


Abstract
The mechanism of human mitochondrial RNA turnover and surveillance is still a matter of debate. We have obtained a cellular model for studying the role of hSuv3p helicase in human mitochondria. Expression of a dominant-negative mutant of the hSUV3 gene which encodes a protein with no ATPase or helicase activity results in perturbations of mtRNA metabolism and enables to study the processing and degradation intermediates which otherwise are difficult to detect because of their short half-lives. The hSuv3p activity was found to be necessary in the regulation of stability of mature, properly formed mRNAs and for removal of the noncoding processing intermediates transcribed from both H and L-strands, including mirror RNAs which represent antisense RNAs transcribed from the opposite DNA strand. Lack of hSuv3p function also resulted in accumulation of aberrant RNA species, molecules with extended poly(A) tails and degradation intermediates truncated predominantly at their 3′-ends. Moreover, we present data indicating that hSuv3p co-purifies with PNPase; this may suggest participation of both proteins in mtRNA metabolism.
INTRODUCTION
RNA degradation plays a key role in controlling the concentration of a given transcript. In addition, prompt removal of aberrant RNA molecules and maturation products is vital for maintaining homeostasis in living cells. In order to ensure proper turnover of RNA and its quality control in different cellular compartments divergent protein complexes have evolved. While their composition for various taxa may be quite different (1–4), all contain exoribonucleases acting either 3′ to 5′ or 5′ to 3′; and helicases which are necessary to unwind double-stranded regions of RNA or act as molecular motors feeding the RNA substrate into the catalytic center of the ribonuclease subunit (5).
In mitochondria control of transcription is usually very simple and post-transcriptional events seem to be the predominant way of regulating gene expression; thus RNA degradation is of particular importance. The first described enzymatic machinery for mitochondrial RNA degradation was the Saccharomyces cerevisiae mitochondrial degradosome or mtEXO. It consists of two subunits: the NTP-dependent RNA helicase related to the DExH/D (Ski2p) superfamily; and the 3′- to 5′-exoribonuclease belonging to the RNR family (RNase II-like), encoded by the nuclear genes SUV3 (YPL029W) (6) and DSS1 (YMR287C) (7), respectively. Activities of both proteins are required for the functioning of the degradosome complex, and the ‘inchworm’ model of its activity has been proposed where depending on ATP hydrolysis the cleft between the two SUV3 domains opens and closes allowing the helicase to move the substrate into the active site of DSS1 ribonuclease (8,9). Inactivation of either SUV3 or DSS1 gene results in respiratory deficiency and decreased stability of the mitochondrial genome. At the molecular level, the loss of the degradosome functions results in overaccumulation of excised introns, accumulation of transcripts with abnormal 5′ and 3′ termini and the appearance of unprocessed high-molecular-weight precursors (10–12). All these phenotypes are consistent with the proposed role of the yeast mitochondrial degradosome in RNA turnover and surveillance.
In contrast to S. cerevisiae, the protein complex in Schizosaccharomyces pombe consisting of SUV3 and DSS1 orthologs (genes rpm2 and rpm1 encoding Pah1p and Par1p, respectively) was found to play only a minor, if any role in RNA degradation: it was proposed to be directly involved in mtRNA processing and was found essential for the generation of 3′-ends of mitochondrial transcripts (13).
A similar protein complex containing orthologs of SUV3 and DSS1, TbSUV3 and TbDSS-1, respectively, was recently described for mitochondria from Trypanosoma (14). The TbDSS-1 exoribonuclease was shown to function in elimination of nonfunctional byproducts of RNA processing and its depletion resulted in aberrant levels of unedited messenger RNAs (mRNAs), edited mRNAs and guide RNAs. Interestingly, TbDSS-1 was suggested to have additional functions in processing of pre-mRNAs and was found to be associated with a large multicomponent protein complex (14–16).
Human mitochondrial RNA turnover, surveillance and processing have remained an unsolved puzzle (17). RNA synthesis in human mitochondria starts at only three points, one for the L strand and two for the H strand, and the resulting large transcripts are subsequently processed into two rRNAs (12S and 16S), 11 polyadenylated mRNAs and 22 transfer RNAs (tRNAs). Mammalian mitochondrial genomes contain no introns, but excised large noncoding regions from the transcribed L-strand must be efficiently degraded (18,19). Human hSuv3p protein seemed to be a good candidate for a subunit of the putative human degradosome: an RNA helicase activity is indispensable for functioning of all known complexes involved in RNA decay and surveillance; and the SUV3 gene was found to be very ancient and its orthologs were found in all eukaryotes and Rhodobacter (20). The human ortholog of the yeast SUV3 gene (hSUV3, SUPV3L1) was identified by homology search and complementary DNA (cDNA) cloning. The protein contains a bona fide mitochondrial leader and it localizes predominantly in the mitochondrial matrix, although a fraction could be found in the nucleus (21). Analysis of the hSUV3 promoter indicates that it is a housekeeping gene (22). hSuv3p protein heterologously expressed in Escherichia coli was found in vitro to be an ATP-dependent multisubstrate helicase, able to unwind double-stranded DNA (dsDNA), double-stranded RNA (dsRNA) and RNA–DNA heteroduplexes (23,24). The details of the hSuv3p mitochondrial function have been missing, except for a single experiment showing the presence of low-molecular-weight truncated ND2 transcripts upon hSUV3 gene silencing (25). Moreover, the fact that the human hSuv3p helicase preferentially unwinds dsDNA (24) and has been found in mitochondrial nucleoids suggested its role in mitochondrial DNA (mtDNA) replication and not necessarily in RNA metabolism (26).
The search for the ribonuclease component of human mitochondrial degradation pathway proved to be difficult. No mammalian orthologs of the yeast DSS1 gene have been found and no RNAse-like genes with a putative mitochondrial signal sequence could be identified (our unpublished data). A number of RNase activities were detected in mammalian mitochondrial extracts, but these could not be identified by proteomic analysis (Chrzanowska-Lightowlers,Z., personal communication). One of the candidates for human mitochondrial exoribonuclease has been polynucleotide phosphorylase (PNPase) which is able to catalyze processive phosphorolytic degradation of RNA from 3′ to 5′; it can also polymerize RNA in a random, template-independent process from 5′ to 3′. PNPases have been conserved in evolution and have been found in bacteria, plants, worms, flies and mammals (27). In E. coli PNPase functions in an RNA degrading complex called degradosome, where it is associated with an RNA helicase, endoribonuclease (ribonuclease E) and enolase (28). It was described as a polyadenylating enzyme in Gram-positive bacteria, cyanobacteria and chloroplasts (29). In plant mitochondria, PNPase is responsible for polyadenylation-dependent degradation of RNAs: particularly of RNA maturation byproducts and of antisense RNAs transcribed from the opposite DNA strand (30,31).
The human PNPase encoded by the nuclear gene PNPT1 has been discovered as an enzyme upregulated in senescent progeroid fibroblasts and in melanoma cells which undergo terminal differentiation (32) and it has been suggested that it acts by degradation of RNAs in the cytosol (33). PNPase was found in mitochondria (34,35), but conflicting data were published on whether PNPase localizes to mitochondria only. While two reports indicated this (34,35), the study of Sarkar and Fisher (36) suggested both mitochondrial and cytosolic localization. The physiological role of PNPase in human mitochondria has been recently a matter of controversy, because two more recent studies found the protein in the mitochondrial intermembrane space (IMS) (35,37). Therefore, the direct participation of PNPase in mitochondrial RNA metabolism was questioned, although the siRNA inhibition of human PNPase expression resulted in the increased abundance of aberrant mtRNAs (38) and elongation of poly(A) tails (38,39). Recently in vitro binding of heterologously expressed hSuv3p and human PNPase was demonstrated (40), but no in vivo data presenting hSuv3p–PNPase interaction have been published to date.
Mutants defective in RNA decay provide a possibility to look into steps of RNA processing which are otherwise impossible to observe because of their high reaction speed and low abundance of intermediates (41). In this paper, we show the construction of a stable human cell line expressing a missense hSuv3p mutant. When expressed, the mutant protein strongly interferes with the native hSuv3p complex and allows detection of otherwise unstable transcripts. The observed phenotypes include accumulation of intergenic noncoding regions transcribed from both strands of mtDNA and perturbations in polyadenylation. Our results suggest that the hSuv3p protein is indeed the key component of RNA metabolism in human mitochondria. We also report co-purification of hSuv3p helicase and PNPase.
MATERIALS AND METHODS
Cell culture and cell manipulations
HeLa or 293 T-Rex cells (Invitrogen) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco) supplemented with 10% fetal bovine serum (Gibco) at 37°C under 5% CO2. The stable, inducible cell lines obtained in this work were cultured by using TET System Approved FBS (Clontech). For cell growth rate assessment 3.5 × 106 of the appropriate cells were plated, induced (or not) after 24 h and detached after the next 24 h. The cells were counted and plated at the same density as previously. Every second day the cells were detached, counted and plated. Part of the detached cells was used for RNA and protein isolation. In the RNA stability assay the cells were split, induced (or not) for 24 h and actinomycin D was added at a concentration of 5 µg/ml. For hSUV3 gene silencing the HeLa cells or 293 T-Rex cells were grown to 40–50% confluence and subjected to stealthRNA (Invitrogen) transfection. Transfections were performed using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer’s recommendations. The stealthRNA specific for hSUV3 silencing with oligo ID HSS110378 (named S1 in the paper), HSS110379 (S2), HSS186162 (S3) and negative control (Stealth™ RNAi Negative Control LO GC, N) were used at the final concentration of 20 nM. 293 T-Rex cells were subjected to a second round of transfection 2 days after first one and cultured for the next 3 days. Cells were collected after 3 days (HeLa) or 5 days (293 T-Rex).
DNA cloning and creation of stable transfected cell lines
DNA fragments encoding either full-length hSuv3p or its form without the 46 N-terminal residues were amplified by polymerase chain reaction (PCR) by using pKK plasmid as a template (20) and cloned into pcDNA5/FRT/TO vector (Invitrogen) to get pRS97 (full length) and pRS68 (truncated form). In order to obtain a DNA construct encoding hSuv3p in which Gly207 was substituted with Val the site-directed mutagenesis was performed on pRS97 and pRS68 as described previously (24) to get pRS98 and pRS100, respectively. To construct DNA plasmids encoding C-terminal TAP protein fusions the DNA fragment encoding TAP tag (42) was PCR amplified and cloned into pcDNA5FRT vector (Invitrogen). Subsequently, DNA fragments encoding PNPase, hSuv3p or its N-terminal 26 aa were cloned in frame with TAP tag encoding DNA to get pcDNA5FRT-PNPTAP, pcDNA5FRT-hSuv3TAP, pcDNA5FRT-mtTAP. Sequences of the used primers are available upon request. The stable inducible cell lines overexpressing hSuv3pWT or hSuv3pG207V were obtained by using the Flp-In™ T-REx™ 293 system (Invitrogen) according to the manufacturer’s instruction. The host cell-line was co-transfected using polyethylenimine (PEI) reagent with 0.2 µg of appropriate construct (pRS68, pRS97, pRS98, pRS100) and 1.8 µg of pOG44 (Invitrogen). Twenty-four hours after transfection, cells were plated and subjected to selection by adding hygromycin B (100 μg/ml) (Invitrogen) and blasticidin (15 μg/ml) (Invitrogen). Selective medium was replaced every 2–3 days. In all presented experiments, expression of the exogenous genes was induced with tetracycline at the concentration of 25 ng/ml. For uninducible cell lines expressing mtTAP (hSuv3p mitochondrial leader with TAP at the C-terminus), hSuv3TAP or PNPTAP identical procedure was applied; however pcDNA5/FRT (Invitrogen) vector was used and cells were not selected with blasticidin.
Western blot analysis
For the hSUV3 gene silencing control, the total protein cell extracts were prepared as described previously (21). Protein concentration was determined by the Bradford method. Twenty micrograms of each total protein extract or an equal amount of each fraction in the case of size exclusion chromatography were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS–PAGE) and transferred to a nitrocellulose membrane (Protran, Whatman GmbH). Western blotting was performed according to standard protocols using the following primary antibodies: PNPase (N-18, sc-49315, Santa Cruz Biotechnology), GAPDH (V-18, sc-20357, Santa Cruz Biotechnology) and hSuv3p described previously (24). Primary antibodies were detected with goat anti-rabbit or rabbit anti-goat peroxidase-conjugated secondary antibodies (Calbiochem) and visualized by enhanced chemiluminescence (Santa Cruz Biotechnology) according to the manufacturer’s instructions.
Immunofluorescence
All procedures were carried out at room temperature. Cells were grown on glass coverslips for 24 h, MitoTracker Red CMXRos (200 nM) (Molecular Probes) was added to the medium and cells were incubated at 37°C for 20 min. After incubation cells were washed two times with medium and three times with phosphate-buffered saline (PBS) and fixed with 3.7% formaldehyde for 25 min. The cells were washed three times with PBS and permeabilized with 0.5% Triton X-100 in 10% FBS/PBS for 15 min. After washing with PBS cells were blocked with PBS solution containing 10% FBS for 30 min. Primary and secondary antibodies were diluted with blocking solution. Cells were incubated in anti-hSuv3p solution (antibody dilution 1:300) for 1 h. Following three washes with PBS, secondary antibodies conjugated with Alexa Fluor 488 were applied at 1:900 (Molecular Probes) for 1 h. Cells were washed two times with PBS, nuclei were stained by 5 min of incubation in Hoechst 33342/PBS solution (1 μg/ml) and washed with PBS. Cells were mounted in ProLong Gold antifade reagent (Invitrogen) and subjected to microscopy analysis. Microscopy was performed on a Nikon Eclipse TE2000-E/C1 confocal microscope with a CFI Plan Apochromat 60.0 × 1.40 oil objective.
RNA isolation and manipulations
Total RNA was isolated by TRI Reagent (Sigma) according to the manufacturer’s instructions. For fractionation of the total RNA the Poly(A) Purist MAG kit (Ambion) was used following the producer’s instruction. Total RNA obtained by TRI Reagent isolation was precipitated by ammonium acetate and ethanol and subjected to two rounds of oligo-dT purification as recommended by the manufacturer. RNA concentration was measured using NanoDrop ND1000 (NanoDrop Technologies).
Northern blots
Northern blot analysis was performed essentially as described previously (43). For standard northern blots 2.5–3 µg of RNA was dissolved in denaturing solution composed of NBC buffer (50 mM boric acid, 1 mM sodium acetate, 5 mM NaOH), 5.5% formaldehyde and 50% formamide, heat-denatured for 5 min at 65°C, mixed with 2 µl of 10 loading dye (15% Ficoll, 0.25% bromophenol blue, 0.25% xylene cyanol in 0.1 M EDTA, pH 8.0) and run on a 1% agarose gel containing formaldehyde (0.9%) using NBC as an electrophoresis buffer. RiboRuler High Range RNA Ladder (Fermentas) was used as a marker. After electrophoresis, RNA was transferred to Nytran-N membrane (Schleicher & Schuell BioScience) by overnight capillary transfer in 10×SSC (1.5 M sodium chloride, 150 mM sodium citrate). The filter was then washed briefly with 2×SSC and RNA was immobilized by ultraviolet-cross-linking. RNA was stained with methylene blue solution (0.02% methylene blue in 0.3 M sodium acetate, pH 5.2). For high-resolution northern blots, 2 µg of RNA was mixed with an equal volume of denaturing loading dye (95% formamide, 20 mM EDTA, 0.25% bromophenol blue, 0.25% xylene cyanol), heat denatured for 5 min at 65°C and run on a 6% polyacrylamide/urea gel in 1× TBE (44). Heat-denaturated pUC19 DNA/MspI (Fermentas) was applied as a marker. After electrophoresis nucleic acids were blotted onto Nytran-N membrane (Schleicher & Schuell BioScience) by semi-dry electrotransfer in 0.5 × TBE buffer using the Transblotter apparatus (Bio-Rad). All hybridizations were performed in PerfectHyb Plus buffer (Sigma). PCR products of the following mtDNA fragments: 3652–4029 (ND1), 4807–5172 (ND2), 10131–10375 (ND3), 10470–10758 (ND4L/4), 7586–7900 (COX2), 904–1307 (12S rRNA) and pUC19 DNA/MspI (Fermentas) were used as templates for preparing probes labeled with [α-32P] dATP (Hartmann Analytic) using HexaLabel DNA Labeling Kit (Fermentas). In the case of strand-specific hybridizations [α-32P] UTP labeled riboprobes were applied. The PCR products containing SP6 or T7 promoter sequences (one on each end) corresponding to: 10470–10758, 10131–10375, 6503–6922, 5587–5891, 4807–5172, 3652-4029 nt in the human mitochondrial genome were used for in vitro transcription. Transcription in vitro was performed by using the T7 transcription kit (Fermentas) and [α-32P] UTP (Hartmann Analytic) following the manufacturer’s instruction. For the analysis of RNA expression corresponding to COX1-tRNATyr intergenic region and tRNATyr, tRNAMet, tRNAGln genes the following oligonucleotides labeled with [γ-32P] ATP (Hartmann Analytic) by T4 polynucleotide kinase (New England Biolabs) were used: cagtgggggtga (RSZ325; intergenic region), tcacccccactg (RSZ326; intergenic region) and tctttagatttacagtccaatgcttcactcagccattttacc (RSZ323; sense tRNATyr), ggtaaaatggctgagtgaagcattggactgtaaatctaaaga (RSZ324; antisense tRNATyr), tagtacgggaagggtataaccaacattttcggggtatggg (RSZ320; sense tRNAMet), cccataccccgaaaatgttggttatacccttcccgtacta (RSZ321; antisense tRNAMet), caaaattctccgtgccacctatcacaccccatccta (RSZ327; sense tRNAGln), taggatggggtgtgataggtggcacggagaattttg (RSZ328; antisense tRNAGln). The levels of other tRNAs were measured using oligonucleotides labeled as above. Sequences of the oligonucleotides are available upon request. Following hybridization, filters were exposed to PhosphorImager screens (Molecular Dynamics). Autoradiograms were obtained by scanning the PhosphorImager screens using a Storm Scanner (Molecular Dynamics) and analyzed with the ImageQuant 5.1 software (Molecular Dynamics). For tRNAs levels assessment (data in Figure 4B) the membranes were stripped and reprobed with two others tRNAs probes. One membrane was used for measurement of levels of three tRNAs. The efficiency of stripping was confirmed by overnight exposition of the membrane to PhosphorImager screens. For standardization the filters were reprobed with a 7SL probe described previously (45) or rRNA was stained with methylene blue.
Figure 4.

Influence of hSuv3pG207V expression on RNAs involved in mitochondrial translation. Changes in steady-state levels of 12S rRNA (A) and analyzed tRNAs (B). In both panels, representative northern blots are shown on the right. Graphs represent the mean of three independent experiments and standard deviation is marked.
Circular reverse transcription PCR (CRT) analysis
The experimental procedure described elsewhere (43) was used with some modifications. In brief, 5 µg of total RNA was circularized with T4 RNA ligase (Fermentas) in a total volume of 15 µl in the presence of DNAse I (Roche). The resulting circular RNA was subjected to standard phenol–chloroform extraction, precipitated with ethanol and resuspended in 10 µl of appropriate primer solution (10 pmol/µl). Reverse transcription was carried out using RevertAid M-MuLV Reverse Transcriptase (Fermentas). The first-strand cDNA (1 µl of the above reaction mixture) was subjected to PCR with external primers. The first PCR product was diluted 100-fold and employed in the second round of PCR by using nested primers. The final PCR products were cloned into pGEM-T Easy Vector (Promega) and the inserts were sequenced using SP6 and/or T7 primers (DNA Sequencing Laboratory IBB PAS). In the case of ND3, ATP6/8 and 12S rRNA analysis the primers described by Tomecki et al. (43) were used. For mirror COX1 analysis the following primers were used: TTCTCGGCCTATCCGGAATG (RSZ300, reverse transcription), CTACCCCGATGCATACACC (RSZ301, first PCR), AGGGTAGACTGTTCAACCTG (RSZ295, first PCR), AACATCCTATCATCTGTAGG (RSZ302, second PCR), TGTGGTCGTTACCTAGAAGG (RSZ303, second PCR).
TAP tag purification and size exclusion chromatography
Mitochondria were isolated as described by Spelbrink et al. (46) with minor modifications. Ten strokes of a tight-fitting pestle were used to homogenize cells. Mitochondria were washed twice with NKM buffer (1 mM Tris–HCl, pH 7.4, 0.13 M NaCl, 5 mM KCl, 2.5 mM MgCl2). To pellet mitochondria the suspension was centrifuged at 12 000g for 1 min. If not otherwise stated the procedure was carried out at 4°C. The final mitochondrial pellet was lysed in lysing buffer [10% glicerol, 50 mM HEPES-KOH pH 8.0, 100 mM KCl, 2 mM EDTA, 1% Triton X-100, 2 mM DTT, 1 mM PMSF, 1× complete protease inhibitor (Roche)] by rotating for 10 min and cleared by centrifugation at 16 100 g for 30 min. The protein extract was mixed with immunoglobulin G (IgG) Sepharose (Amersham) and rotated for 1.5 h. The beads were extensively washed with IPP150 solution (10 mM Tris–HCl pH 8.0, 150 mM NaCl, 0.1% Triton X-100) and then with tobacco etch virus (TEV) cleavage buffer (10 mM Tris–HCl pH 8.0, 150 mM NaCl, 0.5 mM EDTA, 1 mM DTT). Afterwards the beads were resuspended in TEV cleavage buffer containing TEV protease and incubated for 2 h at 18°C. Eluates were collected and analyzed by western blot or used for a second step of chromatography on calmodulin beads (Stratagene). The eluate was adjusted with CaCl2 to 4 mM and incubated with calmodulin beads for 1.5 h at 4°C. The beads were extensively washed with calmodulin binding buffer (10 mM β-mercaptoethanol, 10 mM Tris–HCl pH 8.0, 150 mM NaCl, 0.1% NP-40, 2 mM CaCl2, 1 mM MgOAc, 1 mM imidazole). The bound proteins were heat-eluted with SDS–PAGE loading buffer (50 mM Tris–Cl pH 6.8, 2% SDS, 0.1% bromophenol blue, 10% glycerol, 50 mM β-mercaptoethanol) and analyzed by western blot. For size exclusion chromatography mitochondrial protein lysates obtained from 293 T-Rex cells were separated according to size by using high resolution Superdex 200 10/300 column and AKTA purifier FPLC (GE Healthcare). The column was equilibrated with a solution composed of 10 mM Tris–HCl pH 8.0, 150 mM NaCl.
RESULTS
Development of a cellular model for analyzing the function of the human hSuv3p helicase
In order to determine the role of the hSuv3p helicase in human mitochondria a cell model was constructed. Previous investigations have shown that hSuv3p protein with glycine to valine substitution in position 207, which is located within the Walker A motif (hSuv3pG207V), does not have ATPase or helicase activities (24). Therefore, we assumed that its expression in human cells might lead to dominant-negative effects. Genes encoding the wild-type hSuv3p protein (hSuv3pWT) or mutated hSuv3pG207V were integrated into the genome of human cell line 293 using the FlpIn system, which ensures an induction of the inserted genes by a low tetracycline concentration. A cell line with an integrated empty vector was constructed as an additional control.
A significant overproduction of hSuv3 proteins at similar levels was detected in both 293 hSuv3pWT and 293 hSuv3pG207V cell lines grown for 24 h in medium supplemented with 25 ng/ml tetracycline (Figure 1A). In order to check that the mutation and overproduction does not change the subcellular localization, immunofluorescence analysis was performed 24 h after induction, with uninduced cells used as a control to distinguish the signal from the endo- and exogenous hSuv3p. The results indicate that overproduced hSuv3pG207V protein, as well as hSuv3pWT, is localized in mitochondria (Figure 1B). In contrast to HeLa cells (21) no nuclear localization of hSuv3p protein was observed.
Figure 1.

Expression and subcellular localization of hSuv3pWT and hSuv3pG207V in stable 293 cell lines. (A) Western blot analysis of hSuv3p level in 293 hSuv3pWT or 293 hSuv3pG207V cell lines. Expression of the exogenous genes was induced for 24 h with tetracycline at 25 ng/ml. GAPDH level is shown as the loading control. (B) Subcellular localization of endogenous hSuv3p (uninduced) and overproduced hSuv3pWT or hSuv3pG207V (induced) proteins. Exogenous gene expression was induced for 24 h and cells were subjected to immunofluorescence staining and confocal microscopy. The same microscope settings were used for uninduced and induced cells. Mitochondria were labeled with MitoTracker Red CMX Ros, hSuv3p with primary rabbit anti-hSuv3p antibodies followed by secondary AlexaFluor 488 conjugated antibodies. Nuclei were stained with Hoechst dye. The bar represents 10 μm.
Inhibition of hSuv3p function affects the rate of growth and morphology of cells
In order to evaluate the effect of overexpression of the wild-type and mutated hSuv3p proteins on cell growth, the same numbers of cells were plated and expression of hSuv3pWT or hSuv3pG207V was induced on the next day. Subsequently, the cells were passaged every other day, with the same cell concentrations. Until the third day no difference between the cells overproducing the wild-type or mutated hSuv3p and the cells with an integrated empty vector (Figure 2A) was observed. However, 5 days after induction, a decrease of the growth rate of the cells with hSuv3pG207V was seen, and it was even more pronounced on successive days (Figure 2A). This effect was not observed in cells carrying hSuv3pWT or empty vector (Figure 2A).
Figure 2.

Effect of hSuv3pG207V expression on cell growth rate and cell morphology. (A) Graph representing growth rate of cells with integrated exogenous gene encoding wild-type (WT) or inactive form of hSuv3p (G207V). The growth rate of cells with empty vector integrated is also shown (Vector). Uninduced (–Tet) or induced (+Tet) cells were analyzed. The results represent the mean of three independent experiments. Error bars represent standard deviation. (B) Influence of hSuv3pG207V expression on cell morphology and color of the medium after 5 days of expression.
Moreover, the overproduction of the inactive form of hSuv3p, but not of the wild-type protein, caused a change of cell morphology (Figure 2B, top). In addition, culture medium of cells with hSuv3pG207V turned yellow (Figure 2B, bottom), which suggests a respiration defect similar to observed previously for overproduction of an inactive form of mitochondrial DNA polymerase (47) or silencing of the mitochondrial PNPase (35). This observation was the first indication that hSuv3pG207V expression affects mitochondrial function.
Expression of the inactive form of hSuv3p leads to perturbations in mitochondrial RNA metabolism
In order to analyze the effect of the hSuv3p on mitochondrial RNA metabolism, total RNA isolated after 1, 3 and 5 days of overexpression of the wild-type or inactive form hSuv3p helicase was hybridized with various mitochondrial probes (for ND1, ND2, ND3, ND4/4L and COX2) using the northern-blot technique. Significant perturbations of the level and length of mitochondrial transcripts were found in cells with mutated hSuv3p (Figure 3A and B) when compared to controls already after the first day of induction.
Figure 3.

Expression of hSuv3pG207V affects mitochondrial RNA. Northern blot analysis of RNA isolated from uninduced or induced (+Tet) cells to overproduce wild-type or inactive form of hSuv3p. Impairment of hSuv3p function by expression of its inactive form leads to appearance of heterogeneous RNA (A and B) and affects (A) or not (B) the steady-state level of mRNA. Representative northern blots are shown. The graphs below show the levels of the analyzed mRNAs. Data represent the mean of three independent experiments, standard deviation is marked. The level of 7SL is shown as a loading control. (C) Northern blot analysis of ND3 mRNA in total RNA (Total), unbound (Unbound) and oligo-dT bound RNA fractions (Oligo-dT). A fragment of the membrane corresponding to the methylene blue staining of 18S rRNA is also shown for assessment of the fractionation efficiency.
One to three days of expression of hSuv3pG207V leads to an increase in the levels of some mitochondrial protein-encoding transcripts (ND1, ND2 and ND3) (Figure 3A), but no significant effects were observed for COX2 and ND4/4L mature mRNA (Figure 3B). For each DNA probe, heterogeneous molecules, longer and shorter than the mature mRNAs were observed for all time points (Figure 3). We confirmed that these molecules are indeed RNA and not DNA by RNAseI and DNAseI treatment prior to electrophoresis (data not shown).
In order to check whether the transcripts with inappropriate lengths from cells expressing hSuv3pG207V are polyadenylated, total RNA was fractionated on oligo-dT and northern blots were performed on the same amounts of total RNA and the fractions bound and not bound to oligo-dT (Figure 3C). The oligo-dT bound fraction gave strong hybridization signal from RNA species both shorter and longer than the probed for ND3 mRNA, indicating that these transcripts are indeed polyadenylated (Figure 3C).
The analysis of transcripts involved in mitochondrial translation (12S rRNA and tRNA) revealed that their levels decrease gradually (Figure 4) after induction of mutant hSuv3p expression but not of wild-type form. It is worth noting that the level of each tRNA decreases to the same extent regardless of the location of the particular gene within the mitochondrial genome [i.e. the distance from the transcription initiation site, or whether it is transcribed from the H-strand (Phe, Val, Leu, Ile, Arg and Thr) or L-strand (Ser, Glu and Pro)] (Figure 4B). However, in contrast to mRNA, only mature tRNA molecules were detected, which is consistent with tRNAs being processed by endonucleases.
The observed changes in transcript abundance and the appearance of new RNA species of lower or higher molecular weight than the mature RNAs indicate that hSuv3p protein may participate in RNA degradation and/or processing of the polycistronic transcripts.
Perturbations in RNA metabolism depend on mitochondrial localization of hSuv3pG207V and this effect is equivalent to that observed after transient silencing of the hSUV3 gene
Human hSuv3p was found to be present in small quantities outside mitochondria (21). In order to exclude that the observed perturbations of mitochondrial RNA metabolism were secondary effects of the non-mitochondrial functions of hSuv3p, stable cell lines were obtained with an integrated gene encoding the wild-type or inactive form of the hSuv3p protein, both lacking the mitochondrial targeting signal which is present in the 46 N-terminal amino acids (24). Western blot analysis indicated that both truncated proteins were effectively expressed (Figure 5A). Expression of both the Δ46aa hSuv3pWT protein and the Δ46aa hSuv3pG207V does not have any effects on mitochondrial RNA (Figure 5B). Moreover, no effect of the overproduction of the truncated forms of the hSuv3p protein on the growth rate or morphology of the cells was observed (data not shown).
Figure 5.

Expression of the non-mitochondrially localized hSuv3pWT or hSuv3pG207V does not affect mitochondrial RNA. (A) Western blot analysis of expression of the Δ46hSuv3pWT or Δ46hSuv3pG207V proteins. GAPDH level is shown as the loading control. (B) Northern blot analysis of ND2 and ND3 mRNAs. Cells were induced or not for 24 h, collected and RNA was isolated and analyzed by northern blot. Methylene blue staining of 18S rRNA is shown as loading control. (C and D) Silencing of hSUV3 expression by RNAi. HeLa and 293 cells were untreated (NT) or transfected with siRNAs noncomplementary to hSUV3 mRNA (N) or complementary to hSUV3 mRNA in three different places (S1, S2 and S3). (C) Western blot analysis of hSuv3p levels relative to GAPDH and (D) northern blot analysis of ND2 and ND3 mRNA levels relative to 7SL RNA.
To show that the effects observed after hSuv3pG207V expression are not an artifact due to the presence of large amounts of this protein in mitochondria, ND3 and ND2 transcripts were analyzed after silencing of the endogenous hSUV3 gene by RNAi in 293 and HeLa cell lines. Three siRNAs corresponding to different regions of the hSUV3 gene were used and each of them caused a decrease of the hSuv3p to <10% (Figure 5C). On northern blots we detected the accumulation of ND3 and ND2 mRNAs and the presence of molecules longer and shorter than the analyzed transcript (Figure 5D). The same pattern was observed for all three stealthRNAs, which confirms the specificity of this effect. Moreover, similar effects were observed regardless of whether the hSUV3 gene was silenced in HeLa or 293 cells (Figure 5D).
These results correspond to those obtained for hSuv3pG207V indicating that the expression of the inactive form of hSuv3p is functionally equivalent to transient silencing of the hSUV3 gene.
Perturbation of hSuv3p function stabilizes mature ND3 mRNA
The yeast Suv3 ortholog is a component of the mitochondrial degradosome responsible for RNA degradation. To check whether the human Suv3p helicase is also involved in mtRNA degradation we used a transcriptional inhibitor to analyze the stability of the ND3 transcript in the presence of normal or aberrant hSuv3p helicase. This particular transcript was chosen as it has the shortest half-life of all mitochondrial mRNAs (60 min) (48). The presence of hSuv3pG207V led to significant stabilization of ND3 mRNA indicating that hSuv3p participates in its degradation (Figure 6).
Figure 6.
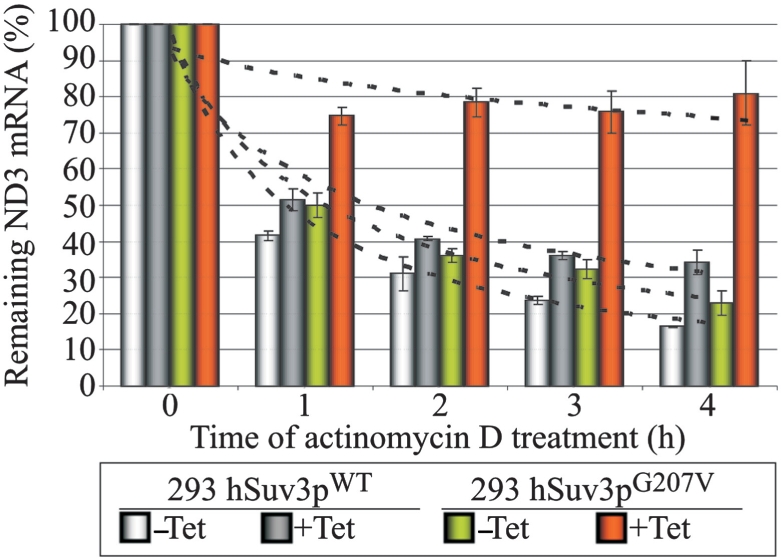
Impairment of hSuv3p function results in ND3 mRNA stabilization. Graph representing the level of ND3 mRNA measured by northern blot in cells untreated (point 0) and treated with the transcription inhibitor actinomycin D. Cells uninduced (–Tet) and induced (+Tet) for 24 h were analyzed. The results represent the mean of three independent experiments, error bars represent standard deviation, trend lines are also marked (dashed lines).
The expression of the catalytically inactive form of hSuv3p leads to the formation of transcripts with inappropriate lengths
In order to precisely analyze the lengths of the mature transcripts in the cells expressing the mutated hSUV3 gene, northern blot analysis was performed using high resolution polyacrylamide gels. Twenty-four hours after induction of hSuv3pG207V expression (Figure 7A) the wild-type length ND3 transcript disappeared and new classes of transcripts were detected: longer by about 20–40 nt and shorter by about 40 nt than the wild-type mature ND3 mRNA (Figure 7A). When the experiment was continued for several more days, the abundance of elongated transcripts decreased, while the level of shortened transcripts increased (Figure 7A).
Figure 7.

Effect of expression of hSuv3pG207V on transcript length. (A) High-resolution northern blot of ND3 mRNA; M, molecular weight marker. (B) Distribution of different classes of truncated clones identified by CRT-PCR. (C) Breaking points of 12S rRNA identified by CRT-PCR shown on a model of 12S rRNA structure. Boxed letters represent the first and the last nucleotide which can be analyzed by the applied assay. Numbers correspond to the nucleotide position in 12S rRNA (954 nt length). In some cases, the precise end of the truncated transcript cannot be identified as the adenine residue may belong to the poly(A) tail. See legend for details. (D) Analysis of the poly(A) tail length assessed by CRT-PCR. Each data point represents a single transcript. Polyadenylated transcripts [poly(A) tail longer than 25 nt] are shown as filled symbols and oligoadenylated ones (tail of 25 nt or less) as empty symbols. In cases where it was impossible to assess the precise length of the tail, because the adenine might belong to the tail or ‘body’ of the transcripts the average number was used. Horizontal lines represent average lengths of oligo- and polyadenylated fractions (E) Schematic representation of the two identified uridinylated transcripts.
The longer transcripts might be due to longer poly(A) tails or represent ND3 mRNA extended by tRNA Arg and/or Gly which arose due to incorrect mitochondrial pre-mRNA processing, whereas the shortened transcripts might represent degradation intermediates.
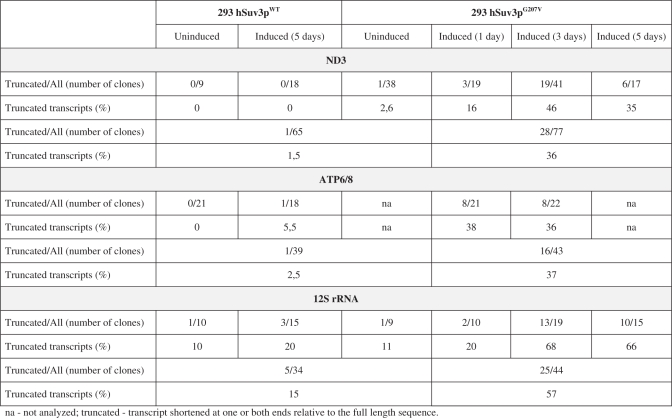
In order to characterize the 5′ and 3′ RNA termini under conditions of altered hSuv3p function we performed sequencing of circular reverse transcription (CRT)-PCR products, selecting for ND3 and ATP6/8 mRNAs and 12S rRNA. An increased number of truncated transcripts were detected (Table 1 ). In control cells, uninduced or overexpressing the wild-type hSuv3p, we detected 1.5%, 2.5% and 15%, of truncated ND3, ATP6/8 and 12S rRNA transcripts, respectively (Table 1). In contrast, in cells expressing the mutated hSuv3p protein these fractions were significantly increased to 36%, 37% and 57%, respectively (Table 1). Thus, the CRT-PCR analysis is in agreement with the results of northern blot analysis (Figures 3A and 4A). Our data indicate that most of the observed transcripts were truncated at the 3′-end only (Figure 7B). Transcripts shortened only on the 5′-end or on both ends were less frequent (Figure 7B).
Table 1.
Summary of the CRT-PCR analysis of the ND3, ATP6/8 mRNA and 12S rRNA
 |
An elevated level of truncated transcripts, mostly on the 3′-end is a further indication that hSuv3p participates in mitochondrial RNA degradation and suggests that it proceeds in the 3′ to 5′ direction. This is in agreement with biochemical data of Shu et al. (23) who demonstrated that the purified hSuv3 protein unwinds double-stranded nucleic acids in the 3′→5′ direction.
We used the model of the 12S rRNA structure available at http://www.rna.ccbb.utexas.edu (49) to propose the site of breakage of the transcript at the 3′-end (Figure 7C). Our analysis of CRT-PCR clones indicated that out of 18 analyzed breakage sites only one was within a double-stranded region while the remaining sites were all in single-stranded regions. Moreover, most of them were grouped at sites located at 3′-end with respect to the double-stranded regions. This is in agreement with the 3′ to 5′ action of the helicase activity of hSuv3p in vitro. We conclude that the lack of this activity in vivo leads to the inability to degrade double-stranded regions.
Lack of hSuv3p activity leads to extension of poly(A) tails
Our sequencing analysis revealed that in cells expressing mutant hSuv3pG207V the poly(A) tails present on mature transcripts were longer than in wild-type cells. For ATP6/8 and ND3 we found 10 and 23 clones, respectively, with a tail longer than 50 nt, whereas in control cells (uninduced and overproducing hSuv3pWT) only one and six such clones were detected, respectively (Figure 7D), with similar numbers of clones analyzed (Table 1). Similarly, 12S rRNA transcripts with poly(A) tails longer than 25 nt were identified in cells with impaired hSuv3p function, but not in the wild-type cells (Figure 7D).
It should be stressed that by sequencing we did not detect any oligo- or polyadenylated ND3 transcripts containing adjacent tRNA molecules which is in agreement with our northern blot analysis (data not shown). The above data indicate that the observed larger bands on high-resolution northern blots (Figure 7A) represent the ND3 mRNA properly excised from its precursor and bearing elongated poly(A) tails, and that the observed elongation is not due to the lack of proper tRNA excision.
We found only two protein-encoding transcripts containing an additional tRNA sequence; interestingly both were polyuridynylated. One of them was an ND3 mRNA truncated at the 5′-end linked to an incomplete tRNAArg at the 3′-end (Figure 7E). The second was an ATP6/8 mRNA truncated at the 3′-end linked to tRNALys at the 5′-end (Figure 7E). Both transcripts were found in cells which had expressed the inactive hSuv3p form for 3 days and it is difficult to determine whether they were formed as a direct effect of hSuv3p function impairment or represent a secondary phenotype.
hSuv3p plays a crucial role in removing noncoding processing intermediates
Both H and L strand of mtDNA are transcribed as long polycistronic transcripts. Most of the mitochondrial mRNAs coding for proteins and rRNAs are transcribed from the H strand. In contrast, the large polycistronic RNA transcribed from the L strand contains only one mRNA for ND6 polypeptide, and eight tRNAs plus long noncoding intergenic regions (50) (Figure 8A). Thus, the heterogeneous long RNA detected on the northern blots shown above (Figures 3, 5B and D) could represent such intergenic regions as the applied probes were complementary to both strands of mtDNA.
Figure 8.

hSuv3p participates in removal of noncoding RNA. (A) Schematic representation of the polycistronic RNA resulting from H- or L-strand transcription and localization of the used riboprobes. The letters on the right refer to the template strand. (B–E) Northern blot analysis using strand-specific riboprobes. RNA isolated from uninduced cells or cells induced for 24 hours was used. (D) Northern blot analysis of the ND2 mirror transcript level in cells untreated (point 0) and treated with transcription inhibitor actinomycin D. Expression of the hSuv3pG207V had been induced for 24 h before actinomycin D was added. Graph on the top shows quantification of the signal by using ImageQuant software.
In order to check whether hSuv3p is involved in the metabolism of the noncoding RNA, we used strand-specific riboprobes corresponding to the ND3 region (L- or H-specific probes) (Figure 8A).
The probe complementary to ND3 mRNA (sense probe) detected only RNA molecules of the size corresponding to mRNA (∼400 nt) and shorter products of its degradation (Figure 8B, L-strand specific ND3 probe, lane 4). The double band is in agreement with the results already shown (Figure 7A). In contrast, the ND3 antisense probe detected molecules in the range from 200 nt to about 4000 nt (Figure 8B), which is in agreement with the hybridization smear on northerns with the use of a double-stranded probe (compare Figure 3). Although some hybridization signal was unspecific (especially from cytoplasmic rRNA), which was shown using RNA derived from 143B rho-zero cells lacking mitochondrial DNA and their parental cell line 143B, this analysis proved that expression of hSuv3pG207V leads to accumulation of noncoding RNA.
To check whether a similar accumulation of undegraded noncoding antisense RNA also takes place for other mtDNA regions, we analyzed transcripts representing regions ND4/4L, COX1, ND1 and ND2. Similarly as in the case of ND3, we detected the dicistronic ND4/4L transcript (∼1.7 knt, Figure 8B) using the sense-detecting probe or noncoding RNAs from ∼200 nt to at least 4000 nt long representing the RNA transcribed from the L strand using the anti-sense detecting probe (Figure 8B). It should, however, be noted that the probe complementary to ND4/4L mRNA also detects small amounts of long RNA, but it does not accumulate to such a high degree as found for the probe for the noncoding RNA.
hSuv3p is involved in the degradation of mirror RNAs
Interestingly, analysis of the remaining mtDNA regions corresponding to COX1, ND1 and ND2 in cells expressing hSuv3pG207V showed not only the appearance of an antisense RNA smear but also a large increase of RNA with the same/similar length to the mRNA transcribed from the H strand (Figure 8C). We have named such noncoding antisense RNA with a similar length to coding sense RNA mirror RNA. This RNA, at least in the case of ND1 and ND2, can also be detected in control cells (Figure 8C) but at a much lower level. This phenomenon was not observed in cells overproducing the wild-type hSuv3p (Figure 8C), which suggests that hSuv3p activity may be required for mirror RNA degradation.
In order to check if mirror RNA stability increases in cells expressing hSuv3pG207V we performed an experiment where we analyzed the changes in mirror ND2 RNA levels after transcription was inhibited. The mirror ND2 transcript was chosen because it can also be detected at low levels in RNA isolated from control cells. The performed analysis indicated that in control cells the mirror ND2 level decreases by about 50% an hour after actinomycin D addition and cannot be detected after 2 h (Figure 8D), while its level in cells expressing hSuv3pG207V does not change during the first hour and the first changes can only be observed 3 h after adding actinomycin D (Figure 8D). This indicates that hSuv3p participates in the degradation of noncoding RNA resulting from L-strand transcription.
In the case of some mRNAs, i.e. ND4/4L and COX1, the expression of hSuv3pG207V and the concomitant increase of antisense RNA leads to the decrease in respective sense mRNA levels (Figure 8B and C). However, this effect was not observed for the other analyzed transcripts. This may be due to in vivo annealing of sense RNA to its antisense counterpart and subsequent degradation by a yet unknown activity. It remains to be seen if this is only the collateral phenotype of our experimental setup, or if it may play a role in mitochondrial gene expression.
So far, we have demonstrated that human hSuv3p helicase is necessary for degradation of noncoding RNA resulting from L strand transcription. To see whether hSuv3p is also involved in eliminating noncoding RNA transcribed from the H strand, we have analyzed by strand-specific probes the region encompassing four tRNA genes: tRNA Tyr, Cys, Asn and Ala (Figure 8A). These tRNAs are generated by L-strand transcription. In the cells expressing inactive hSuv3p we found a considerable increase in the amount of 300–350-nt-long RNA transcribed from the H strand (Figure 8E), its length corresponds to the sum of the lengths of tRNA Tyr, Cys, Asn and Ala (305 nt). We suggest that this transcript corresponds to the H-strand transcribed mirror of tRNA Tyr, Cys, Asn and Ala (see also Figure 9C). This experiment indicates that hSuv3p is involved in removal of noncoding RNA transcribed from the H strand, similarly to the removal of L-strand intermediates as has been shown above.
Figure 9.

Mapping the ends of mirror transcripts. (A) Schematic map of the COX1 region of mtDNA. Boxed arrows represent coding sequences. Processing points identified by CRT-PCR are marked. (B) Analysis of the poly(A) tail length of the COX1 mirror transcript assessed by CRT-PCR. Symbols represent single transcripts. RNA isolated from 293 hSuv3pG207V cells uninduced and induced for 24 h was used. Horizontal lines represent average lengths of oligo- and polyadenylated fractions (C) Northern blot analysis confirming the CRT-PCR analysis of the COX1 mirror transcript. Strand-specific probes were used. (D) Northern blot analysis of the 3′-end of the ND2 mirror transcript. Strand-specific probes were used. See text for details and Figures 8A and 9A for a schematic representation of the polycistronic transcripts.
Mirror RNAs can be generated by proper excision of tRNA molecules
Inhibition of hSuv3p function in hSuv3pG207V cells allows studying mtRNA processing in details which is otherwise difficult because of the very short half-life of intermediates. Mirror RNAs are almost undetectable under normal conditions as they are very rapidly degraded. Taking advantage of their abundance in hSuv3pG207V cells we decided to characterize them further. The region corresponding to the COX1 gene was chosen for this analysis. The COX1 mRNA is transcribed from the H strand while on the opposite L-strand this sequence is flanked by two tRNA genes: Tyr and Ser (Figures 8A and 9A). In addition we analyzed the ND2 region.
Analysis of the COX1 region
We analyzed the ends of the COX1 mirror transcripts by sequencing products of CRT-PCR from hSuv3pG207V cells either not induced or induced for 1 day. As expected, a higher proportion of full-length mirror COX1 was found in RNA isolated after induction of mutated protein: among 26 clones 21 (80%) and 18 (69%), respectively, had 5′- and 3′-ends as predicted from the position of tRNAs in the primary transcript, which define the processing site (Figure 9A). No clones representing transcripts extended beyond these points were identified. This indicates that the mirror RNA of COX1 is the product of correct excision of natural native tRNATyr and tRNASer from the polycistronic RNA transcribed from the L strand. In the uninduced cells more truncated transcripts, especially at the 3′-end, were found: 84% clones (21/26) had the 5′-end and only 40% had the 3′-end without truncation.
Among all truncated transcripts the identified truncations were mainly from the 3′-end (62.5%) whereas the transcripts truncated at the 5′-end or both the 5′ and 3′-end were found in 13% and 24.5% clones, respectively.
A similar result was obtained for ND3 and ATP6/8 mRNAs and 12S rRNA (Figure 7B). The obtained data indicate that the degradation of both functional (mRNA, rRNA) and noncoding RNAs occurs mainly in the 3′→5′ direction.
Moreover, data obtained from the CRT-PCR analysis indicate that mirror RNAs are polyadenylated (Figure 9B). Similarly to normal RNAs (Figure 7D), expression of hSuv3pG207V leads to elongation of poly(A) tails of mirror COX1 (Figure 9B). The detection of polyadenylated transcripts resulting from L-strand transcription is in agreement with our analysis of the RNA fraction bound to oligo-dT (Figure 3C) and data obtained by other authors (51).
The CRT-PCR method is not fully quantitative so we decided to confirm our sequencing results by northern blots using two complementary single-stranded probes detecting the 12-nt intergenic region between the COX1 and tRNATyr genes (Figure 9A). Only the probe targeting RNAs transcribed from the L strand recognized molecules corresponding in size to the COX1 mirror RNA (compare Figure 9C and Figure 8C) and only in the case of cells expressing hSuv3pG207V (Figure 9C). As described above, this RNA was found in uninduced cells using CRT-PCR, but its level is probably too low for detection using the northern technique with the short oligonucleotide probe. Thus, our hybridization results for cells expressing hSuv3pG207V confirm that the majority of mirror COX1 RNA contains the 12-nt region adjacent to the 5′-end of tRNATyr.
On the other hand, the probe detecting the 12-nt intergenic COX1-tRNATyr region transcribed from the H strand does not bind to any RNA, except for a low level of unspecific hybridization to cytoplasmic rRNA (Figure 9C). The intergenic RNA fragment is not present in the correctly processed COX1 mRNA (38). Therefore, our results indicate that the expression of mutated hSuv3p does not affect COX1 mRNA maturation.
To verify that the mirror COX1 transcript is not elongated by tRNATyr (see Figure 9A) we used two probes detecting this tRNA in either L- or H-strand-derived transcripts. The probe recognizing L-strand-derived transcripts detected only native tRNATyr, whereas the other probe detected noncoding RNA ∼300–350 nt long (Figure 9C), the same size as seen previously using a riboprobe detecting mirror RNA. This RNA represents sequences between tRNA Trp and COX1 genes transcribed from the H strand (Figure 8A and E). This result indicates that tRNATyr is not a component of the L-strand transcribed mirror COX1 which is fully in agreement with our sequencing data.
Analysis of the ND2 region
To confirm that mirror RNAs are generated by a standard tRNA excision pathway, we performed a similar experiment for the mirror ND2. The ND2 gene is preceded by the tRNAMet gene (both RNAs transcribed from the H strand), which is adjacent to the tRNAGln gene (transcribed from the L strand) (Figure 8A). Hybridization with a probe complementary to tRNAMet detected only this tRNA (Figure 9D), whereas the probe complementary to the antisense tRNAMet detected an RNA with the size corresponding to mirror RNA previously detected by the L-strand-specific riboprobe (compare Figure 9D with Figure 8C). These data show that the mirror ND2 has at its 3′-end the antisense tRNAMet sequence.
As expected for the mirror ND2 formation by excision of natural tRNA (tRNAGln), the probe complementary to tRNAGln did not detect any other RNA molecules than tRNA (Figure 9D) even after prolonged exposure (data not shown). The probe corresponding to tRNAGln but complementary to the L strand recognizes a short RNA molecule, which accumulates in cells expressing hSuv3pG207V (Figure 9D). This RNA corresponds to RNA encoded on the L strand between tRNAMet and tRNAIle (Figure 8A) which is released by the excision of those tRNAs from the primary transcript. These RNA molecules are longer than tRNAGln probably due to polyadenylation [not detected for full-length tRNAs (51)].
Our results show that the processing of RNA transcribed from the L-strand follows the canonical way of tRNA excision. Moreover, they also indicate that when hSuv3p function is disturbed, tRNA excision is correct, but 3′→5′ degradation of processing intermediates is perturbed.
Human Suv3 helicase co-purifies with PNPase
So far, the search for a ribonuclease responsible for mitochondrial RNA decay has not been successful. However, it was found that silencing of PNPase affects mitochondrial RNA metabolism (38,39). Therefore, we decided to check whether hSuv3p may interact with PNPase. For this purpose, we obtained a 293 cell line expressing constitutively the hSuv3p protein fused to a TAP tag (protein A and calmodulin-binding protein) at the C-terminus. As a control, we obtained a stable cell line expressing the TAP tag fused to the mitochondrial leader of hSuv3p. Both fusion proteins localize to mitochondria (data not shown). Mitochondrial extracts from such cells were subjected to affinity chromatography on IgG Sepharose and purified proteins were eluted by cutting off the TAP tag with TEV protease. PNPase presence in the eluate was checked by western blot analysis. PNPase was only present in the eluate where the cell extract was derived from cells expressing hSuv3pTAP (Figure 10A) and was not observed in the control eluate (Figure 10A).
Figure 10.

Co-purification of hSuv3p with PNPase. The mtTAP, hSuv3TAP and PNPTAP correspond to the name of the cell line and the type of expressed TAP fusion protein. The mtTAP is a short polypeptide which probably causes its low stability and lower expression than hSuv3TAP. (A and B) Western blot analysis of PNPase presence in total (Total), mitochondrial (Mito) lysates and elution (Elution) fraction obtained after one- (A) or two-step (B) chromatography. The mtTAP and hSuv3TAP fusion proteins were detected because the secondary antibodies used for PNPase detection bind directly to the A protein, a TAP tag component. (C) Western blot analysis of the hSuv3p presence in total (Total), mitochondrial (Mito) lysates and elution (Elution) fraction. The PNPTAP fusion protein was detected because the rabbit primary anti-hSuv3p antibodies bind directly to the A protein, a TAP tag component. (D) Western blot analysis of hSuv3p and PNPase presence in fractions obtained by size exclusion chromatography. The migration of the marker proteins is shown. Fractions in which hSuv3p and PNPase migrate together are boxed.
Even after two-step affinity chromatography PNPase was still present in the eluate when hSuv3p was TAP-tagged, whereas it was not present in the control purification (Figure 10B). To confirm these results, we performed a purification using cells with stable expression of the C-terminal PNPTAP fusion and hSuv3p presence was analyzed by western blots. The hSuv3p was present in the eluate from cells expressing PNPTAP but not from control cells (Figure 10C). This shows that both proteins co-purify and thus suggests they may interact in vivo.
To see whether the whole fraction of hSuv3p may interact with PNPase we analyzed the mitochondrial lysate from 293 cells by gel filtration chromatography and the obtained fractions were tested by western blot using antibodies recognizing PNPase or hSuv3p. Two fractions of hSuv3p were distinguished; one comigrating with PNPase (fractions 3–10) and one free from PNPase (fractions 11–14) (Figure 10D). PNPase is not detected in fractions 11–14 even after a very long exposure. PNPase migrates in fractions 3–10 which encompass proteins between 232 and 440 kDa. This is in agreement with the analysis of PNPase migration in BN-PAGE (35) and with the size of the PNPase-hSuv3p complex obtained in vitro by Wang et al. (40). Our results suggest that a fraction of both proteins could interact in vivo.
DISCUSSION
The results presented in this paper show that the human nuclear-encoded ATP-dependent helicase hSuv3p is a major player in mitochondrial RNA metabolism; it participates in regulation of the half-life of properly formed mature mRNAs, in RNA surveillance, i.e. removal of aberrant RNAs and in decay of processing intermediates derived from transcripts synthesized from the L strand or H strand of mitochondrial DNA. These conclusions come from experiments based on a stable cell line expressing in an inducible manner the missense mutant G207V of hSuv3p which leads to impairment of the function of the endogenous protein. Such an approach provides several experimental advantages over transient gene silencing by siRNA: upon induction all cells express the dominant negative mutant protein, while siRNA transfection efficiency may vary from experiment to experiment. In addition, the very low concentration of the inducer does not interfere with mitochondrial metabolism. Moreover, the inducible system allows studying the function of proteins necessary for cell survival and gives a better chance to observe primary effects in contrast to uninducible systems when compensatory mechanisms may mask the primary effects. Finally, the stable cell line allows the continuation of experiments for much longer than in the case of siRNA transfection.
If the hSuv3p protein participates in the coding RNA decay system, perturbation of its activity should lead to the increased stability of such RNA. Indeed, for the ND3 transcript, which has the shortest half-life among mitochondrial mRNAs, we observed a decrease of the rate of mature mRNA clearance in the mutant cell line compared to controls within 4 h after arresting mitochondrial transcription with actinomycin D. The increase of steady-state mRNA level for ND3, as well as other transcripts, 1–3 days after induction of mutant hSuv3p are in agreement with these results. When our experiments were continued for up to 9 days we could observe progressive decay of mRNA levels, but we assume this is a secondary effect resulting from a general deregulation of mitochondrial nucleic acid metabolism. In conclusion, our results indicate that the hSuv3 protein is indeed involved in the decay of properly formed mRNA molecules.
In contrast to the observed increased stability of mature protein-coding transcripts, all tested tRNA species showed significantly lower abundance upon expression of the hSuv3p mutant. This may indicate that tRNA turnover is accomplished by a different enzymatic system, but more research is needed to explain this observation.
A function of RNA surveillance systems is the removal of physiologically normal, but not further required processing intermediates. The unique feature of mammalian mitochondrial gene expression is generation of two large transcripts encompassing the full L strand and H strand, which subsequently undergo processing. In contrast to the densely packed H-strand-derived transcript, the entire L-strand-derived RNA encodes only one polypeptide ND6 and 8 tRNA molecules. Therefore, the majority of L-strand-derived RNA represents intergenic regions, which after excision are very quickly degraded and usually escape detection. Using H-strand-specific probes we have shown that the expression of the mutant hSuv3p protein leads to a very strong accumulation of the processing intermediates generated by transcription of the L strand. Our results lead to the conclusion that the hSuv3p protein is necessary for the proper degradation of processing intermediates in human mitochondria. In this respect, it resembles the yeast mitochondrial degradosome, where the knockout of the ySUV3 gene results in strong accumulation of excised introns.
Our experimental setup provides a possibility to look closely into the details of this process, and constitutes a valuable tool for further studies of the activities that participate in it. One of the characteristic features of these processing intermediates detected in flagranti is so-called mirror RNAs; i.e. RNA molecules that result from transcription and processing of the same DNA region, but originating from the complementary DNA strand. RNA processing in mammalian mitochondria depends in most cases on punctuation of protein-coding sequences by tRNA sequences. Our data have shown that in the case of mirror RNAs, the processing machinery properly recognizes flanking tRNA molecules and produces in this way the mirror RNA which is subsequently polyadenylated and degraded. There is an open question if such mirror RNAs play a physiological role, for example, in mtDNA replication or translational control. Our data indicate that the increased level of a given antisense RNA correlates with the lower level of some of the respective functional RNAs (COX1, ND4/4L). This fact may suggest a regulatory role for mirror RNAs, in addition it seems that another, not yet described degradation system is responsible for the observed decay of ‘sense’ RNA.
The second function of a surveillance system is the degradation of aberrantly formed RNAs. We reasoned that the inhibition of hSuv3p function may result in the increased abundance of such molecules. We performed sequencing of re-ligated RNA molecules which enables to analyze both 3′- and 5′-ends of mitochondrial RNAs. We were able to detect the accumulation of truncated transcripts which, taking into account our other data, most likely represent degradation intermediates. Interestingly, in two isolated cases we have found polyuridinylated RNA. Although their frequency is low, they were identified only in RNA isolated from cells with impaired hSuv3p function. Moreover, they are the only identified transcripts which were composed of mRNA and tRNA and therefore represent the products of improper processing of the polycistronic precursor. We are not able to distinguish whether they appear as a direct or indirect consequence of the lack of hSuv3p function but it may suggest that polyuridinylation is used for tagging and removing aberrantly processed transcripts. It should be noted that Slomovic and Schuster (38) have shown an increased amount of improperly matured polyuridinylated COX1 transcript in cells with silenced PNPase.
Until now, functions of mitochondrial complexes containing SUV3 orthologs have only been described for two yeast species and Trypanosoma. The SUV3/DSS1 mitochondrial degradosome from S. cerevisiae has been postulated to participate in RNA surveillance, but not in RNA processing (10). In contrast, the protein complex from S. pombe, containing orthologs of Suv3 and Dss1 proteins, respectively Pah1p and Par1p, was recently postulated to be a mitochondrial processosome: inactivation of any of the two subunits resulted in complete block of RNA processing, with a variety of truncations and extensions at 3′- and 5′-ends of mRNAs. The most characteristic phenotype of these mutants was the complete lack of properly formed mature mRNAs. Thus, the S. pombe Pah1p protein was suggested to play a minor role, if any, in RNA decay (13).
Do the observed aberrant forms of mtRNA represent products of mitochondrial RNA metabolism caught in flagranti due to impaired degradation or do they reflect hSuv3p participation in RNA processing? Our hybridization data and sequence analysis indicate that the aberrant forms exist in the control cells, but their abundance is much lower. Thus, we favor the hypothesis that using our experimental system we only increase their abundance and that the hSuv3p protein may not play a direct role in RNA processing. In addition, we detect a significant amount of mature, properly formed mRNA, and our sequencing analysis revealed only two cases of intermediates containing an unprocessed tRNA molecule on either end. Therefore, it seems that the human hSuv3p function differs from that in the S. pombe processosome, and is rather functionally similar to the S. cerevisiae degradosome. Since the majority of truncations occurred at the 3′-end we suggest that the hSuv3p-dependent degradation pathway operates in the 3′-5′ direction. This resemblance of functions has been conserved in evolution despite the fact that no human ortholog of yeast DSS1 gene has been detected.
The presence of PNPase in the mitochondrial IMS has been an unsolved puzzle: what is the role, if any, of this enzyme in mitochondrial RNA metabolism, and what are the molecular partners of PNPase? How can PNPase residing in the IMS be involved in processing and possibly polyadenylation of mitochondrial RNAs which are localized in the matrix? Conflicting results were presented when PNPase expression was downregulated by using RNA interference: one study showed that lowering the PNPase levels did not affect mitochondrial morphology or the consumption of oxygen (39), the other study showed that PNPase knockdown results in the reduction of mitochondrial membrane potential, fragmentation of mitochondria, significant lowering of ATP level and lower levels of activity of respiratory complexes (35). In addition, several laboratories studied mitochondrial RNA metabolism in mammalian cells depleted for PNPase: Nagaike et al. (39) reported the presence of longer poly(A) tails of some mitochondrial transcripts upon RNA interference, but the steady-state levels of the mRNAs remained unchanged. Similarly, Chen et al. (35) did not detect any changes in mtRNA abundance. On the other hand, in PNPase knockdown cells, Slomovic and Schuster (38) reported abolishment of stable polyadenylation and the presence of an unprocessed 9 nt sequence at the 5′-end of COX1 mRNA, while poly(A) tails of ND5 and ND3 transcripts increased in length. Nevertheless, the authors suggested that these phenotypes are secondary effects, because they considered unlikely the direct action of IMS-localized PNPase on RNA metabolism in the matrix. The possible link between polyadenylation and degradation in mitochondrial RNA was therefore missing (17).
Our data show that TAP-tagged hSuv3p co-purifies with PNPase, and TAP-tagged PNPase co-purifies with hSuv3p. This is in agreement with the data of Wang et al., (40) who recently demonstrated that the two proteins form a stable and active complex in vitro. Our result suggests in vivo interaction of those proteins although we cannot exclude the possibility that the observed interaction occurs after the lysis of mitochondria, in which case the intermembrane-residing PNPase would react with matrix-residing hSuv3p. More research is needed to resolve this issue, especially in the context of the well-documented localization of PNPase in the IMS. Our gel filtration analysis of mitochondrial proteins revealed that hSuv3p and PNPase elute in two overlapping peaks, which suggests that only a relatively small portion of the two proteins may form a complex. Therefore, in vivo only a fraction of PNPase could enter the mitochondrial matrix and form a stable complex with the hSuv3p protein, which then could participate in mtRNA surveillance. This suggestion is in contrast to Slomovic and Schuster (38), who favored the model of indirect influence of PNPase silencing on mtRNA metabolism.
Data presented in this paper on lengthening of poly(A) tails upon hSuv3p inhibition are in agreement with those of others (38,39), who reported elongation of poly(A) tails of mitochondrial mRNAs upon transient PNPase silencing. We consider this a further indication that both proteins may cooperate in vivo. Nevertheless, the effects of the hSuv3p mutant on mtRNA metabolism observed by us are stronger than those published due to the PNPase silencing: in particular no accumulation of the L-strand-derived processing intermediates has been reported previously in cells with a lower level of PNPase. It is possible that PNPase silencing using siRNA was not sufficiently strong to inhibit its activity in the mitochondrial matrix. The other possibility is that similarly to bacterial systems, the mammalian mitochondrial degradosome contains yet another ribonuclease which has escaped identification to date. Clearly, more research is needed to solve this problem.
FUNDING
The Polish National Centre for Research and Development (NR13004704); the Polish Ministry for Science and Higher Education (NN301014333); Polish Mitochondrial Network MitoNet; and Faculty of Biology, University of Warsaw intramural grants (BW179112, BW179107).
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
The authors thank Prof Johannes Spelbrink for 143B and 143B rho0 cells; Dr. Monika Hejnowicz, Dr. Leszek Lipinski and Dr. Rafal Tomecki for helpful discussions; Dr. Andrzej Dziembowski for helpful comments on TAP purification and Aleksander Chlebowski for his help with the manuscript.
REFERENCES
- 1.Houseley J, Tollervey D. The many pathways of RNA degradation. Cell. 2009;136:763–776. doi: 10.1016/j.cell.2009.01.019. [DOI] [PubMed] [Google Scholar]
- 2.Carpousis AJ. The RNA degradosome of Escherichia coli: an mRNA-degrading machine assembled on RNase E. Annu. Rev. Microbiol. 2007;61:71–87. doi: 10.1146/annurev.micro.61.080706.093440. [DOI] [PubMed] [Google Scholar]
- 3.Parker R, Song H. The enzymes and control of eukaryotic mRNA turnover. Nat. Struct. Mol. Biol. 2004;11:121–127. doi: 10.1038/nsmb724. [DOI] [PubMed] [Google Scholar]
- 4.Gagliardi D, Stepien PP, Temperley RJ, Lightowlers RN, Chrzanowska-Lightowlers ZM. Messenger RNA stability in mitochondria: different means to an end. Trends Genet. 2004;20:260–267. doi: 10.1016/j.tig.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 5.Linder P. Dead-box proteins: a family affair–active and passive players in RNP-remodeling. Nucleic Acids Res. 2006;34:4168–4180. doi: 10.1093/nar/gkl468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stepien PP, Margossian SP, Landsman D, Butow RA. The yeast nuclear gene suv3 affecting mitochondrial post-transcriptional processes encodes a putative ATP-dependent RNA helicase. Proc. Natl Acad. Sci. USA. 1992;89:6813–6817. doi: 10.1073/pnas.89.15.6813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dmochowska A, Golik P, Stepien PP. The novel nuclear gene DSS-1 of Saccharomyces cerevisiae is necessary for mitochondrial biogenesis. Curr. Genet. 1995;28:108–112. doi: 10.1007/BF00315775. [DOI] [PubMed] [Google Scholar]
- 8.Malecki M, Jedrzejczak R, Stepien PP, Golik P. In vitro reconstitution and characterization of the yeast mitochondrial degradosome complex unravels tight functional interdependence. J. Mol. Biol. 2007;372:23–36. doi: 10.1016/j.jmb.2007.06.074. [DOI] [PubMed] [Google Scholar]
- 9.Rogowska AT, Puchta O, Czarnecka AM, Kaniak A, Stepien PP, Golik P. Balance between transcription and RNA degradation is vital for Saccharomyces cerevisiae mitochondria: reduced transcription rescues the phenotype of deficient RNA degradation. Mol. Biol. Cell. 2006;17:1184–1193. doi: 10.1091/mbc.E05-08-0796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dziembowski A, Piwowarski J, Hoser R, Minczuk M, Dmochowska A, Siep M, van der Spek H, Grivell L, Stepien PP. The yeast mitochondrial degradosome. Its composition, interplay between RNA helicase and RNase activities and the role in mitochondrial RNA metabolism. J. Biol. Chem. 2003;278:1603–1611. doi: 10.1074/jbc.M208287200. [DOI] [PubMed] [Google Scholar]
- 11.Dziembowski A, Malewicz M, Minczuk M, Golik P, Dmochowska A, Stepien PP. The yeast nuclear gene DSS1, which codes for a putative RNase II, is necessary for the function of the mitochondrial degradosome in processing and turnover of RNA. Mol. Gen. Genet. 1998;260:108–114. doi: 10.1007/s004380050876. [DOI] [PubMed] [Google Scholar]
- 12.Margossian SP, Li H, Zassenhaus HP, Butow RA. The DExH box protein Suv3p is a component of a yeast mitochondrial 3′-to-5′ exoribonuclease that suppresses group I intron toxicity. Cell. 1996;84:199–209. doi: 10.1016/s0092-8674(00)80975-7. [DOI] [PubMed] [Google Scholar]
- 13.Hoffmann B, Nickel J, Speer F, Schafer B. The 3′ ends of mature transcripts are generated by a processosome complex in fission yeast mitochondria. J. Mol. Biol. 2008;377:1024–1037. doi: 10.1016/j.jmb.2008.01.038. [DOI] [PubMed] [Google Scholar]
- 14.Mattiacio JL, Read LK. Evidence for a degradosome-like complex in the mitochondria of Trypanosoma brucei. FEBS Lett. 2009;583:2333–2338. doi: 10.1016/j.febslet.2009.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mattiacio JL, Read LK. Roles for TbDSS-1 in RNA surveillance and decay of maturation by-products from the 12S rRNA locus. Nucleic Acids Res. 2008;36:319–329. doi: 10.1093/nar/gkm690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Penschow JL, Sleve DA, Ryan CM, Read LK. TbDSS-1, an essential Trypanosoma brucei exoribonuclease homolog that has pleiotropic effects on mitochondrial RNA metabolism. Eukaryot. Cell. 2004;3:1206–1216. doi: 10.1128/EC.3.5.1206-1216.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bobrowicz AJ, Lightowlers RN, Chrzanowska-Lightowlers Z. Polyadenylation and degradation of mRNA in mammalian mitochondria: a missing link? Biochem. Soc. Trans. 2008;36:517–519. doi: 10.1042/BST0360517. [DOI] [PubMed] [Google Scholar]
- 18.Falkenberg M, Larsson NG, Gustafsson CM. DNA replication and transcription in mammalian mitochondria. Annu. Rev. Biochem. 2007;76:679–699. doi: 10.1146/annurev.biochem.76.060305.152028. [DOI] [PubMed] [Google Scholar]
- 19.Fernandez-Silva P, Enriquez JA, Montoya J. Replication and transcription of mammalian mitochondrial DNA. Exp. Physiol. 2003;88:41–56. doi: 10.1113/eph8802514. [DOI] [PubMed] [Google Scholar]
- 20.Dmochowska A, Kalita K, Krawczyk M, Golik P, Mroczek K, Lazowska J, Stepien PP, Bartnik E. A human putative Suv3-like RNA helicase is conserved between Rhodobacter and all eukaryotes. Acta Biochim. Pol. 1999;46:155–162. [PubMed] [Google Scholar]
- 21.Szczesny RJ, Obriot H, Paczkowska A, Jedrzejczak R, Dmochowska A, Bartnik E, Formstecher P, Polakowska R, Stepien PP. Down-regulation of human RNA/DNA helicase SUV3 induces apoptosis by a caspase- and AIF-dependent pathway. Biol. Cell. 2007;99:323–332. doi: 10.1042/BC20060108. [DOI] [PubMed] [Google Scholar]
- 22.Minczuk M, Lilpop J, Boros J, Stepien PP. The 5′ region of the human hSUV3 gene encoding mitochondrial DNA and RNA helicase: promoter characterization and alternative pre-mRNA splicing. Biochim. Biophys. Acta. 2005;1729:81–87. doi: 10.1016/j.bbaexp.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 23.Shu Z, Vijayakumar S, Chen CF, Chen PL, Lee WH. Purified human SUV3p exhibits multiple-substrate unwinding activity upon conformational change. Biochemistry. 2004;43:4781–4790. doi: 10.1021/bi0356449. [DOI] [PubMed] [Google Scholar]
- 24.Minczuk M, Piwowarski J, Papworth MA, Awiszus K, Schalinski S, Dziembowski A, Dmochowska A, Bartnik E, Tokatlidis K, Stepien PP, et al. Localisation of the human hSuv3p helicase in the mitochondrial matrix and its preferential unwinding of dsDNA. Nucleic Acids Res. 2002;30:5074–5086. doi: 10.1093/nar/gkf647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Khidr L, Wu G, Davila A, Procaccio V, Wallace D, Lee WH. Role of SUV3 helicase in maintaining mitochondrial homeostasis in human cells. J. Biol. Chem. 2008;283:27064–27073. doi: 10.1074/jbc.M802991200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bogenhagen DF, Rousseau D, Burke S. The layered structure of human mitochondrial DNA nucleoids. J. Biol. Chem. 2008;283:3665–3675. doi: 10.1074/jbc.M708444200. [DOI] [PubMed] [Google Scholar]
- 27.Lin-Chao S, Chiou NT, Schuster G. The PNPase, exosome and RNA helicases as the building components of evolutionarily-conserved RNA degradation machines. J. Biomed. Sci. 2007;14:523–532. doi: 10.1007/s11373-007-9178-y. [DOI] [PubMed] [Google Scholar]
- 28.Marcaida MJ, DePristo MA, Chandran V, Carpousis AJ, Luisi BF. The RNA degradosome: life in the fast lane of adaptive molecular evolution. Trends Biochem. Sci. 2006;31:359–365. doi: 10.1016/j.tibs.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 29.Portnoy V, Palnizky G, Yehudai-Resheff S, Glaser F, Schuster G. Analysis of the human polynucleotide phosphorylase (PNPase) reveals differences in RNA binding and response to phosphate compared to its bacterial and chloroplast counterparts. RNA. 2008;14:297–309. doi: 10.1261/rna.698108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Holec S, Lange H, Kuhn K, Alioua M, Borner T, Gagliardi D. Relaxed transcription in Arabidopsis mitochondria is counterbalanced by RNA stability control mediated by polyadenylation and polynucleotide phosphorylase. Mol. Cell Biol. 2006;26:2869–2876. doi: 10.1128/MCB.26.7.2869-2876.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Perrin R, Lange H, Grienenberger JM, Gagliardi D. AtmtPNPase is required for multiple aspects of the 18S rRNA metabolism in Arabidopsis thaliana mitochondria. Nucleic Acids Res. 2004;32:5174–5182. doi: 10.1093/nar/gkh852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leszczyniecka M, Kang DC, Sarkar D, Su ZZ, Holmes M, Valerie K, Fisher PB. Identification and cloning of human polynucleotide phosphorylase, hPNPase old-35, in the context of terminal differentiation and cellular senescence. Proc. Natl Acad. Sci. USA. 2002;99:16636–16641. doi: 10.1073/pnas.252643699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sarkar D, Park ES, Emdad L, Randolph A, Valerie K, Fisher PB. Defining the domains of human polynucleotide phosphorylase (hPNPaseOLD-35) mediating cellular senescence. Mol. Cell Biol. 2005;25:7333–7343. doi: 10.1128/MCB.25.16.7333-7343.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Piwowarski J, Grzechnik P, Dziembowski A, Dmochowska A, Minczuk M, Stepien PP. Human polynucleotide phosphorylase, hPNPase, is localized in mitochondria. J. Mol. Biol. 2003;329:853–857. doi: 10.1016/s0022-2836(03)00528-x. [DOI] [PubMed] [Google Scholar]
- 35.Chen HW, Rainey RN, Balatoni CE, Dawson DW, Troke JJ, Wasiak S, Hong JS, McBride HM, Koehler CM, Teitell MA, et al. Mammalian polynucleotide phosphorylase is an intermembrane space RNase that maintains mitochondrial homeostasis. Mol. Cell Biol. 2006;26:8475–8487. doi: 10.1128/MCB.01002-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sarkar D, Fisher PB. Polynucleotide phosphorylase: an evolutionary conserved gene with an expanding repertoire of functions. Pharmacol. Ther. 2006;112:243–263. doi: 10.1016/j.pharmthera.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 37.Rainey RN, Glavin JD, Chen HW, French SW, Teitell MA, Koehler CM. A new function in translocation for the mitochondrial i-AAA protease Yme1: import of polynucleotide phosphorylase into the intermembrane space. Mol. Cell Biol. 2006;26:8488–8497. doi: 10.1128/MCB.01006-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Slomovic S, Schuster G. Stable PNPase RNAi silencing: its effect on the processing and adenylation of human mitochondrial RNA. RNA. 2008;14:310–323. doi: 10.1261/rna.697308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nagaike T, Suzuki T, Katoh T, Ueda T. Human mitochondrial mRNAs are stabilized with polyadenylation regulated by mitochondria-specific poly(A) polymerase and polynucleotide phosphorylase. J. Biol. Chem. 2005;280:19721–19727. doi: 10.1074/jbc.M500804200. [DOI] [PubMed] [Google Scholar]
- 40.Wang DD, Shu Z, Lieser SA, Chen PL, Lee WH. Human mitochondrial SUV3 and polynucleotide phosphorylase form a 330-kDa heteropentamer to cooperatively degrade double-stranded RNA with a 3'-to-5' directionality. J. Biol. Chem. 2009;284:20812–20821. doi: 10.1074/jbc.M109.009605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Preker P, Nielsen J, Kammler S, Lykke-Andersen S, Christensen MS, Mapendano CK, Schierup MH, Jensen TH. RNA exosome depletion reveals transcription upstream of active human promoters. Science. 2008;322:1851–1854. doi: 10.1126/science.1164096. [DOI] [PubMed] [Google Scholar]
- 42.Rigaut G, Shevchenko A, Rutz B, Wilm M, Mann M, Seraphin B. A generic protein purification method for protein complex characterization and proteome exploration. Nat. Biotechnol. 1999;17:1030–1032. doi: 10.1038/13732. [DOI] [PubMed] [Google Scholar]
- 43.Tomecki R, Dmochowska A, Gewartowski K, Dziembowski A, Stepien PP. Identification of a novel human nuclear-encoded mitochondrial poly(A) polymerase. Nucleic Acids Res. 2004;32:6001–6014. doi: 10.1093/nar/gkh923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sambrook J, Russell DW. Molecular Cloning: A Laboratory Manual. 3rd edn. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2001. [Google Scholar]
- 45.Piechota J, Szczesny R, Wolanin K, Chlebowski A, Bartnik E. Nuclear and mitochondrial genome responses in HeLa cells treated with inhibitors of mitochondrial DNA expression. Acta Biochim. Pol. 2006;53:485–495. [PubMed] [Google Scholar]
- 46.Spelbrink JN, Toivonen JM, Hakkaart GA, Kurkela JM, Cooper HM, Lehtinen SK, Lecrenier N, Back JW, Speijer D, Foury F, et al. In vivo functional analysis of the human mitochondrial DNA polymerase POLG expressed in cultured human cells. J. Biol. Chem. 2000;275:24818–24828. doi: 10.1074/jbc.M000559200. [DOI] [PubMed] [Google Scholar]
- 47.Jazayeri M, Andreyev A, Will Y, Ward M, Anderson CM, Clevenger W. Inducible expression of a dominant negative DNA polymerase-gamma depletes mitochondrial DNA and produces a rho0 phenotype. J. Biol. Chem. 2003;278:9823–9830. doi: 10.1074/jbc.m211730200. [DOI] [PubMed] [Google Scholar]
- 48.Piechota J, Tomecki R, Gewartowski K, Szczesny R, Dmochowska A, Kudla M, Dybczynska L, Stepien PP, Bartnik E. Differential stability of mitochondrial mRNA in HeLa cells. Acta Biochim. Pol. 2006;53:157–168. [PubMed] [Google Scholar]
- 49.Cannone JJ, Subramanian S, Schnare MN, Collett JR, D'S;ouza LM, Du Y, Feng B, Lin N, Madabusi LV, Muller KM, et al. The comparative RNA web (CRW) site: an online database of comparative sequence and structure information for ribosomal, intron, and other RNAs. BMC Bioinformatics. 2002;3:2. doi: 10.1186/1471-2105-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, et al. Sequence and organization of the human mitochondrial genome. Nature. 1981;290:457–465. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- 51.Slomovic S, Laufer D, Geiger D, Schuster G. Polyadenylation and degradation of human mitochondrial RNA: the prokaryotic past leaves its mark. Mol. Cell Biol. 2005;25:6427–6435. doi: 10.1128/MCB.25.15.6427-6435.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]