

Abstract
Before a comparative diagnostic trial is carried out, maximum sample sizes for the diseased group and the nondiseased group need to be obtained to achieve a nominal power to detect a meaningful difference in diagnostic accuracy. Sample size calculation depends on the variance of the statistic of interest, which is the difference between receiver operating characteristic summary measures of 2 medical diagnostic tests. To obtain an appropriate value for the variance, one often has to assume an arbitrary parametric model and the associated parameter values for the 2 groups of subjects under 2 tests to be compared. It becomes more tedious to do so when the same subject undergoes 2 different tests because the correlation is then involved in modeling the test outcomes. The calculated variance based on incorrectly specified parametric models may be smaller than the true one, which will subsequently result in smaller maximum sample sizes, leaving the study underpowered. In this paper, we develop a nonparametric adaptive method for comparative diagnostic trials to update the sample sizes using interim data, while allowing early stopping during interim analyses. We show that the proposed method maintains the nominal power and type I error rate through theoretical proofs and simulation studies.
Keywords: Diagnostic accuracy, Error spending function, ROC, Sensitivity, Specificity
1. INTRODUCTION
When a new medical diagnostic test is developed, trials are carried out to compare the diagnostic accuracy of the new test with some existing one. In these comparative diagnostic trials, it is of interest to investigate the difference between summary measures of receiver operating characteristic (ROC) curves for the diagnostic tests. Common ROC summary measures include the area under the ROC curve (AUC), partial area under the ROC curve (pAUC), and sensitivities at a certain specificity. Wieand and others (1989) introduce a general family of ROC summary statistics, hereafter referred to as the Δ-statistic, for comparing the accuracy of 2 diagnostic tests. Their statistics include all aforementioned common summary measures.
Due to both ethical and cost concerns, it is important that a comparative diagnostic trial is terminated, should one test be proved to be more accurate than the other. Mazumdar and Liu (2003) propose a parametric group sequential method to allow early termination of diagnostic trials. Tang and others (2008) discuss a general nonparametric sequential ROC method that can be implemented with popular group sequential design (GSD) methods such as the O'Brien–Fleming test, Pocock test, and a more flexible error spending approach (Lan and DeMets, 1983). Detailed discussion on GSDs is provided in Jennison and Turnbull (2000).
Planning a sequential diagnostic trial requires calculating maximum sample sizes for the diseased group and the nondiseased group to meet a prespecified power and to maintain a specified type I error rate. Often, parametric distributions are assumed for test outcomes from 2 groups of subjects under consideration, and power calculations are made using variances under this assumed model. In many situations, it is difficult to assume a proper parametric model, let alone to specify the values of the parameters in the model, especially when correlation parameters are involved due to repeated measurements on the same subjects. For instance, under a popular binormal model assumption, one needs to specify 2 separate bivariate normal distributions, each of which consists of 2 mean parameters, 2 variance parameters, and a correlation coefficient. As a result, a total of 10 parameters are needed to calculate the variance of the estimated difference of the AUCs or pAUCs or the sensitivities at some specificity between the 2 tests. Therefore, even if a correct parametric model is specified, when some of these 10 nuisance parameters are incorrectly specified, the calculated variance will differ from the true one. For instance, when the assumed correlation parameters are much smaller than the true ones, the calculated variance becomes incorrectly smaller, which subsequently results in smaller maximum sample sizes. A study based on these sizes will not achieve the desired power. There has been scant discussion on how to adaptively estimate sample sizes in comparative diagnostic trials. Wu and others (2008) propose a 2-stage method to recalculate the sample sizes by assuming bivariate binormal distributions for test outcomes. Their method is sensitive to distributional assumptions and, moreover, does not allow early stopping of the trial should statistically significant evidence be found against the null hypothesis.
In the clinical trial literature, several methods have been proposed to both recalculate sample sizes and allow early stopping during interim analyses. Denne and Jennison (2000) and Proschan and others (2006) introduce adaptive approaches to use internal pilot data to update sample sizes. Although the method in Denne and Jennison (2000) is applicable in small samples, calculation of critical boundary values is based on t-statistics and thus nontrivial. The adaptive approach in Proschan and others (2006) based on z-statistics is simpler to use and performs well for large sample sizes. They obtain a variance estimate from internal pilot data and then update the variance to recalculate sample sizes.
In this paper, we propose a nonparametric group sequential method by combining the sequential Δ-statistic with the adaptive method of Proschan and others (2006) and the error spending approach (Lan and DeMets, 1983) in comparative diagnostic trials. Good logistics for the adaptive method reside in diagnostic trials. For instance, biomarker results are quickly available once the markers are assayed. Patients’ true disease status is often in the record when they are accrued in the trial. These avoid delay in obtaining valid data for comparing biomarkers during interim analysis. However, test statistics involved in diagnostic biomarker trials are more complicated than many statistics in clinical trials. It is unclear whether adapting the aforementioned methods in diagnostic trials is able to maintain the desired error size and power. We will investigate theoretical and finite sample properties of the proposed method.
In Section 2, we give a brief introduction to GSD and adaptive sample size recalculation. We also briefly introduce the Δ-statistic and its asymptotic resemblance to a Brownian motion process. In Section 3, we develop an adaptive nonparametric method. Our method recalculates the sample sizes using internal pilot data to ensure sufficient power and also allows early termination during interim looks. The method is particularly useful when the same subject is diagnosed with 2 different tests, which is a common practice in diagnostic studies in order to minimize confounding effect due to different characteristics among subjects (Hanley and McNeil, 1982). Section 3.3 shows the large sample property of the proposed method. In Section 4, a method to determine the initial sample sizes used in the adaptive procedures is introduced and its drawback is illustrated. In Section 5, we present simulation results for the finite sample performance of our method with regard to the specified power and the nominal type I error rate for AUC and pAUC comparisons. Section 6 illustrates the application of our method in a cancer diagnostic trial. Discussion is in Section 7.
2. SOME BACKGROUND
In this section, we will briefly introduce GSD, adaptive sample size calculation, and the Δ-statistic.
2.1. Group sequential design
We consider a general group sequential sampling plan with maximum K analyses. An error spending function f(τ),τ ∈ [0,1], is chosen to determine the boundaries of the kth analysis, k = 1,…,K. To be an error spending function, f(τ) must be increasing and satisfy f(0) = 0 and f(1) = α. We consider 4 boundaries − ∞ < ak ≤ bk ≤ ck ≤ dk < ∞ at each of the K analyses, with at least one inequality before the Kth stage. A test statistic wk for comparing 2 diagnostic tests is calculated using all available data at the kth stage and is compared with stopping boundaries. If wk ≤ ak, bk < wk < ck, or wk ≥ dk, before the final stage, then the trial is stopped earlier without accruing more subjects. We would decide that diagnostic test 1 is inferior, approximately equivalent, or superior to test 2, respectively, depending on which boundary is reached. Otherwise, the study accrues sufficient subjects to proceed to analysis k + 1. The trial eventually stops at the Kth stage if not so before or at the K − 1th stage. In practice, the boundaries are usually set to ak = bk = ck = dk for one-sided tests and ak = bk < ck = dk for two-sided tests.
2.2. Adaptive sample size calculation
At the planning stage of a trial, maximum sample sizes are required to achieve the desired power to detect a meaningful alternative. Emerson and others (2007) provide a detailed description for calculating such sample sizes in clinical trials. Given a specific sequential design with K maximum number of interim analyses for a single sample, the maximal number NK of sampling units needed is given by NK = δαβ2V/Δa2, where Δa is the value under the alternative hypothesis to be detected with statistical power β in a level α hypothesis test, V is the variance due to a sampling unit, and δαβ is the design alternative in some standardized version of the test. Provided that the value of δαβ is specific to the chosen stopping rule in a GSD, the sample size is given in a two-sided test by
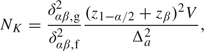 |
(2.1) |
where δαβ,g2/δαβ,f2 is the sample size ratio of a sequential design to the fixed sample design. The ratio, often referred to as the sample size inflation factor, is a fixed number given some specific design. Proschan (2004) introduces the concept of internal pilot data that often refers of available data in an ongoing trial. With the internal pilot data, the variance estimate 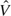 is calculated to update maximum sample size,
is calculated to update maximum sample size, 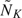 :
:
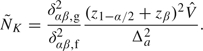 |
(2.2) |
Sometimes the updated maximum sample sizes may be lower than the original ones. If this happens, Proschan (2004) recommends setting the final sample sizes equal to max(, NK) = NK because a sufficient budget has been set aside for accruing NK subjects.
2.3. Δ-Statistic
In a prototypical comparative diagnostic trial, 2 diagnostic tests are conducted on M diseased subjects and N nondiseased subjects. We denote the measurements from test ℓ (ℓ = 1,2) on the ith diseased subject as Xℓi, where i = 1,…,M, and the measurements on the jth nondiseased subject as Yℓj, where j = 1,…,N. Define the joint cumulative survival functions (X1i,X2i) ∼ F(x1,x2) for the diseased population with marginal survival functions Xℓi ∼ Fℓ(x). Similarly, define (Y1j,Y2j) ∼ G(y1,y2) for the nondiseased population with marginal survival functions Yℓj ∼ Gℓ(y). Without loss of generality, we assume that measurements tend to be larger for the diseased than for the nondiseased. At each threshold c, a pair of sensitivity (Se) and specificity (Sp) is thus given by
The ROC curve for the ℓth test is a plot of Se versus 1 − Sp for the threshold c in ( − ∞, + ∞). 1 − Sp is also known as false-positive rate (FPR). The ROC curve for test ℓ is defined as ROCℓ(u) = Fℓ{Gℓ − 1(u)}, where u is in [0,1].
Wieand and others (1989) introduce a Δ-statistic based on the weighted AUC 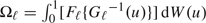 , with some probability measure W(u) for u ∈ (0,1). The difference between the 2 weighted areas becomes Δ = Ω1 − Ω2. Substituting the empirical survival functions
, with some probability measure W(u) for u ∈ (0,1). The difference between the 2 weighted areas becomes Δ = Ω1 − Ω2. Substituting the empirical survival functions 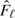 and
and 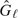 for Fℓ and Gℓ, respectively, the Δ-statistic is given by
for Fℓ and Gℓ, respectively, the Δ-statistic is given by
| (2.3) |
When W(u) = u for 0 < u < 1, 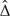 compares the AUCs of 2 tests; when W(u) = u for 0 < u1 ≤ u ≤ u2 ≤ 1, and 0 otherwise, compares pAUCs between FPRs u1 and u2; and when W(u) is a point mass at u0, compares sensitivities at a given level of specificity u0. Borrowing from results in Tang and others (2008), the asymptotic variance of takes the form
compares the AUCs of 2 tests; when W(u) = u for 0 < u1 ≤ u ≤ u2 ≤ 1, and 0 otherwise, compares pAUCs between FPRs u1 and u2; and when W(u) is a point mass at u0, compares sensitivities at a given level of specificity u0. Borrowing from results in Tang and others (2008), the asymptotic variance of takes the form
| (2.4) |
where vX and vY are
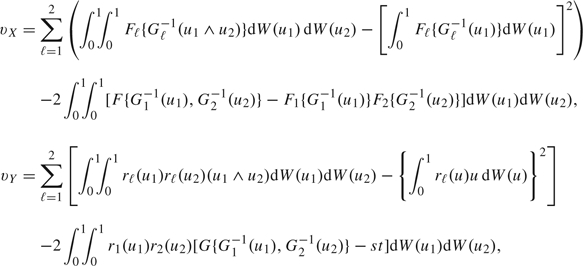 |
with rℓ(u) = Fℓ′{Gℓ − 1(u)}/Gℓ′{Gℓ − 1(u)}. Here, the variance, VΔ, contributed by a diseased subject is 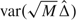 , given by
, given by
| (2.5) |
where λ = M/N. In the next section, we develop the sequential version of this statistic and combine it with the aforementioned GSD and adaptive sample size calculation.
3. ADAPTIVE SEQUENTIAL METHOD
The purpose of this section was to combine the concepts in Section 2 and introduce an adaptive method in sequential diagnostic trials. We define the following symbols for the kth stage of a GSD with a maximum K analyses, k = 1,…,K:
mk, nk are the numbers of available observations for diseased and nondiseased groups, respectively,
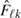 ,
, 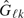 are respective empirical survival functions,
are respective empirical survival functions,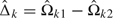 , where
, where 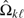 is the ℓth empirical weighted AUC (wAUC),
is the ℓth empirical weighted AUC (wAUC),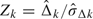 , where
, where 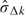 estimates σΔ from available data at kth look,
estimates σΔ from available data at kth look,Ik = 1/σΔk, statistical information, consequently, Ik ≤ Ik + 1,k = 1,…,K,
τk = Ik/IK.
Define 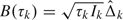 , which is an asymptotically unbiased estimator for
, which is an asymptotically unbiased estimator for 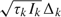 , with asymptotic variance var(B(τk)) = τk. B(τk) behaves asymptotically like a Brownian motion process with drift parameter
, with asymptotic variance var(B(τk)) = τk. B(τk) behaves asymptotically like a Brownian motion process with drift parameter 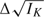 (Tang and others, 2008). Therefore, the sequential Δ-statistic has an independent increments structure and can be easily adapted to general GSDs.
(Tang and others, 2008). Therefore, the sequential Δ-statistic has an independent increments structure and can be easily adapted to general GSDs.
Pilot data nonparametrically estimate the variance of the Δ-statistic from (2.4) to determine the maximum sample sizes. In the absence of pilot data, estimation of the initial maximum sample sizes can be obtained in several ways. One way is to assume parametric forms for Fℓ, Gℓ, and the bivariate survival functions, F(x1,x2) and G(y1,y2), and substitute them into (2.4). Thus, given specified β and α, sample sizes can be calculated from (2.1). Alternative ways are described in Section 4.
3.1. Sample size recalculation
The dependence of sample sizes on prespecified values of correlation parameters can be reduced by updating the sample sizes at the interim analysis. Although the variance of the Δ-statistic is derived using asymptotic results, previous simulation studies in Tang and others (2008) demonstrate that the variance estimation has excellent finite sample performance for sample sizes as small as 50.
Before the trial is conducted, (4.1) in Section 4 can be used to obtain the initial maximum sample sizes, MK, for the diseased and, NK, for the nondiseased. As the trial is carried out sequentially, available data at the first interim analysis serve as internal pilot data for sample size reestimation. The estimates of vX and vY are given by (2.4) and are subsequently used to recalculate new maximum sample sizes, say 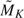 and for the 2 groups, respectively. Analogous to (2.2), given a hypothesized value Δa, , and are
and for the 2 groups, respectively. Analogous to (2.2), given a hypothesized value Δa, , and are
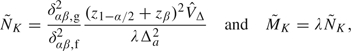 |
(3.1) |
where Δ estimates VΔ from available data at the first look. By following Proschan (2004), setting the final sample sizes equal to max(, NK), and max(,MK) guarantees that the original number of observed subjects N1 will not exceed 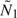 based on updated sample sizes.
based on updated sample sizes.
3.2. Stopping rule
Based on the new sample sizes, and , the fraction τ1 of the maximum information spent at the first analysis is given by τ1 = σΔ12/σΔK2, where σΔK2 is the maximum variance at the final stage of analysis. It follows from the variance expression in (2.4) that τ1 has a simplified form as
Since the same allocation ratio λ between the diseased and the nondiseased is maintained at each analysis throughout the trial, we can also obtain the fraction τ1 by using τ1 = N1/. The type I error rate spent at the first analysis is π1 = f(τ1), and the boundary values are determined by the inverse function of the standard normal distribution function, Φ. For instance, in the example of common two-sided tests of equal weighted AUCs, where − ∞ < a1 = b1 < c1 = d1 < ∞, we have − a1 = d1 = Φ − 1(1 − π1/2). We use the test outcomes on the first M1 diseased subjects and N1 nondiseased subjects to compute the empirical survival functions 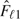 and
and 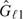 and the wAUC estimator
and the wAUC estimator 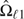 . The estimates are used to compare ROC curves using interim contrast 1, its standard error σΔ1, and the interim standardized statistic
. The estimates are used to compare ROC curves using interim contrast 1, its standard error σΔ1, and the interim standardized statistic 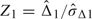 .
.
At the time of the kth analysis, we have diagnostic test data available on the first mk diseased subjects and the first nk nondiseased subjects, allowing us to calculate the standardized test statistic Zk. The type I error rate spent at the kth analysis is given by
where τk* = Mk/. The boundary values (ak,bk,ck,dk) at the kth analysis are then computed to maintain the overall type I error rate α. For example, in a two-sided hypothesis test with − ∞ < ak = bk < ck = dk < ∞, we would choose stopping boundaries to ensure
| (3.2) |
If Zk ≤ ak, or Zk ≥ dk, the study is stopped without accruing more subjects. Otherwise, more subjects are recruited for the next analysis. At the final look if ZK is within the boundaries, we will conclude no significant evidence against the null.
3.3. Large sample property
In this section, we discuss the reason that our adaptive procedure is able to control the specified type I error rate and maintain the desired power. According to the proof of Theorem 1 in Tang and others (2008), the convergence of empirical ROC curves, 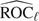 , ℓ = 1,2, is given by
, ℓ = 1,2, is given by
converges in distribution to
| (3.3) |
where 𝕌1,ℓ and 𝕌2,ℓ, ℓ = 1,2, are limiting Gaussian processes. Asymptotically, (3.3) is equivalent to
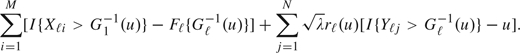 |
Thus, the Δ-statistic is asymptotically equivalent to the summation of
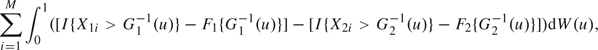 |
(3.4) |
and
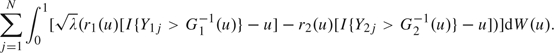 |
(3.5) |
Denote (3.4) as ∑i = 1MWi and (3.5) as ∑j = 1NVj. We see that i.i.d. random variables Wis are independent of i.i.d. random variables Vjs. Based on the result 11.1 in Proschan and others (2006), it follows that estimating the nuisance variance in (2.4) provides no information of the sequentially estimated Δ-statistic. This suggests that we can look at data during the interim analysis as though the recalculated sample sizes have been fixed before the trial. These updated sample sizes give sufficient power, and the error spending function in (3.2) controls type I error rate as the maximum error spent is restricted to be the specified level α.
4. INITIAL SAMPLE SIZE DETERMINATION AND THE EFFECT OF CORRELATION ON POWER
This section gives a brief overview of sample size calculation from hypothesized AUC values and demonstrates that misspecified parameter values might lead to huge loss of power. Various authors have proposed methods to obtain maximum sample sizes without having to tediously guess specific parameter values when comparing the AUCs of 2 tests. For uncorrelated test results, Hanley and McNeil (1982) propose a conservative approach to calculating sample sizes from negative exponential models when comparing 2 AUCs. The advantage of using negative exponential models is that the variance of the estimated difference of AUCs can be calculated solely from specified AUC values under the null and the alternative. Since the resulting variance is larger than that under normal or gamma distributions, subsequent sample sizes are thus larger under negative exponential models than the other 2 models. For correlated test results, a nice method for determining the initial sample sizes is provided in section 8.3.4 of Pepe (2003). Instead of specifying parameters for test results, her method only requires specifying 2 ROC curves and their correlation parameter. Pepe (2003) also suggests assuming the correlation between ROC curves is 0, which yields conservative sample sizes. Another way to obtain conservative sample sizes is introduced by Tang and others (2008) based on the assumption of negative exponential distributions. Maximum sample sizes, MK, for the diseased and, NK, for the nondiseased can be obtained for the O'Brien–Fleming test, the triangular test, and the Pocock test (see Jennison and Turnbull, 2000, for detailed description of these methods) by
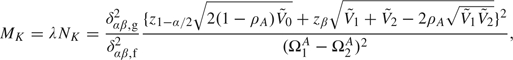 |
(4.1) |
where ρA denotes the correlation between 2 AUCs, and 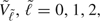 is derived from the hypothesized AUC values, Ω1A, Ω2A, and Ω0A = (Ω1A + Ω2A)/2, and is given by
is derived from the hypothesized AUC values, Ω1A, Ω2A, and Ω0A = (Ω1A + Ω2A)/2, and is given by
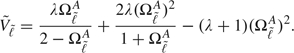 |
Equation (4.1) includes the method in Hanley and McNeil (1982) as a special case when test results are not correlated and no interim looks are conducted, that is when λ = 1, ρA = 0 and δαβ,g2/δαβ,f2 = 1. However, the sample size formula in (4.1) still requires a parameter measuring the correlation between 2 estimated AUCs. Specifying such a parameter may not be trivial due to unknown correlation structure between the AUCs, and misspecification of the parameter may substantially adversely affect the power to detect a meaningful difference in diagnostic accuracy. As recommended in Pepe (2003), conservative sample sizes may be obtained by assuming that ρA = 0.
Figure 1 illustrates the effect of the correlation coefficient ρA on actual powers. We used K = 3, M3 = N3 = 300, α = 0.05, and various pairs of AUCs. The actual power of the test from (4.1) was then plotted against various values of ρA in the range of [ − 1,1]. The figure shows that the actual power varies substantially as the value of ρA varies. Apparently, the study may be severely under (over)- powered if the true value of ρA is much less (larger) than the one specified. Both circumstances are undesirable because the former is unable to detect with adequate power the minimal relevant difference between AUCs and the latter unnecessarily increases the sample sizes actually needed for the study. Furthermore, if one is interested in comparing pAUCs or other ROC summary measures other than AUCs, there are no explicit variance formulas that only utilize the hypothesized values of the ROC summary measures. One has to specify the values of individual parameters in the underlying distributions. It is thus appropriate to use the proposed adaptive method to recalculate sample sizes using internal pilot data.
Fig. 1.

Actual power for testing equals AUCs when the correlation parameter ρA varies from – 1 to 1 in (4.1): solid line, ΩA1 = 0.70, ΩA2 = 0.75; dashed line, ΩA1 = 0.75, ΩA2 = 0.80; dotted line, ΩA1 = 0.70, ΩA2 = 0.80.
5. SIMULATION STUDIES
We simulated the performance of our adaptive group sequential design method (thereafter referred as AGSD) for comparing AUCs and pAUC, for K = 2,3 under 3 parametric models. Performance was evaluated in terms of actual type I error rate and actual power under simulated data. We used the error spending function by Kim and DeMets (1992) with f(τ) = min(ατ,α) to determine the boundaries at each analyses. We also applied the regular GSD by Tang and others (2008), which does not update the original sample sizes, and investigated its simulated powers when some parameter was misspecified.
The test outcomes from the 2 diagnostic tests were simulated, respectively, from 3 parametric models, the bivariate normal (Binorm), bivariate lognormal (Bilog), and bivariate exponential (Biexp). The null hypothesis of equal AUCs or equal pAUCs were set to be false under the alternative with a nominal power of 80%. The bivariate normal models have the forms of (X1,X2) ∼ N{(μ1,μ2),Σ} and (Y1,Y2) ∼ N{(0,0),Σ}, where diagonal elements of Σ are 1s and off-diagonal elements of Σ are correlation parameter ρs. Mean parameters μ1 and μ2 were computed according to specified AUC or pAUC values. We specified 3 pairs of AUCs, (0.70,0.75), (0.75,0.80), and (0.70,0.80) with ρ = 0.3 under two-sided alternative hypotheses. The bivariate lognormal models have the forms of exp(X1,X2) and exp(Y1,Y2) for the diseased and nondiseased subjects, respectively. The AUCs under the lognormal models are the same as under the binormal models since ROC curves are invariant to monotone transformations. For comparing pAUCs in the range of FPRs, (0,0.6), we specified 3 pairs of particular values, (0.30,0.35), (0.35,0.40), and (0.30,0.40), under two-sided alternative hypotheses. The bivariate lognormal models are given by exponential transformation on (X1,X2) and (Y1,Y2), respectively.
The bivariate exponential random variables were generated with a distribution in Gumbel (1960), which has the form of H(x,y) = H1(x)H2(y)[1 + 4ρ{1 − H1(x)}{1 − H2(y)}], where ρ ∈ [ − 0.25,0.25], and was set to be 0.10 for the simulation. Bivariate exponential data were generated with the marginal survival functions exp( − βℓ1x) and exp( − βℓ2y) for diseased and nondiseased subjects, respectively, where ℓ = 1,2, representing the type of tests. In the simulation, we set β11 = β21 = 1. β12 and β22 were chosen according to the AUC or pAUC values.
We simulated 1000 data sets for each pair of AUCs or pAUCs under the 3 aforementioned model assumptions. We conducted sequential analyses for K = 2 and K = 3. For each simulated data set, the numbers of available observations in each group at the first interim analysis were M1 = N1 = MK/K, where the initial sample sizes MK were determined by (4.1) with a misspecified correlation ρA = 0.85. The initial sample sizes range from 40 to 163 subjects per group for various AUCs or pAUCs. At the first look, we updated sample sizes and from available observations by substituting nonparametric variance estimates in (3.1). Comparing with initial sample sizes, average updated sample sizes increase from 30% to around 200%. We specified the error spending function to be f(τ) = max(τα,α) with α = 0.05. The critical values at the first look were then calculated by using 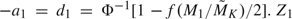 . Z1 was compared with these critical values. If Z1 ≤ a1, or Z1 ≥ d1, then we rejected the null hypothesis of equal AUCs in favor of the alternative. Otherwise, we simulated
. Z1 was compared with these critical values. If Z1 ≤ a1, or Z1 ≥ d1, then we rejected the null hypothesis of equal AUCs in favor of the alternative. Otherwise, we simulated 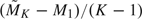 more observations to proceed to the second analysis. At the second look, Z2 was calculated and compared with critical values b2 and c2 obtained from (3.2). If Z2 ≤ a2, or Z2 ≥ d2, we stopped without simulating more observations. Otherwise, for K = 2, we would fail to reject the null. For K = 3, we would continue to simulate
more observations to proceed to the second analysis. At the second look, Z2 was calculated and compared with critical values b2 and c2 obtained from (3.2). If Z2 ≤ a2, or Z2 ≥ d2, we stopped without simulating more observations. Otherwise, for K = 2, we would fail to reject the null. For K = 3, we would continue to simulate 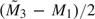 more observations and compare Z3 with critical values given by the error spending function. It was then decided whether to reject the null for the simulated data set. For K = 2 or 3, we calculated how many times out of 1000 that the null hypothesis was rejected during either the interim analyses or the final analysis and obtained the simulated powers. We also conducted simulation studies using GSD and calculated its simulated powers. Unlike the AGSD method, which updated sample sizes, the GSD method kept the original maximum sample sizes throughout the simulation. The results are presented in Table 1. It is clear that AGSD maintains the specified power, while GSD is underpowered due to misspecified sample sizes.
more observations and compare Z3 with critical values given by the error spending function. It was then decided whether to reject the null for the simulated data set. For K = 2 or 3, we calculated how many times out of 1000 that the null hypothesis was rejected during either the interim analyses or the final analysis and obtained the simulated powers. We also conducted simulation studies using GSD and calculated its simulated powers. Unlike the AGSD method, which updated sample sizes, the GSD method kept the original maximum sample sizes throughout the simulation. The results are presented in Table 1. It is clear that AGSD maintains the specified power, while GSD is underpowered due to misspecified sample sizes.
Table 1.
Simulated powers (in %) with the nominal level 80% in the GSDs
| Comparing AUCs | |||||||||
| Two looks (K = 2) |
Three looks (K = 3) |
||||||||
| AGSD |
GSD |
AGSD |
GSD |
||||||
| Ω1\Ω2 | 0.70 | 0.75 | 0.70 | 0.75 | 0.70 | 0.75 | 0.70 | 0.75 | |
| Binorm | 0.75 | 82.10 | NA | 55.40 | NA | 80.70 | NA | 53.90 | NA |
| 0.80 | 82.50 | 81.70 | 63.00 | 58.60 | 81.00 | 79.10 | 63.90 | 57.60 | |
| Bilog | 0.75 | 79.70 | NA | 55.80 | NA | 80.10 | NA | 57.90 | NA |
| 0.80 | 80.50 | 78.10 | 68.40 | 60.50 | 81.00 | 78.50 | 63.70 | 57.50 | |
| Biexp | 0.75 | 79.90 | NA | 74.10 | NA | 78.80 | NA | 72.60 | NA |
| 0.80 | 78.20 | 79.40 | 66.50 | 65.80 | 78.20 | 78.40 | 58.10 | 76.10 | |
| Comparing PAUCs | |||||||||
| Ω1\Ω2 | 0.30 | 0.35 | 0.30 | 0.35 | 0.30 | 0.35 | 0.30 | 0.35 | |
| Binorm | 0.35 | 79.30 | NA | 55.10 | NA | 77.10 | NA | 57.90 | NA |
| 0.40 | 75.60 | 79.40 | 68.60 | 60.20 | 81.50 | 77.50 | 52.00 | 59.20 | |
| Bilog | 0.35 | 80.00 | NA | 58.70 | NA | 78.60 | NA | 57.30 | NA |
| 0.40 | 82.50 | 77.90 | 67.40 | 61.90 | 79.30 | 78.30 | 67.60 | 61.80 | |
| Biexp | 0.35 | 81.50 | NA | 67.70 | NA | 79.70 | NA | 66.60 | NA |
| 0.40 | 82.10 | 80.60 | 48.20 | 62.40 | 76.20 | 79.00 | 46.90 | 71.50 | |
The rejection rate with 1000 realizations. The 95 prediction interval is (80.0% ± 2.48%). NA, not applicable.
We also evaluated the performance of our method on controlling the type I error rate. We again simulated 2000 test results for each pair of AUCs or pAUCs under the 3 aforementioned parametric distributions, the bivariate normal (Binorm), bivariate lognormal (Bilog), and bivariate exponential (Biexp). The null hypotheses of equal AUCs or equal pAUCs were set to be true. We used AUCs (0.70, 0.75, and 0.80) and pAUCs (0.30, 0.35, and 0.40). In the simulations, Δ1 under the alternative was set to be 0.05. The correlation coefficient ρ was set to be 0.3. We used a misspecified correlation ρA in (4.1) to compute initial maximum sample sizes. We used type I error rate 0.05 and power 0.8 in the simulation. The simulation settings were the same as those for power calculation, except that the null was true. Our method was applied to the simulated data sets, and rejection rates were calculated from the number of rejections out of 1000 data sets under each setting. The results are presented in Table 2. As can be seen from the table, our method is able to control the overall type I error rate as all rejection rates are close to the nominal level.
Table 2.
Simulated type I errors (in %) with the nominal level 5% in the GSDs
| Comparing AUCs |
||||||
| Two looks (K = 2) |
Three looks (K = 3) |
|||||
| Ω1(Ω2) |
Binorm |
Bilog |
Biexp |
Binorm |
Bilog |
Biexp |
| 0.70 | 5.05 | 5.80 | 5.30 | 5.10 | 5.60 | 5.00 |
| 0.75 | 5.30 | 5.15 | 5.30 | 5.55 | 5.45 | 4.70 |
| 0.80 | 5.55 | 4.65 | 5.40 | 5.10 | 4.80 | 5.45 |
| Comparing PAUCs | ||||||
| 0.30 | 5.40 | 5.20 | 4.80 | 5.15 | 5.30 | 5.00 |
| 0.35 | 5.60 | 5.35 | 5.40 | 5.65 | 5.75 | 5.55 |
| 0.40 | 4.55 | 5.95 | 5.25 | 5.50 | 5.70 | 5.95 |
The rejection rate with 2000 realizations. The 95% prediction interval is (5.00% ± 0.95%).
6. AN EXAMPLE
In this section, we illustrate the application of our method in a cancer diagnostic trial described in Lloyd (1998). The data were collected by taking measurements of a reference biomarker and 6 newly developed biomarkers on blood samples of 135 cancer patients and 218 noncancer patients. These markers are indexed from A to G. We redesigned part of the trial with the proposed method for a comparison between the reference marker A and biomarker E with 3 looks. Because of insufficient knowledge about the trial, we assume M3 = 135 and N3 = 218 were calculated to achieve a prespecified power for a contrast Δa = 0.1 between AUCs. At the first look, we accrued data on first 45 cancer patients and 73 noncancer patients and calculated the interim contrast 1 = 0.0045, Δ = 0.1526, 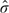 Δ1 = 0.0583, and the interim normalized statistic = 0.0770. The estimate Δ was used in (2.1) to obtain the updated sample sizes,
Δ1 = 0.0583, and the interim normalized statistic = 0.0770. The estimate Δ was used in (2.1) to obtain the updated sample sizes, 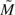 3 = 109 and
3 = 109 and 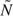 3 = 175. As these updated sizes are smaller than the originally planned ones, the original sample sizes were used for the study. Since Z1 fell within the boundary (a1 = − 2.3940,d1 = 2.3940), we continued with the second look at 45 more cancer patients and 73 more noncancer patients. We calculated interim contrast 2 = 0.0346, its standard error Δ2 = 0.0327, and
3 = 175. As these updated sizes are smaller than the originally planned ones, the original sample sizes were used for the study. Since Z1 fell within the boundary (a1 = − 2.3940,d1 = 2.3940), we continued with the second look at 45 more cancer patients and 73 more noncancer patients. We calculated interim contrast 2 = 0.0346, its standard error Δ2 = 0.0327, and 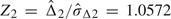 from accumulated 90 cancer patients and 146 noncancer patients. Now Z2 was still within the boundary (a2 = − 2.2937,c2 = 2.2937). When the trial was continued to the third look with accruing all patients, the statistic Z3 = 2.9782 was outside the boundary (a3 = − 2.1999,d3 = 2.1999). Thus, at the end of the trial, we came to a conclusion that 2 biomarkers have different diagnostic accuracy regarding their AUCs.
from accumulated 90 cancer patients and 146 noncancer patients. Now Z2 was still within the boundary (a2 = − 2.2937,c2 = 2.2937). When the trial was continued to the third look with accruing all patients, the statistic Z3 = 2.9782 was outside the boundary (a3 = − 2.1999,d3 = 2.1999). Thus, at the end of the trial, we came to a conclusion that 2 biomarkers have different diagnostic accuracy regarding their AUCs.
7. DISCUSSION
Sample size and power calculation for a study often involve certain parameters whose values need to be specified at the planning stage of the study. With a fixed sample size computed based on the specified values, the power of the study can be substantially affected if these values differ from the true values of the parameters. To complicate the issue, however, it can be quite challenging to verify the specifications at the the planning stage of the study. A remedy to this problem is to use internal pilot data to reexamine these assumptions and update the sample sizes accordingly so that the desired power can be maintained for the study.
Comparing the accuracy of 2 diagnostic tests, parametrically or nonparametrically, in terms of their ROC summary measures usually involves 2 bivariate distributions, 1 for the cases and 1 for controls. The power required for these studies depends on quite a few nuisance parameters whose values need to be specified. To relax such dependence, the present paper proposed an adaptive group sequential approach to designing such studies. In this approach, initial maximum sample sizes are computed using an approximate formula that only requires specification of the between-test correlation coefficient. At the first interim analysis, maximum sample sizes are updated using the Δ-statistic whose variance is estimated from the interim data. Stopping boundaries are determined using the updated sample size and a proper error spending function. Our simulation studies show that the proposed adaptive design maintains the desired power without scarifying the nominal type I error rate.
Diagnostic biomarker studies are of several different design types, including cohort studies with both definitive tests and biomarkers measured for all subjects in a cohort with definitive tests done before measuring biomarkers (Pepe and others, 2001), and a recently introduced nested case–control studies by Pepe and others (2008). Definitive tests are often invasive and costly. In some cohort studies, definitive test results are already in the record and assaying biomarkers is of low cost, the proposed design may be carried out with just 2 looks, with the first look updating sample sizes. Otherwise, we recommend more than 2 looks in the proposed sequential design to minimize the number of subjects who undergo definitive tests by possibly stopping the trial earlier.
The present paper only examines the issue of reestimating the variance of the Δ-statistic adjusting for sample size. Using the interim data, other assumptions at the planning stage of the study can also be reexamined. For example, we can utilize the interim data to evaluate whether the AUC difference to be detected is reasonable or whether the case-to-control allocation ratio need to be changed. All these evaluations may lead to reestimation of the sample sizes.
FUNDING
Intramural Research Program of the National Institute of Child Health and Human Development, National Institutes of Health to A.L.
Acknowledgments
The authors would like to thank an associate editor and 2 referees for their constructive comments and suggestions. The authors are thankful to Dr. Nell Sedransk for reading an earlier draft and providing valuable comments. The authors would also like to thank Prof. Chris Lloyd for generously sharing the cancer biomarker data. The opinions expressed are those of the author and not necessarily of the National Institutes of Health. Conflict of Interest: None declared.
References
- Denne JS, Jennison C. A group sequential t-test with updating of sample size. Biometrika. 2000;87:125–134. [Google Scholar]
- Emerson SS, Kittelson JM, Gillen DL. Frequentist evaluation of group sequential clinical trial designs. Statistics in Medicine. 2007;26:5047–5080. doi: 10.1002/sim.2901. [DOI] [PubMed] [Google Scholar]
- Gumbel EJ. Bivariate exponential distributions. Journal of the American Statistical Association. 1960;55:698–707. [Google Scholar]
- Hanley J, McNeil B. The meaning and use of the area under a receiver operating characteristic (ROC) curve. Radiology. 1982;143:29–36. doi: 10.1148/radiology.143.1.7063747. [DOI] [PubMed] [Google Scholar]
- Jennison C, Turnbull B. Group Sequential Methods with Applications to Clinical Trials. New York: Chapman and Hall; 2000. [Google Scholar]
- Kim K, DeMets D. Sample size determination for group sequential clinical trials with immediate response. Statistics in Medicine. 1992;11:1391–1399. doi: 10.1002/sim.4780111012. [DOI] [PubMed] [Google Scholar]
- Lan KKG, DeMets DL. Discrete sequential boundaries for clinical trials. Biometrika. 1983;70:659–663. [Google Scholar]
- Lloyd CJ. Using smoothed receiver operating characteristic curves to summarize and compare diagnostic systems. Journal of the American Statistical Association. 1998;93:1356–1364. [Google Scholar]
- Mazumdar M, Liu A. Group sequential design for diagnostic accuracy studies. Statistics in Medicine. 2003;22:727–739. doi: 10.1002/sim.1386. [DOI] [PubMed] [Google Scholar]
- Pepe MS. The Statistical Evaluation of Medical Tests for Classification and Prediction. Oxford, UK: Oxford University Press; 2003. [Google Scholar]
- Pepe MS, Etzioni R, Feng Z, Potter J, Thompson M, Thornquist M, Winget M, Yasui Y. Phases of biomarker development for early detection of cancer. Journal of the National Cancer Institute. 2001;93:1054–1061. doi: 10.1093/jnci/93.14.1054. [DOI] [PubMed] [Google Scholar]
- Pepe MS, Feng Z, Janes H, Bossuyt PM, Potter JD. Pivotal evaluation of the accuracy of a biomarker used for classification or prediction: standards for study design. Vol. 100. Journal of the National Cancer Institute; 2008. pp. 1432–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proschan MA. Two-stage sample size re-estimation based on nuisance parameter: a review. Journal of Biopharmaceutical Statistics. 2004;15:559–574. doi: 10.1081/BIP-200062852. [DOI] [PubMed] [Google Scholar]
- Proschan MA, Lan KKG, Wittes JT. Statistical Monitoring of Clinical Trials: A Unified Approach. New York: Springer; 2006. [Google Scholar]
- Tang L, Emerson SS, Zhou X-H. Nonparametric and semiparametric group sequential methods for comparing accuracy of diagnostic tests. Biometrics. 2008;64:1137–1145. doi: 10.1111/j.1541-0420.2008.01000.x. [DOI] [PubMed] [Google Scholar]
- Wieand S, Gail MH, James BR, James KL. A family of nonparametric statistics for comparing diagnostic markers with paired or unpaired data. Biometrika. 1989;76:585–592. [Google Scholar]
- Wu C, Liu A, Yu KF. An adaptive approach to designing comparative diagnostic accuracy studies. Journal of Biopharmaceutical Statistics. 2008;18:116–125. doi: 10.1080/10543400701668282. [DOI] [PubMed] [Google Scholar]