

Abstract
Recent studies have established an important role of Th17 in induction of autoimmune diseases. We have found that although IL-17 receptor A (IL-17RA) −/− mice were resistant to experimental autoimmune encephalomyelitis (EAE), a small number of them developed milder clinical signs of this autoimmune disease. In addition, blockade of TGFβ in IL-17RA−/− mice resulted in much more severe clinical signs of EAE and significantly increased parenchymal lymphocyte infiltration in the central nervous system (CNS). Furthermore, the number of autoreactive Th1 cells was greatly increased in the inflamed spinal cord of IL-17RA−/− mice. These data support a role of IL-17RA-independent mechanisms in causing autoimmunity and its regulation by TGFβ.
Introduction
Recent studies have established a critical role of Th17 cells in the initiation of EAE. Autoreactive Th17 cells readily transfer EAE in an adoptive transfer model (1). Mice developed attenuated EAE when injected with anti-IL-17 antibodies (1–3), whereas IL-17 −/− mice are resistant to EAE (4). Key promoters of Th17 lineages such as IL-6, IL-1, IL21 and IL-23 are all required for the development of EAE (5–12). TGFβ1, a factor that is important for Th17 differentiation in mice, is also critical for the initiation of EAE (13, 14). Factors inhibiting Th17 cells such as IL-25 and IL27 inhibit EAE (15) (16, 17). In addition, Act1, a molecule within the IL-17 signal transduction pathway is required for EAE (18, 19). Therefore, autoreactive Th17 cells are important for EAE.
Th17 cells make both IL-17 and IL-17F (1, 20). Recently, an IL-17 IL-17F heterodimer was found to be expressed in Th17 cells together with IL-17 and IL-17F homodimers (21, 22). IL-17 binds and signals through IL-17RA (23). IL-17F also functions through IL-17RA because a specific anti-IL17RA antibody neutralizes its function (24) and IL17RA −/− cells do not respond to IL17F in vitro (25). Therefore, studying IL-17RA−/− mice should allow us to assess the effect of the combined loss of function of IL-17 and IL-17F.
Despite the appreciation of a critical role of Th17 cells in causing CNS autoimmunity, the pathology of a Th17-dominated inflammation in experimental models differs from that in most clinical specimens (26). Therefore, it is important to study a model system when IL-17 function is deficient. In this study, we have found that the IL-17RA is important for the development of EAE and a small number of IL-17RA −/− mice developed very mild clinical signs of EAE. In addition, autoreactive Th1 responses were drastically dampened in IL-17RA −/− mice. Interestingly, systemically blockade of TGF β in IL-17RA −/− mice resulted in exacerbated EAE and disease severity correlated with increased Th1 responses. These data suggest IL-17RA independent EAE is likely mediated by Th1 immune response and is suppressed by TGFβ.
Materials and Methods
Mice
The IL-17RA−/− mice were obtained from Dr. J. J. Peschon (Amgen, Seattle, WA) as previously described (27) and bred in the University of Pittsburgh Animal Care Facility. The IL-17RA−/− mice were backcrossed to C57BL/6 mice for more than 10 generations. The IL-17RA−/− mice were generated by crossing IL-17RA+/− (het) with IL-17RA−/−. Littermate IL-17RA+/− and wild type C57BL/6 mice were used as controls. All mice were housed under specific pathogen-free conditions under a protocol approved by the Institutional Animal Care and Use Committee.
EAE induction
Mice were used for EAE induction by subcutaneous injection of 150 μg MOG35–55 (MEVGWYRSPFSRVVHLYRNGK) peptide in incomplete Freund’s adjuvant and 500 μg of heat-inactivated M. tuberculosis. 100 ng of pertussis toxin was injected intravenously on days 0 and 2. To block TGFβ systemically in vivo, mice were injected i.p. at 5 days and 9 days post-EAE elicitation with 400 μg of anti-TGFβ antibody (clone 1D11) or 400μg of rat IgG (ICN Biomedicals, Inc.). Mice were monitored for clinical signs of EAE, scored as follows: 0, normal; 1, flaccid tail; 2, hind limb weakness or abnormal gait with poor ability to right from supine; 3, partial hind limb paralysis; 4, both hind limbs paralyzed with or without forelimb paralysis and incontinence; 5, moribund state. The experiments were performed in accordance with the regulation of the Institutional Animal Care and Use Committee of University of Pittsburgh.
Central nervous system (CNS) cell isolation and antibody staining
To isolate cells from the CNS, mice were deeply anesthetized and perfused intracardially with cold RPMI 1640 medium. Brain and spinal cord cell suspensions were incubated with 1 mg/ml collagenase II (Sigma-Aldrich) at 37°C for 20 min, then resuspended in 30% Percoll (GE Healthcare), which was then layered over 70% Percoll. Mononuclear cells were separated from the interface after centrifugation at 600 g for 20 min and washed in PBS. The total number of cells derived from each animal was determined using a haemocytometer. CNS-derived inflammatory cells were incubated with anti-CD45, anti-CD11b and anti-CD4 antibodies for 15 min on ice in Hank’s buffer (containing 1% FCS). Flow cytometric analysis was performed using a FACS flow cytometer (Becton Dickinson).
Ex-vivo analysis of spleen cells, lymph node cells, and CNS cells by intracellular cytokine staining
Splenocytes, lymphocytes and CNS infiltrating leukocytes were cultured with or without MOG35–55 peptide at 50μg/ml for 18 h. Brefeldin A was added for the last 4 h at 10μg/ml. Cells were transferred to a V-bottom plate, stained with anti-CD4 in Hank’s buffer (containing 1% FCS), then fixed with 2% formaldehyde, which was followed by permeabilization with 0.5% saponin. The cells were subsequently stained with anti-IL-17 and anti-IFNγ antibody (eBioscience). Flow cytometric analysis was performed using a flow cytometer (Becton Dickinson).
Histological analysis
Mice were deeply anesthetized and perfused intracardially with cold PBS. Brains and spinal cords were removed and freshly frozen. For immunohistochemistry, sections (6μm) were fixed with ice cold acetone for 10 min and air-dried, then blocked in PBS with 3% BSA for 30 min at room temperature. The sections were then incubated with primary biotinylated anti-CD4 and anti-CD11b antibody (eBioscience) for 1h at room temperature, followed by being incubated with biotinylated rabbit anti-rat Ig and streptavidin-conjugated alkaline phosphatase for 1 h at room temperature. The sections were subsequently stained with Histomark RED Phosphatase Substrate Kit (KPL, Inc) and counterstained with hematoxylin.
Results
IL-17 receptor signaling is important for developing and sustaining EAE
Blockade of IL-17 has been shown to dampen EAE severity (1, 2). IL-17−/− and IL-17F−/− mice are all partially resistant to EAE (25). Since both IL-17 and IL-17f are expressed in Th17 cells, we decided to determine if blocking the function of both cytokines could totally eliminate EAE. We therefore induced EAE in IL-17RA −/− mice because both IL-17a and IL-17f utilize this receptor (20). IL-17RA −/− mice developed EAE with much lower incidence (about 25%) and reduced peak clinical scores compared to wild type mice (figure 1). In addition, the disease is delayed and shortened in IL-17RA −/− mice that succumbed to EAE (figure 1). Therefore, IL-17RA signaling is important for initiating, as well as sustaining EAE.
Figure 1.
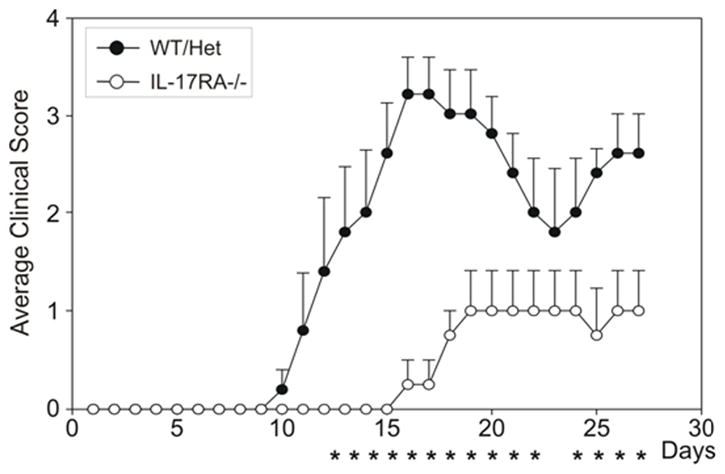
Ameliorated EAE in IL-17RA −/− mice. Wild type mice and IL-17RA −/− mice that had been backcrossed onto C57BL/6 background were immunized with MOG35–55 peptide to elicit EAE. Clinical signs and scores were recorded as described in Materials and Methods. Data are the average clinical scores from mice that have shown clinical signs of EAE. Data are representative of five independent experiments. *. P<0.05 determined by Mann-Whitney statistics.
IL-17 signaling is required for the generation of Th1 cells and accumulation of CD4+ T cells in spinal cords
In order to understand the cellular mechanisms for the severe reduction of EAE scores in IL-17RA −/− mice, we decided to determine the Th1 and Th17 responses against MOG35–55 during early phase of EAE. We found that IL-17RA is not required for the generation of Th17 cells because we observed similar numbers of MOG-specific Th17 cells in both wild type mice and IL-17RA −/− mice (figure 2a, c). Similarly, T cell proliferative responses to MOG35–55 were not significantly altered in IL-17RA−/− mice compared to controls (supplementary figure 1). Interestingly, the number of MOG-specific Th1 cells was severely reduced in IL-17RA −/− mice compared to those in wild type mice (figure 2b, d). TNFα is made by both Th1 cells and Th17 cells. We have found that MOG-specific TNFα-producing T cells were reduced in IL-17RA −/− mice compared to wild type controls (supplementary figure 2). This is consistent with a reduced Th1 response in IL-17RA −/− mice. Moreover, we have examined other cytokines such as IL-6, IL-4, and IL-10 and did not detect any difference between wild type and IL-17RA deficient mice (data not shown). Consistent with the finding in draining lymph nodes, a significantly reduced number of CD4+ T cells were harvested from the CNS of IL-17RA −/− mice compared to those from wild type mice (figure 2g). Although the frequencies of Th1 and Th17 cells were similar to controls the absolute number of infiltrating Th1 and Th17 cells diminished in the CNS of IL-17RA −/− mice compared to controls. Therefore, IL-17 signaling is required for the generation of MOG-specific Th1 cells and CNS inflammation.
Figure 2.

MOG-specific Th1 responses were reduced in IL-17RA −/− mice. Wild type mice (WT), IL-17RA +/− (Het), and IL-17RA −/− littermates were immunized with MOG35–55 peptide to elicit EAE. At ten days post-immunization, lymphocytes were isolated from draining lymph nodes, spleens, and CNS and stimulated with MOG35–55 peptide as described in Materials and Methods. Frequencies of CD4+ T cells that made IL-17 (A, C, E) or IFNγ (B, D, F) among total lymphocytes were determined by intracellular cytokine staining assay. The number of CD4+ T cells infiltrating CNS (G) was calculated based on flow cytometry and cell count. Six IL-17RA −/− and six wild type and IL-17RA +/− mice were used. Student T test was performed.
The accumulation of CD4+ T cells and macrophages in the CNS is a hallmark pathological change in EAE and correlates well with EAE disease scores at the early phase of the disease. We therefore decided to determine the nature of the pathological changes in the spinal cords of IL-17RA −/− mice that developed EAE. At the acute phase of EAE, massive macrophage infiltration was observed in the spinal cord white matter of wild type mice (figure 3a and b ). Extensive parenchymal infiltration could be observed during the peak EAE in wild type mice. In contrast, very few macrophages were observed in the spinal cord of IL-17RA −/− mice (figure 3c). Even in IL-17RA −/− mice that developed severe acute disease such as score 3, the number of infiltrating macrophages were reduced (figure 3d). In majority of the IL-17RA −/− mice, the infiltration was in meninges instead of parenchyma. The CD4+ T cells followed the same pattern as macrophages in the histochemical analyses (data not shown). Therefore, IL-17 receptor signaling is important for the accumulation of macrophages and CD4+ T cells in the inflamed spinal cord during EAE.
Figure 3.

Reduced CNS stroma infiltration by leukocytes in IL-17RA −/− mice. Wild type mice (WT), IL-17RA +/− (Het), and IL-17RA −/− littermates were immunized with MOG35–55 peptide to elicit EAE. At 15 days post-immunization, immuno-histochemistry was done as described in Materials and Methods to determine the degree of leukocyte infiltration using anti-CD11b antibody.
Blockade of TGFβ increased anti-MOG Th1 responses and resulted in severe EAE in IL-17RA −/− mice
The reduction of autoreactive Th1 cells might account for some of the reduced clinical signs of EAE in IL-17RA−/− mice. Because TGFβ is critical for suppressing the generation of Th1 cells, blockade of TGFβ should therefore reverse the effect of IL-17RA deficiency on the levels of Th1 cells and lead to more severe EAE. To test this idea, we systemically blocked TGFβ in IL-17RA −/− mice during the induction of EAE and then examined the frequencies of autoreactive Th1 and Th17 cells before the disease onset. This treatment resulted in increased MOG-specific Th1 responses in IL-17RA −/− mice to a level similar to that seen in wild type mice (figure 4b and d). In contrast, the number of autoreactive Th17 cells remained not significantly changed in either wild type or IL-17RA −/− mice after TGFβ treatment (figure 4a and c). Although there were not significant changes in levels of TGFβ mRNA in IL-17RA −/− mice compared to controls (supplementary figure 3), TGFβ blockade resulted in much more significant increases in Th1 cells in IL-17RA −/− mice than in controls. Similar to Th1 cells, TNFα-producing cells also increased after TGFβ blockade (supplementary figure 2). Therefore, these data suggest that, in vivo, TGFβ specifically suppresses autoreactive Th1 responses.
Figure 4.

Blockade of TGFβ recovered normal Th1 responses against the MOG peptide in IL-17RA −/− mice. Wild type mice (WT), IL-17RA +/− (Het), and IL-17RA −/− littermates were immunized with MOG35–55 peptide to elicit EAE. Anti-TGFβ antibodies were administered at day 5 and day 9. At ten days post-immunization, lymphocytes were isolated from draining lymph nodes and stimulated with MOG35–55 peptide as described in Materials and Methods. Frequencies of CD4+ T cells that made IL-17 (A) (C) or IFN-γ (B) (D) among total lymphocytes were determined by intracellular cytokine staining assay. More than seven mice were used in each experimental group. Student T test was performed.
Consistent with elevated levels of Th1 cells during the EAE induction, injection of anti-TGFβ antibodies systemically resulted in more severe clinical signs of EAE in IL-17RA −/− mice (figure 5 and supplementary figure 4). At the peak of disease, clinical scores were much reduced in IL-17RA−/− mice compared to wild type mice (figure 5). In contrast, clinical scores in IL-17RA−/− mice treated with anti-TGFβ were elevated to levels similar to wild type mice (figure 5). In addition, the incidence of EAE in IL-17RA −/− mice was increased from about 25% to about 45% after anti-TGFβ treatment. Therefore, TGFβ functionally suppresses IL-17R signaling-independent EAE, likely through antagonizing autoimmune MOG-specific Th1 responses.
Figure 5.

Blockade of TGFβ exacerbated EAE in IL-17RA −/− mice. Wild type mice (WT), IL-17RA +/− (Het), and IL-17RA −/− littermates were immunized with MOG35–55 peptide to elicit EAE. Anti-TGFβ antibodies were administered at day 5 and day 9. Clinical signs and scores were recorded as described in Materials and Methods. Data were the average clinical scores from mice that have shown clinical signs of EAE. Each point represents the mean of 5–8 animals and vertical lines are s.e.m. Where no error bar appears, the s.e.m = 0. * p < 0.05 compared to the IL-17RA−/− group. Statistical analysis was performed using one-way analysis of variance test (ANOVA) followed by Tukey’s post hoc t-test.
IL-17 signaling independent disease showed the typical EAE pathology
Blockade of IFNγ function was shown to induce exacerbated EAE. However the pathology of this EAE is quite different from classic EAE and chronic inflammation in multiple sclerosis. The non-classic EAE is dominated by neutrophils and Th17 cells (26, 28). To determine the nature of inflammation in IL-17RA−/− mice, we examined the histopathology of the spinal cords. In striking contrast to IL-17RA −/− mice not treated with anti-TGFβ antibodies, increased macrophage infiltration was observed in IL-17RA −/− mice treated with anti-TGFβ antibodies (figure 6a to d ). In addition, while infiltration in IL-17RA −/− mice was restricted to the meninges and subpial white matter, extensive parenchymal infiltration by macrophages (figure 6a to d ) and CD4+ T cells (data not shown) was observed in IL-17RA−/− mice. The number of infiltrating CD4+ T cells was also increased to a similar level as that in wild type mice (figure 6e). The percentage of neutrophils, a marker for acute inflammation and non-classic EAE (29), was examined to help determine the nature of inflammation. Consistent with a Th1-driven inflammation, the neutrophils were not different between wild type mice and IL-17RA−/− mice (figure 6f). Therefore, blockade of TGFβ resulted in severe inflammation mediated by macrophage and CD4+ T cells.
Figure 6.

Severe CNS inflammation in IL-17RA −/− mice treated with anti-TGFβ antibodies. Wild type mice (WT), IL-17RA +/− (Het), and IL-17RA −/− littermates were immunized with MOG35–55 peptide to elicit EAE. Anti-TGFβ antibodies were administered at day 5 and day 9. At 24 days post-immunization, immuno-histochemistry was done as described in Materials and Methods to determine the degree of leukocyte infiltration using anti-CD11b antibody. (A) IL-17RA +/− injected with control Ig, peak clinical score 4. (B) IL-17RA +/− injected with anti-TGFβ, peak clinical score 4. (C) IL-17RA −/− injected with control Ig, peak clinical score 1. (D) IL-17RA −/− injected with anti-TGFβ, peak clinical score 2. Wild type mice (WT), IL-17RA +/− (Het), and IL-17RA −/− littermates were immunized with MOG35–55 peptide to elicit EAE. Anti-TGFβ antibodies were administered at day 5 and day 9. Mice developed EAE were used around day 20 after the induction of EAE. The number of CD4+ T cells infiltrating CNS was calculated based on flow cytometry and cell count (E). CNS infiltrating cells were analyzed for Gr-1 and CD11b positive cells (F). CNS infiltrating lymphocytes were stimulated for 16 hours with MOG35–55 peptide, and then cells making IFNγ or IL-17 were analyzed by flow cytometry (G). The result was the average of data obtained from 4 independent experiments.
To further characterize the nature of inflammation in IL-17RA−/− mice, we examined the percentage of MOG-specific CD4+ T cells that make IFNγ and IL17 in wild type and IL17RA−/− mice that have been treated with anti-TGFβ to induce severe EAE. Compared with wild type mice, MOG-specific Th1 cells were drastically increased in IL-17RA−/− mice that developed severe EAE after anti-TGFβ treatment (figure 6g). In contrast, MOG-specific Th17 levels showed a modest but not statistically significant increase in IL-17RA −/− mice compared to controls. These data support the idea of Th1-mediated EAE in the absence of IL-17 signaling.
TGFβ inhibits both IFNγ production and the expression of CCR5 on Th1 cells
To examine the role of TGFβ in the generation of Th1 and Th17 cells, we cultured naive CD4+ T cells in the presence of TGFβ. As reported, TGFβ promoted Th17 generation (data not shown), but potently inhibited the production of IFNγ in Th1 cells (figure 7a). We further examined whether TGFβ displayed any inhibitory effect on CCR5 expression by quantitative PCR, which showed low levels of CCR5 expression after the TGFβ treatment (Figure 7b). Therefore, TGFβ inhibits both the generation of autoreactive Th1 cells and their migration to inflamed tissues.
Figure 7.

Effects of TGFβ on IFNγ production and CCR5 expression. CD4+ T cells were cultured under Th1 conditions in the absence or presence of TGFβ (3 ng/ml) for 4 days and then restimulated for 4 hours with plate-bound anti-mouse CD3 (10 μg/ml or 2 μg/ml) or alternatively with PMA (5 μg/m) and ionomycin (1 μg/ml). A) Intracellular staining using anti-IFNγ mAb. The lower panel shows specific inhibition of IFNγ production with TGFβ. B) qPCR assessment of CCR5 expression from Control (■) and TGFβ (□) cultures. RQ, relative quantity calculated by the Step one real time PCR software.
Discussion
In this study, we found the resistance to EAE due to the lack of the IL-17RA signaling can be reversed by blockade of TGFβ. Besides blocking the function of Th17 cells, IL-17RA deficiency also led to a reduced number of Th1 cells. Therefore, these results suggest that Th1 cells or other IL-17RA-independent mechanisms play a role in causing EAE clinical signs. Consistent with this idea, blockade of TGFβ significantly increased the autoimmune Th1 responses and the severity of EAE in IL-17RA −/− mice. Therefore, our results support the idea that the IL-17RA-independent autoimmune responses are required for EAE and these responses are suppressed by TGFβ.
Consistent with our study, previous studies also support our finding that IL-17 signaling is required for the generation of Th1 cells in vivo (30, 31). Our data supports the notion that the diminished Th1 responses very likely contributed to the resistance to EAE in IL-17RA −/− mice. The reason why the anti-MOG Th1 response is reduced in IL-17RA deficient mice remains elusive. It is known that IL-17 can induce IL-6 production by tissue stromal cells and endothelial cells (23). Because IL-6 is an very important cytokine inhibiting the development of regulatory T cells, it is possible that the lack of IL-17 signaling leads to a reduction of IL-6 in inflamed tissues or the peripheral lymphoid system, leading to diminished deterrence to the development of regulatory T cells. We have tested this possibility in vitro. We cultured CD4+CD25− T cells with TGFβ1 to induce differentiation of regulatory T cell and examined the effect of IL-17 on this process. We found that addition of IL-17 to the regulatory T cell culture drastically reduced the number of foxp3+ T cells (supplementary figure 5). Blockade of IL-6 rendered IL-17 ineffective (supplementary figure 5). We have further found that the effect of IL-17 is dependent on the presence of non-T cells because IL-17 did not suppress the generation of regulatory T cells when T cells alone were stimulated with plate bound anti-CD3 and anti-CD28 (data not shown). Therefore, IL-17 antagnizes the function of TGFβ by inducing IL-6 in splenocytes or tissue stromal cells. These data suggest that one possible mechanism that IL-17 promotes Th1 responses is through inhibiting the generation of regulatory T cells.
Recent studies revealed an interesting role of TGFβ in the generation of Th17 cells at least in the murine system (13, 14, 32–34). This function of TGFβ affects only naive CD4+ T cells but not effector/memory T cells. In fact, long term culture human Th17 cells in TGFβ diminished the production of IL-17 in these cells (35). Therefore, although TGFβ is important in establishing Th17 cells, it is generally suppressing the function of effector T cells. This notion is consistent with a well established role of TGFβ in suppressing autoimmunity (36). Administration of TGFβ was shown to suppress the development of EAE (37). In addition, blockade of TGFβ by antibodies were shown to worsen EAE (37). Our study further established in vivo that TGFβ drastically suppresses Th1 responses and their CNS infiltration in the absence of IL-17 signaling. Blockade of TGFβ significantly augmented Th1 responses and modestly increased Th17 responses. Collectively, these studies support a role of TGFβ in suppressing the effector function of autoreactive T cells, particularly Th1 cells.
It is worth pointing out that although TGF-β blockade increased EAE incidence from 20% to 45% in IL-17RA −/− mice, this was still two-fold less than that of wild type mice. It remains to be elucidated why half of the mice were absolutely dependent on IL-17 for their development of EAE. This result suggests that there are additional functions of IL-17 that are critical for CNS inflammation. These functions may include their abilities to induce chemokine production and to cause disruption of tight junctions of blood-brain barrier endothelial cells (2, 38, 39).
Although many recent studies strongly support a critical role of IL-17 in initiating as well as sustaining EAE (1–4), Th17 response alone was unable to sustain EAE (13). Therefore, IL-17RA-independent mechanisms, particularly pathogenic Th1 cells, likely contribute to causing EAE, a notion supported by our results. The role of Th1 in causing EAE is, however, controversial. There is a wealth of evidence supporting the role of Th1 cells in EAE. IFNγ exacerbated Multiple Sclerosis in human clinical trials (40). In addition, in vitro generated Th1 cell lines can transfer diseases to naive animals (41). Moreover, T-bet, a master regulator for Th1 lineage and a factor dispensable for the generation of Th17 cells, is critically required for EAE (42, 43). Contradictory to these results, IFNγ, the hallmark cytokine for Th1 cells, is not required for the generation of EAE (44). The lack of IFNγ function even exacerbated EAE (44). Although the CNS autoimmune disease in IFNγ −/− mice was quite severe, histophathological studies revealed that it belongs to a non-classic form of EAE (26). The non-classic from of EAE is defined by brain-targeted neuronal deficit (28), pathological changes dominated by neutrophil infiltration (28, 29), and recently, Th17 prevalence (26). Classic EAE is mediated by dominant Th1 responses with the help of Th17 and the immune attack focuses on the spinal cord (26). Our data support the idea that classic EAE can develop, although with reduced efficiency, in the total absence of IL-17, and possibly also IL-17F, signaling. Moreover, out data showed that TGFβ negatively regulates classic EAE in this setting.
One of the many functions of Th1 responses is likely to promote a switch from acute inflammation dominated by neutrophils to a chronic inflammation dominated by T cells and macrophages. The fact that the lack of IFNγ led to a neutrophils dominated CNS inflammation supports this idea (29). Therefore, Th1 and Th17 lineage cooperate to initiate EAE. How these two lineages play out in later diseases remain to be elucidated.
The cytokines produced by Th1 cells such as TNF-α and IFN-γ are likely involved in IL-17RA-independent CNS inflammation. However, cytokines produced by Th17 cells such as IL-22 (45) and IL21 (10, 11) can also contribute to the pathology in IL-17RA −/− mice. The contribution of individual cytokines will be determined in the future.
Supplementary Material
Acknowledgments
The authors thank Drs. Penny Morel and Lisa Borghesi for critical reading and Ms. Elizabeth Chen, Mr. David Stratton, and Ms. Christen Shiber for helping preparing the manuscript.
Footnotes
This work is partly supported by National Institutes of Health (NIH) grant no. AI063496 to B. Lu. B. Lu is supported by NIH grant no. 1 K01 AR048854. Y. Zhao was partly supported by NIH grant no. P30-AR47372.
References
- 1.Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, Wang Y, Hood L, Zhu Z, Tian Q, Dong C. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–1141. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Uyttenhove C, Van Snick J. Development of an anti-IL-17A auto-vaccine that prevents experimental auto-immune encephalomyelitis. Eur J Immunol. 2006;36:2868–2874. doi: 10.1002/eji.200636662. [DOI] [PubMed] [Google Scholar]
- 4.Komiyama Y, Nakae S, Matsuki T, Nambu A, Ishigame H, Kakuta S, Sudo K, Iwakura Y. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J Immunol. 2006;177:566–573. doi: 10.4049/jimmunol.177.1.566. [DOI] [PubMed] [Google Scholar]
- 5.Infante-Duarte C, Horton HF, Byrne MC, Kamradt T. Microbial lipopeptides induce the production of IL-17 in Th cells. J Immunol. 2000;165:6107–6115. doi: 10.4049/jimmunol.165.11.6107. [DOI] [PubMed] [Google Scholar]
- 6.Samoilova EB, Horton JL, Hilliard B, Liu TS, Chen Y. IL-6-deficient mice are resistant to experimental autoimmune encephalomyelitis: roles of IL-6 in the activation and differentiation of autoreactive T cells. J Immunol. 1998;161:6480–6486. [PubMed] [Google Scholar]
- 7.Eugster HP, Frei K, Kopf M, Lassmann H, Fontana A. IL-6-deficient mice resist myelin oligodendrocyte glycoprotein-induced autoimmune encephalomyelitis. Eur J Immunol. 1998;28:2178–2187. doi: 10.1002/(SICI)1521-4141(199807)28:07<2178::AID-IMMU2178>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 8.Okuda Y, Sakoda S, Bernard CC, Fujimura H, Saeki Y, Kishimoto T, Yanagihara T. IL-6-deficient mice are resistant to the induction of experimental autoimmune encephalomyelitis provoked by myelin oligodendrocyte glycoprotein. Int Immunol. 1998;10:703–708. doi: 10.1093/intimm/10.5.703. [DOI] [PubMed] [Google Scholar]
- 9.Sutton C, Brereton C, Keogh B, Mills KH, Lavelle EC. A crucial role for interleukin (IL)-1 in the induction of IL-17-producing T cells that mediate autoimmune encephalomyelitis. J Exp Med. 2006;203:1685–1691. doi: 10.1084/jem.20060285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Korn T, Bettelli E, Gao W, Awasthi A, Jager A, Strom TB, Oukka M, Kuchroo VK. IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature. 2007;448:484–487. doi: 10.1038/nature05970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nurieva R, Yang XO, Martinez G, Zhang Y, Panopoulos AD, Ma L, Schluns K, Tian Q, Watowich SS, Jetten AM, Dong C. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature. 2007;448:480–483. doi: 10.1038/nature05969. [DOI] [PubMed] [Google Scholar]
- 12.Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, Lucian L, To W, Kwan S, Churakova T, Zurawski S, Wiekowski M, Lira SA, Gorman D, Kastelein RA, Sedgwick JD. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–748. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- 13.Veldhoen M, Hocking RJ, Flavell RA, Stockinger B. Signals mediated by transforming growth factor-beta initiate autoimmune encephalomyelitis, but chronic inflammation is needed to sustain disease. Nat Immunol. 2006;7:1151–1156. doi: 10.1038/ni1391. [DOI] [PubMed] [Google Scholar]
- 14.Li MO, Wan YY, Flavell RA. T cell-produced transforming growth factor-beta1 controls T cell tolerance and regulates Th1- and Th17-cell differentiation. Immunity. 2007;26:579–591. doi: 10.1016/j.immuni.2007.03.014. [DOI] [PubMed] [Google Scholar]
- 15.Kleinschek MA, Owyang AM, Joyce-Shaikh B, Langrish CL, Chen Y, Gorman DM, Blumenschein WM, McClanahan T, Brombacher F, Hurst SD, Kastelein RA, Cua DJ. IL-25 regulates Th17 function in autoimmune inflammation. J Exp Med. 2007;204:161–170. doi: 10.1084/jem.20061738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Batten M, Li J, Yi S, Kljavin NM, Danilenko DM, Lucas S, Lee J, de Sauvage FJ, Ghilardi N. Interleukin 27 limits autoimmune encephalomyelitis by suppressing the development of interleukin 17-producing T cells. Nat Immunol. 2006;7:929–936. doi: 10.1038/ni1375. [DOI] [PubMed] [Google Scholar]
- 17.Stumhofer JS, Laurence A, Wilson EH, Huang E, Tato CM, Johnson LM, Villarino AV, Huang Q, Yoshimura A, Sehy D, Saris CJ, O’Shea JJ, Hennighausen L, Ernst M, Hunter CA. Interleukin 27 negatively regulates the development of interleukin 17-producing T helper cells during chronic inflammation of the central nervous system. Nat Immunol. 2006;7:937–945. doi: 10.1038/ni1376. [DOI] [PubMed] [Google Scholar]
- 18.Chang SH, Park H, Dong C. Act1 adaptor protein is an immediate and essential signaling component of interleukin-17 receptor. J Biol Chem. 2006;281:35603–35607. doi: 10.1074/jbc.C600256200. [DOI] [PubMed] [Google Scholar]
- 19.Qian Y, Liu C, Hartupee J, Altuntas CZ, Gulen MF, Jane-Wit D, Xiao J, Lu Y, Giltiay N, Liu J, Kordula T, Zhang QW, Vallance B, Swaidani S, Aronica M, Tuohy VK, Hamilton T, Li X. The adaptor Act1 is required for interleukin 17-dependent signaling associated with autoimmune and inflammatory disease. Nat Immunol. 2007;8:247–256. doi: 10.1038/ni1439. [DOI] [PubMed] [Google Scholar]
- 20.Kolls JK, Linden A. Interleukin-17 family members and inflammation. Immunity. 2004;21:467–476. doi: 10.1016/j.immuni.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 21.Wright JF, Guo Y, Quazi A, Luxenberg DP, Bennett F, Ross JF, Qiu Y, Whitters MJ, Tomkinson KN, Dunussi-Joannopoulos K, Carreno BM, Collins M, Wolfman NM. Identification of an interleukin 17F/17A heterodimer in activated human CD4+ T cells. J Biol Chem. 2007;282:13447–13455. doi: 10.1074/jbc.M700499200. [DOI] [PubMed] [Google Scholar]
- 22.Chang SH, Dong C. A novel heterodimeric cytokine consisting of IL-17 and IL-17F regulates inflammatory responses. Cell Res. 2007;17:435–440. doi: 10.1038/cr.2007.35. [DOI] [PubMed] [Google Scholar]
- 23.Yao Z, Fanslow WC, Seldin MF, Rousseau AM, Painter SL, Comeau MR, Cohen JI, Spriggs MK. Herpesvirus Saimiri encodes a new cytokine, IL-17, which binds to a novel cytokine receptor. Immunity. 1995;3:811–821. doi: 10.1016/1074-7613(95)90070-5. [DOI] [PubMed] [Google Scholar]
- 24.McAllister F, Henry A, Kreindler JL, Dubin PJ, Ulrich L, Steele C, Finder JD, Pilewski JM, Carreno BM, Goldman SJ, Pirhonen J, Kolls JK. Role of IL-17A, IL-17F, and the IL-17 receptor in regulating growth-related oncogene-alpha and granulocyte colony-stimulating factor in bronchial epithelium: implications for airway inflammation in cystic fibrosis. J Immunol. 2005;175:404–412. doi: 10.4049/jimmunol.175.1.404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang XO, Chang SH, Park H, Nurieva R, Shah B, Acero L, Wang YH, Schluns KS, Broaddus RR, Zhu Z, Dong C. Regulation of inflammatory responses by IL-17F. J Exp Med. 2008;14:14. doi: 10.1084/jem.20071978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stromnes IM, Cerretti LM, Liggitt D, Harris RA, Goverman JM. Differential regulation of central nervous system autoimmunity by T(H)1 and T(H)17 cells. Nat Med. 2008;14:337–342. doi: 10.1038/nm1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ye P, Rodriguez FH, Kanaly S, Stocking KL, Schurr J, Schwarzenberger P, Oliver P, Huang W, Zhang P, Zhang J, Shellito JE, Bagby GJ, Nelson S, Charrier K, Peschon JJ, Kolls JK. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J Exp Med. 2001;194:519–527. doi: 10.1084/jem.194.4.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wensky AK, Furtado GC, Marcondes MC, Chen S, Manfra D, Lira SA, Zagzag D, Lafaille JJ. IFN-gamma determines distinct clinical outcomes in autoimmune encephalomyelitis. J Immunol. 2005;174:1416–1423. doi: 10.4049/jimmunol.174.3.1416. [DOI] [PubMed] [Google Scholar]
- 29.Tran EH, Prince EN, Owens T. IFN-gamma shapes immune invasion of the central nervous system via regulation of chemokines. J Immunol. 2000;164:2759–2768. doi: 10.4049/jimmunol.164.5.2759. [DOI] [PubMed] [Google Scholar]
- 30.Nakae S, Komiyama Y, Nambu A, Sudo K, Iwase M, Homma I, Sekikawa K, Asano M, Iwakura Y. Antigen-specific T cell sensitization is impaired in IL-17-deficient mice, causing suppression of allergic cellular and humoral responses. Immunity. 2002;17:375–387. doi: 10.1016/s1074-7613(02)00391-6. [DOI] [PubMed] [Google Scholar]
- 31.Lubberts E, Schwarzenberger P, Huang W, Schurr JR, Peschon JJ, van den Berg WB, Kolls JK. Requirement of IL-17 receptor signaling in radiation-resistant cells in the joint for full progression of destructive synovitis. J Immunol. 2005;175:3360–3368. doi: 10.4049/jimmunol.175.5.3360. [DOI] [PubMed] [Google Scholar]
- 32.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 33.Mangan PR, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO, Hatton RD, Wahl SM, Schoeb TR, Weaver CT. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 34.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 35.Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F. Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat Immunol. 2007;8:942–949. doi: 10.1038/ni1496. [DOI] [PubMed] [Google Scholar]
- 36.Gorelik L, Flavell RA. Transforming growth factor-beta in T-cell biology. Nat Rev Immunol. 2002;2:46–53. doi: 10.1038/nri704. [DOI] [PubMed] [Google Scholar]
- 37.Santambrogio L, Hochwald GM, Saxena B, Leu CH, Martz JE, Carlino JA, Ruddle NH, Palladino MA, Gold LI, Thorbecke GJ. Studies on the mechanisms by which transforming growth factor-beta (TGF-beta) protects against allergic encephalomyelitis. Antagonism between TGF-beta and tumor necrosis factor. J Immunol. 1993;151:1116–1127. [PubMed] [Google Scholar]
- 38.Shen F, Hu Z, Goswami J, Gaffen SL. Identification of common transcriptional regulatory elements in interleukin-17 target genes. J Biol Chem. 2006;281:24138–24148. doi: 10.1074/jbc.M604597200. [DOI] [PubMed] [Google Scholar]
- 39.Kebir H, Kreymborg K, Ifergan I, Dodelet-Devillers A, Cayrol R, Bernard M, Giuliani F, Arbour N, Becher B, Prat A. Human T(H)17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat Med. 2007;13:1173–1175. doi: 10.1038/nm1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Panitch HS, Bever CT., Jr Clinical trials of interferons in multiple sclerosis. What have we learned? J Neuroimmunol. 1993;46:155–164. doi: 10.1016/0165-5728(93)90245-t. [DOI] [PubMed] [Google Scholar]
- 41.Lees JR, Archambault AS, Russell JH. T-cell trafficking competence is required for CNS invasion. J Neuroimmunol. 2006;177:1–10. doi: 10.1016/j.jneuroim.2006.05.024. [DOI] [PubMed] [Google Scholar]
- 42.Bettelli E, Sullivan B, Szabo SJ, Sobel RA, Glimcher LH, Kuchroo VK. Loss of T-bet, but not STAT1, prevents the development of experimental autoimmune encephalomyelitis. J Exp Med. 2004;200:79–87. doi: 10.1084/jem.20031819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lovett-Racke AE, Rocchini AE, Choy J, Northrop SC, Hussain RZ, Ratts RB, Sikder D, Racke MK. Silencing T-bet defines a critical role in the differentiation of autoreactive T lymphocytes. Immunity. 2004;21:719–731. doi: 10.1016/j.immuni.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 44.Chu CQ, Wittmer S, Dalton DK, Haynes L, Swain SL. Failure to suppress the expansion of the activated CD4 T cell population in interferon gamma-deficient mice leads to exacerbation of experimental autoimmune encephalomyelitis. J Exp Med. 2000;192:123–128. doi: 10.1084/jem.192.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kreymborg K, Etzensperger R, Dumoutier L, Haak S, Rebollo A, Buch T, Heppner FL, Renauld JC, Becher B. IL-22 is expressed by Th17 cells in an IL-23-dependent fashion, but not required for the development of autoimmune encephalomyelitis. J Immunol. 2007;179:8098–8104. doi: 10.4049/jimmunol.179.12.8098. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.