

Abstract
The peptide self-assembly mimic (PSAM) from the outer surface protein A (OspA) can form highly stable but soluble β-rich self-assembly-like structures similar to those formed by native amyloid-forming peptides. However, unlike amyloids that predominantly form insoluble aggregates, PSAMs are highly water-soluble. Here, we characterize the conformations of these soluble β-sheet-rich assemblies. We simulate PSAMs with different-sized β-sheets in the presence and absence of end-capping proteins using all-atom explicit-solvent molecular dynamics, comparing the structural stability, conformational dynamics, and association force. Structural and free-energy comparisons among β-sheets with different numbers of layers and sequences indicate that in similarity to amyloids, the intersheet side chain-side chain interactions and hydrogen bonds combined with intrasheet salt bridges are the major driving forces in stabilizing the overall structural organization. A detailed structural analysis shows that in similarity to amyloid fibrils, all wild-type and mutated PSAM structures display twisted and bent β-sheets to some extent, implying that a twisted and bent β-sheet is a general motif of β-rich assemblies. Thus, our studies indicate that soluble β-sheet-rich peptide self-assemblies can provide good amyloid mimics, and as such confirm on the atomic scale that they are excellent systems for amyloid studies. These results provide further insight into the usefulness of such mimics for nanostructure design.
Introduction
The self-assembly of protein/peptide building blocks, as well as DNA and RNA segments, into well-defined three-dimensional nanostructures and supramolecules is a common process in natural systems (1–4). β-rich self-assemblies have attracted considerable interest in the fields of material, biological, and nanotechnological sciences because they constitute a major structural class of polypeptides (5). These well-known structural motifs are found in ordered amyloid fibrils associated with many human neurodegenerative diseases, including Alzheimer's, Parkinson's, and type II diabetes (6–9). Amyloid fibrils contain a predominant cross-β-sheet core structure. Apart from the cross-β-sheet motif and favorable tight intersheet van der Waals (VDW) packing, little is known about the high-resolution structure of amyloid aggregates of different sizes and conformations. This is primarily because the fibrils are insoluble and thus it is very difficult to obtain the structure using conventional biophysical techniques such as x-ray crystallography and solution NMR (10–13).
To overcome this insolubility problem, a number of groups (5,10,14,15) have developed a host-guest strategy that transforms amyloid-forming peptide fragments into water-soluble proteins. These soluble assemblies are highly relevant to the native amyloid fibrils; they serve as model systems and thus are useful for obtaining detailed structures and their biophysical properties. Takano and co-workers (14) determined the atomic-level structure of Aβ28–42 in aqueous solution by fusing two Aβ sequences (Aβ10–24 and Aβ28–42) at three positions of the C-terminal region of ribonuclease HII from a hyperthermophile (Tk-RNase HII). A ThT-binding analysis revealed that Tk-RNase HII1–174–Aβ10–24 did not aggregate, whereas Tk-RNase HII1–174–Aβ28–42 aggregated rapidly. This suggests that the Aβ C-terminal region, including residues 28–42, is important for fiber formation (14). Hosia and co-workers (16) used two different sequences—a turn motif (YNGK) and a less rigid sequence (AAAK)—to link KFFE, one of the shortest amyloid fibril-forming peptides. Electron microscopy results showed different aggregation behaviors for KFFE-AAAK-KFFE and KFFE-YNGK-KFFE: KFFE-AAAK-KFFE forms abundant and thick fibrils, whereas KFFE-YNGK-KFFE does not form fibrils, suggesting design strategies for altering the tendency of a peptide to form amyloid. Nelson et al. (17) crystallized a seven-residue fragment (GNNQQNY) from the yeast protein Sup35 using x-ray microcrystallography. The structure clearly shows a cross-β spine architecture with a dry interface formed by interdigitated side-chain “steric zippers” between the layers without β-sheet twisting. However, subsequent explicit solvent molecular-dynamics (MD) simulations from the crystal structure indicated that the cross-β spine architecture is twisted in the absence of crystal packing (18–20).
Recently, the Koide group (5,10,21) engineered a single-layer β-sheet (SLB) segment of the outer surface protein A (OspA) from Borrelia burgdorferi by covalently linking several β-hairpin units. They capped the β-sheet edges of the SLB by two globular domains to enhance solubility by blocking the infinite extension along the long axis and infinite lamination of the β-sheet layers. By using this “link-and-cap” strategy, Koide et al. successfully determined the atomic structures of the peptide self-assembly mimic (PSAM) using x-ray crystallography and solution NMR spectroscopy.
We previously used modeling and MD simulations to predict native and soluble amyloid oligomers with distinct structural morphologies, sizes, and sequences (19,22–25). The structures of these oligomers display relatively high stability. In this study, motivated by the experimental work of Koide's group (5,10,21), we focus on the peptide self-assembly mimic to probe on the atomic scale whether soluble β-sheet-rich peptide self-assemblies can provide good amyloid mimics presenting similar conformational properties. We use the sequence of the Koide laboratory's peptide and construct various self-assembly models containing the PSAM end-capping globular protein domains (Fig. 1). Since PSAM contains a high content of β-sheet structure that is composed of several repetitive β-hairpin building blocks, we also use this β-hairpin to computationally design different β-sheet-rich structures of different sizes lacking end-capping domains. By comparing and characterizing the β-sheets with and without these end caps, we can examine the effects of the end-capping proteins on the structural stability, conformational dynamics, and lateral association of β-sheet assemblies. Further, two types of mutations are engineered, creating and destroying aromatic ladders, to examine the effect of hydrophobic packing on the structural stability of the preformed β-sheets. Finally, a comparison of the PSAM assemblies with amyloid structures suggests that twisting and bending in the β-sheet-rich assemblies are observed in both assemblies. This study provides detailed structural information about soluble β-sheet-rich self-assemblies and correlates it with that of insoluble amyloid aggregates.
Figure 1.
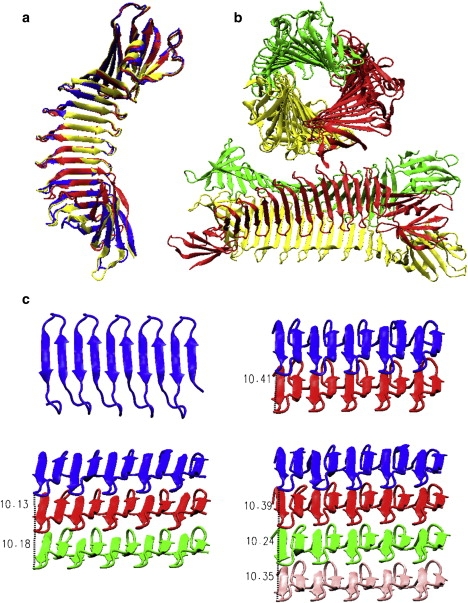
(a) Superimposition of three monomeric-single-layer PSAM structures with different numbers of copies of β-hairpin units. PDB codes: 2HKD (blue), 2AF5 (red), and 2FKG (yellow). (b) A trimeric structure (PDB code: 2FKJ) is packed by three single-layer PSAMs along a threefold axis. (c) Cartoon of the β-sheet structures without the end-capping proteins; each sheet has five copies of β-hairpin units. The intersheet distance of multiple-layer assemblies is ∼10.5 Å.
Materials and Methods
PSAM structural models
Three models of the monomeric wild-type PSAMs (Protein Data Base (PDB) codes: 2AF5, 2FKG, and 2HKD) and one of the trimeric wild-type PSAM (PDB code: 2FKJ) derived from the SLB of OspA of Makabe et al. (10) were used as initial configurations. In this article, the term “wild-type” indicates PSAM constructs that contain β-hairpin copies of the original amino acid sequence (see below), although all PSAM proteins are mutant forms of OspA. These structures, which are capped by big, bulky, globular domains at the N-/C-terminals that prevent fibril growth, have a relatively flat β-sheet in the middle comprised of different numbers of covalently bonded copies of the β-hairpin building blocks, as shown in Fig. 1. Each β-hairpin building block has a 23-residue sequence of Leu1-Ser-Glu-Lys-Lys-Ile6-Thr-Arg-Ala-Asp10-Lys-Ser-Ser-Thr-Glu15-Glu-Lys-Phe-Asn-Glu20-Lys-Gly-Glu23. Four residues (Arg8-Ala9-Asp10-Lys11) form a turn connecting two antiparallel β-strands. For the monomeric PSAM structures, 2AF5 contains two β-hairpin building blocks (residues 132–177), whereas both 2FKG and 2HKD contain three building blocks (residues 132–200) in the middle of the assembly. In the 2FKJ crystal, two identical PSAMs and one distinct PSAM were packed to form a trimeric structure (PDB code: 2FKJ). Each monomer contains five β-hairpin building blocks (residues 132–246), and this PSAM is predominantly monomeric in solution (10).
β-sheet models
To examine the effect of end-capping proteins on the conformation and dynamics of PSAMs, we constructed flat β-sheet structures using a different number of copies of the β-hairpin building block (residues 128–150 in the 2AF5 structure), without the capping proteins. Five and 10 repetitive neighboring building blocks were covalently bonded to yield two SLBs. Two, three, and four copies of the same β-sheet containing five hairpins were then stacked on top of each other in the lateral direction to form two, three, and four layers, respectively, with an initial interlayer distance of ∼10.5 Å (Fig. 1 c). Each model has an acetylated N-terminal and an amidated C-terminal to avoid terminal charges. Note that the β-sheet models (i.e., single- and multiple-layer models) differ from the experimental PSAM structures in the number of β-hairpins, number of β-sheets, and absence of end caps. All-atom MD simulations in the presence of explicit solvent and ions probed the structural stability of these β-sheet structures as indicated by their capacity to retain the initial organization and their conformational dynamics. All starting structures of the mutants were built from the wild-type β-sheet models by replacing the side chains of the targeted residues, but without changing the backbone conformations and side-chain orientations. The structure of the designed mutant was first minimized for 500 steps using the steepest-descent algorithm, and the backbone of the protein was restrained before it was subjected to the system setup and production runs. The MD simulation protocol is described in the Supporting Material.
Results and Discussion
Conformational dynamics of β-rich assemblies with end caps
The overall structural stability of wild-type PSAMs is measured by the root mean-square deviations (RMSDs) of the backbone atoms relative to their initial energy-minimized structures. Fig. S1 b presents the backbone RMSDs of four different PSAMs with end caps from their starting structures. All monomeric PSAM structures (PDB codes: 2AF5, 2FKG, and 2HKD) experienced large structural deviation and fluctuations with averaged RMSD values between 5 and 6 Å. However, as compared to whole structures, the RMSD values measured only for the β-hairpin building block regions were dramatically reduced to 1.5–2.0 Å (Fig. S1 c), consistent with experimental measurements (10). Snapshots of the monomeric PSAMs taken from the simulations at 1 ns intervals were superimposed on top of each other. It can be clearly seen that overall the PSAM structures remained very stable during the 40 ns simulations, although the two end edges displayed structural fluctuation to some extent (Fig. 2), consistent with the radius of gyration (Rg; Fig. S1 a), and the overall size of the monomeric PSAMs did not experience a large change. The residue-based root mean-square fluctuation (RMSF) of the backbones from the average structure during MD simulations was calculated to assess the local dynamics and flexibility of each residue. The RMSF profiles show that the residues at the β-hairpin region (residues 105–150 for 2AF5, and residues 105–173 for 2FKG and 2HKD) have relatively lower fluctuations than residues at the edges (data not shown). Taken together, the results indicate that the relatively large structural fluctuation comes mainly from the two edges of the assemblies, and the β-hairpin secondary structures are well conserved without obvious bending or twisting.
Figure 2.
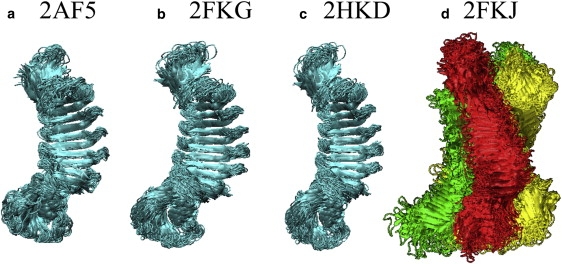
Superimposition of 40 structures taken every nanosecond from the MD trajectories for monomeric (a) 2AF5, (b) 2FKG, and (c) 2HKD structures, and (d) trimeric 2FKJ structures.
Protein fibrils often consist of three or more β-sheets associated in the lateral direction. We therefore performed additional MD simulations to investigate the stability of a trimeric β-sheet structure with end caps (PDB code: 2FKJ), in which each sheet contains five covalently bonded β-hairpin building blocks. The trimeric structure was very stable, displaying an Rg of ∼41.4 ± 0.2 Å. The trimeric structure also had a relatively smaller structural deviation than the monomeric structures (Fig. S1 b). A similar trend was also observed in the RMSF profile (data not shown). Both the RMSD and RMSF consistently show that the trimeric structure has a higher stability than the monomeric structures, mainly because the trimeric structure has a much larger interface involving side chain-side chain interactions. In similarity to amyloid fibril growth in the lateral direction, the trimeric PSAM enhances its stability as it progresses along a screw axis.
Conformational dynamics of single-layer β-rich assemblies without end caps
Simulations of the PSAMs with end caps show that the β-hairpins embedded in all monomeric and trimeric self-assembly mimics display small structural changes and fluctuations, and preserve their secondary structure, demonstrating that they are very versatile building blocks that can be used to construct stable β-rich assemblies. Thus, we modeled two SLBs without end caps (one with five and the other with 10 identical β-hairpins). Each β-hairpin was covalently bonded to the other two units at the N- and C-termini. The conformational dynamics of the monomers of these SLBs are quantitatively measured by the backbone RMSD relative to the initial structure. As can be seen in Fig. S2 a, for both the five- and 10-β-hairpin sheets, the RMSDs rose quickly to plateaus of 5.1 and 10.7 Å at the first 5 ns, followed by small fluctuations during the rest of the simulations, indicating that the longer sheet experiences larger structural deviation than the shorter sheet.
To further identify the origin of the structural deviation, we calculated the local flexibility induced by each residue by the RMSF. Fig. S2 b shows that the RMSF profiles of both single β-sheets have a similar symmetrical shape. The most flexible regions of the β-sheets are the edge residues of the N- and C-termini and the loop residues connecting the β-strands, whereas the most rigid regions are the central residues of the β-strands. The fluctuation difference between the edge and central residues becomes even more pronounced for the longer β-sheet. It can be seen that for the 10-β-hairpin sheet, the smallest fluctuation occurred at the central residues of Glu107-Glu108-Lys109, Ser117-Glu118-Lys119, and Ser127-Ser128-Thr129, as confirmed by visual inspection of the MD trajectories in Fig. 3. By superimposing β-sheet structures at every nanosecond, we can see that both the five- and 10-β-hairpin sheets displayed a twisted and bent structure as the simulations proceeded, although they initially had a flat shape without curvature. Structural twisting and bending gradually spread from the central residues to the edge residues with stepwise increasing displacements from their original positions. Thus, the long sheet experiences a larger conformational change than the short one, in agreement with the RMSD, and the edge residue is more flexible than the central residue, in agreement with the RMSF. It should be noted that although the whole β-sheets were twisted and bent, the secondary structure of the antiparallel β-sheet, as calculated by the Kabsch and Sander algorithm (26), was well preserved during the 20 ns simulations (Fig. S2 c).
Figure 3.
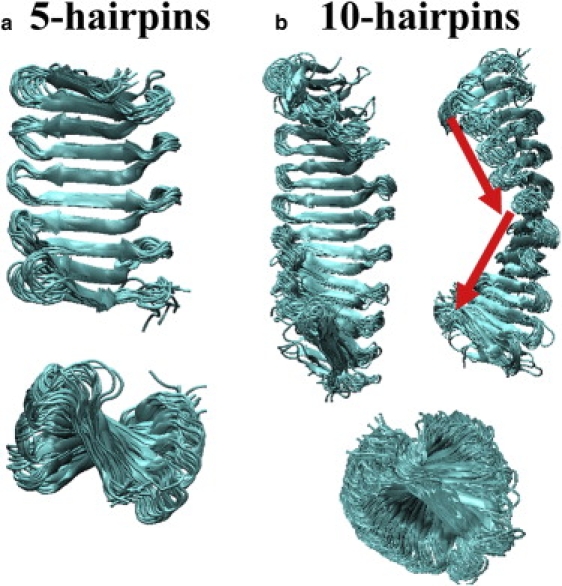
Superimposition of 20 structures taken every nanosecond from the MD trajectories for SLBs consisting of (a) five and (b) 10 repetitive β-hairpins. All structures display a bent and twisted conformation from the top and side views.
Since the SLB is completely exposed to water, the conformational change of the β-sheet can be attributed to two types of interactions: sheet-water and intrasheet. The sheet-water interactions are estimated by the number of hydrogen bonds between the β-sheet and the waters, whereas the intrasheet interactions are calculated by backbone and side-chain contacts consisting of backbone hydrogen bonds and side-chain contacts (19). Fig. S3 shows that the intrasheet interactions (hydrogen bonds and side-chain contacts) were largely maintained for the simulated period of time for both single β-sheet models. However, the hydrogen bonds between the waters and the five-hairpin β-sheet decreased by ∼20%. It is interesting that, for the 10-hairpin β-sheet, the number of hydrogen bonds between the sheet and water molecules dramatically decreased by ∼50% during the first 5 ns, then fluctuated between 5 and 11 ns, and eventually leveled off to ∼90% of the initial value at 20 ns. A correlation of the intra- and intersheet interactions with the conformational change of the single β-sheet suggests that when the single β-sheet experiences structural twisting and bending, the water is reorganized around the sheet, causing the breakage and reformation of the hydrogen bonds between the sheet and the water molecules. A larger conformational change requires a longer time to reorganize the water molecules. Taken together, these results indicate that a flat SLB is not an energetically favorable conformation in solution. The twisted and bent sheet appears more stable (22,23,25), and the structural twisting and bending is strongly mediated by the waters.
We examined the effect of end caps on the structural dynamics of the β-hairpins for both experimental PSAM assemblies with end caps and simulated β-sheet models without end caps. The average backbone RMSDs of the β-hairpin building blocks in four PSAM assemblies from the last 10 ns were 1.5 ± 0.2 (2AF5, two β-hairpins), 1.8 ± 0.2 (2FKG, three β-hairpins), 1.9 ± 0.3 (2HKD, three β-hairpins), and 2.5 ± 0.2 (2FKJ, five β-hairpins for each layer), which are much smaller than the RMSDs of 5.1 ± 0.3 and 10.7± 0.3 for the modeled five-β-hairpin and 10-β-hairpin sheets without end caps (Table 1 and Fig. S1). A comparison of the structural stability between single-layer β-rich assemblies with and without end caps suggests that the introduction of the end-capped protein at the N-/C-termini can enhance the stability of assemblies.
Table 1.
Details of the simulation systems
| Simulations |
Number of β-hairpin copies per sheet |
All backbone RMSD (Å) |
β-hairpin RMSD (Å) |
Time (ns) |
|---|---|---|---|---|
| PSAMs with end-capped proteins | ||||
| 2AF5 | 2 | 5.5 ± 0.9 | 1.5 ± 0.2 | 40 |
| 2FKG | 3 | 6.0 ± 0.7 | 1.8 ± 0.2 | 40 |
| 2HKD | 3 | 5.7 ± 0.6 | 1.9 ± 0.3 | 40 |
| 2FKJ | 5 | 4.7 ± 0.3 | 2.5 ± 0.2 | 40 |
| PSAMs without end-capped proteins | ||||
| Single-layer sheet | 5 | 5.1 ± 0.3 | 20 | |
| Single-layer sheet | 10 | 10.7± 0.3 | 20 | |
| Two-layer sheet | 5 | 4.1 ± 0.3 | 20 | |
| Three-layer sheet | 5 | 5.9 ± 0.2 | 20 | |
| Four-layer sheet | 5 | 7.3 ± 0.5 | 20 | |
| Four-layer sheet, Phe→Ala | 5 | 7.3 ± 0.2 | 20 | |
| Four-layer sheet, Lys→Tyr | 5 | 6.5 ± 0.4 | 20 | |
RMSDs are averaged from the last 10 ns.
Structural stability of multiple-layer β-sheets without end caps
To investigate to what extent the number of the β-sheets affects their structural dynamics, we computationally designed two-, three-, and four-layer β-sheets without end caps, with each sheet consisting of five repetitive β-hairpins packed in parallel. In simulations, a poorly designed initial model will dissociate or break, whereas a well-designed model will retain a stable structure during nanosecond-scale simulations. Analysis of the RMSDs for all multiple-β-sheet models showed that the two β-sheets (averaged RMSD = 4.1 ± 0.3 Å) exhibited smaller structural deviation than the three and four β-sheets, with an increase of averaged RMSDs of 5.9 ± 0.2 and 7.3 ± 0.5 Å, respectively (Fig. 4 a and Table 1). Nonetheless, the large RMSDs alone for the three and four β-sheets do not necessarily imply unstable structures. Visual inspection of the MD trajectories showed that all multiple β-sheet models were able to maintain the overall structural integrity without sheet dissociation. To further confirm the structural organization of the β-sheets, interstrand (dstrand) and intersheet (dsheet) distances were used to characterize the sheet-sheet associations. The dstrand is calculated by averaging the mass center distance between each residue in one sheet and the respective in-register residue in the adjacent sheet, whereas dsheet is calculated by averaging the mass center distance between sheets (19). The MD trajectories of all β-sheet models displayed a quick increase in the intersheet distances at the first 5 ns, consistent with the concomitant RMSD increase, and finally stabilized at plateaus of 14.4 ± 0.2 Å for the two sheets, 14.2 ± 0.2 Å and 14.1 ± 0.1 Å for the three sheets, and 13.8 ± 0.2 Å, 13.2 ± 0.2 Å, and 13.5 ± 0.2 Å for the four sheets at 20 ns, respectively (Fig. S4 a). The intersheet distance slightly decreased as the number of β-sheets increased due to the geometrical strain induced by the β-sheets.
Figure 4.
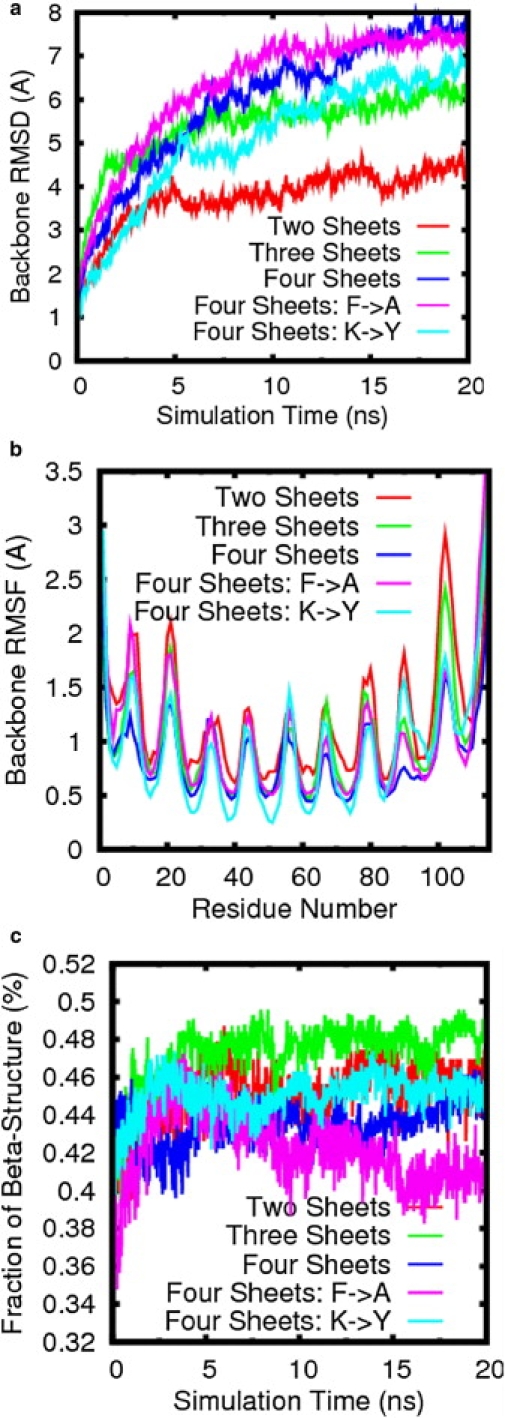
Structural characterization of multiple-layer β-sheets without end-capped proteins for wild-type and mutated sequences (Phe→Ala or Lys→Tyr). The time evolution of the (a) backbone RMSD, (b) backbone RMSF, and (c) β-strand population is shown.
Since the residues at both sheet edges are more exposed, they are easily attacked by water molecules. Especially for the two-layer β-sheets, a comparison of the results in Fig. S5 a and Fig. S6 a shows that the water molecules can unzip the parallel-packed β-sheets and pull apart interacting edge residues at the interface, leading to a greater interstrand distance between the two interacting edges. However, water molecules did not penetrate into the central region through the unprotected ends of the β-sheets. For the three-layer and four-layer β-sheets, similar results were observed with a large interstrand distance between the edge residues and a small interstrand distance between the central residues (Fig. S5, b and c). However, overall, the interactions between water molecules and these residues, regardless of their positions, did not show a significant difference as displayed in the three- and four-layer β-sheets (Fig. S6, b and c), which indicates that the majority of the residues in the β-sheets are still well protected from hydration. This is because a two-layer β-sheet structure has only one dehydrated interface, whereas three- and four-layer β-sheets have two and three dehydrated interfaces, respectively, and apparently can withstand the disassociation by water. The residue-based RMSFs consistently showed that all multiple β-sheets display small RMSFs for the central residues and large RMSFs for the edge and loop residues, indicating that the residues at the termini or in the loop region are more flexible than the residues in the central region (Fig. 4 b). A comparison of the RMSD, sheet-sheet distance, and secondary structure, and a visual inspection of the MD trajectories indicate that although all multiple-layer β-sheets exhibit structural deviation to some extent with respect to their initial perfect flat structures, they are still able to maintain the whole and secondary structures without disassociation. The structural deviation that arises from the accumulation of small distortions within each β-sheet and distance increase between β-sheets has little effect on the overall parallel β-sheet packing.
Inter- and intrasheet interactions
To further quantitate the driving force underlying the β-sheet association, we calculated and averaged the interaction energy between β-sheets by using the generalized Born with simple switching method (27) in the CHARMM program for the last 10-ns simulations (Fig. 5), where Ei(i+1) represents the interaction energy between sheet i and its neighboring sheet i + 1. The interaction energy remained roughly constant for each of the systems during the last 10 ns, indicating that the systems were in equilibrium. The averaged interaction energies for the 10 ns were −304.3, −656.5, and −736.6 kcal/mol for two-, three-, and four-layer β-sheets, respectively. The four-layer β-sheets had the lowest interaction energy, whereas the two-layer β-sheet system had relatively large but still favorable (i.e., negative) free energy.
Figure 5.
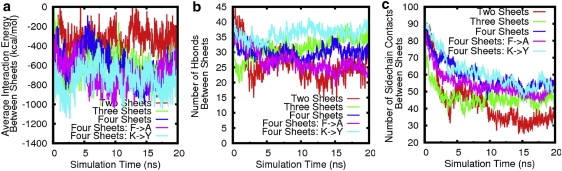
(a) Interaction energies between β-sheets for all β-sheet models without end-capped proteins. The time evolution of (b) hydrogen bonds and (c) side-chain contacts between neighboring β-sheets is shown. All of the values are normalized by the number of a pair of neighboring β-sheets.
Hydrogen bonds and side chain-side chain interactions between β-sheets play important roles in stabilizing the structures and preventing water penetration. Fig. 5, b and c, show the time evolution of hydrogen bonds and side-chain contacts between neighboring β-sheets. It can be seen in Fig. 5 b that for all multiple-layer β-sheet models, the number of hydrogen bonds between neighboring β-sheets remained approximately constant, whereas the number of side-chain contacts dropped in the first 10 ns and then remained constant for the rest of the simulations. The three- and four-sheet structures had a larger number of hydrogen bonds and side-chain contacts than the two-sheet structure. It is of interest to compare the contributions of side-chain interactions and hydrogen bonds with the association force between β-sheets. The average numbers of side-chain contacts from the last 10 ns were 33.6, 45.4, and 53.9, whereas the corresponding hydrogen bonds were 23.5, 33.4, and 29.8 for two-, three-, and four-sheet structures, respectively. The averaged side-chain interactions between neighboring β-sheets (as calculated by the same algorithm used for the intersheet interaction) from the last 10 ns were −197.2, −334.9, and −345.0 kcal/mol for two-, three-, and four-layer β-sheets, respectively. A comparison of the side-chain interactions with the averaged intersheet interactions indicates that side-chain interactions contribute ∼64%, 51%, and 47% to the two-, three-, and four-layer β-sheet associations, respectively. Thus, side-chain contacts contribute roughly equally to hydrogen bonds in β-sheet association. The residues at the edges were easily attacked by water molecules, leading to the loss of those interactions to some extent. Nonetheless, despite the loss of the contacts at the edges, many polar interactions (Ser2, Glu3, Lys5, Thr7, Lys11, Ser13, Glu15, Lys17, and Thr21 in one sheet versus Lys4, Arg8, Asp10, Thr14, and Glu16 in the other) showed good conservation of those contacts at the interface.
Within the β-sheet, the intrasheet salt bridges of Asp10+i∗23-Lys11+i∗23 (i = 0–4, where i represents the i+1st β-hairpin unit) helped to enhance the turn stability, whereas the salt bridges of Lys5+i∗23-Glu15+i∗23 (i = 0–4), Lys17+i∗23-Glu3+i∗23 (i = 0–4), Lys28+i∗23-Glu15+i∗23 (i = 0–3), and Lys17+i∗23-Glu26+i∗23 (i = 0–3) improved the β-strand association within the same β-sheet. In similarity to the intersheet interactions, for all wild-type models, all of the salt bridges (Asp10+i∗23I-Lys11+i∗23II, Lys4+i∗23I-Glu3+i∗23II, Glu16+i∗23I-Lys17+i∗23II, i = 0–4) between the β-sheets were well retained throughout the simulations. The attractive electrostatic interactions contributed by these salt bridges also assist in maintaining the β-sheet secondary structure. As expected, a comparison of the inter- and intrasheet interactions for all three systems suggests that between the sheets, side chain-side chain interactions are dominant forces in stabilizing the organization of the β-sheets, whereas within the sheets, the driving forces in association and stabilization of the secondary and sheet structures are salt bridges. Thus, it can be reasonably expected that more stable structures will be obtained if more β-strands are added within a β-sheet or more β-sheets are associated in the lateral direction.
Effect of the aromatic ladder on the structural twist and bend of β-sheets
Biancalana et al. (21) observed that a Tyr aromatic ladder can stabilize and rigidify the PSAM in timescales of microseconds and less. To computationally examine the effect of an aromatic ladder on the overall structural stability and on the twisting and bending characteristics of β-sheets, we performed two sets of mutation simulations on the four-layer β-sheet models with the most favorable intersheet energy. In the first simulation, an aromatic ladder was created by substituting Lys residues of 4, 17, 27, 40, 50, 63, 73, 86, 96, and 109 with Tyr residues, and in the second one an aromatic ladder was eliminated by substituting Phe residues of 18, 41, 64, 87, and 110 with Ala residues.
As compared to the wild-type, the Tyr ladder enhanced the structural stability of the four-layer β-sheets, resulting in a decrease in RMSD and interaction energy. The stacked ladders restricted the mobility of the aromatic side chains as well as their neighboring residues. The sheet-sheet interaction energies, averaged over the last 10 ns, were −720.7 and −764.2 kcal/mol for four-layer β-sheets without and with Tyr ladders (Fig. 5 a). It can be seen that the sheet-sheet interactions become more favorable when the Tyr ladders are created, due to the increased side-chain contacts and hydrogen bonds between the adjacent β-sheets (Fig. 5, b and c).
On the other hand, deletion of the Phe ladder in the four-layer β-sheets had little effect on the overall structural organization. A structural and energetic analysis showed that both the wild-type and mutant had comparable RMSDs (7.3 Å for wild-type versus 7.3 Å for mutant) and sheet-sheet interactions (−720.7 kcal/mol for wild-type versus −717.6 kcal/mol for mutant). Visual inspection of the MD trajectory showed that although the four-layer β-sheet mutant was able to maintain its parallel packing integrity, the two top layers in the mutant tended to rotate by ∼13° with respect to the other two bottom layers, leading to β-strands that were not in-register between neighboring β-sheets.
The different effects of creating a Tyr ladder and deleting a Phe ladder on the four-layer β-sheet can be attributed to the positions of the mutated residues. Unlike created Tyr residues that reside in the middle of a β-strand region, deleted Phe residues are located in a boundary region between a β-strand and a loop of the β-hairpin unit; consequently, their interactions with the surrounding residues typically are not similar to those with the residues in the β-strand region. Thus, replacing the Phe with Ala does not serve to truly dissect the contributions of aromatic ladders to the β-sheet structure and stability. In similarity to amyloid fibrils, only an aromatic ladder occurring in the β-strand region (e.g., a Tyr ladder) can be considered as an aromatic cross-strand ladder, consistent with the observation by Biancalana et al. (21). Of interest, all mutants displayed different degrees of twist of β-sheet assemblies (Fig. 6), indicating that the cross-β filaments may be intrinsically chiral.
Figure 6.
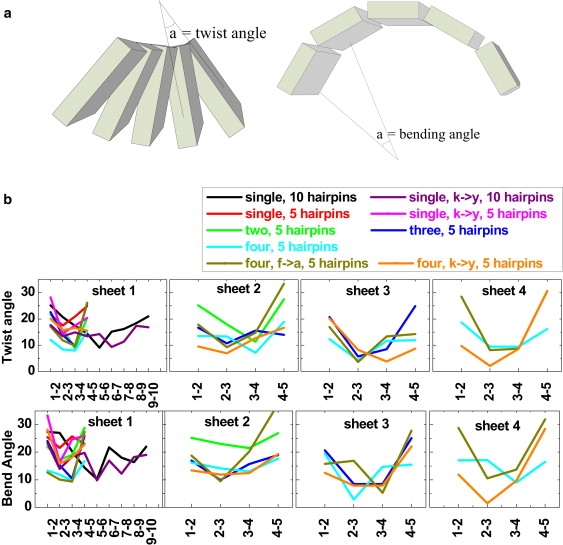
(a) Schematic definition of twist and bend angles. (b) Relative orientation between adjacent β-hairpin units measured by the twist and bend angles for all wild-type and mutated β-sheet models.
Twisting and bending structural characteristics of β-sheet models
Although all structures were initially constructed from a flat β-sheet (Fig. 1), they all present different degrees of twist and bend between strands of the same sheet depending on the number of layers in the assembly. We quantitatively analyzed the relative orientation (twisting and bending) of adjacent β-hairpin units for all β-sheet models, as shown in Fig. 6. We measured the twist angle between adjacent β-hairpin units of the same β-sheet by averaging over all angles between two vectors connecting the first Cα atom to the last Cα atom in the same β-strand portion, where 0° represents no twisting between adjacent β-hairpin units. As for the bend angle, we define a plane for each β-hairpin unit, with the corresponding vector normal to each of these planes. The bend angle is defined by the angle of neighboring normal vectors, where 0° represents a perfect flat plane (Fig. 6 a). A comparison of the structural variation as measured by the twist and bend angles reveals the following: First, all β-sheet models exhibit twisting and bending (18,19,28). Visual inspection of the structures along the five MD trajectories revealed that the bending mostly occurred at the flexible loops connecting the β-strands. In similarity to the twist angle of other amyloidogenic peptides (22,23,25,29), when we exclude the edge residues of each sheet that display large distortion, the average twist angle is ∼11.5°. Second, β-strands and sheets that are largely exposed to solvent display larger twisting and bending than those protected from water access. It can be seen that the twisting and bending angles of adjacent β-hairpin units in the middle of β-sheets underwent relatively smaller strand-strand orientation than those of adjacent β-hairpin units at the edge sheets, i.e., there was a general trend of large twisting and bending angles of edge pairs and small angles of center pairs (i.e., a “U”-shape for the bending angle). This is not surprising; it indicates that the twisting and bending of the β-structure result from the competition between protein-protein and protein-water interactions (30,31). Third, the aromatic ladder can also rigidify and flatten these β-sheets to some extent. Structural bending and twisting could be a generic feature of β-sheets, optimizing a combination of factors such as side-chain contacts, VDW interactions, and protein-solvent interactions.
Structural comparison with β-sheet-rich amyloid fibrils
The twisted β-sheets are not unique to PSAM assemblies. A number of simulation studies have shown that many amyloid fibrils, such as Alzheimer's Aβ (22,32); GNNQQNY from the yeast prion protein (20,31); the human islet amyloid polypeptide IAPP22-27 (NFGAIL), KFFE, KVVE, KLLE, and KAAE (33,34); the human calcitonin hormone (residues 15–19, DFNKF); and NHVTLSQ from human β2-microglobulin (25) display twisted and bent structures rather than idealized flat structures. Twisted β-sheets are a common structural feature of amyloid organization and other protein and peptide assemblies that contain β-sheets (35,36). Twisted and bent structures can minimize the exposure of hydrophobic residues to the solvent and increase the VDW interactions via side-chain contacts, leading to greater structural stability. Multiple β-sheet associations can offer different assembly pathways, leading to heterogeneous structures with distinct sizes and morphologies (e.g., rod, linear, annular, or sphere), and enhancing the overall stability of the assemblies.
A β-strand-turn-β-strand motif similar to that used in our simulations has been observed in amyloid protofilaments of Aβ, human CA150 (37), and the β2-microglobulin (38,39). The net bend angle of 180° brings the two β-sheets into contact through side chain-side chain interactions (40). As compared with an extended conformation, the β-strand-turn-β-strand motif induces more tightly packed residues in a limited space, leading to more intramolecular interactions via hydrophobic interactions, hydrogen bonds, steric zippers, and salt bridges, which are absent in a single-strand peptide. Similarly to the PSAM peptide, buried intrasheet salt bridges at the loop region are also observed between Asp23 and Lys28 in Aβ (41,42) and between Glu7 and Arg24 in human CA150 peptide (37), further contributing to structural stability. Thus, a loop-induced motif could be inherently stable because of these intramotif interactions.
Conclusions
β-sheet-rich motifs are important structural templates in protein and peptide self-assemblies. The β-hairpin PSAM designed by Koide and co-workers (5,10,21) has a high β-sheet content similar to that of amyloid fibrils, but also has high solubility. PSAMs provide a model system for structural and quantitative thermodynamic studies of β-sheet-rich assemblies because they overcome the fundamental difficulties of insolubility and recalcitrance to crystallization of amyloid aggregates associated with neurodegenerative diseases. Koide and his colleagues covalently linked the β-hairpins to create single sheets, and stabilized the systems by end-capping the PSAMs with bulky protein domains. In this study, we performed a series of MD simulations to characterize the conformations, dynamics, and stability of PSAMs in the presence and absence of end-capping proteins. In the simulations of the PSAMs with the end-capping proteins, all models, including single-layer and three-layer β-sheets, were very stable, primarily due to the end-capping proteins, which packed against the edges and restricted the motion of the β-sheets. In the simulations without end-capping proteins, multiple β-sheet assemblies (two-, three-, and four-layer sheets) displayed relatively smaller structural deviations than the single β-sheet assemblies. In parallel, a comparison of the results from the wild-type and sequences with mutated aromatic residues suggests that the intersheet hydrogen bonds and side-chain packing are the driving forces in associating these β-sheets, whereas the intrasheet salt bridges provide subsequent stabilization to maintain the β-hairpin motif. The introduction of the conformationally restricted aromatic ladders in the β-strand region appears to provide an additional stabilizing force for multiple β-sheets. The simulation data also indicate that all of these assemblies adopt twisted and bent structures, similar to amyloid fibrils. The twisting and bending are mediated by both peptide-peptide and peptide-water interactions. Our structural and energetic analyses, combined with prior knowledge of amyloid fibrils and β-sheets in globular proteins, point to twisting and bending as intrinsic features of β-sheet-rich assemblies. Although the simulated models do not exhaustively cover all packing possibilities of β-sheet-rich assemblies, they represent a typical set of likely β-sheet structures optimized by the maximal overlap of side-chain contact, hydrophobic interactions, and hydrogen bonds. We conclude that the soluble PSAMs appear to be a good mimic of the insoluble amyloid aggregates. In addition, it should be noted that, similarly to multiple structural morphologies in amyloid oligomers, our parallel and in-register β-sheet models do not exclude other possible β-sheet organizations, such as antiparallel, mixed parallel/antiparallel packed, and unregistered packed organizations.
Supporting Material
MD simulation protocol and seven figures are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(09)01574-4.
Supporting Material
Acknowledgments
We thank Dr. Shohei Koide for his valuable comments and suggestions. This study utilized the high-performance Biowulf PC cluster at the Ohio Supercomputer Center.
This work was supported by the American Chemical Society Petroleum Research Fund (48188-G5) and 3M Non-Tenured Faculty Award (J. Zheng). This project was funded in whole or in part with federal funds from the U.S. National Cancer Institute, National Institute of Health, under contract number HHSN261200800001E. This research was supported (in part) by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. government.
Contributor Information
Ruth Nussinov, Email: ruthnu@helix.nih.gov.
Jie Zheng, Email: zhengj@uakron.edu.
References
- 1.Hill D.J., Mio M.J., Moore J.S. A field guide to foldamers. Chem. Rev. 2001;101:3893–4012. doi: 10.1021/cr990120t. [DOI] [PubMed] [Google Scholar]
- 2.Alemán C., Zanuy D., Nussinov R. Concepts and schemes for the re-engineering of physical protein modules: generating nanodevices via targeted replacements with constrained amino acids. Phys. Biol. 2006;3:S54–S62. doi: 10.1088/1478-3975/3/1/S06. [DOI] [PubMed] [Google Scholar]
- 3.Schafmeister C.E., Brown Z.Z., Gupta S. Shape-programmable macromolecules. Acc. Chem. Res. 2008;41:1387–1398. doi: 10.1021/ar700283y. [DOI] [PubMed] [Google Scholar]
- 4.McCullagh M., Prytkova T., Schatz G.C. Modeling self-assembly processes driven by nonbonded interactions in soft materials. J. Phys. Chem. B. 2008;112:10388–10398. doi: 10.1021/jp803192u. [DOI] [PubMed] [Google Scholar]
- 5.Makabe K., Biancalana M., Koide S. High-resolution structure of a self-assembly-competent form of a hydrophobic peptide captured in a soluble β-sheet scaffold. J. Mol. Biol. 2008;378:459–467. doi: 10.1016/j.jmb.2008.02.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dobson C.M. Experimental investigation of protein folding and misfolding. Methods. 2004;34:4–14. doi: 10.1016/j.ymeth.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 7.Lamour Y. Alzheimer's disease: a review of recent findings. Biomed. Pharmacother. 1994;48:312–318. doi: 10.1016/0753-3322(94)90177-5. [DOI] [PubMed] [Google Scholar]
- 8.Lashuel H.A., Lansbury P.T. Are amyloid diseases caused by protein aggregates that mimic bacterial pore-forming toxins? Q. Rev. Biophys. 2006;39:167–201. doi: 10.1017/S0033583506004422. [DOI] [PubMed] [Google Scholar]
- 9.Selkoe D.J. Alzheimer's disease: genes, proteins, and therapy. Physiol. Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 10.Makabe K., McElheny D., Koide S. Atomic structures of peptide self-assembly mimics. Proc. Natl. Acad. Sci. USA. 2006;103:17753–17758. doi: 10.1073/pnas.0606690103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Benseny-Cases N., Cócera M., Cladera J. Conversion of non-fibrillar β-sheet oligomers into amyloid fibrils in Alzheimer's disease amyloid peptide aggregation. Biochem. Biophys. Res. Commun. 2007;361:916–921. doi: 10.1016/j.bbrc.2007.07.082. [DOI] [PubMed] [Google Scholar]
- 12.Fändrich M., Zandomeneghi G., Diekmann S. Apomyoglobin reveals a random-nucleation mechanism in amyloid protofibril formation. Acta Histochem. 2006;108:215–219. doi: 10.1016/j.acthis.2006.03.012. [DOI] [PubMed] [Google Scholar]
- 13.Guo Z., Eisenberg D. The structure of a fibril-forming sequence, NNQQNY, in the context of a globular fold. Protein Sci. 2008;17:1617–1623. doi: 10.1110/ps.036368.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Takano K., Endo S., Kanaya S. Structure of amyloid β fragments in aqueous environments. FEBS J. 2006;273:150–158. doi: 10.1111/j.1742-4658.2005.05051.x. [DOI] [PubMed] [Google Scholar]
- 15.Stott K., Blackburn J.M., Perutz M. Incorporation of glutamine repeats makes protein oligomerize: implications for neurodegenerative diseases. Proc. Natl. Acad. Sci. USA. 1995;92:6509–6513. doi: 10.1073/pnas.92.14.6509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hosia W., Bark N., Tjernberg L. Folding into a β-hairpin can prevent amyloid fibril formation. Biochemistry. 2004;43:4655–4661. doi: 10.1021/bi036248t. [DOI] [PubMed] [Google Scholar]
- 17.Nelson R., Sawaya M.R., Eisenberg D. Structure of the cross-β spine of amyloid-like fibrils. Nature. 2005;435:773–778. doi: 10.1038/nature03680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Esposito L., Pedone C., Vitagliano L. Molecular dynamics analyses of cross-β-spine steric zipper models: β-sheet twisting and aggregation. Proc. Natl. Acad. Sci. USA. 2006;103:11533–11538. doi: 10.1073/pnas.0602345103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zheng J., Ma B., Nussinov R. Structural stability and dynamics of an amyloid-forming peptide GNNQQNY from the yeast prion sup-35. Biophys. J. 2006;91:824–833. doi: 10.1529/biophysj.106.083246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang J., Tan C., Luo R. All-atom computer simulations of amyloid fibrils disaggregation. Biophys. J. 2008;95:5037–5047. doi: 10.1529/biophysj.108.131672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Biancalana M., Makabe K., Koide S. Aromatic cross-strand ladders control the structure and stability of β-rich peptide self-assembly mimics. J. Mol. Biol. 2008;383:205–213. doi: 10.1016/j.jmb.2008.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zheng J., Jang H., Nussinov R. Modeling the Alzheimer Aβ 17–42 fibril architecture: tight intermolecular sheet-sheet association and intramolecular hydrated cavities. Biophys. J. 2007;93:3046–3057. doi: 10.1529/biophysj.107.110700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zheng J., Jang H., Nussinov R. Annular structures as intermediates in fibril formation of Alzheimer Aβ 17–42. J. Phys. Chem. B. 2008;112:6856–6865. doi: 10.1021/jp711335b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zheng J., Ma B., Nussinov R. Molecular dynamics simulations of Alzheimer Aβ40 elongation and lateral association. Front. Biosci. 2008;13:3919–3930. doi: 10.2741/2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zheng J., Jang H., Nussinov R. β2-microglobulin amyloid fragment organization and morphology and its comparison to Aβ suggests that amyloid aggregation pathways are sequence specific. Biochemistry. 2008;47:2497–2509. doi: 10.1021/bi7019194. [DOI] [PubMed] [Google Scholar]
- 26.Kabsch W., Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 27.Im W., Lee M.S., Brooks C.L. Generalized Born model with a simple smoothing function. J. Comput. Chem. 2003;24:1691–1702. doi: 10.1002/jcc.10321. [DOI] [PubMed] [Google Scholar]
- 28.Takeda T., Klimov D.K. Replica exchange simulations of the thermodynamics of Aβ fibril growth. Biophys. J. 2009;96:442–452. doi: 10.1016/j.bpj.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chebaro Y., Mousseau N., Derreumaux P. Structures and thermodynamics of Alzheimer's amyloid-β Aβ (16–35) monomer and dimer by replica exchange molecular dynamics simulations: implication for full-length Aβ fibrillation. J. Phys. Chem. B. 2009;113:7668–7675. doi: 10.1021/jp900425e. [DOI] [PubMed] [Google Scholar]
- 30.Periole X., Rampioni A., Mark A.E. Factors that affect the degree of twist in β-sheet structures: a molecular dynamics simulation study of a cross-β filament of the GNNQQNY peptide. J. Phys. Chem. B. 2009;113:1728–1737. doi: 10.1021/jp8078259. [DOI] [PubMed] [Google Scholar]
- 31.Berryman J.T., Radford S.E., Harris S.A. Thermodynamic description of polymorphism in Q- and N-rich peptide aggregates revealed by atomistic simulation. Biophys. J. 2009;97:1–11. doi: 10.1016/j.bpj.2009.03.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bu Z., Shi Y., Tycko R. Molecular alignment within β-sheets in Aβ (14–23) fibrils: solid-state NMR experiments and theoretical predictions. Biophys. J. 2007;92:594–602. doi: 10.1529/biophysj.106.091017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wei G., Mousseau N., Derreumaux P. Sampling the self-assembly pathways of KFFE hexamers. Biophys. J. 2004;87:3648–3656. doi: 10.1529/biophysj.104.047688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bellesia G., Shea J.-E. What determines the structure and stability of KFFE monomers, dimers, and protofibrils? Biophys. J. 2009;96:875–886. doi: 10.1016/j.bpj.2008.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zheng J., Ma B., Nussinov R. Consensus features in amyloid fibrils: sheet-sheet recognition via a (polar or nonpolar) zipper structure. Phys. Biol. 2006;3:P1–P4. doi: 10.1088/1478-3975/3/3/P01. [DOI] [PubMed] [Google Scholar]
- 36.Tsai H.-H., Gunasekaran K., Nussinov R. Sequence and structure analysis of parallel β helices: implication for constructing amyloid structural models. Structure. 2006;14:1059–1072. doi: 10.1016/j.str.2006.03.015. [DOI] [PubMed] [Google Scholar]
- 37.Ferguson N., Becker J., Fersht A.R. General structural motifs of amyloid protofilaments. Proc. Natl. Acad. Sci. USA. 2006;103:16248–16253. doi: 10.1073/pnas.0607815103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Iwata K., Fujiwara T., Goto Y. 3D structure of amyloid protofilaments of β2-microglobulin fragment probed by solid-state NMR. Proc. Natl. Acad. Sci. USA. 2006;103:18119–18124. doi: 10.1073/pnas.0607180103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sasahara K., Yagi H., Goto Y. Heat-triggered conversion of protofibrils into mature amyloid fibrils of β2-microglobulin. Biochemistry. 2007;46:3286–3293. doi: 10.1021/bi602403v. [DOI] [PubMed] [Google Scholar]
- 40.Naito A., Kawamura I. Solid-state NMR as a method to reveal structure and membrane-interaction of amyloidogenic proteins and peptides. Biochim. Biophys. Acta. 2007;1768:1900–1912. doi: 10.1016/j.bbamem.2007.03.025. [DOI] [PubMed] [Google Scholar]
- 41.Ma B., Nussinov R. Stabilities and conformations of Alzheimer's β-amyloid peptide oligomers (Aβ 16–22, Aβ 16–35, and Aβ 10–35): sequence effects. Proc. Natl. Acad. Sci. USA. 2002;99:14126–14131. doi: 10.1073/pnas.212206899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Petkova A.T., Yau W.M., Tycko R. Experimental constraints on quaternary structure in Alzheimer's β-amyloid fibrils. Biochemistry. 2006;45:498–512. doi: 10.1021/bi051952q. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.