

Abstract
Stem cells are an emerging strategy for treatment of myocardial infarction, limited however to postinjury intervention. Preventive stem cell-based therapy to augment stress tolerance has yet to be considered for lifelong protection. Here, pluripotent stem cells were microsurgically introduced at the blastocyst stage of murine embryo development to ensure stochastic integration and sustained organ contribution. Engineered chimera displayed excess in body weight due to increased fat deposits, but were otherwise devoid of obesity-related morbidity. Remarkably, and in sharp contrast to susceptible nonchimeric offspring, chimera was resistant to myocardial infarction induced by permanent coronary occlusion. Infarcted nonchimeric adult hearts demonstrated progressive deterioration in ejection fraction, while age-matched 12–14-months-old chimera recovered from equivalent ischemic insult to regain within one-month preocclusion contractile performance. Electrical remodeling and ventricular enlargement with fibrosis, prominent in failing nonchimera, were averted in the chimeric cohort characterized by an increased stem cell load in adipose tissue and upregulated markers of biogenesis Ki67, c-Kit, and stem cell antigen-1 in the myocardium. Favorable outcome in infarcted chimera translated into an overall benefit in workload capacity and survival. Thus, prenatal stem cell transplant yields a cardioprotective phenotype in adulthood, expanding cell-based indications beyond traditional postinjury applications to include pre-emptive therapy.
Keywords: Blastocyst, Chimera, Embryonic stem cells, Heart disease, Prenatal therapy, Prevention
Introduction
Stem cell technology provides an emerging platform for a regenerative approach with the goal of halting or reversing progression of myocardial injury [1–3]. Endogenous cell-based, self-repair mechanisms have been increasingly recognized as a natural process for tissue homeostasis [4]. Fundamental to heart muscle rejuvenation is cardiomyocyte renewal through recruitment of progenitor pools within the body [5, 6], as recently validated by a demonstrated continuous turnover during lifespan [7]. Notably, stem cell contribution to postnatal heart has been documented by the self/nonself chimerism characteristic of patients following allogeneic transplantation [8–10]. Furthermore, increase in innate stem cell load within failing hearts contributes to a regenerative response and involves ongoing derivation of cardiomyocytes from circulating or resident progenitors [11, 12]. However, in the context of large-scale destruction following ischemic injury, the regenerative response beyond the homeostatic need is limited in its ability to salvage a deteriorating myocardium [13]. Delivery of exogenous embryonic stem cells following myocardial infarction offers a strategy to boost cell-mediated repair postinjury [14–17], but does not confer immunity against ischemic insult.
In this regard, a pre-emptive intervention that could engineer stress tolerance for the infarcted myocardium would offer the earliest possible prevention to tissue at risk avoiding the anticipated progression towards heart failure. Indeed, cell-based engineering of a resistant phenotype would provide an alternative to traditional postinjury approaches. To this end, embryonic stem cells delivered as early as in perinatal stages of embryo development have been recently demonstrated to achieve intrauterine rescue of an otherwise fatal congenital cardiac defect [18]. However, the delayed impact of prenatal stem cell transplantation on adult disease tolerance is so far unknown.
Here, embryonic stem cell delivery into a developing host embryo was tested to determine whether bioengineered chimeric tissue influences adult tolerance to ischemic heart disease associated with debilitating organ failure. Chimera was generated through microinjection of embryonic stem cells into preimplantation blastocysts, and cardiac stress tolerance evaluated in middle-age infarcted offspring. In contrast to vulnerable nonchimera counterparts, engineered chimera sustained contractile function with no structural remodeling following ischemic insult. The beneficial effect of chimeric tissue in the adult heart under imposed stress provides the initial evidence of preventive regenerative medicine in the setting of myocardial infarction implemented through prophylactic intervention.
Materials and Methods
Chimera Production
Blastocysts collected at 3.5 days postcoitum, from C57BL/6J mice, were injected with ROSA26 embryonic stem cells, constitutively expressing the lacZ reporter transgene. Injected blastocysts were reimplanted into pseudo-pregnant surrogates to allow for full-term development and production of adult chimera [19].
Physiological Parameters
Body size parameters, waist-height index, and body mass index, were calculated as follows: waist-height index = maximum waist circumference (cm)/height (length from the nose to the toe, m); body mass index = body weight (kg)/height2 (m2). Following 1-week of acclimatization, blood pressure was measured by automated tail-cuff recording (Columbus Instruments, Columbus, OH, http://www.colinst.com) in awake and restrained mice [20]. Systolic and diastolic values were derived from 10 sequential recordings. Whole blood glucose levels were measured by tail blood sampling (OneTouch Ultra, Lifescan, Milpitas, CA, http://www.lifescan.com) at nonfasting and fasting (16-hour-long overnight fasting) conditions. In glucose tolerance test, blood glucose was measured before and after (15, 30, 60, 120 minutes) intraperitoneal injection of glucose (2 g/kg) in fasting mice [21].
Adipose Tissue
Adipose tissue was removed from subcutaneous inguinal and abdominal regions under 5% isoflurane terminal anesthesia. Minced tissue was digested using 0.1% collagenase containing 1 mg/ml of collagenase type I (Worthington Biochemical, Lakewood, NJ, http://www.worthington-biochem.com) in phosphate buffered saline at 37°C for 90 minutes. Digested cells were filtered through 100-μm nylon cell strainers (BD Biosciences, San Jose, CA, http://www.bdbiosciences.com) and centrifuged at 400 g for 5 minutes. The resulting pellet was treated with red blood cell lysis buffer for 5 minutes at room temperature. Cultured cells were suspended in high-glucose Dulbecco’s modified Eagle’s medium with 20% fetal bovine serum, 100 U/ml penicillin, 100 g/ml streptomycin, and 2 mM L-glutamine (Invitrogen, Carlsbad, CA, http://www.invitrogen.com). After 24 hours, washes removed nonadherent cells. After 72 hours, adherent cells were dislodged and quantified using a hemocytometer.
Cardiac Function and Structure
In anesthetized mice (1.5% isoflurane), left ventricular function and structure were quantified by trans-thoracic echocardiography (15L8 transducer, Sequoia 512, Acuson, Mountain View, CA, http://www.medical.siemens.com). Left ventricular fractional shortening (% FS) was calculated as [(LVDd − LVDs)/LVDd] × 100, where LVDd is left ventricular end-diastolic dimension (mm) and LVDs, left ventricular end-systolic dimension (mm). Velocity of left ventricular circumferential shortening was calculated as [(LVDd − LVDs)/LVDd]/Et, where Et is ejection time determined from the actual pulsed-wave Doppler tracings [22]. Cardiac output was determined by the product of the aortic root cross-section area, the velocity time integral measured from the peak trans-aortic Doppler tracing, and heart rate. Ejection fraction (%) was calculated as [(LVVd − LVVs)/LVVd] × 100, where LVVd is left ventricular end-diastolic volume (μL) and LVVs, left ventricular end-systolic volume (μl). Left ventricular weight (mg) was derived as [(LVDd + IVST + PWT)3 − LVDd3 × 1.055, where IVST is interventricular septum thickness (mm), and PWT, posterior wall thickness (mm). In vivo hemodynamics in mechanically ventilated mice was measured by a 1.4-Fr pressure-volume catheter (SPR-839, MPVS-400, Millar Instruments, Houston, TX, http://www.millarinstruments.com) following carotid artery cannulation and advancement across the aortic valve for left ventricular catheterization [22].
Myocardial Infarction
Male, 12–14-months-old, nonchimeric and chimeric mice underwent myocardial infarction through a minimally invasive procedure with permanent ligation of the left anterior coronary artery and closure of the thorax within 20 minutes. Following intubation and mechanical ventilation (Mini Vent 845, Hugo Sacks Electronik, March-Hugstetten, http://www.hugo-sachs.de), the left coronary artery was ligated with a 9-0 suture. Upon complete coronary occlusion, left ventricular wall color change was immediately visualized. Onset of electrical abnormality consistent with an acute injury pattern was monitored using 4-limb lead electrocardiography (MP150, Biopac, Goleta, CA, http://www.biopac.com/). The extent of acute cardiac insult was determined by echocardiography at mid-ventricular transverse section to quantify the percentage of akinetic left ventricular perimeter, a validated surrogate of myocardial infarction size [23, 24]. Infarcted mice were followed for up to 1-month with multiple cardiac and systemic end-points prospectively assessed.
Histology
Autopsy tissue was prepared for cryosections, and cut at 7 μm. Cell proliferation was evaluated with the Ki67 antibody (1:500 clone MIB-1, Diagnostic Biosystems, Pleasanton, CA, http://www.dbiosys.com) in combination with cardiac sarcomeric α-actinin (1:500, Sigma-Aldrich, St. Louis, MO, http://www.sigmaaldrich.com). Myocardial scar formation and interstitial fibrosis was documented using phosphotungstic acid hematoxylin and Masson’s trichrome-stain, and progenitor populations visualized with c-Kit antibody (1:200, R&D systems, Minneapolis, MN) and stem cell antigen-1 (Sca-1) antibody (1:150, BD Biosciences), and quantified in random field selected sections by direct visualization (Zeiss LSM image browser, Thornwood, NY, http://www.zeiss.com) or automated analysis (MetaMorph, Visitron Universal Imaging, Union City, CA, http://www.molecular-devices.com) as described [25, 26]. Nuclei were stained with 4′,6′-diamidino-2-phenylindole (Molecular Probes). Images were taken using laser confocal microscopy (Zeiss LSM 510 Axiovert).
Systemic Effects
Lung congestion, exercise tolerance, and survivorship were collectively used to assess the systemic effects of myocardial infarction, and the impact of prenatal therapy. The treadmill exercise protocol (Columbus Instruments) included a stepwise increase in incline and velocity at 3-minute intervals. Workload (J), a composite parameter incorporating time, speed and incline, was calculated as the sum of kinetic (Ek = [m × v2]/2) and potential (Ep = m × g × v × t × sinθ) energy, where m represents animal mass, v treadmill velocity, g acceleration due to gravity, t elapsed time at a protocol level, and θ angle of incline [27].
Statistical Analysis
Data are presented as means ± SEM. Student’s t test or nonparametric test were used to evaluate significance (JMP 6, SAS, Cary, NC, http://www.sas.com). A p value < .05 was predetermined.
Results
Chimeric Offspring Produced from Mosaic Embryos Integrate Exogenous Stem Cell-Derived Blastomeres
Embryonic stem cells, previously established to successfully treat ischemic heart disease postinjury [28, 29], were here transplanted into a recipient embryo at the earliest stage of development to generate stochastic integration throughout differentiating lineages. Using microinjection, ~15–20 embryonic stem cells, obtained from the ROSA26 line constitutively expressing the LacZ reporter, were placed into the blastocoele of a C57BL/6J embryo collected at 3.5 days post coitum to create viable mosaic blastocysts (Fig. 1A). Surgical transfer of derived blastocysts into the uterus of pseudopregnant females yielded full-term chimera offspring (Fig. 1A). Within adult offspring, tissues derived from ROSA26 embryonic stem cells were traced by the β-galactosidase transgene and demonstrated expression patterns determined by lineage specification of progenitor pools at early stages of development. The chimeric heart contained ROSA26 stem cell-derived tissue consistent with the embryonic patterning of ventricles, which created normal development patterns characterized by wedge-shaped patches of labeled progeny (Fig. 1B). On average, chimeric incorporation ranged between 5% and 20% of labeled stem cell-derived tissue, consistent throughout postnatal life as verified by comparing tail clipping and postmortem analysis. In this way, transplantation of pluripotent stem cells into preimplantation blastocysts generated a mosaic embryo with sustained engraftment of stem cell-derived tissue in adult chimeric offspring.
Figure 1.

Production of sustained chimerism. (A): Chimera was generated through injection of ROSA26 embryonic stem cells into native blastocysts, followed by reimplantation of the engineered chimeric blastocyst into pseudo-pregnant surrogates. Tissues containing ROSA26 progeny, expressing the lacZ reporter transgene, were systemically detected in adult chimera by β-galactosidase expression (black staining), including cardiomyocytes contributing to the right and left heart ventricle (B).
Adult Chimera are Obese with No Evidence of Risk Factors for Cardiac Disease
Although born with no apparent abnormalities, chimera grew significantly larger in body size compared with nonchimeric controls (Fig. 2A–2E). With age, the difference in body weights progressively increased between chimeric and nonchimeric cohorts. While at 1-month, nonchimera and chimera was similar at 16.2 ± 1.4 g (n = 10) and 16.6 ± 1.2 g (n = 3) respectively, following 2 months of age cohorts displayed divergent weight growth dynamics with 22.3 ± 2.5 g for non-chimera (n = 20) in contrast to 33.3 ± 3.3 g for chimera (n = 3, p < .05). By 6 months, nonchimera was at 30.4 ± 0.7 g (n = 16), while chimera reached 38.0 ± 0.5 g (n = 7, p < .05; Fig. 2C). Ultimately, by 8 months, chimera was 1.5 times heavier than nonchimera, at 45.6 ± 2.3 g (n = 7) and 30.7 ± 1.2 g (n = 24; p < .05), respectively. Noticeably, chimera demonstrated central obesity characterized by a pear-shaped body habitus (Fig. 2A), associated with dominant depots of subcutaneous and visceral fat (Fig. 2B). A significant increase in the waist-height index characterized chimera, i.e., 58.5 ± 3.9 cm/m for nonchimera (n = 10) versus 70.3 ± 6.6 cm/m for chimera (n = 4, p < .05; Fig. 2D). Accordingly, the body-mass index was at 1.79 ± 0.05 kg/m2 for nonchimera (n = 10), but was significantly increased to 2.16 ± 0.06 kg/m2 in chimera (n = 4, p < .05; Fig. 2E). Despite the abundance of adipose tissue in chimera, blood pressure was similar between cohorts (Fig. 2F). Diastolic pressure was 63 ± 3 mmHg and 62 ± 2 mmHg, and systolic pressure was 129 ± 4 mmHg and 135 ± 3 mmHg in nonchimera (n = 5) and chimera (n = 7), respectively (p > .05; Fig. 2F). Furthermore, fasting blood glucose was 97 ± 6 mg/dl (n = 10) in nonchimera and 117 ± 10 mg/dl in chimera (n = 7; p > .05), and nonfasting blood glucose was also similar at 164 ± 10 mg/dl in nonchimera (n = 11) and 164 ± 14 mg/dl in chimera (n = 8, p > .05). Glucose tolerance test was indistinguishable between nonchimera and chimera (Fig. 2G). Thus, despite divergent body weights, the overall baseline cardiac disease risk profiles were similar between cohorts.
Figure 2.

Adult chimera baseline phenotype. Compared with age-matched nonchimeric controls, chimeric mice (A) demonstrated central obesity with significant fat deposits (B) and increased body weight (C), waist circumference (D), and body mass index (E). Obesity in chimera was not associated with hypertension (F) or glucose intolerance (G). †p < .05 versus nonchimeric controls. Abbreviations: s, subcutaneous fat; v, visceral fat.
Chimera Demonstrate Cardioprotective Phenotype Following Myocardial Infarction
Throughout the one year follow-up, nonchimera, and chimera demonstrated similar cardiac performance on serial prospective echocardiography (Fig. 3A) with essentially equal values recorded between cohorts for %FS (Fig. 3B) as well as cardiac output (Fig. 3C). On catheterization, end-diastolic and peak systolic left ventricular pressures were also indistinguishable in nonchimera versus chimera (Fig. 3D). Similarly, no significant difference was detected in cardiac structure, including left ventricular dimensions (Fig. 3E), left ventricular weights (Fig. 3F) or coronary artery anatomy (Fig. 3G–3H). In addition, there was no significant difference between cohorts in electrocardiographic profiles, including heart rates in the awake state (nonchimera: 716.4 ± 21.4 beats/minute, n =5; chimera: 701.3 ± 9.9 beats/minute, n = 7). Upon permanent ligation of the left anterior coronary artery in sex-matched middle-aged nonchimera and chimera cohorts, electrocardiography demonstrated ST elevation within 20 minutes following definitive surgical ligation (Fig. 4A). This acute injury pattern was concordant with a significant decrease in cardiac contractile function monitored by echocardiography (Fig. 4B–4D). In both cohorts, akinetic regions developed at the anteroseptal wall of the left ventricle in response to complete coronary occlusion (Fig. 4C). The initial area of akinesis, a noninvasive surrogate marker of infarct size [23, 24], was indistinguishable (p > 0.05) between the two cohorts at 20 minutes postligation with 27.8% ± 5.1% in control (n = 4) and 28.4% ± 8.6% in chimera (n = 3), and persisted in both groups for 2 days (p > .05; Fig. 4B). Ejection fraction equally decreased from 68.1% ± 4.9% to 43.2% ± 7.5% in nonchimera (n = 7, p < .05) and from 69.7% ± 5.1% to 44.0% ± 5.6% in chimera (n = 6) within 20 minutes postligation (p < .05; Fig. 4D). Likewise, %FS significantly dropped from 50.9% ± 1.4% at pre to 28.6% ± 4.7% at 20 minutes postligation in nonchimera (n = 5, p < .05) and from 54.1% ± 2.5% at pre to 35.6% ± 3.8% at 20 minutes postligation in chimera (n = 6, p < .05), with no difference between cohorts in this acute phase postligation (p = .30, Fig. 4C). Despite comparable acute ischemic insult, middle-age nonchimeric hearts deteriorated with further decline in systolic function, in contrast to chimera that displayed progressive recovery of cardiac performance (Fig. 4C). Ejection fraction—an indicator of global contractile performance that incorporates the hyperkinetic response of noninfarcted areas in infarcted hearts, significantly recovered within 1–2 weeks of follow-up postligation in chimera compared to nonchimera cohort (Fig. 4D), albeit parameters reflecting regional activity demonstrated a delayed recovery (not illustrated). While infarcted nonchimeric hearts remained akinetic in the anterior wall throughout the 1-month follow-up (38.1% ± 7.7%, n = 4), infarcted chimera recovered to essentially normokinesis within this timeframe (3.4% ± 1.4%, n = 5, p < .05; Fig. 4E). Indeed, at this time-point, ejection fraction demonstrated sustained divergence between cohorts, with nonchimera at 27.8% ± 5.2% (n = 5) versus chimera at 63.5% ± 4.6% (n = 4, p < .05; Fig. 4D). Moreover, %FS was significantly different with 21.4% ± 3.3% for nonchimera (n = 5) and 47.4% ± 4.9% for chimera (n = 4) at 1-month postligation (p < .05; Fig. 4C and 4F). The velocity of circumferential shortening was at 4.39 ± 0.67 circumference/s for nonchimera (n = 5), but improved to 9.67 ± 0.25 circumference/s for chimera (n = 4, p < .05; Fig. 4G). The QT interval, representing ventricular depolarization and repolarization, was prolonged in nonchimera hearts, but not in chimera (Fig. 4H). On average, the change in QT interval duration (ΔQT) before and 1-month after ischemic insult was 25.1 ± 5.4 ms for nonchimera (n = 5) and 3.2 ± 6.3 ms for chimera (n = 4, p < .05; Fig. 4H), indicating that the electrical instability of nonchimera was averted in chimera. Thus, in contrast to the vulnerable nonchimera, matched chimera demonstrated a capacity to recover cardiac contractile and electrical function postinfarction.
Figure 3.

Equivalent baseline cardiac function and structure in nonchimera and chimera. (A): At 6-month and 1-year follow-up, M-mode echo-cardiography demonstrated similar systolic (solid yellow arrow) and diastolic (dotted yellow arrow) wall motion in nonchimera controls (left) and chimera (right), with equivalent fractional shortening (B) and cardiac output (C). (D): Left ventricular catheterization measured indistinguishable end-diastolic and peak systolic pressures. Left ventricular dimensions (E) and left ventricular weights (F) were not significantly different in non-chimera controls versus chimera at 6 months and 1-year follow-up. Representative cross-sections of anterior and lateral walls indicate similar anatomy and distribution of coronary arteries and respective branches in control (G) and chimeric (H) hearts. Abbreviations: BW, body weight; LV, left ventricular/ventricle; RV, right ventricle.
Figure 4.

Disparity in cardiac recovery between chimera versus nonchimera following permanent coronary ligation. At 20 minutes postligation, controls and chimera displayed equivalent ST elevation injury pattern on electrocardiography ([A], arrow), comparable anterior wall akiness (B), and similar drop in ejection fraction on prospective serial echocardiography (M-mode in [C] and B-mode in [D]). The initial area of akinesis, a marker of regional dysfunction (B), and ejection fraction decline, reflecting global contractile compromise (D), persisted in both groups over the following two days. Prospectively, however, nonchimera showed progressive decline in contractile function (C, D, F, G), while chimera recovered global (D) and regional (E, F, G) contractility and electrical performance (H). Scale bar indicates 2 mm in (C). *p < .05 versus preinfarction (Pre-MI), with 95% confidence interval indicated by dotted lines; †p < .05 versus infarcted nonchimera. Abbreviations: Ao, aorta; cir/s, circumferences/s; LA, left atrium; LVDd (dotted line in [C]), left ventricular end-diastolic dimension; LVDs (solid line in [C]), left ventricular end-systolic dimension; LVVd, left ventricular end-diastolic volume; LVVs, left ventricular end-systolic volume; MI, myocardial infarction; post, posterior wall of the left ventricle; sep, interventricular septal wall; Vcf, velocity of circumferential shortening.
Preserved Cardiac Structure in Chimera Postinfarction
Contractile and electrical deterioration prominent in infarcted nonchimeric hearts was accompanied by maladaptive structural remodeling, a sensitive prognostic indicator of poor outcome (Fig. 5). Within 1-month following myocardial infarction, the left ventricles of nonchimeric hearts were significantly dilated with prominent wall thinning, and advanced cardiomegaly was visualized by echocardiography (Fig. 5A–5C). The left ventricle was estimated in nonchimeric hearts at 107.3 ± 2.0 mg preinfarction, but increased to 149.1 ± 24.8 mg postinfarction (n = 6, p < .05; Fig. 5D). Autopsy revealed advanced ischemic myopathy with broad aneurysm formation (Fig. 5E), extensive myocardial scarring (Fig. 5F) and significant pulmonary congestion (Fig. 5H), typical of malignant progression towards overt heart failure. In contrast, infarcted chimera avoided cardiomegaly, with left ventricles at 100.0 ± 4.5 mg pre and 120.7 ± 13.0 mg postinfarction (n = 5, Fig. 5D). Moreover, neither aneurysmal wall thinning (Fig. 5E) nor prominent scar formation (Fig. 5G) was observed, and retrograde pulmonary congestion was absent (Fig. 5H). Thus, pre-emptive transplantation of stem cells at the embryonic stage halted development of cardiomyopathy traits in the adult chimera, averting progression of disease following severe ischemic insult.
Figure 5.

Chimera hearts are resistant to maladaptive remodeling. One month after MI, structural remodeling in nonchimeric hearts underlying ischemic cardiomyopathy included wall thinning of the infarcted area (interventricular septal wall thickness, IVST, [A]), left ventricular dilatation (B, C), and increase of left ventricular weight (D). Autopsy demonstrated upon gross inspection that infarcted nonchimeric hearts developed cardiomegaly with aneurysm formation (E), validated by wall thinning within the scared (phosphotungstic acid hematoxylin, red) left ventricle upon histological examination (F), with resulting lung congestion (H). In contrast, infarcted chimera hearts demonstrated structural parameters similar to preinfarcted values (A–D), without gross dilation (E), wall thinning or prominent scarring (G), and absence of pulmonary edema (H). Scale bar indicates 3 mm in (E). *p < .05 versus preinfarction (Pre), with 95% confidence interval indicated by dotted lines; †p < .05 versus infarcted nonchimera. Abbreviations: A, aneurysm; ant, anterior-wall of the left ventricle; LA, left atrium; late, lateral-wall of the left ventricle; LV, left ventricle; LVDd, left ventricular end-diastolic dimension; LVVd, left ventricular end-diastolic volume; MI, myocardial infaction; post, posterior-wall of the left ventricle; MI, myocardial infarction; RA, right atrium, RCA, right coronary artery; late, lateral-wall of the left ventricle; RV, right ventricle; s, ligation suture of the left anterior descending artery; sep, septal-wall of the left ventricle.
Ischemic Injury Activates Tissue Repair and Reduces Pathologic Fibrosis in Chimera
Nonchimera responded to infarction with moderate upregulation of cells positive for Ki67 compared to chimeric infarcted hearts characterized by a pronounced increase in expression of this lineage-independent cell proliferation marker (Fig. 6A–6B). On average, Ki67 expression increased in nonchimera from 2.4% ± 0.4% pre to 4.9% ± 0.6% postinfarction (n = 5, p < .05), in contrast to a prominent surge observed in chimera from 2.1% ± 0.3% pre to 18.5% ± 2.4% postinfarction (n = 5, p < .05). Such induction corresponded to an up-regulation of mesenchymal stem cells within visceral and subcutaneous adipose deposits, with chimera containing two to three orders of magnitude higher load over nonchimera following ischemic challenge. Within infarcted chimeric hearts, the stem cell factor receptor, c-Kit, demonstrated only a moderate progenitor increase in nonchimera, i.e., from 0.5% ± 0.4% and 2.3% ± 0.5% (n = 5), but robustly expanded in chimera from 1.3% ± 0.5% to 8.5% ± 1.0% (n = 5), pre and postinfarction, respectively (p < .05; Fig. 6C–6D). This distinctive response pattern was validated with demonstration of the cardiac stem cell marker Sca-1 significantly increased in chimera from 1.6% ± 0.2% pre to 8.0% ± 2.3% postinfarction (n = 5; p < .05), whereas the biomarker expression profile in nonchimera essentially did not change from 0.7% ± 0.7% to 1.3% ± 0.7% preinfarction and postinfarction (n = 5; p > .05), respectively (Fig. 6D–6E). Activation of the tissue repair process inversely correlated with fibrosis, prominent in failing postinfarcted nonchimeric hearts but marginal in chimera counterparts (Fig. 6F). On average, trichrome staining of fibrotic tissue involved 9.7% ± 0.7% of nonchimeric (n = 14), in contrast to 2.7% ± 0.4% of chimeric (n = 18, p < .05) myocardium (Fig. 6F). Thus, a pronounced cellular response with antifibrotic outcome differentiated the vulnerable nonchimera from the resistant chimeric phenotype in the setting of ischemic insult.
Figure 6.

Upregulated markers of cardiac biogenesis and blunted fibrosis in infarcted chimera. Under the stress of permanent coronary ligation (MI), infarcted chimera displayed increased mitotic rate in the myocardium (Ki67, [A]; arrow heads in [B]), associated with a marked upregulation of progenitor (c-Kit, [C]) and cardiac stem cell (stem cell antigen-1, Sca-1, [E]) biomarkers, compared with nonchimera counterparts. (D): Arrow heads indicate c-kit and Sca-1 double positive cells. *p < .05 versus preinfarction (Pre), with 95% confidence interval indicated by dotted lines; †p < .05 versus infarcted nonchimera. (F): Myopathic nonchimeric hearts developed advanced fibrosis blunted in chimera (blue in Masson’s trichrome-staining). Abbreviations: Actinin, sarcomeric α-actinin; DAPI, 4′,6′-diamidino-2-phenylindole; MI, myocardial infarction; Sca-1, stem cell antigen-1.
Chimera Achieve Increased Workload and Overall Survivorship Postinfarction
Beyond enhanced cardioprotection, chimera displayed a favorable disease course postinfarction while nonchimera developed malignant heart failure with reduced exercise capacity after 1-month of coronary occlusion (Fig. 7A–7B). Ultimately, progression of disease halved the infarcted nonchimeric cohort (Fig. 7C). In contrast, infarcted chimera halted the development of debilitating systemic symptoms, with superior exercise workload capacity and stress test performance, as well as increased survival compared to nonchimeric counterparts at 1-month of follow-up. Thus, stem cell transplant into preimplantation embryo yielded a myocardial infarction-tolerant adult phenotype mitigating the clinically relevant endpoints of the heart failure syndrome.
Figure 7.
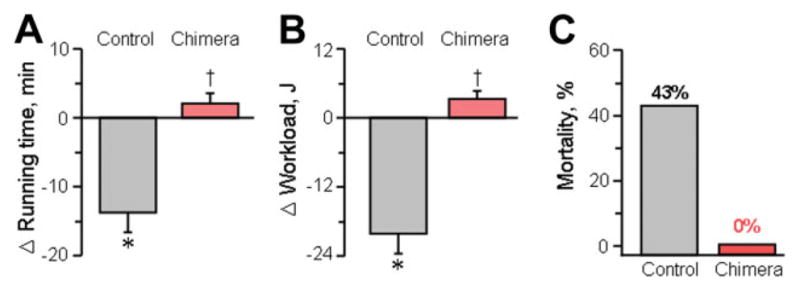
Workload and survival benefit in chimera. Chimera displayed favorable systemic outcomes including a higher exercise capacity ([A] running time; [B] workload) evaluated by treadmill and no mortality (C) within 1-month postinfarction. *p < .05 versus pre-infarction; †p < .05 versus nonchimera.
Discussion
This study provides first evidence that transplantation of embryonic stem cells into preimplantation blastocyst yields a myocardial infarction-resistant adult phenotype. Integration of pluripotent donor cells into early-stage host embryos secured viable mosaicism. Cardiac chimerism patterned by stochastic contribution within a healthy prenatal environment ensured longitudinal sustainability throughout adult age. Exposure to severe ischemic insult unmasked a cardioprotective responsiveness that distinguished resilient chimera from vulnerable nonchimera. Contractile insufficiency, electrical remodeling and cardiomegaly, all prominent in the infarcted nonchimera, precipitated poor exercise capacity and ultimately high mortality. In contrast, prearranged chimera tolerated permanent coronary occlusion, and demonstrated preserved cardiac function and structure with marked benefit in physical performance and survival on postinfarction follow-up. Pre-emptive stem cell-based intervention in utero thus provides a strategy to engineer tolerance, and prevent incidence of life-threatening organ failure in the adult. In this way, prenatal transplantation of embryonic stem cells expands the scope of traditional retrospective therapy to the previously unexplored prospective protection.
Although conventional postinjury treatment and resulting in situ engraftment of embryonic stem cells within the damaged myocardium produces benefit through a pleiotropic regenerative response with the goal of restoring tissue loss [30–33], it was unknown whether anticipatory chimerism could prevent ischemia-induced heart muscle dysfunction. Spontaneous cardiac chimerism originating from progenitor pools contributes to the repopulation of <1% of the cells each year in adult tissue [7, 34, 35]. The limited expansion of heart parenchyma, while maintaining functional homeostasis, remains largely insufficient to achieve an adequate reparative response in cases of severe injury [35]. Despite the insufficient rate of heart rejuvenation in the setting of acute disease, such as myocardial infarction, the continuous turnover accumulated over a lifespan contributes up to 50% of the tissue mass and validates the essential role of acquired chimerism [7]. The ability to trace the site of progenitor cell origin within the adult chimeric tissue, recognizing the limitations of current biomarkers, is confounded by the ongoing self-renewal process that simultaneously draws from multiple undetermined sources [7].
Herein, by coercing chimerism through blastocyst transplantation, a developing mosaic embryo enabled a privileged early and robust (5–20%) contribution to cardiogenesis with spontaneous incorporation into nascent ventricles, in line with the recent demonstration that chimera offspring do not display a preferential tissue allocation [36]. Delivery of exogenous embryonic stem cells into the blastocoel of an early embryo joined native to non-native progeny to yield composite, chimeric blastocysts with a finite blastomere number. In the context of the developing embryo, microsurgical transfers allow effective and sustained integration of independent sources of pluripotent progenitors that are in principle fully competent to differentiate into all lineages [37–39]. In view of the physical restrictions imposed by the inner cell mass, the dual population of coexisting native and implanted progenitor cells creates the opportunity for competitive selection and titration of overall chimerism to meet developmental requirements [40–42]. Predetermined chimerism through embryo manipulation allowed assessment of the therapeutic impact of prenatal enrichment with non-native progenitors upon cardiac stress challenge in the adulthood. The pattern of tissue formation optimized to accommodate the high-demand of initial cardiogenesis reinforced a failure-safe blueprint that was maintained in offspring throughout lifespan. The selection for stress-tolerant progeny and complementary cellular interactions, not available to nonchimeric counterparts, created thereby a phenotype resilient to otherwise debilitating disease.
Although the full spectrum of contributing cell types and their origin remains to be defined, the favorable outcome in infarcted chimera corresponded to reparative features of cardiac biogenesis characterized by increased mitotic activity and tissue-specific progenitor cell load associated with reduced fibrosis. Moreover, the surplus of adipose tissue that developed in chimera contained elevated levels of nonhematopoietic mesenchymal progenitor pools, in line with the therapeutic potential of adult stem cells in the setting of myocardial infarction [3, 31, 43]. Although there has been clinical evidence for acute cardiac tolerance in obesity, central obesity—as displayed by chimera generated herein—has been linked to progressive cardiomyopathy and increased risk of mortality mediated by left ventricular dysfunction [44–46]. Engineered chimera presented here underscore the acquired tolerance in the context of obesity and severe ischemic insult.
In summary, cell-based therapies have not been previously established for prevention of cardiac disease. In utero therapy has been introduced three decades ago for reconstruction of congenital defects, whereby the current practice of surgery for selected prenatal cardiac lesions provides postnatal survival advantage [47, 48]. Beyond reconstructive surgery, stem cell transplantation in prenatal development could offer an innovative approach for primary prevention, which has previously been applied to model systems to correct congenital anomaly [49–53]. With preimplantation intervention to pattern chimeric tissue sustained throughout life-span, the present engineering offers the first proof-of-principle for prevention of adult cardiac stress intolerance to ischemic injury through pre-emptive cell-based intervention.
Acknowledgments
We thank Dr. Robert Benezra and the Transgenic Core Facility at Memorial Sloan-Kettering Cancer Center for support with initial studies and chimera production. We also thank Jonathan J Nesbitt and Lois A Rowe for superb technical support throughout the study. This work was supported by the National Institutes of Health, American Society for Clinical Pharmacology and Therapeutics, Marriott Heart Disease Research Program, Marriott Foundation, Ted Nash Long Life Foundation, Ralph Wilson Medical Research Foundation, and Mayo Clinic Clinician-Investigator Program.
Footnotes
Disclosure of Potential Conflicts of Interest
The authors indicate no potential conflicts of interest.
Author contributions: S.Y.: conception and design, provision of study material, collection and assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript; T.J.N.: conception and design, provision of study material, collection and assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript; A.B.: conception and design, provision of study material, collection and assembly of data, data analysis and interpretation, final approval of manuscript; R.J.C.D.: provision of study material, collection and assembly of data, final approval of manuscript; D.F.: conception and design, provision of study material, data analysis and interpretation, administrative support, manuscript writing, final approval of manuscript; AT: conception and design, data analysis and interpretation, administrative support, manuscript writing, final approval of manuscript.
References
- 1.Segers VF, Lee RT. Stem-cell therapy for cardiac disease. Nature. 2008;451:937–942. doi: 10.1038/nature06800. [DOI] [PubMed] [Google Scholar]
- 2.Wu SM, Chien KR, Mummery C. Origins and fates of cardiovascular progenitor cells. Cell. 2008;132:537–543. doi: 10.1016/j.cell.2008.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dimmeler S, Leri A. Aging and disease as modifiers of efficacy of cell therapy. Circ Res. 2008;102:1319–1330. doi: 10.1161/CIRCRESAHA.108.175943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Laird DJ, von Andrian UH, Wagers AJ. Stem cell trafficking in tissue development, growth, and disease. Cell. 2008;132:612–630. doi: 10.1016/j.cell.2008.01.041. [DOI] [PubMed] [Google Scholar]
- 5.Anversa P, Kajstura J, Leri A, et al. Life and death of cardiac stem cells: A paradigm shift in cardiac biology. Circulation. 2006;113:1451–1463. doi: 10.1161/CIRCULATIONAHA.105.595181. [DOI] [PubMed] [Google Scholar]
- 6.Torella D, Ellison GM, Méndez-Ferrer S, et al. Resident human cardiac stem cells: Role in cardiac cellular homeostasis and potential for myocardial regeneration. Nat Clin Pract Cardiovasc Med. 2006;3(Suppl 1):S8–S13. doi: 10.1038/ncpcardio0409. [DOI] [PubMed] [Google Scholar]
- 7.Bergmann O, Bhardwaj RD, Bernard S, et al. Evidence for cardiomyocyte renewal in humans. Science. 2009;324:98–102. doi: 10.1126/science.1164680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Quaini F, Urbanek K, Beltrami AP, et al. Chimerism of the transplanted heart. N Engl J Med. 2002;346:5–15. doi: 10.1056/NEJMoa012081. [DOI] [PubMed] [Google Scholar]
- 9.Kajstura J, Hosoda T, Bearzi C, et al. The human heart: A self-renewing organ. Clin Translat Sci. 2008;1:80–86. doi: 10.1111/j.1752-8062.2008.00030.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deb A, Wang S, Skelding KA, et al. Bone marrow-derived cardiomyocytes are present in adult human heart: A study of gender-mismatched bone marrow transplantation patients. Circulation. 2003;107:1247–1249. doi: 10.1161/01.cir.0000061910.39145.f0. [DOI] [PubMed] [Google Scholar]
- 11.Kubo H, Jaleel N, Kumarapeli A, et al. Increased cardiac myocyte progenitors in failing human hearts. Circulation. 2008;118:649–657. doi: 10.1161/CIRCULATIONAHA.107.761031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rupp S, Koyanagi M, Iwasaki M, et al. Characterization of long-term endogenous cardiac repair in children after heart transplantation. Eur Heart J. 2008;29:1867–1872. doi: 10.1093/eurheartj/ehn223. [DOI] [PubMed] [Google Scholar]
- 13.Urbanek K, Torella D, Sheikh F, et al. Myocardial regeneration by activation of multipotent cardiac stem cells in ischemic heart failure. Proc Natl Acad Sci USA. 2005;102:8692–8697. doi: 10.1073/pnas.0500169102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kolossov E, Bostani T, Roell W, et al. Engraftment of engineered ES cell-derived cardiomyocytes but not BM cells restores contractile function to the infarcted myocardium. J Exp Med. 2006;203:2315–2327. doi: 10.1084/jem.20061469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Behfar A, Perez-Terzic C, Faustino RS, et al. Cardiopoietic programming of embryonic stem cells for tumor-free heart repair. J Exp Med. 2007;204:405–420. doi: 10.1084/jem.20061916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Laflamme MA, Chen KY, Naumova AV, et al. Cardiomyocytes derived from human embryonic stem cells in pro-survival factors enhance function of infarcted rat hearts. Nat Biotechnol. 2007;25:1015–1024. doi: 10.1038/nbt1327. [DOI] [PubMed] [Google Scholar]
- 17.van Laake LW, Passier R, Monshouwer-Kloots J, et al. Human embryonic stem cell-derived cardiomyocytes survive and mature in the mouse heart and transiently improve function after myocardial infarction. Stem Cell Res. 2007;1:9–24. doi: 10.1016/j.scr.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 18.Fraidenraich D, Stillwell E, Romero E, et al. Rescue of cardiac defects in id knockout embryos by injection of embryonic stem cells. Science. 2004;306:247–252. doi: 10.1126/science.1102612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Conner DA. Chimeric mouse production by microinjection. Curr Protoc Mol Biol. 2001;23:7.1–7.20. doi: 10.1002/0471142727.mb2307s53. [DOI] [PubMed] [Google Scholar]
- 20.Kane GC, Behfar A, Dyer RB, et al. KCNJ11 gene knockout of the Kir62 KAtp channel causes maladaptive remodeling and heart failure in hypertension. Hum Mol Genet. 2006;15:2285–2297. doi: 10.1093/hmg/ddl154. [DOI] [PubMed] [Google Scholar]
- 21.Kane GC, Behfar A, Yamada S, et al. ATP-sensitive K+ channel knockout compromises the metabolic benefit of exercise training, resulting in cardiac deficits. Diabetes. 2004;53(Suppl 3):S169–S175. doi: 10.2337/diabetes.53.suppl_3.s169. [DOI] [PubMed] [Google Scholar]
- 22.Yamada S, Kane GC, Behfar A, et al. Protection conferred by myocardial ATP-sensitive K+ channels in pressure overload-induced congestive heart failure revealed in KCNJ11 Kir6.2-null mutant. J Physiol. 2006;577:1053–1165. doi: 10.1113/jphysiol.2006.119511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Santos L, Mello AF, Antonio EL, et al. Determination of myocardial infarction size in rats by echocardiography and tetrazolium staining: Correlation, agreements, and simplifications. Braz J Med Biol Res. 2008;41:199–201. doi: 10.1590/s0100-879x2008005000007. [DOI] [PubMed] [Google Scholar]
- 24.Fishbein MC, Meerbaum S, Rit J, et al. Early phase acute myocardial infarct size quantification: Validation of the triphenyl tetrazolium chloride tissue enzyme staining technique. Am Heart J. 1981;101:593–600. doi: 10.1016/0002-8703(81)90226-x. [DOI] [PubMed] [Google Scholar]
- 25.Nelson TJ, Faustino RS, Chiriac A, et al. CXCR4+/FLK-1+ biomarkers select a cardiopoietic lineage from embryonic stem cells. Stem Cells. 2008;26:1464–1473. doi: 10.1634/stemcells.2007-0808. [DOI] [PubMed] [Google Scholar]
- 26.Yamada S, Nelson TJ, Crespo R, et al. Embryonic stem cell therapy of heart failure in genetic cardiomyopathy. Stem Cells. 2008;26:2644–2653. doi: 10.1634/stemcells.2008-0187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zingman LV, Hodgson DM, Bast PH, et al. Kir6.2 is required for adaptation to stress. Proc Natl Acad Sci USA. 2002;99:13278–13283. doi: 10.1073/pnas.212315199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hodgson DM, Behfar A, Zingman LV, et al. Stable benefit of embryonic stem cell therapy in myocardial infarction. Am J Physiol Heart Circ Physiol. 2004;287:H471–H479. doi: 10.1152/ajpheart.01247.2003. [DOI] [PubMed] [Google Scholar]
- 29.Nelson TJ, Ge ZD, Van Orman J, et al. Improved cardiac function in infarcted mice after treatment with pluripotent embryonic stem cells. Anat Rec A Discov Mol Cell Evol Biol. 2006;288:1216–1224. doi: 10.1002/ar.a.20388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Srivastava D, Ivey KN. Potential of stem-cell-based therapies for heart disease. Nature. 2006;441:1097–1099. doi: 10.1038/nature04961. [DOI] [PubMed] [Google Scholar]
- 31.Passier R, van Laake LW, Mummery CL. Stem-cell-based therapy and lessons from the heart. Nature. 2008;453:322–329. doi: 10.1038/nature07040. [DOI] [PubMed] [Google Scholar]
- 32.Pucéat M, Ballis A. Embryonic stem cells: From bench to bedside. Clin Pharmacol Ther. 2007;82:337–339. doi: 10.1038/sj.clpt.6100298. [DOI] [PubMed] [Google Scholar]
- 33.Behfar A, Faustino RS, Arrell DK, et al. Guided stem cell cardiopoiesis: Discovery and translation. J Mol Cell Cardiol. 2008;45:523–529. doi: 10.1016/j.yjmcc.2008.09.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kajstura J, Urbanek K, Rota M, et al. Cardiac stem cells and myocardial disease. J Mol Cell Cardiol. 2008;45:505–513. doi: 10.1016/j.yjmcc.2008.05.025. [DOI] [PubMed] [Google Scholar]
- 35.Hsieh PC, Segers VF, Davis ME, et al. Evidence from a genetic fate-mapping study that stem cells refresh adult mammalian cardiomyocytes after injury. Nat Med. 2007;13:970–974. doi: 10.1038/nm1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stillwell E, Vitale J, Zhao Q, et al. Blastocyst injection of wild type embryonic stem cells induces global corrections in mdx mice. Plos One. 2009;4:e4759. doi: 10.1371/journal.pone.0004759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wood SA, Allen ND, Rossant J, et al. Non-injection methods for the production of embryonic stem cell-embryo chimaeras. Nature. 1993;365:87–89. doi: 10.1038/365087a0. [DOI] [PubMed] [Google Scholar]
- 38.Tam PP, Rossant J. Mouse embryonic chimeras: Tools for studying mammalian development. Development. 2003;130:6155–6163. doi: 10.1242/dev.00893. [DOI] [PubMed] [Google Scholar]
- 39.Breault DT, Min IM, Carlone DL, et al. Generation of mTert-GFP mice as a model to identify and study tissue progenitor cells. Proc Natl Acad Sci USA. 2008;105:10420–10425. doi: 10.1073/pnas.0804800105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Roeder I, Kamminga LM, Braesel K, et al. Competitive clonal hematopoiesis in mouse chimeras explained by a stochastic model of stem cell organization. Blood. 2005;105:609–616. doi: 10.1182/blood-2004-01-0282. [DOI] [PubMed] [Google Scholar]
- 41.Blau HM, Brazelton TR, Weimann JM. The evolving concept of a stem cell: Entity or function? Cell. 2001;105:829–841. doi: 10.1016/s0092-8674(01)00409-3. [DOI] [PubMed] [Google Scholar]
- 42.Chien KR, Domian IJ, Parker KK. Cardiogenesis and the complex biology of regenerative cardiovascular medicine. Science. 2008;322:1494–1497. doi: 10.1126/science.1163267. [DOI] [PubMed] [Google Scholar]
- 43.Christoforou N, Gearhart JD. Stem cells and their potential in cell-based cardiac therapies. Prog Cardiovasc Dis. 2007;49:396–413. doi: 10.1016/j.pcad.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 44.Wong C, Marwick TH. Obesity cardiomyopathy: Pathogenesis and pathophysiology. Nat Clin Pract Cardiovasc Med. 2007;4:436–443. doi: 10.1038/ncpcardio0943. [DOI] [PubMed] [Google Scholar]
- 45.Ammar KA, Redfield MM, Mahoney DW, et al. Central obesity: Association with left ventricular dysfunction and mortality in the community. Am Heart J. 2008;156:975–981. doi: 10.1016/j.ahj.2008.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thakker GD, Frangogiannis NG, Zymek PT, et al. Increased myocardial susceptibility to repetitive ischemia with high-fat diet-induced obesity. Obesity. 2008;16:2593–2600. doi: 10.1038/oby.2008.414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Glatz JA, Tabbutt S, Gaynor JW, et al. Hypoplastic left heart syndrome with atrial level restriction in the era of prenatal diagnosis. Ann Thorac Surg. 2007;84:1633–1638. doi: 10.1016/j.athoracsur.2007.06.061. [DOI] [PubMed] [Google Scholar]
- 48.Marshall AC, Levine J, Morash D, et al. Results of in utero atrial septoplasty in fetuses with hypoplastic left heart syndrome. Prenat Diagn. 2008;28:1023–1028. doi: 10.1002/pd.2114. [DOI] [PubMed] [Google Scholar]
- 49.Zanjani ED, Flake AW, Rice H, et al. Long-term repopulating ability of xenogeneic transplanted human fetal liver hematopoietic stem cells in sheep. J Clin Invest. 1994;93:1051–1055. doi: 10.1172/JCI117054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liechty KW, MacKenzie TC, Shaaban AF, et al. Human mesenchymal stem cells engraft and demonstrate site-specific differentiation after in utero transplantation in sheep. Nat Med. 2000;6:1282–1286. doi: 10.1038/81395. [DOI] [PubMed] [Google Scholar]
- 51.Airey JA, Almeida-Porada G, Colletti EJ, et al. Human mesenchymal stem cells form Purkinje fibers in fetal sheep heart. Circulation. 2004;109:1401–1407. doi: 10.1161/01.CIR.0000124222.16321.26. [DOI] [PubMed] [Google Scholar]
- 52.Fraidenraich D, Benezra R. Embryonic stem cells prevent developmental cardiac defects in mice. Nat Clin Pract Cardiovasc Med. 2006;3(Suppl 1):S14–S17. doi: 10.1038/ncpcardio0402. [DOI] [PubMed] [Google Scholar]
- 53.Chino T, Tamai K, Yamazaki T, et al. Bone marrow cell transfer into fetal circulation can ameliorate genetic skin diseases by providing fibroblasts to the skin and inducing immune tolerance. Am J Pathol. 2008;173:803–814. doi: 10.2353/ajpath.2008.070977. [DOI] [PMC free article] [PubMed] [Google Scholar]