al. Myocardial steatosis is an independent predictor of diastolic dysfunction in type 2 diabetes mellitus. J Am Coll Cardiol. 2008;52:1793–9. doi: 10.1016/j.jacc.2008.07.062. [DOI] [PubMed] [Google Scholar]
- 6.How OJ, Aasum E, Severson DL, Chan WY, Essop MF, Larsen TS. Increased myocardial oxygen consumption reduces cardiac efficiency in diabetic mice. Diabetes. 2006;55:466–73. doi: 10.2337/diabetes.55.02.06.db05-1164. [DOI] [PubMed] [Google Scholar]
- 7.Boudina S, Sena S, Theobald H, et al. Mitochondrial energetics in the heart in obesity-related diabetes: direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes. 2007;56:2457–66. doi: 10.2337/db07-0481. [DOI] [PubMed] [Google Scholar]
- 8.Garnier A, Fortin D, Zoll J, et al. Coordinated changes in mitochondrial function and biogenesis in healthy and diseased human skeletal muscle. Faseb J. 2005;19:43–52. doi: 10.1096/fj.04-2173com. [DOI] [PubMed] [Google Scholar]
- 9.Anderson EJ, Lustig ME, Boyle KE, et al. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J Clin Invest. 2009 doi: 10.1172/JCI37048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seppet E, Eimre M, Peet N, et al. Compartmentation of energy metabolism in atrial myocardium of patients undergoing cardiac surgery. Mol Cell Biochem. 2005;270:49–61. doi: 10.1007/s11010-005-3780-y. [DOI] [PubMed] [Google Scholar]
- 11.Anderson EJ, Yamazaki H, Neufer PD. Induction of endogenous uncoupling protein 3 suppresses mitochondrial oxidant emission during fatty acid-supported respiration. J Biol Chem. 2007;282:31257–66. doi: 10.1074/jbc.M706129200. [DOI] [PubMed] [Google Scholar]
- 12.Anderson EJ, Neufer PD. Type II skeletal myofibers possess unique properties that potentiate mitochondrial H(2)O(2) generation. Am J Physiol Cell Physiol. 2006;290:C844–51. doi: 10.1152/ajpcell.00402.2005. [DOI] [PubMed] [Google Scholar]
- 13.Madrazo JA, Kelly DP. The PPAR trio: regulators of myocardial energy metabolism in health and disease. J Mol Cell Cardiol. 2008;44:968–75. doi: 10.1016/j.yjmcc.2008.03.021. [DOI] [PubMed] [Google Scholar]
- 14.Saks VA, Vasil'eva E, Belikova Yu O, et al. Retarded diffusion of ADP in cardiomyocytes: possible role of mitochondrial outer membrane and creatine kinase in cellular regulation of oxidative phosphorylation. Biochim Biophys Acta. 1993;1144:134–48. doi: 10.1016/0005-2728(93)90166-d. [DOI] [PubMed] [Google Scholar]
- 15.Seppet EK, Eimre M, Anmann T, et al. Structure-function relationships in the regulation of energy transfer between mitochondria and ATPases in cardiac cells. Exp Clin Cardiol. 2006;11:189–94. [PMC free article] [PubMed] [Google Scholar]
- 16.Digerness SB, Reddy WJ. The malate-aspartate shuttle in heart mitochondria. J Mol Cell Cardiol. 1976;8:779–85. doi: 10.1016/0022-2828(76)90084-5. [DOI] [PubMed] [Google Scholar]
- 17.Bauer C, Von Korff RW. Variation in endogenous substrates and pyruvate metabolism in isolated heart mitochondria of several species. Biochim Biophys Acta. 1967;131:280–7. doi: 10.1016/0005-2728(67)90141-7. [DOI] [PubMed] [Google Scholar]
- 18.Dandona P, Thusu K, Cook S, et al. Oxidative damage to DNA in diabetes mellitus. Lancet. 1996;347:444–5. doi: 10.1016/s0140-6736(96)90013-6. [DOI] [PubMed] [Google Scholar]
- 19.Severson DL. Diabetic cardiomyopathy: recent evidence from mouse models of type 1 and type 2 diabetes. Can J Physiol Pharmacol. 2004;82:813–23. doi: 10.1139/y04-065. [DOI] [PubMed] [Google Scholar]
- 20.Lopaschuk GD. Malonyl CoA control of fatty acid oxidation in the diabetic rat heart. Adv Exp Med Biol. 2001;498:155–65. doi: 10.1007/978-1-4615-1321-6_21. [DOI] [PubMed] [Google Scholar]
- 21.Liu L, Zhang Y, Chen N, Shi X, Tsang B, Yu YH. Upregulation of myocellular DGAT1 augments triglyceride synthesis in skeletal muscle and protects against fat-induced insulin resistance. J Clin Invest. 2007;117:1679–89. doi: 10.1172/JCI30565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ussher JR, Koves TR, Jaswal JS, et al. Insulin-stimulated cardiac glucose oxidation is increased in high fat diet-induced obese mice lacking malonyl CoA decarboxylase. Diabetes. 2009 doi: 10.2337/db09-0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Herrero P, Peterson LR, McGill JB, et al. Increased myocardial fatty acid metabolism in patients with type 1 diabetes mellitus. J Am Coll Cardiol. 2006;47:598–604. doi: 10.1016/j.jacc.2005.09.030. [DOI] [PubMed] [Google Scholar]
- 24.Young ME, Guthrie PH, Razeghi P, et al. Impaired long-chain fatty acid oxidation and contractile dysfunction in the obese Zucker rat heart. Diabetes. 2002;51:2587–95. doi: 10.2337/diabetes.51.8.2587. [DOI] [PubMed] [Google Scholar]
- 25.Boudina S, Bugger H, Sena S, et al. Contribution of impaired myocardial insulin signaling to mitochondrial dysfunction and oxidative stress in the heart. Circulation. 2009;119:1272–83. doi: 10.1161/CIRCULATIONAHA.108.792101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Choksi KB, Boylston WH, Rabek JP, Widger WR, Papaconstantinou J. Oxidatively damaged proteins of heart mitochondrial electron transport complexes. Biochim Biophys Acta. 2004;1688:95–101. doi: 10.1016/j.bbadis.2003.11.007. [DOI] [PubMed] [Google Scholar]