


Abstract
Intercellular exchange of protein and RNA-containing microparticles is an increasingly important mode of cell–cell communication. Here we investigate if mesenchymal stem cells (MSCs) known for secreting therapeutic paracrine factors also secrete RNA-containing microparticles. We observed that human embryonic stem cell (hESC)-derived MSC conditioned medium contained small RNAs (less than 300 nt) encapsulated in cholesterol-rich phospholipid vesicles as evidenced by their RNase sensitivity only in the presence of a sodium dodecyl sulfate-based cell lysis buffer, phospholipase A2 and a chelator of cholesterol, cyclodextrin and the restoration of their lower than expected density by detergent or phospholipase A2 treatment. MicroRNAs (miRNAs) such as hsa-let-7b and hsa-let-7g were present in a high precursor (pre)- to mature miRNA ratio by microarray analysis and quantitative reverse transcription–polymerase chain reaction. The pre-miRNAs were cleaved to mature miRNA by RNase III in vitro. High performance liquid chromatography-purified RNA-containing vesicles have a hydrodynamic radius of 55–65 nm and were readily taken up by H9C2 cardiomyocytes. This study suggests that MSCs could facilitate miRNA-mediated intercellular communication by secreting microparticles enriched for pre-miRNA.
INTRODUCTION
The proper functioning of a multicellular organism requires an intricate intercellular communication system to ensure proper coordination among different cell and tissue types. Cell–cell communication in most mammalian systems is highly sophisticated and involves multiple modes of communication. These include the use of electrical impulses in the neural tissues, small chemicals such as amines and steroids, peptides, lipids and gases. Increasingly, intercellular exchange of microparticles is seen as an important mode of intercellular communication (1). Microparticles are lipid vesicles that are <1 μm in diameter, and are secreted by cells and platelets (2–6). They have been implicated in health and diseases (7). Many cell types are known to secrete microparticles and these include some cancer cells (3), neurons (8) and many of the vascular and hematopoietic cell types such as endothelial cells, dendritic cells and B cells (9).
Mesenchymal stem cells (MSCs), derived from adult bone marrow, have emerged as one of the most promising stem cell types for treating cardiovascular disease (10), and MSC is known to exert its therapeutic effects via secreted trophic factors (11–14). It is therefore possible that MSCs also secrete microparticles. To test this hypothesis, highly proliferative MSCs derived from human embryonic stem cells (hESCs) were used, as these cells are amenable to large-scale production of secretion in the form of conditioned medium (CM) (15). As recent reports have demonstrated that some microparticles contain RNA (16–19), we specifically investigated if hESC-derived MSCs secrete RNA-containing microparticles.
We were able to extract RNA from culture medium conditioned by hESC–MSCs. The extracted RNAs were small and were less than 300 nt. These RNAs were resistant to RNase unless the CMs were pretreated with a cell lysis buffer, cyclodextrin, a chelator of cholesterol and phospholipase A2, indicating that these RNAs were encapsulated in a cholesterol-rich, phospholipid vesicle. The RNAs in the CM have a much lower than expected buoyant density of 1.11–1.15 g/ml on sucrose density gradient. Treatment of the CM with a cell lysis buffer or a mixture of cyclodextrin and phospholipase A2 increased the buoyant density of the RNA to 1.15–1.18 g/ml and 1.13–1.16 g/ml, respectively. Microarray analysis for the presence of microRNAs (miRNAs) revealed that the secreted RNA contained many miRNAs that were essentially a subset of those in MSCs. Quantitative reverse transcription–polymerase chain reaction (qRT–PCR) analysis of two randomly selected miRNAs further revealed that they were present predominantly in the precursor (pre)-miRNA form and could be cleaved by RNase III to form the mature miRNA. Fractionation by size exclusion using high performance liquid chromatography (HPLC) resolved the secretion into several fractions; one of which contained a homogenously sized population of microparticles with a hydrodynamic radius of 55–65 nm. Only this fraction contained RNA, and pre-miRNAs were enriched in this HPLC-purified microparticles. When these purified microparticles were labeled with a lipid membrane-intercalating dye, the microparticles were taken up by the H9C2 rat cardiomyocytes.
MATERIALS AND METHODS
Preparation of CM
As described previously (15), hESC-derived HuES9.E1 MSCs were used (20) for the production of MSC secretion in the form of CM. To harvest MSC secretion, 80% of the confluent HuES9.E1 cultures were washed with phoshate-buffered saline (PBS), transferred to a chemically defined, serum-free culture medium for an overnight incubation, washed with PBS and cultured in fresh, chemically defined, serum-free culture medium for 3 days. The CM which contained MSC secretion was collected, clarified by centrifugation, concentrated 50 times using 100-kDa MW cutoff ultrafiltration membranes (Satorius) and sterilized by filtration through a 220-nm filter.
Isolation and detection of RNA
RNA was isolated from CM by adding three volumes of Trizol LS (Invitrogen) to one volume of CM and completing the extraction according to the manufacturer’s protocol. Total RNA and small RNAs from MSC were purified using Trizol (Invitrogen) and mirVana™ miRNA Isolation Kit (AppliedBiosystems), respectively. RNA was quantitated using Quant-iTTM RiboGreen® RNA Assay Kit (Invitrogen) and resolved on 1.5% agarose gel containing 8% glyoxal or 15% Novex® Tris–borate–EDTA (TBE)–urea gels (Invitrogen). The separated RNA in the gels was visualized by ethidium bromide (EB) staining. For some experiments, CM was pre-incubated with equal volume of PBS, cell lysis buffer (Biovision), 40 mM cyclodextrin (Sigma-Aldrich) or 4000 U/ml phospholipase A2 (Sigma-Aldrich) at 37°C for 30 min followed by addition of one-tenth volume of a 1-mg/ml RNase A (Roche). These mixtures were then incubated for another 5 min at 37°C. The RNAs were extracted with Trizol LS and separated on the TBE–urea gels followed by EB staining.
Sucrose gradient density equilibrium centrifugation
For sucrose gradient density equilibrium centrifugation, 14 sucrose solutions with concentrations from 22.8% to 60% were prepared and layered sequentially in a SW60 ultracentrifuge tube (Beckman Coulter, Inc.) starting with the most concentrated solution. CM, or pretreated CM, or RNA MW markers (RiboRuler™ Low and high Range RNA Ladder, Fermentas) were loaded on top before ultracentrifugation for 16.5 h at 200 000g, 4°C in a SW60Ti rotor (Beckman Coulter, Inc.). After centrifugation, the gradients were removed from the top in 13 fractions, and the density of each fraction was calculated by weighing a fixed volume of each fraction. Some of the CM samples were pretreated with an equal volume of lysis buffer (Biovision) or phospholipase A2 and cyclodextrin at a final concentration of 2000 U/ml or 40 mM, respectively, before being loaded on sucrose gradient density equilibrium centrifugation. The lysis buffer was added to CM in a 1:1 volume ratio with a cocktail of protease inhibitor (Halt Protease Inhibitor Cocktail, EDTA-Free, Thermo Scientific). The mixture was incubated for 30 min at room temperature with gentle shaking. RNA was extracted and assayed.
Protein analysis
Analysis of proteins by western blot hybridization was performed using standard protocols. Briefly, proteins were denatured, separated on 4–12% polyacrylamide gels, electroblotted onto a nitrocellulose membrane and probed with antibodies against human CD9, Alix (Santa Cruz Biotechnology, Santa Cruz, CA, USA) or DICER and AGO2 (Abcam, Inc., Cambridge, MA, USA) followed by horseradish peroxidase-coupled secondary antibodies against the primary antibody (Santa Cruz Biotechnology). The blot was then incubated with a chemiluminescent HRP substrate and exposed to an X-ray film.
Microarray assay and data process
The microarray experiments were performed by LC Sciences (www.LCsciences.com) with small RNA preparation from two independently prepared samples of MSC or CM. The microarray hybridizaton was performed in duplicate. In the first experiment, MSC RNA samples were labeled with Cy5, and CM RNA samples were labeled with Cy3. In the second experiment, the dyes were swopped. The RNAs were labeled with Cy5 or Cy3 and hybridized with the preloaded probes. The data in each experiment were analyzed using standard data analysis that includes the determination of detectable signals, calculation of signal intensities and calculation of differential ratios. First, the background was subtracted. Background is determined using a regression-based background mapping method. The regression was performed on 5–25% of the lowest intensity data points excluding blank spots. Raw data matrices were then subtracted by the background matrix. With background subtraction, Cy3/Cy5 channel normalization was carried out using a LOWESS (locally weighted regression) method on the background-subtracted data. The normalization removed system-related variations, such as sample amount variations, different labeling dyes and signal gain differences of scanners so that biological variations can be faithfully revealed. A transcript was listed as detectable if it met at least two conditions: signal intensity higher than 3× (background SD) and spot CV < 0.5. CV was calculated by (SD)/(signal intensity). When repeating probes are found on an array, a transcript is listed as detectable only if the signals from at least 50% of the repeating probes are above detection level. After the normalization, the P-values of the difference between Cy3 and Cy5 signals were calculated and differences were significant if P < 0.01.
Analysis of mature or pre-miRNA by qRT–PCR
To detect mature miRNA in MSC or CM, small RNAs of 10 ng were converted into cDNA and resuspended in 15 µl using TaqMan microRNA Reverse Transcription Kit (Applied Biosystems) according to manufacturer’s protocol. PCR reaction for each RNA sample was performed in triplicate, with 1 µl cDNA and 10 µl of TaqMan 2X Universal PCR Master Mix (No AmpErase® UNG, Applied Biosystems) in 20 µl reaction volume in a StepOne Plus Realtime PCR system (Applied Biosystems). The thermal cycling parameters were one cycle of 95°C of 10 min followed by 40 cycles of 95°C of 15 s and then one cycle of 60°C of 60 s. The primers used were TaqMan primers, hsa-let-7b (Cat AB Assay ID_000378, Applied Biosystems) and hsa-let-7 g (Cat AB Assay ID_000383, Applied Biosystems).
For qRT–PCR detection of pre-miRNAs, the primers were designed using Primer Express ® Software Version 3.0 (Applied Biosystems) as previously described (21). In order to convert to cDNA, 10 ng of the RNA sample was denatured with 10 pmol of the lower primer in a 12-µl reaction volume at 80°C for 5 min. The RNA and lower primer were then annealed at 60°C for 5 min and then cooled to room temperature. RNase inhibitor, 5× reaction buffer, dNTPs, DTT and the Thermoscript reverse transcriptase (Invitrogen) were added, as recommended by the manufacturer. The reaction mixture was incubated for 45 min at 60°C followed by 5 min incubation at 85°C. PCR was then performed using one-tenth of the reaction mixture containing cDNA with a SYBR green PCR Master Mix (Applied Biosystems) in 20 µl reaction volume. The PCR parameters were one cycle of 15 s at 95°C and 40 cycles of 1 min at 60°C. PCR reactions were performed in triplicate. Primer sequences for hsa-let-7b pre-miRNA (accession number MI0000063, http://microrna.sanger.ac.uk) were 5′TGAGGTAGTAGGTTGTGTGGT3′ and 5′GGAAGGCAGTAGGTTGTATAG3′; and for hsa-let-7g pre-miRNA (accession number MI0000137, http://microrna.sanger.ac.uk) 5′GTAGTAGTTTGTACAGTTTGAGGGT3′ and 5′GGCAGTGGCCTGTACAGT3′. The primer sets for primary hsa-let-7b and hsa-let-7g were 5′GCCAGGCAGGGGCTGGTGCTGG3′, 5′GGAAGGCAGTAGGTTGTATAG3′ and 5′TCCTGTCTCAAGTGCATCCTGAAG3′, 5′GGCAGTGGCCTGTACAGT3′, respectively.
To test whether the pre-miRNAs can be processed into mature miRNA in vitro, the RNAs were treated with RNase III. Briefly, 100 ng RNAs from MSC or CM were incubated with 1 U RNase III (Applied Biosystems) in 10 µl volume for 30 min at 37°C, and then heated at 85°C for 20 min. One-tenth volume of the RNase III-treated reaction mixture or without treated was converted into cDNA using primers for mature miRNA or pre-miRNAs as mentioned above.
HPLC fractionation and dynamic light scattering analysis
The instrument setup consisted of a liquid chromatography system with a binary pump, an auto injector, a thermostated column oven and a UV–visible detector operated by the Class VP software from Shimadzu Corporation (Kyoto, Japan). The chromatography columns used were TSK Guard column SWXL, 6 × 40 mm and TSK gel G4000 SWXL, 7.8 × 300 mm from Tosoh Corporation (Tokyo, Japan). The following detectors, Dawn 8 (light scattering), Optilab (refractive index) and QELS (dynamic light scattering) were connected in series following the UV–visible detector. The last three detectors were from Wyatt Technology Corporation (CA, USA) and were operated by the ASTRA software. The components of the sample were separated by size exclusion, i.e. the larger molecules will elute before the smaller molecules. The eluent buffer used was 20 mM phosphate buffer with 150 mM of NaCl at pH 7.2. This buffer was filtered through a pore size of 0.1 μm and degassed for 15 min before use. The chromatography system was equilibrated at a flow rate of 0.5 ml/min until the signal in Dawn 8 stabilized at ∼0.3 detector voltage units. The UV–visible detector was set at 220 nm and the column was oven equilibrated to 25°C. The elution mode was isocratic and the run time was 40 min. The volume of sample injected ranged from 50 µl to 100 µl. The percent area of the exosome peak versus all other peaks was integrated from the UV–visible detector. The hydrodynamic radius, Rh, was computed by the QELS and Dawn 8 detectors. The highest count rate (in Hertzs) at the peak apex was taken as the Rh. Peaks of the separated components visualized at 220 nm were collected as fractions for further characterization studies.
Uptake of labeled microparticles by H9C2 cardiomyocytes
The H9C2 cells were cultured in high-glucose Dulbecco's; modified Eagle medium (DMEM) medium containing 10% fetal calf serum, 1% glutamine–penicillin–streptomycin and 1% sodium pyruvate (all from Invitrogen). For microparticle labeling, 50 µl of 3 µg HPLC-purified microparticles or PBS was mixed with 0.2 µl of fluorescent labeling dye PKH67 (Sigma-Aldrich) at room temperature for 5 min. The labeled mixtures were dialyzed in PBS for 24 h using D-Tube Dialyzer (6–8 kDa) (Novagen). After that, one volume of the labeled mixtures and four volumes of culture medium were added into H9C2 cells in 48-well plate. After another 2-h incubation, the cells were fixed in 4% paraformaldehyde (Sigma-Aldrich) for 10 min and stained with DAPI (300 nM) (Roche) for 5 min. The cell images were taken at 400× magnification using Olympus IX-71 microscope and DP-70 camera. The images were merged with DP manager software. The scale bar is 200 µm.
Statistical analysis
Statistical significance was analyzed using Student’s t-test.
RESULTS
Presence of RNA in the secretion of MSCs
To determine if MSCs secrete RNA, MSC CM was extracted for RNA using standard RNA extraction methodology. A typical CM was prepared by collecting a chemically defined medium that had been conditioned by MSC for 3 days and then concentrating the medium 50 times with a 100-kDa filter. The average RNA yield was 5–6 µg RNA/mg protein. When resolved on either a denaturing glyoxal agarose gel or 15% TBE–urea gel, it was observed that the RNA contained mainly small RNAs of less than 300 nt with undetectable levels of 18S and 28S ribosomal RNAs (Figure 1).
Figure 1.

Resolving secreted RNAs by size on agarose and polyacrylamide gels. (A) Cellular or secreted RNAs were separated on a 1.5% glyoxal agarose gel. (B) Cellular or secreted RNAs were separated on a 15% TBE–urea gel. The MW markers used were denatured 10-bp DNA ladder, high-range RNA ladder (0.2–6 kb) or low-range RNA ladder (0.1–1 kb).
Secreted RNAs were small and susceptible to RNase A only in the presence of a sodium dodecyl sulfate-based cell lysis buffer, cyclodextrin or phospholipase A2
Recent reports of RNA secretion have demonstrated that these RNAs are secreted in phospholipid vesicles known as exosomes (16–19). Therefore, to determine if the secreted RNAs in the CM were also in phospholipid vesicles, CM was pretreated with RNase A, or RNase A in the presence of a SDS-based cell lysis buffer, cyclodextrin or phospholipase A2 before RNA extraction. RNase A treatment alone did not reduce RNA yield or affect the size distribution of RNA in the CM (Figure 2A). However, RNase A treatment in the presence of a SDS-based cell lysis buffer, cyclodextrin or phospholipase A2 either completely or partially degraded the RNA in the CM. The size distribution of the secreted RNAs was also not altered when the extraction was performed in the presence of RNAse inhibitors, or in the presence of a SDS-based cell lysis buffer, cyclodextrin or phospholipase A2 without RNase (Figure 2B). Together, these observations suggested that the RNA was protected from RNase activity by a cholesterol-rich phospholipid membrane unless the membrane was first dissolved by a SDS-based cell lysis buffer, cyclodextrin that chelates and extracts cholesterol or degradation by an phospholipid degrading enzyme, phospholipase A2.
Figure 2.

Secreted RNAs were sequestered in phospholipid vesicles. (A) Untreated CM or CM pretreated with SDS-based lysis buffer, cyclodextrin or phospholipase A2 was incubated with RNase A. After incubation, the CMs were extracted for RNA and the RNAs were resolved on a 15% TBE–urea gel. (B) RNA was extracted from CM without RNase inhibitor (lane 2), with RNase inhibitor (lane 3) or CM pretreated with cyclodextrin (lane 4), SDS-based lysis buffer (lane 5) or phospholipase A2 (lane 6).
Secreted RNAs have a buoyant density of 1.11–1.15 g/ml
If secreted RNAs were encapsulated in lipid vesicles, they should be precipitated by ultracentrifugation and would be expected to have a flotation density of lipid vesicles in the range of 1.10–1.18 g/ml (22,23). By centrifuging the CM first at 10 000g for 30 min and then ultracentrifuging the supernatant at 200 000g for 2 h followed by RNA extraction from the supernatant and pellet of each centrifugation, we detected RNA in the supernatant after the 10 000g centrifugation and enriched in the pellet after 200 000g ultracentrifugation (Figure 3A). Fractionation of the CM into 13 fractions by sucrose density gradient equilibrium ultracentrifugation revealed that unlike purified RNA molecular weight (MW) markers with a buoyant density of 1.15–1.18 g/ml, the secreted RNAs have a lower buoyant density of 1.11–1.15 g/ml (Figure 3B). This buoyant density was increased after the CM was treated with a combination of phospholipase A2 and cyclodextrin (1.13–1.16 g/ml) or an SDS-based lysis buffer (1.15–1.18 g/ml) (Figure 3B and C). Together, these observations confirmed that the secreted RNAs were encapsulated in cholesterol-rich phospholipid vesicles.
Figure 3.

Precipitation of RNA-containing vesicles or flotation density of RNA in a sucrose gradient density. (A) The CM was centrifuged at 10 000g for 30 min. An aliquot of the supernatant was then ultracentrifuged at 200 000g for 2 h at 4°C in a SW60Ti rotor (Beckman Coulter, Inc.). RNA was extracted from the supernatant (S) and the pellet (P) after each centrifugation, and the RNAs were resuspended in TE. The volume of each RNA sample was adjusted to the volume used for the centrifugation and equal volume of each fraction was then resolved on a 15% TBE–urea gel. (B) CM treated with PBS, phospholipase A2 and cyclodextrin or SDS-based lysis buffer were loaded on a preformed sucrose density gradient as described in ‘Materials and Methods’ section and ultracentrifuged for 16.5 h at 200 000g at 4°C. The gradients were removed from the top in 13 fractions and the density of each fraction was calculated by weighing a fixed volume of each fraction. The fractions were extracted for RNA, and the RNA yield for each fraction was assayed. The RNA concentration was then normalized to the highest concentration in each gradient. RNA from each fraction in a gradient was resolved on a 15% TBE–urea gel. CM treated with PBS is indicated in upper panel, CM treated with phospholipase A2 and cyclodextrin in middle panel and CM treated with SDS-based lysis buffer in lower panel. (C) The relative RNA yield for each fraction was plotted against the density of the fraction.
Secreted RNA contained miRNAs
As the secreted RNAs were of less than 300 nt, we tested the RNAs for the presence of miRNAs by performing microarray hybridization. MSC expressed at least 151 miRNAs including some passenger miRNA sequences denoted as miRNA* (Table 1). Sixty miRNAs were detected in the CM. Of these, 45 were also found in MSCs. The remaining 15 were not detected in MSCs and they were probably masked by the relative high abundance of the other miRNA species in the MSCs (see ‘The relative abundance of miRNAs in the CM and MSCs’ in Supplementary Material). A most notable observation was that 9 of the 13 members in one of the most highly conserved and developmentally important human let-7 family (24,25) were expressed in MSCs. They were hsa-let-7a, hsa-let-7b, hsa-let-7c, hsa-let-7d, hsa-let-7e, hsa-let-7f, hsa-let-7g, hsa-let-7i and hsa-miR-98, but not hsa-let-7j, hsa-let-7k and hsa-miR-202. Of these, only hsa-let-7a, hsa-let-7b, hsa-let-7c and hsa-let-7d were detected in the CM. The passenger miRNA sequences of hsa-let-7b and hsa-let-7d were also detected in the CM and not detectable in MSCs. These differences suggested that secretion of miRNAs including passenger miRNA sequences is a selective, and not a random process by MSCs. The microarray analysis also revealed the presence of miRNA-923, and miRNA-923 has recently been confirmed as a degradative product of ribosomal RNA (http://microrna.sanger.ac.uk/cgi-bin/sequences/mirna_entry.pl?acc=MI0005715). This suggested that while the secretion did not contain intact ribosomal RNA, it contained degraded ribosomal RNA and possibly degraded mRNA.
Table 1.
Detection of 166 miRNA in MSC and CM as determined by microarray
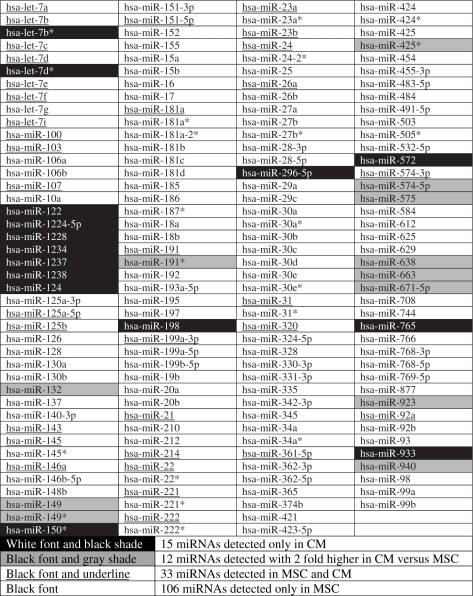 |
Two independent samples from MSC or CM were analyzed in duplicate. A transcript was listed as detectable if it met at least two conditions: signal intensity higher than 3 × (background SD) and spot CV < 0.5. CV was calculated by (SD)/(signal intensity). When repeating probes are present on an array, a transcript is listed as detectable only if the signals from at least 50% of the repeating probes are above detection level. Small RNA was prepared from MSC and CM, and hybridized to a microarray chip containing 723 probes, which detects miRNA transcripts listed in Sanger miRBase Release 10.1.
Secreted RNA contained pre-miRNA
The detection of passenger miRNA sequences by microarray analysis suggested that either pre-miRNAs or passenger miRNAs, which are normally degraded, were present in both CM and their cell source. The microarray analysis also suggested that passenger miRNA sequences for some of the miRNAs were present at a relatively higher level in the CM than in their cell source. To evaluate these observations, we determined the ratio of pre-miRNA and its mature miRNA levels for two members of the let-7 family, namely hsa-let-7b and hsa-let-7g by qRT–PCR. The ratios of pre- to mature miRNA level for hsa-let-7b and hsa-let-7g were determined before and after treatment with RNase III to cleave any pre-miRNAs from approximately a 70 nt RNA species to approximately a 22 nt mature miRNA. We observed that RNase III treatment caused a preferential loss of RNA in the 50- to 100-nt region (Figure 4A). Before RNase III treatment, the ratio of pre-miRNA to mature miRNA for hsa-let-7b and hsa-let-7g in the secretion after normalizing to that in MSC were 169.1 ± 12.1 (P < 0.0001) and 1077.8 ± 123.51 (P < 0.0001), respectively (Figure 4B). After RNase III treatment, the ratio of pre- to mature miRNA was reduced for hsa-let-7b and hsa-let-7g in both CM and MSC (Figure 4B). The decrease in these ratios was due to a decrease in pre-miRNA level coupled with a concomitant increase in mature miRNA level as reflected by the respective increases and decreases in Ct values (Figure 4C). To determine if pri-miRNA was present in the secretion, RT–PCR was performed using primers specific for hsa-let-7b and hsa-let-7g pri-miRNAs (Figure 4D). Hsa-let-7b and hsa-let-7g pri-miRNAs were not detected in the secretion. As mature miRNAs are biologically functional only when they are associated with RNA-induced silencing complex (RISC), we examined the possibility of association of the secreted mature miRNAs with RISC by assaying for the presence of Dicer and Ago2, the main components of RISC by western blot hybridization (Figure 4E). Both the proteins were present in MSC but not detected in the secretion. Together, these observations suggested that both hsa-let-7b and hsa-let-7g miRNAs were present as mature and pre-miRNAs in both CM and MSCs but were secreted predominantly as pre-miRNA, and these pre-miRNAs can be cleaved by RNase III to form mature miRNAs. The secreted mature miRNAs were not associated with RISC as the major RISC protein components were not present in the secretion.
Figure 4.

Cleavage of pre-miRNA to mature miRNA by RNase III. (A) Extracted RNA was treated with RNase III and separated with untreated RNA on the 15% TBE–urea gel. (B) The ratio of pre-/mature miRNA for hsa-let-7b and hsa-let-7g in CM and MSC was determined by qRT–PCR and normalized to that in untreated RNA from MSC. (C) Hsa-let-7b and hsa-let-7g pre- and mature miRNA level before and after RNase III treatment was determined by qRT–PCR. The relative pre- and mature miRNA level was measured by their Ct value. (D) Genomic DNA from MSC and cDNA from the secreted RNA were amplified using primers specific for either hsa-let-7b or hsa-let-7g pri-miRNA. The expected sizes for hsa-let-7b and hsa-let-7g pri-miRNA amplified PCR products were 139 and 145 bp, respectively. (E) Cell lysate from MSCs and MSC CM were analyzed by western blot hybridization and probed with antibodies against Dicer, Ago2, CD9 and Actin.
Purification of homogenously sized microparticles by size exclusion on HPLC
As the miRNAs were encapsulated in lipid vesicles, we hypothesized that these vesicles could be isolated by size exclusion on a HPLC. Fractionation of the CM and NCM revealed the presence of four fractions unique to the CM, suggesting that these were secreted by the MSCs (Figure 5A). Fractions 2–4 contained proteins with decreasing MW with the highest MW in Fraction 2 and the lowest in Fraction 4, while Fraction 1 contained proteins with MW range that encompassed those in Fractions 2–4. RNA was present only in the fastest eluting Fraction 1 (Figure 5B). Two proteins commonly associated with exosomes, CD9 (20 Kd) and Alix (100 Kd), were found almost exclusively in this fraction in spite of their large size difference (Figure 5B). Together, these suggested that the RNA was present in phospholipid vesicles known as exosomes (19). Unlike RNA extracted from CM, the RNA in the HPLC-purified microparticles had a narrower MW range between 50 and 100 nt instead between 10 and 300 nt range of RNA extracted from CM, suggesting that pre-miRNAs might be further enriched in this fraction. The ratio of pre- to mature miRNA for hsa-let-7b and hsa-let-7g as determined by qRT–PCR was significantly increased by a factor of 44.55 ± 5.05 (P = 0.0001) and 11.19 ± 2.01 (P = 0.0009), respectively (Figure 5C). The RNAs in the HPLC-purified fraction had a buoyant density of 1.11–1.15 g/ml (Figure 5D). The size of the particles in the first fraction was sufficiently homogenous such that their hydrodynamic radius was 55–65 nm as determined by dynamic light scattering. The particle sizes in the other fraction were too heterogeneous to be determined.
Figure 5.

Enrichment of pre-miRNA in HPLC-purified cardioprotective microparticles. (A) CM and NCM were fractionated by HPLC into eight fractions, F1–F8, based on UV absorbance at 220 nm. Each of the fractions was then analyzed by dynamic light scattering and the signal output in voltage was then calculated according to the manufacturer’s protocol to calculate particle size. (B) Fractions F1–F8 were extracted for RNA and the extractions were analyzed on a 15% TBE–urea gel. Protein from CM and fractions F1–F4 were analyzed by western blot hybridization and probed with antibodies against CD9 and Alix. (C) RNA extracted from F1 fraction and unfractionated CM was assayed for pre- and mature forms of hsa-let-7b and hsa-let-7g miRNA by qRT–PCR. The relative ratio of pre- and mature forms for hsa-let-7b and hsa-let-7g miRNA in the F1 fraction was normalized to that in the CM. (D) The HPLC F1 was further fractionated into 13 fractions on a sucrose density gradient. Each of the fractions was extracted for RNA and resolved on a 15% TBE–urea gel.
Uptake of phospholipid microparticles by H9C2 cardiomyocytes
Since miRNAs exert their biological effects intracellularly, we investigate if the HPLC-purified RNA-containing microparticles could be taken up by H9C2 cardiomyocytes. The purified microparticles were labeled with a fluorescent phospholipid membrane-intercalating dye PKH67 and fed to H9C2 cardiomyocytes. Within 2 h, the fluorescent label was observed in the cytoplasm but not nucleus of the cells, suggesting that the HPLC-purified microparticles can be taken up intracellularly into the cytoplasm (Figure 6).
Figure 6.

HPLC-purified cardioprotective microparticles can be taken by H9C2 cardiomyocytes. The HPLC-purified cardioprotective microparticles (HPLC F1) or an equivalent volume of PBS was labeled with PKH67, a lipid membrane-intercalating dye. The labeled fraction was added to the culture medium of H9C2 cardiomyocytes. After 2 h, H9C2 cells were fixed, counterstained with DAPI and analyzed by Olympus fluorescent microscopy. The images were taken at 400X magnifications at room temperature and were merged by Olympus DP manager software. (A–C) H9C2 cells incubated with PKH67-labeled HPLC F1 (FITC). (D–F) H9C2 cells incubated with PKH67-labeled PBS. The scale bar is 200 µm.
DISCUSSION
The therapeutic effects of MSC transplantation on cardiovascular tissue repair have been attributed in part to its paracrine secretion (11–14). In support of this hypothesis, we have previously demonstrated that secretion from hESC-derived MSCs was cardioprotective in both pig and animal models of myocardial ischemia and reperfusion injury (26). Since microparticles are increasingly found in the secretion of many cell types with potentially important functions in health and diseases, we investigated if MSC secretion also contains microparticles. As microparticles have also been shown to contain RNAs, we specifically investigate if MSCs secrete RNA-containing microparticles. Here we characterized a chemically defined culture medium conditioned by hESC-derived MSCs (15) and observed that this CM contained RNA encapsulated in cholesterol-rich phospholipid vesicles as evidenced by their susceptibility to RNase digestion only in the presence of a SDS-based lysis buffer, a cholesterol chelator, cyclodextrin and phospholipase A2. Consistent with this, these RNAs also have a buoyant density of a lipid vesicle, i.e. 1.11–1.15 g/ml, and this density was sensitive to SDS, phospholipase A2 and cyclodextrin. Unlike cellular RNAs, these secreted RNAs were mainly small RNAs with no detectable 18S or 28S RNAs or RNAs greater than 500 nt, suggesting that secretion of these RNAs was not a random cellular process but a selective process that targets small RNAs such as miRNAs and, most possibly, other small non-coding RNAs (27) or RNA degradative intermediates, but not the more abundant intact mRNAs or ribosomal RNAs present in cells.
The recent reports of RNA secretion in microparticles have led to the postulation that these vesicles of RNAs could serve as vehicles of genetic communication between cells (19,28). It has been shown that mRNAs in these secreted RNAs could be translated into proteins after taken up by the cells. However, the physiological relevance of such mRNAs to cellular functions remains to be determined as mRNAs constitute a very small proportion of the secreted RNAs. Instead, most of the secreted RNAs are small RNAs, and mature miRNAs of 17–22 nt were implicated in the composition of these small RNAs (19,28). However, it is unlikely that these secreted mature miRNAs play any biological functions as these miRNAs were not associated with RISC, and being mature miRNA, they can no longer be loaded into RISC to effect their functions. Incorporation of mature miRNA into RISC is mediated through the loading of pre-miRNA into RISC followed by cleavage of pre-miRNA by Dicer (29–31), whereupon the guide or mature miRNAs are then incorporated into the RISC while the passenger miRNAs are discarded (32–34).
Our observations that MSCs preferentially secrete miRNA in the precursor instead of the mature form and that these pre-miRNAs were enriched in microparticles, possibly exosomes, that were readily taken up by cells suggest that there are important physiological functions underlying this secretion. They suggest that MSC can potentially exert miRNA-mediated biological effects on other cells through the secretion of pre-miRNA in microparticles. However, the biological function of secreted pre-miRNAs is presently difficult to assess at this time as we still cannot accurately predict or determine the targets of miRNAs. As such, the physiological relevance of hsa-let-7b and hsa-let-7g pre-miRNA remains to be determined.
In conclusion, our study demonstrated that the paracrine secretion of MSCs contains RNAs besides proteins and lipids. Most of this secreted RNAs were encapsulated in phospholipid vesicles, and these vesicles can be purified as a population of homogenously sized particles by size exclusion on a HPLC. These particles contained small RNAs that included miRNAs and also some degraded ribosomal RNA intermediates. The miRNAs consisted mainly of biologically functional pre-miRNAs. As the microparticles can be readily taken up by cells, our findings implicate a novel intercellular role for miRNA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Agency for Science and Technology-Biomedical Research Council (A*STAR). Funding for open access charge: A*STAR.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We gratefully acknowledge Kong Meng Hoi and Eddy Tan, Bioprocessing and Technology Institute, for helping in the purification of the HPLC fractions, and Jayanthi Padmanabhan, Bioprocessing and Technology Institute, for technical assistance in preparing the secretion.
REFERENCES
- 1.Hugel B, Martinez MC, Kunzelmann C, Freyssinet JM. Membrane microparticles: two sides of the coin. Physiology (Bethesda) 2005;20:22–27. doi: 10.1152/physiol.00029.2004. [DOI] [PubMed] [Google Scholar]
- 2.Chironi GN, Boulanger CM, Simon A, Dignat-George F, Freyssinet JM, Tedgui A. Endothelial microparticles in diseases. Cell Tissue Res. 2009;335:143–151. doi: 10.1007/s00441-008-0710-9. [DOI] [PubMed] [Google Scholar]
- 3.Castellana D, Kunzelmann C, Freyssinet JM. Pathophysiologic significance of procoagulant microvesicles in cancer disease and progression. Hamostaseologie. 2009;29:51–57. [PubMed] [Google Scholar]
- 4.Burnier L, Fontana P, Kwak BR, Angelillo-Scherrer A. Cell-derived microparticles in haemostasis and vascular medicine. Thromb. Haemost. 2009;101:439–451. [PubMed] [Google Scholar]
- 5.Alijotas-Reig J, Palacio-Garcia C, Vilardell-Tarres M. Circulating microparticles, lupus anticoagulant and recurrent miscarriages. Eur. J. Obstet. Gynecol. Reprod. Biol. 2009;145:22–26. doi: 10.1016/j.ejogrb.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 6.Aharon A, Katzenell S, Tamari T, Brenner B. Microparticles bearing tissue factor and tissue factor pathway inhibitor in gestational vascular complications. J. Thromb. Haemost. 2009;7:1047–1050. doi: 10.1111/j.1538-7836.2009.03342.x. [DOI] [PubMed] [Google Scholar]
- 7.Aharon A, Brenner B. Microparticles, thrombosis and cancer. Best Pract. Res. Clin. Haematol. 2009;22:61–69. doi: 10.1016/j.beha.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 8.Marzesco AM, Janich P, Wilsch-Brauninger M, Dubreuil V, Langenfeld K, Corbeil D, Huttner WB. Release of extracellular membrane particles carrying the stem cell marker prominin-1 (CD133) from neural progenitors and other epithelial cells. J. Cell Sci. 2005;118:2849–2858. doi: 10.1242/jcs.02439. [DOI] [PubMed] [Google Scholar]
- 9.Shet AS. Characterizing blood microparticles: technical aspects and challenges. Vasc. Health Risk Manag. 2008;4:769–774. doi: 10.2147/vhrm.s955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pittenger MF, Martin BJ. Mesenchymal stem cells and their potential as cardiac therapeutics. Circ. Res. 2004;95:9–20. doi: 10.1161/01.RES.0000135902.99383.6f. [DOI] [PubMed] [Google Scholar]
- 11.Caplan AI, Dennis JE. Mesenchymal stem cells as trophic mediators. J. Cell Biochem. 2006;98:1076–1084. doi: 10.1002/jcb.20886. [DOI] [PubMed] [Google Scholar]
- 12.Gnecchi M, He H, Liang OD, Melo LG, Morello F, Mu H, Noiseux N, Zhang L, Pratt RE, Ingwall JS, et al. Paracrine action accounts for marked protection of ischemic heart by Akt-modified mesenchymal stem cells. Nat. Med. 2005;11:367–368. doi: 10.1038/nm0405-367. [DOI] [PubMed] [Google Scholar]
- 13.Gnecchi M, He H, Noiseux N, Liang OD, Zhang L, Morello F, Mu H, Melo LG, Pratt RE, Ingwall JS, et al. Evidence supporting paracrine hypothesis for Akt-modified mesenchymal stem cell-mediated cardiac protection and functional improvement. FASEB J. 2006;20:661–669. doi: 10.1096/fj.05-5211com. [DOI] [PubMed] [Google Scholar]
- 14.Schafer R, Northoff H. Cardioprotection and cardiac regeneration by mesenchymal stem cells. Panminerva Med. 2008;50:31–39. [PubMed] [Google Scholar]
- 15.Sze SK, de Kleijn DP, Lai RC, Khia Way Tan E, Zhao H, Yeo KS, Low TY, Lian Q, Lee CN, Mitchell W, et al. Elucidating the secretion proteome of human embryonic stem cell-derived mesenchymal stem cells. Mol. Cell Proteomics. 2007;6:1680–1689. doi: 10.1074/mcp.M600393-MCP200. [DOI] [PubMed] [Google Scholar]
- 16.Rosell R, Wei J, Taron M. Circulating microRNA signatures of tumor-derived exosomes for early diagnosis of non-small-cell lung cancer. Clin. Lung Cancer. 2009;10:8–9. doi: 10.3816/CLC.2009.n.001. [DOI] [PubMed] [Google Scholar]
- 17.Taylor DD, Gercel-Taylor C. MicroRNA signatures of tumor-derived exosomes as diagnostic biomarkers of ovarian cancer. Gynecol. Oncol. 2008;110:13–21. doi: 10.1016/j.ygyno.2008.04.033. [DOI] [PubMed] [Google Scholar]
- 18.Hunter MP, Ismail N, Zhang X, Aguda BD, Lee EJ, Yu L, Xiao T, Schafer J, Lee ML, Schmittgen TD, et al. Detection of microRNA expression in human peripheral blood microvesicles. PLoS ONE. 2008;3:e3694. doi: 10.1371/journal.pone.0003694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007;9:654–659. doi: 10.1038/ncb1596. [DOI] [PubMed] [Google Scholar]
- 20.Lian Q, Lye E, Suan Yeo K, Khia Way Tan E, Salto-Tellez M, Liu TM, Palanisamy N, El Oakley RM, Lee EH, Lim B, et al. Derivation of clinically compliant MSCs from CD105+, CD24- differentiated human ESCs. Stem Cells. 2007;25:425–436. doi: 10.1634/stemcells.2006-0420. [DOI] [PubMed] [Google Scholar]
- 21.Schmittgen TD, Jiang J, Liu Q, Yang L. A high-throughput method to monitor the expression of microRNA precursors. Nucleic Acids Res. 2004;32:e43. doi: 10.1093/nar/gnh040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Raposo G, Nijman HW, Stoorvogel W, Liejendekker R, Harding CV, Melief CJ, Geuze HJ. B lymphocytes secrete antigen-presenting vesicles. J. Exp. Med. 1996;183:1161–1172. doi: 10.1084/jem.183.3.1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thery C, Amigorena S, Raposo G, Clayton A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol. 2006 doi: 10.1002/0471143030.cb0322s30. Chapter 3, Unit 3.22. [DOI] [PubMed] [Google Scholar]
- 24.Jerome T, Laurie P, Louis B, Pierre C. Enjoy the silence: the story of let-7 microRNA and cancer. Curr. Genomics. 2007;8:229–233. doi: 10.2174/138920207781386933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roush S, Slack FJ. The let-7 family of microRNAs. Trends Cell Biol. 2008;18:505–516. doi: 10.1016/j.tcb.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 26.Timmers L, Lim SK, Arslan F, Armstrong JS, Hoefler IE, Doevendans PA, Piek JJ, El Oakley RM, Choo A, Lee CN, et al. Reduction of myocardial infarct size by human mesenchymal stem cell conditioned medium. Stem Cell Res. 2007;1:129–137. doi: 10.1016/j.scr.2008.02.002. [DOI] [PubMed] [Google Scholar]
- 27.Mattick JS, Makunin IV. Small regulatory RNAs in mammals. Hum. Mol. Genet. 2005;14:R121–R132. doi: 10.1093/hmg/ddi101. [DOI] [PubMed] [Google Scholar]
- 28.Smalheiser NR. Exosomal transfer of proteins and RNAs at synapses in the nervous system. Biol. Direct. 2007;2:35. doi: 10.1186/1745-6150-2-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chendrimada TP, Gregory RI, Kumaraswamy E, Norman J, Cooch N, Nishikura K, Shiekhattar R. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature. 2005;436:740–744. doi: 10.1038/nature03868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gregory RI, Chendrimada TP, Cooch N, Shiekhattar R. Human RISC couples microRNA biogenesis and posttranscriptional gene silencing. Cell. 2005;123:631–640. doi: 10.1016/j.cell.2005.10.022. [DOI] [PubMed] [Google Scholar]
- 31.Maniataki E, Mourelatos Z. A human, ATP-independent, RISC assembly machine fueled by pre-miRNA. Genes Dev. 2005;19:2979–2990. doi: 10.1101/gad.1384005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hutvagner G, McLachlan J, Pasquinelli AE, Balint E, Tuschl T, Zamore PD. A cellular function for the RNA-interference enzyme Dicer in the maturation of the let-7 small temporal RNA. Science. 2001;293:834–838. doi: 10.1126/science.1062961. [DOI] [PubMed] [Google Scholar]
- 33.Ketting RF, Fischer SE, Bernstein E, Sijen T, Hannon GJ, Plasterk RH. Dicer functions in RNA interference and in synthesis of small RNA involved in developmental timing in C. elegans. Genes Dev. 2001;15:2654–2659. doi: 10.1101/gad.927801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jaskiewicz L, Filipowicz W. Role of Dicer in posttranscriptional RNA silencing. Curr. Top. Microbiol. Immunol. 2008;320:77–97. doi: 10.1007/978-3-540-75157-1_4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.