

Abstract
Intravenous immunoglobulin (IVIg) is used increasingly in the management of patients with neurological conditions. The efficacy and safety of IVIg treatment in chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) and Guillain–Barré syndrome (GBS) have been established clearly in randomized controlled trials and summarized in Cochrane systematic reviews. However, questions remain regarding the dose, timing and duration of IVIg treatment in both disorders. Reports about successful IVIg treatment in other neurological conditions exist, but its use remains investigational. IVIg has been shown to be efficacious as second-line therapy in patients with dermatomyositis and suggested to be of benefit in some patients with polymyositis. In patients with inclusion body myositis, IVIg was not shown to be effective. IVIg is also a treatment option in exacerbations of myasthenia gravis. Studies with IVIg in patients with Alzheimer's disease have reported increased plasma anti-Aβ antibody titres associated with decreased Aβ peptide levels in the cerebrospinal fluid following IVIg treatment. These changes at the molecular level were accompanied by improved cognitive function, and large-scale randomized trials are under way.
Keywords: Alzheimer's disease, autoimmune myopathy, chronic inflammatory demyelinating polyradiculoneuropathy, Guillain–Barré syndrome, intravenous immunoglobulin
Introduction
In many institutions, neurological diseases have become responsible for more use of intravenous immunoglobulin (IVIg) than any other acquired diseases. Following Paul Imbach's observation that IVIg is effective in the treatment of thrombocytopenia [1], its use was tested in chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) in the Netherlands. Benefits were reported initially in observational studies [2], which were established later by randomized controlled trials, summarized in a Cochrane systematic review and confirmed in the recently completed Immune Globulin Intravenous CIDP Efficacy (ICE) trial [3,4]. The ICE trial, a large randomized, double-blind, placebo-controlled, response conditional cross-over trial of IVIg in 117 patients with CIDP [4], led to the registration of IVIg (Gamunex) for CIDP in the United States and Canada. The latest information and remaining questions about IVIg in CIDP were discussed in a presentation by Dr Norman Latov.
Paradoxically for Guillain–Barré syndrome (GBS), the EMEA have authorized the use of IVIg in Europe, but the Food and Drug Administration (FDA) have not authorized it in the United States, although it is used widely there. It was again in the Netherlands where IVIg was first tested for its efficacy in GBS, and the first randomized controlled trials showed similar efficacy to plasma exchange [5–7]. In his presentation, Dr David Cornblath summarized the evidence for the use of IVIg in GBS, now drawn from several randomized controlled trials summarized in a Cochrane review [8]. Despite the results from these trials, there remains a need for more research to determine the efficacy of IVIg in disease variants and to perform dose-ranging studies, especially of a second IVIg dose in patients who do not begin to improve within a reasonable time after the first dose.
Most patients with GBS are now being treated with IVIg, but because they receive only one course, GBS does not account for large usage of IVIg. On the other hand, the smaller number of patients with chronic inflammatory neuropathies, who receive long-term repeated IVIg treatment, account for a high proportion of neurology department budgets. This includes not only CIDP but the related condition of multi-focal motor neuropathy (MMN), where a response to IVIg but not to any other treatment can be seen in more than three-quarters of patients [9].
There are other peripheral neuropathies in which there are reports of the efficacy of IVIg. These include diabetic amyotrophy [10], vasculitic peripheral neuropathy [11] and painful sensory neuropathy associated with Sjögren's syndrome [12]. The evidence for these conditions has been insufficient to earn a recommendation for the use of IVIg from national or international guidelines [13–15].
The possible use of IVIg has been explored for a wide range of neurological conditions besides peripheral neuropathies. It was shown that IVIg is clinically beneficial and reduces complement deposition in a randomized trial in dermatomyositis [16], as presented by Dr Marinos Dalakas. Based upon this evidence, IVIg was included in guidelines for managing corticosteroid-resistant disease [13–15].
No difference in efficacy between IVIg and plasma exchange for treating exacerbations of myasthenia gravis was shown in randomized trials: the evidence has been summarized in a Cochrane review [17]. Consequently, IVIg has been accepted as a treatment option for such exacerbations in national and international guidelines [13–15]. Considerable effort has been devoted to exploring a possible role for IVIg in multiple sclerosis, with negative results in secondary progressive disease and conflicting, but eventually negative, results in relapsing–remitting disease [18,19]. Anecdotal reports of benefit from IVIg have included its use in neuromyotonia and paraneoplastic syndromes [20,21], some forms of encephalitis [22], childhood treatment-resistant epilepsy [23] and narcolepsy [24].
The concept of testing IVIg as a possible treatment for Alzheimer's disease (AD) followed from observations that IVIg contained measurable quantities of anti-amyloid-beta (Aβ) antibodies [25], which are deficient in the blood [26] and spinal fluid [27] of patients with AD. The development of IVIg as an investigational treatment for AD was outlined by Dr Marc Weksler and the results of two open-label phase I studies [25,28] and a recently completed placebo-controlled phase II trial were presented. If an ongoing US phase III pivotal study confirms that IVIg slows progression of AD and is well tolerated, the future needs of millions of AD patients worldwide could exceed the supplies of IVIg available for all indications. Strategies will have to be developed to address this issue, which may include drawing upon novel alternative sources of pooled human immunoglobulin and platforms for generating mixed human monoclonal antibodies. Pursuing these and other strategies may become of paramount importance as neurological and non-neurological indications for IVIg continue to grow in the 21st century.
CIDP, presented by Norman Latov
CIDP is an autoimmune disease that targets the myelin sheaths of the peripheral nerves, leading to weakness, sensory loss and impairment of gait and coordination. There is no definitive test for CIDP, and in most patients diagnosis is based on the clinical presentation and demonstration of demyelinating abnormalities in electrodiagnostic studies. Numerous diagnostic criteria are available, which work well in both practice and research studies.
Given that CIDP is a treatable and potentially reversible disease, there is a pressing need for research into biomarkers that would enable the development of more reliable diagnostic tests [29,30].
The treatment often preferred for CIDP is IVIg, based on its demonstrated efficacy and safety, as confirmed by the recently published ICE study [4]. In this study, patients received an initial loading dose of 2 g/kg IVIg, followed by 1 g/kg at 3-week intervals, with those exhibiting improvement continuing treatment for up to 24 weeks. Improvement, as measured by the inflammatory neuropathy cause and treatment (INCAT) disability score, was seen in 54% of the patients in the treatment arm, compared with 21% of those receiving placebo (P = 0·002).
The initial dose used in the ICE study (2 g/kg) was similar to that used in practice. This dose was shown to be more effective than 1 g/kg or 0·25 g/kg in a previous trial [31], although higher doses were not examined. The initial dose is usually given over one or several days, depending on tolerability or convenience. Patients who do not respond to an initial dose may respond to subsequent doses, as was seen in the ICE trial and an earlier, smaller study [32]. In the ICE study, 44% of responders improved by 3 weeks after the initial treatment, and an additional 50% of patients responded only after a second dose of 1 g/kg at week 3, as measured at week 6 of the study [33]. However, it is not known whether even more patients would have improved if additional treatments had been given, as patients who did not show improvement, including those who were stable, were crossed-over at week 6. In clinical practice, initial responses have been seen up to 3 months into the treatment, and stabilization of previously progressive disease is considered to be a positive response. Additional studies are therefore needed to explore the full potential of IVIg therapy in these patients.
IVIg responsive patients in the ICE trial were treated with 1 g/kg every 3 weeks for up to 24 weeks, with the responsive patients re-randomized to continue treatment or placebo in phase 2 of the study for an additional 24 weeks. Continued improvement was observed in some patients at up to 32 weeks into the study [33]. Approximately 50% of the responders in the first phase of the study suffered a relapse during phase 2 when switched to placebo. Given the goal of achieving maximal improvement, a reasonable strategy would be to continue treatment until the improvement plateaus, before stopping to see whether additional treatments are still needed. Discontinuing the treatments prior to that point would risk leaving the patient with less than optimal function, although one study noted that patients in remission may continue to improve after the treatments were discontinued [34].
For purposes of the trial, patients in the ICE study were maintained on doses of 1 g/kg every 3 weeks. In practice, however, after initial treatment, follow-up doses of 0·5 g/kg every 2 weeks, 1 g/kg every 3 weeks or 2 g/kg every 4 weeks are used commonly, depending on individual preference.
Most patients who relapse require long-term maintenance therapy. The alternative, to treat only after a relapse, puts the patient in danger of developing irreversible axonal damage and increasing debility secondary to accumulated injuries [35]. A retrospective analysis of CIDP patients treated with different doses of IVIg showed that maintenance doses and schedules vary significantly between patients, arguing for individualized dosing that is determined empirically [36]. However, maintaining less than optimal levels of IVIg may result in further deterioration, so that dosing should be directed at maintaining maximal function [37].
CIDP is a treatable disease whose manifestations can be prevented by early diagnosis and treatment with IVIg. Additional efforts are needed, however, to develop more reliable diagnostic tests, establish optimal treatment regimens and increase awareness of this condition.
GBS in children, presented by David R. Cornblath
GBS is an autoimmune disorder of the peripheral nervous system. GBS in children and adults shares many features, but has several important differences. In both adults and children, GBS consists of four major subtypes: acute inflammatory demyelinating polyneuropathy (AIDP); acute motor axonal neuropathy (AMAN); acute motor and sensory axonal neuropathy (AMSAN); and Fisher syndrome. The subtypes can be differentiated by clinical, electrophysiological and pathological findings [38,39].
The incidence of GBS in children up to age 18 years is approximately one per 100 000/year, compared with approximately two per 100 000/year in adults. The incidence is lower in young children, while in adults there is an increasing incidence of GBS with advancing years [40]. In the United States and western Europe, AIDP is the most common GBS subtype, while in Asia and Latin America axonal forms are more frequent [41,42].
Diagnosis of GBS is made in the setting of the classic clinical scenario of a monophasic illness reaching a nadir within 4 weeks with symmetric weakness and sensory loss, areflexia and elevated cerebrospinal fluid (CSF) protein without pleocytosis [38,39]. Children present slightly differently. Leg or back pain occurs in the majority of children and disease progression is much faster, with 80% reaching nadir within 2 weeks [43]. While approximately 30% of adults require assisted ventilation, this is needed in only about 15% of children. In children, recovery is usually better and the mortality lower. Presumed antecedent inciting events, such as infections, occur in up to 80% of children [38,39].
While initial data on the pathology and pathogenesis of GBS were obtained primarily from adults, more recent studies have involved all age groups. Molecular mimicry probably plays an important role in the pathogenesis. Infection with a pathological agent such as Campylobacter jejuni leads to the formation of cross-reacting antibodies. In AIDP, such cross-reacting anti-myelin or anti-ganglioside antibodies attack Schwann cell surface epitopes of motor and sensory fibres. Subsequent complement activation and macrophage infiltration leads to multi-focal inflammatory demyelination with conduction failure and secondary axonal degeneration. AMAN and AMSAN are characterized by axonal/nodal antibody binding, complement activation, macrophage attachment at nodes, opening of the peri-axonal space and macrophage infiltration in motor axons in AMAN, or in motor and sensory axons in AMSAN [38,39]. In severe cases, secondary axonal degeneration is observed.
In controlled clinical trials in GBS patients, which have included only small numbers of those less than 18 years old, plasma exchange or the use of IVIg has been shown to be beneficial [44]. Supportive care is critical at all ages. Studies performed specifically in children have been smaller than those in adults, but the results of trials in children with severe GBS support the beneficial effect of IVIg seen in adults [45,46].
Children with mild GBS (i.e. patients able to walk 5 m unaided) who received IVIg 0·5 g/kg/day over 2 days had significantly faster improvement (median 8 days) compared with patients receiving supportive treatment (median 32 days) (Table 1) [47]. In the same trial, children with severe GBS (i.e. unable to walk 5 m unaided) were randomized to receive 1 g/kg IVIg over 2 days or 0·4 g/kg IVIg over 5 days. While recovery did not differ significantly between the children treated for 2 days versus 5 days, relapses of the disease were found to be more frequent in the children treated with the shorter course and higher dose, indicating that longer treatment may be more beneficial [47]. Although the results are encouraging, due to the small number of patients involved, further studies are needed to confirm the results in children.
Table 1.
Distribution of disability scores at randomization, at the nadir of the disease, and 4 weeks after randomization in the early treatment study [47]. In group A, pediatric patients were randomized initially for no treatment (four of the children were treated with intravenous immunoglobulin (IVIg) in the later course because of loss of independent walking). In group B, pediatric patients were randomized to IVIg.
| No. of children |
||||||
|---|---|---|---|---|---|---|
| At randomization |
At nadir |
4 weeks after randomization |
||||
| Score | Group A | Group B | Group A | Group B | Group A | Group B |
| Normal | 0 | 0 | 0 | 0 | 0 | 3 |
| Able to run | 0 | 1 | 0 | 0 | 1 | 6 |
| Walks 5 m unaided | 7 | 12 | 2 | 8 | 3 | 3 |
| Walks with aid | 0 | 1 | 2 | 3 | 1 | 2 |
| No walking, lifts legs | 0 | 0 | 1 | 0 | 1 | 0 |
| Not able to lift legs | 0 | 0 | 1 | 3 | 1 | 0 |
| Artificial ventilation | 0 | 0 | 1 | 0 | 0 | 0 |
| n | 7 | 14 | 7 | 14 | 7 | 14 |
| Median score | 2 | 2 | 3 | 2 | 2 | 1 |
| 95% CI | 2–2 | 1–3 | 2–6 | 2–5 | 1–5 | 0–3 |
| P* | 0·80 | 0·25 | 0·025 | |||
Reproduced with permission from [47], copyright© 2005 by the AAP. CI: confidence interval.
Mann–Whitney test.
Other aspects of GBS therapy remain unresolved and require future research. Additional primary treatments are needed, as up to 20% of patients with GBS die or are unable to walk after 1 year. Treatments to enhance nerve regeneration and to improve function in existing but partially repaired nerves are also required. The Inflammatory Neuropathy Consortium of the Peripheral Nerve Society defined a need for trials of IVIg treatment in mild GBS and Fisher syndrome, an IVIg dose-finding study in GBS and studies on the use of complement inhibitors and sodium channel blockers. Administration of a second IVIg dose in GBS patients who are still bed-bound 2 weeks after the first course may be beneficial and also requires further investigation.
IVIg for the treatment of inflammatory muscular disorders, presented by Marinos C. Dalakas
The autoimmune myopathies are rare inflammatory diseases characterized by muscle weakness, which is usually proximal, painless and of insidious onset. The three groups of autoimmune myopathies are dermatomyositis (DM), polymyositis (PM) and inclusion body myositis (IBM) [48]. Several small controlled trials with high-dose IVIg have been conducted in patients with DM and IBM; however, no controlled studies in patients with PM have been carried out so far, due to the difficulty in obtaining large enough numbers of patients with this rare condition [49].
DM is an inflammatory disease, affecting skin and muscle and causing varying degrees of muscle weakness, ranging from mild to severe. In this condition, prominent inflammation is observed usually at the periphery of the fascicle, leading to atrophy of the fibres around the fascicle. DM is characterized by complement-mediated microangiopathy that begins with complement activation in the periphery that leads eventually to the formation of membrane attack complexes (MACs), which are deposited on the capillaries causing destruction of capillaries [50]. A number of cytokines and chemokines are thought to be in involved in the process.
The therapeutic approach to DM treatment usually involves steroids or immunosuppressants, such as azathioprine, mycophenolate mofetil, methotrexate, cyclophosphamide or cyclosporine. If these are not sufficient to control the disease, other alternatives include polyclonal IVIgs or monoclonal antibodies. In DM, IVIg is thought to work by inhibiting complement consumption and intercepting membrane attack complexes, suppressing cytokines, adhesion molecules and fibrogenetic factors, and altering biologically relevant immunoregulatory or tissues remodelling genes [50].
A double-blind, randomized, placebo-controlled study was conducted in DM patients who were resistant or partially responsive to conventional therapies [16]. The patients continued to receive the same low doses of prednisone and were assigned randomly to receive one infusion of IVIg (2 g/kg body weight) or placebo per month for 3 months, with the option of crossing-over to the alternative therapy for 3 more months. The results showed that IVIg is very effective in improving both muscle strength and skin rash. The clinical benefit, which was impressive in patients with early disease, was associated with improvements in the muscle cytoarchitecture. Quantitative histological studies in repeated muscle biopsies showed a statistically significant increase in the size of muscle fibres and the number of capillaries with normalization of the capillary diameter. Resolution of the aberrant immunopathological parameters, including interception of complement activation products and down-regulation of T cells, intercellular adhesion molecule-1 (ICAM-I), vascular cell adhesion molecule (VCAM), transforming growth factor (TGF)-β and major histocompatibility complex (MHC)-I molecules, was also noted [51,52]. Further, a number of immunoregulatory and structural genes were modified in patient muscle biopsies after therapy. Based on these positive clinical findings, IVIg in combination with prednisone was recommended recently by the European Federation of Neurological Societies (EFNS) as a second-line treatment for patients with DM [13].
While DM is a complement-mediated vasculopathy, PM is mediated by T cells. Controlled studies of IVIg in this disease have not been performed, and only one study has examined long-term outcomes in PM patients. This study showed that 25 of 35 patients (71%) responded to treatment with high-dose IVIg in combination with other immuno suppressants after 6 months of treatment. After 51 months, 50% of the responders were still responding well, with 12 patients in full remission without any medication [53].
IBM is a progressive inflammatory skeletal muscle disease that presents with a distinctive pattern of weakness in the wrist and finger flexors and quadriceps muscles. It is characterized by inflammatory cells surrounding myofibres and rimmed vacuoles [54]. In addition to inflammatory processes caused by CD8+ T cell cytotoxicity, a degenerative process is also involved. Numerous medications, such as steroids, immunosuppressants, radiation and interferon (IFN)-β, have been tried for the treatment of this disease, mostly unsuccessfully.
The first study of IVIg treatment in IBM patients showed only marginal benefits. However, an improvement in the ability to swallow was observed, implying mild regional benefits [55]. In another study with 37 IBM patients, IVIg treatment combined with prednisone did not have a significant benefit [56].
It is interesting to note that IVIg modified certain immunoregulatory and structural genes in the muscles of DM patients who responded to IVIg therapy. For example, the expression of the chemokine Mig/CXCL9 gene was up-regulated after IVIg treatment in the muscles of patients with DM, but not in patients with IBM, who did not respond well to IVIg therapy. Expression of the anosmin-1/KAL-1 gene, which encodes a protein involved in fibrosis, was reduced after IVIg therapy in DM patients, but was unchanged in IBM patients (Fig. 1) [57]. These results suggest that some molecules such as anosmin-1/KAL-1 may be a marker of response to IVIg therapy, but additional markers need to be explored.
Fig. 1.
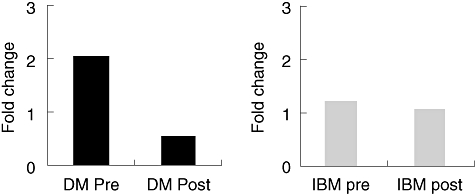
Anosmin-1 (KAL-1) is down-regulated in dermatomyositis (DM) but not inclusion body myositis (IBM) after intravenous immunoglobulin. Microarray results.
IVIg in AD, presented by Marc Weksler
AD is the most common neurodegenerative disorder leading to dementia and irreversible loss of neurones. The pathological hallmarks of AD are extracellular accumulation of Aβ peptides, 40–42 amino acid fragments of the beta-amyloid precursor protein (APP), as senile plaques and intracellular neurofibrillary tangles composed of tau proteins.
A mouse model for AD, the platelet-derived growth factor-driven APP (PDAPP) transgenic mouse, was developed in 1995 by overexpression of human mutant APP [58]. These mice develop many of the pathological hallmarks of AD, including Aβ deposits, neuritic plaques, synaptic loss and impaired memory. Mouse models for AD have since led to a better understanding of the disease and have facilitated the investigation of treatment options.
The first immunotherapeutic approach to AD showed that active immunization of these transgenic mice with Aβ peptides inhibits the formation and promotes the clearance of Aβ plaques [59,60]. Subsequent studies demonstrated that Aβ immunization also reduces cognitive dysfunction. Aβ vaccination was investigated in a mouse model, where mice develop learning deficits as amyloid accumulates. At an age when untreated transgenic mice show memory deficits, the Aβ-vaccinated transgenic mice showed cognitive performance superior to that of the control transgenic mice and performed almost as well as non-transgenic mice in a water-maze test [61].
These promising results led to clinical studies of active immunization in humans with AD [62,63]. These studies, however, were complicated by the development of meningoencephalitis in 6% of the patients treated with vaccine AN1792 in a phase II clinical trial [62,63]. Furthermore, only 20% of the patients immunized with AN1792 developed a twofold increase in anti-Aβ antibodies.
However, progress was made with the discovery that peripheral administration of antibodies against Aβ peptide could reduce amyloid burden to a similar extent as active immunization [64]. These results were found despite the relatively modest serum levels of the anti-Aβ antibodies that were administered, and the small percentage of these antibodies that crossed the blood–brain barrier and entered the central nervous system. Passive immunization had the advantage that the potentially harmful activation of host T cells could be avoided.
Based on the finding that externally administered antibodies were able to protect PDAPP mice from AD, it was hypothesized that high titres of natural anti-Aβ antibodies may protect humans from AD, while low levels may predispose certain individuals to the development of AD. Studies have found reduced levels of anti-Aβ antibodies both in the serum [26] and CSF [27] of patients with AD. These results were confirmed in a mouse AD model, where anti-Aβ antibody levels were measured at various ages [65]. The level of anti-Aβ antibody fell significantly at the age of 9 months, at the age when amyloid plaques started to appear in the brain of the mice, and was persistently low thereafter. The observed effect was not due to a general deterioration of the immune response, as immunoglobulin levels were generally found to be higher in the older compared with the younger mice. Autoantibody-decorated plaques were found frequently in patients with AD and patients with low antibody-levels were shown to harbour more diffuse plaques than patients with high levels (Fig. 2) [66]. Autoantibodies against Aβ may therefore be important for maintaining plaque homeostasis.
Fig. 2.
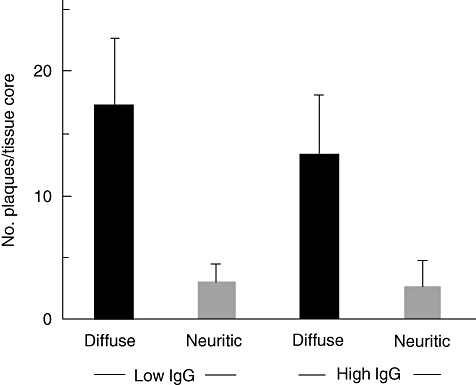
High serum amyloid-beta (Aβ)-autoantibody levels are associated with decreased diffuse plaques in Alzheimer's disease (AD). AD patients were allocated to a group with high plaque labelling index [high immunoglobulin (Ig)G] and one with low plaque labelling index (low IgG). Patients with low plaque scores (low IgG) harboured more diffuse plaques, but not neuritic plaques than patients with high plaque scores (high IgG). Figure reproduced with permission from [66].
IVIg has been shown to contain autoantibodies against many states of Aβ peptide aggregation including monomers, oligomers and fibrils and may therefore have a distinct advantage over monoclonal anti-Aβ until the precise pathogenic state(s) of the Aβ peptide is known [67]. The potential benefits of IVIg were first shown in five AD patients [68]. Although the small sample size limited conclusions, this study paved the way for further studies employing an open-label dose-ranging trial, including eight patients with mild AD. IVIg was added to approved AD therapies for 6 months, discontinued, and then resumed for another 9 months. IVIg infusion increased the level of plasma anti-Aβ antibody titres, which was associated with an increase in Aβ peptide levels in the serum and a decrease in Aβ peptide levels in the CSF [28]. Cognitive function, as measured by Mini-Mental State Evaluation (MMSE), showed improvement in six of eight patients after 6 months of IVIg therapy. When IVIg treatment was stopped, Aβ peptide levels in the CSF receded to their pretreatment levels, but improved again when treatment was reinitiated after 3 months (Fig. 3). Follow-up to 30 months has now been performed, with all patients having stabilized in their disease progression.
Fig. 3.
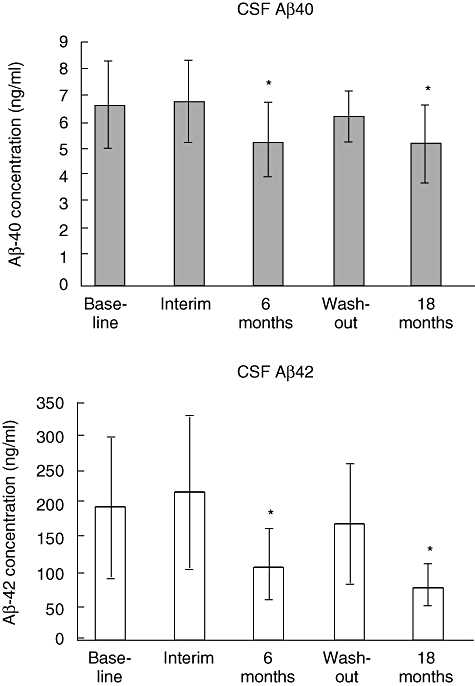
Cerebrospinal fluid (CSF) amyloid-beta (Aβ) decreases during intravenous immunoglobulin (IVIg) treatment. Both CSF Aβ40 and Aβ42 levels are decreased significantly following IVIg treatment for 6 months. When IVIg treatment was stopped (wash-out), Aβ peptide levels in the CSF receded to their pretreatment levels, but improved again when treatment was reinitiated after 3 months. *Significant difference from baseline by t-test at P< 0·005.
Subsequently, a phase II study was performed in three groups of eight patients with mild to moderate AD (MMSE 14–26) who were assigned randomly to groups receiving either 0·4 g IVIg/kg/month, 0·8 g IVIg/kg/month or placebo (saline) [69]. The levels of anti-Aβ antibodies and of Aβ 40/42 peptides in plasma and CSF samples were quantified by enzyme-linked immunosorbent assay (ELISA). Cognitive and behavioural assessments, using the Alzheimer's Disease Assessment Scale-cognitive subscale (ADAS-cog), Clinical Global Impression of Change (CGIC), modified Mini-Mental State (3MS) and activities of daily living (ADL) scales, were performed before and after 3, 6 and 9 months of infusion therapy. Cerebral glucose uptake was measured by positron emission tomography (PET) scanning after 18-fluorodeoxyglucose injection before and 6 months after infusion therapy.
IVIg infusions increased anti-Aβ antibody and Aβ 40/42 peptides in plasma and decreased Aβ 40/42 peptides in CSF compared with values prior to treatment. While glucose uptake in AD vulnerable regions decreased by 6–8% in untreated patients, uptake remained stable in IVIg-treated patients (unpublished data).
Large-scale studies are under way to determine the role of IVIg in the treatment of AD. If positive, this would lead to a further increase in the demand for immunoglobulin therapeutics. Alternative sources of immunoglobulins may have to be considered to satisfy patient needs, which may include polyclonal recombinant antibodies or the production of artificial human antibodies in transgenic animals.
Summary
IVIg is used increasingly in neurological diseases. While its efficacy and safety in CIDP and GBS have been demonstrated clearly, questions remain regarding the dose, timing and duration of IVIg treatment in both disorders. Further studies are required to establish its efficacy firmly in other inflammatory neuro muscular disorders. In AD, large-scale randomized trials are under way, and the results of these studies are awaited eagerly.
Acknowledgments
The authors would like to thank nspm ltd for providing medical writing services, with financial support through an unrestricted educational grant from CSL Behring AG.
Disclosures
RH has received honoraria for speaking or consultancies from Baxter, CSL Behring, Genzyme, Kedrion, LFB, Octapharma and Talecris. RH also served on the steering committee of the Talecris ICE trial. MCD has received honoraria for speaking or consultancies from Baxter, CSL Behring, Octapharma and Talecris and served in the steering committee of Talecris's ICE trial.
DC has been a consultant for Merck, Pfizer, Mitshubishi Pharma, Sangamo, Sanofi-Aventis, Bristol-Myers Squibb, Eisai, Octapharma, Sun Pharma, Acorda, DP Clinical, Exelixis, Geron, Johnson & Johnson, Genyzme, Cebix, Abbott, CSL Behring, Bionevia. DC has been on the Data Safety Monitoring Board for Pfizer, Schwarz Biosciences, Avigen, FoldRx, Johnson & Johnson, GlaxoSmithKline. DC has been involved in Technology Licensing for Abbott, Johnson & Johnson, Sanofi-Aventis.
NL has acted as a paid consultant to Quest Diagnostics, CSL Behring, Talecris Biopharmaceuticals, and Octapharma, and received research funding from Talecris Biopharmaceuticals.
MEW has received honorarium and travel expenses to the Interlaken Symposium. MEW has also received honoraria for speaking and research support for clinical studies of IVIg therapy in patients with Alzheimer's disease from Baxter Bioscience.
NR declared that he has no conflicts of interest.
References
- 1.Imbach P, Barandun S, d'Apuzzo V, et al. High-dose intravenous gammaglobulin for idiopathic thrombocytopenic purpura in childhood. Lancet. 1981;1:1228–31. doi: 10.1016/s0140-6736(81)92400-4. [DOI] [PubMed] [Google Scholar]
- 2.Vermeulen M, van der Meché FGA, Speelman JD, et al. Plasma and gammaglobulin infusion in chronic inflammatory polyneuropathy. J Neurol Sci. 1985;70:317–26. doi: 10.1016/0022-510x(85)90173-x. [DOI] [PubMed] [Google Scholar]
- 3.Eftimov F, Winer JB, Vermeulen M, et al. Intravenous immunoglobulin for chronic inflammatory demyelinating polyradiculoneuropathy [update of Cochrane Database Syst Rev 2002;(2):CD001797. Cochrane Database Syst Rev. 2009;1:CD001797. doi: 10.1002/14651858.CD001797.pub2. [DOI] [PubMed] [Google Scholar]
- 4.Hughes RA, Donofrio P, Bril V, et al. Intravenous immune globulin (10% caprylate-chromatography purified) for the treatment of chronic inflammatory demyelinating polyradiculoneuropathy (ICE study): a randomised placebo-controlled trial. Lancet Neurol. 2008;7:136–44. doi: 10.1016/S1474-4422(07)70329-0. [DOI] [PubMed] [Google Scholar]
- 5.Kleyweg RP, van der Meché FGA, Meulstee J. Treatment of Guillain–Barré Syndrome with high dose gammaglobulin. Neurology. 1988;38:1639–42. doi: 10.1212/wnl.38.10.1639. [DOI] [PubMed] [Google Scholar]
- 6.van der Meché FG, Schmitz PI. A randomized trial comparing intravenous immune globulin and plasma exchange in Guillain–Barré syndrome. Dutch Guillain–Barré Study Group. N Engl J Med. 1992;326:1123–9. doi: 10.1056/NEJM199204233261705. [DOI] [PubMed] [Google Scholar]
- 7.Plasma Exchange/Sandoglobulin Guillain–Barré Syndrome Trial Group. Randomised trial of plasma exchange, intravenous immunoglobulin, and combined treatments in Guillain–Barré syndrome. Lancet. 1997;349:225–30. [PubMed] [Google Scholar]
- 8.Hughes RC, Raphaël J-C, Swan AV, van Doorn PA. Intravenous immunoglobulin for Guillain–Barré syndrome. Cochrane Database Syst Rev. 2006;1:CD002063. doi: 10.1002/14651858.CD002063.pub3. [DOI] [PubMed] [Google Scholar]
- 9.van Schaik IN, Van den Berg LH, de Haan R. Intravenous immunoglobulin for multifocal motor neuropathy. Cochrane Database Syst Rev. 2005;2:CD004429. doi: 10.1002/14651858.CD004429.pub2. [DOI] [PubMed] [Google Scholar]
- 10.Krendel DA, Costigan DA, Hopkins LC. Successful treatment of neuropathies in patients with diabetes mellitus. Arch Neurol. 1995;52:1053–61. doi: 10.1001/archneur.1995.00540350039015. [DOI] [PubMed] [Google Scholar]
- 11.Levy Y, Uziel Y, Zandman G, et al. Response of vasculitic peripheral neuropathy to intravenous immunoglobulin. Ann NY Acad Sci. 2005;1051:779–86. doi: 10.1196/annals.1361.121. [DOI] [PubMed] [Google Scholar]
- 12.Kizawa M, Mori K, Iijima M, et al. Intravenous immunoglobulin treatment in painful sensory neuropathy without sensory ataxia associated with Sjogren's syndrome. J Neurol Neurosurg Psychiatry. 2006;77:967–9. doi: 10.1136/jnnp.2005.084533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Elovaara I, Apostolski S, van Doorn P, et al. EFNS guidelines for the use of intravenous immunoglobulin in treatment of neurological diseases: EFNS task force on the use of intravenous immunoglobulin in treatment of neurological diseases. Eur J Neurol. 2008;15:893–908. doi: 10.1111/j.1468-1331.2008.02246.x. [DOI] [PubMed] [Google Scholar]
- 14.Feasby T, Banwell B, Benstead T, et al. Guidelines on the use of intravenous immune globulin for neurologic conditions. Transfus Med Rev. 2007;21:S57–107. doi: 10.1016/j.tmrv.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 15.Lin HH, Spies JM, Lu JL, Pollard JD. Effective treatment of experimental autoimmune neuritis with human immunoglobulin. J Neurol Sci. 2007;256:61–7. doi: 10.1016/j.jns.2007.02.017. [DOI] [PubMed] [Google Scholar]
- 16.Dalakas MC, Illa I, Dambrosia JM, et al. A controlled trial of high-dose intravenous immune globulin infusions as treatment for dermatomyositis. N Engl J Med. 1993;329:1993–2000. doi: 10.1056/NEJM199312303292704. [DOI] [PubMed] [Google Scholar]
- 17.Gajdos P, Chevret S, Toyka K. Intravenous immunoglobulin for myasthenia gravis. Cochrane Database Syst Rev. 2003;2:CD002277. doi: 10.1002/14651858.CD002277. [DOI] [PubMed] [Google Scholar]
- 18.Fazekas F, Lublin FD, Li D, et al. Intravenous immunoglobulin in relapsing–remitting multiple sclerosis: a dose-finding trial. Neurology. 2008;71:265–71. doi: 10.1212/01.wnl.0000318281.98220.6f. [DOI] [PubMed] [Google Scholar]
- 19.Sorensen PS, Fazekas F, Lee M. Intravenous immunoglobulin G for the treatment of relapsing–remitting multiple-sclerosis: a meta-analysis. Eur J Neurol. 2002;9:557–63. doi: 10.1046/j.1468-1331.2002.00501.x. [DOI] [PubMed] [Google Scholar]
- 20.Counsell CE, McLeod M, Grant R. Reversal of subacute paraneoplastic cerebellar syndrome with intravenous immunoglobulin. Neurology. 1994;44:1184–5. doi: 10.1212/wnl.44.6.1184. [DOI] [PubMed] [Google Scholar]
- 21.Uchuya M, Graus F, Vega F, et al. Intravenous immunoglobulin treatment in paraneoplastic neurological syndromes with antineuronal autoantibodies. J Neurol Neurosurg Psychiatry. 1996;60:388–92. doi: 10.1136/jnnp.60.4.388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sahlas DJ, Miller SP, Guerin M, et al. Treatment of acute disseminated encephalomyelitis with intravenous immunoglobulin. Neurology. 2000;54:1370–2. doi: 10.1212/wnl.54.6.1370. [DOI] [PubMed] [Google Scholar]
- 23.Hart YM, Cortez M, Andermann F, et al. Medical treatment of Rasmussen's syndrome (chronic encephalitis and epilepsy): effect of high-dose steroids or immunoglobulins in 19 patients. Neurology. 1994;44:1030–6. doi: 10.1212/wnl.44.6.1030. [DOI] [PubMed] [Google Scholar]
- 24.Dauvilliers Y, Carlander B, Rivier F, et al. Successful management of cataplexy with intravenous immunoglobulins at narcolepsy onset. Ann Neurol. 2004;56:905–8. doi: 10.1002/ana.20339. [DOI] [PubMed] [Google Scholar]
- 25.Dodel RC, Du Y, Depboylu C, et al. Intravenous immunoglobulins containing antibodies against beta-amyloid for the treatment of Alzheimer's disease. J Neurol Neurosurg Psychiatry. 2004;75:1472–4. doi: 10.1136/jnnp.2003.033399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weksler ME, Relkin N, Turkenich R, et al. Patients with Alzheimer disease have lower levels of serum anti-amyloid peptide antibodies than healthy elderly individuals. Exp Gerontol. 2002;37:943–8. doi: 10.1016/s0531-5565(02)00029-3. [DOI] [PubMed] [Google Scholar]
- 27.Du Y, Dodel R, Hampel H, et al. Reduced levels of amyloid beta-peptide antibody in Alzheimer disease. Neurology. 2001;57:801–5. doi: 10.1212/wnl.57.5.801. [DOI] [PubMed] [Google Scholar]
- 28.Relkin N, Szabo P, Adamiak B, et al. 18-Month study of intravenous immunoglobulin for treatment of mild Alzheimer disease. Neurobiol Aging. 2008 doi: 10.1016/j.neurobiolaging.2007.12.021. E-publication ahead of print. [DOI] [PubMed] [Google Scholar]
- 29.Renaud S, Hays AP, Brannagan TH, et al. Gene expression profiling in chronic inflammatory demyelinating polyneuropathy. J Neuroimmunol. 2005;159:203–14. doi: 10.1016/j.jneuroim.2004.10.021. [DOI] [PubMed] [Google Scholar]
- 30.Sommer C, Koch S, Lammens M, et al. Macrophage clustering as a diagnostic marker in sural nerve biopsies of patients with CIDP. Neurology. 2005;65:1924–9. doi: 10.1212/01.wnl.0000188879.19900.b7. [DOI] [PubMed] [Google Scholar]
- 31.Kubori T, Mezaki T, Kaji T, et al. The clinical usefulness of high dose intravenous immunoglobulin therapy for chronic inflammatory demyelinating polyneuropathy and multifocal motor neuropathy. No To Shinkeil. 1999;51:127–35. In Japanese. [PubMed] [Google Scholar]
- 32.Mendell JR, Barohn RJ, Freimer ML, et al. Randomized controlled trial of IVIg in untreated chronic inflammatory demyelinating polyradiculoneuroapthy. Neurology. 2001;56:445–9. doi: 10.1212/wnl.56.4.445. [DOI] [PubMed] [Google Scholar]
- 33.Latov N, Brill V, Katzberg H, et al. Timing of clinical response to immune globulin intravenous, 10% caprylate/chromatography purified (IGIV-C) in chronic inflammatory demyelinating polyneuropathy (CIDP) Ann Neurol. 2008;64(Suppl. 12):S8. Abstract M–18. [Google Scholar]
- 34.Hahn AF, Bolton CF, Zochode D, Feasby TE. Intravenous immunoglobulin treatment in chronic inflammatory demyelinating polyneuropathy. A double blind, placebo-controlled, cross over study. Brain. 1996;119:1067–77. doi: 10.1093/brain/119.4.1067. [DOI] [PubMed] [Google Scholar]
- 35.Harbo T, Anderse H, Jakobsen J. Length dependent weakness and electrophysiologic signs of secondary axonal loss in chronic inflammatory demyelinating polyradiculoneuropathy. Muscle Nerve. 2008;38:1036–45. doi: 10.1002/mus.21000. [DOI] [PubMed] [Google Scholar]
- 36.Rajabally YA, Seow H, Wilson P. Dose of intravenous immunoglobulins in chronic inflammatory demyelinating polyneuropathy. J Peripher Nerv Syst. 2006;11:325–9. doi: 10.1111/j.1529-8027.2006.00105.x. [DOI] [PubMed] [Google Scholar]
- 37.Vucic S, Black K, Baldassari LE, et al. Long-term effects of intravenous immunoglobulin in CIDP. Clin Neurophys. 2007;118:1980–4. doi: 10.1016/j.clinph.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 38.Hughes RA, Cornblath DR. Guillain–Barré syndrome. Lancet. 2005;366:1653–66. doi: 10.1016/S0140-6736(05)67665-9. [DOI] [PubMed] [Google Scholar]
- 39.van Doorn PA, Ruts L, Jacobs BC. Clinical features, pathogenesis, and treatment of Guillain–Barré syndrome. Lancet Neurol. 2008;7:939–50. doi: 10.1016/S1474-4422(08)70215-1. [DOI] [PubMed] [Google Scholar]
- 40.The Italian Guillain–Barré Study Group. The prognosis and main prognostic indicators of Guillain-Barré syndrome. A multicentre prospective study of 297 patients. Brain. 1996;119:2053–61. [PubMed] [Google Scholar]
- 41.Hadden RD, Cornblath DR, Hughes RA, et al. Electrophysiological classification of Guillain–Barré syndrome: clinical associations and outcome. Plasma Exchange/Sandoglobulin Guillain–Barré Syndrome Trial Group. Ann Neurol. 1998;44:780–8. doi: 10.1002/ana.410440512. [DOI] [PubMed] [Google Scholar]
- 42.McKhann GM, Cornblath DR, Ho T, et al. Clinical and electrophysiological aspects of acute paralytic disease of children and young adults in northern China. Lancet. 1991;338:593–7. doi: 10.1016/0140-6736(91)90606-p. [DOI] [PubMed] [Google Scholar]
- 43.Bradshaw DY, Jones HR., Jr Guillain–Barré syndrome in children: clinical course, electrodiagnosis, and prognosis. Muscle Nerve. 1992;15:500–6. doi: 10.1002/mus.880150415. [DOI] [PubMed] [Google Scholar]
- 44.Hughes RA, Raphaël JC, Swan AV, van Doorn PA. Intravenous immunoglobulin for Guillain–Barré syndrome. Cochrane Database Syst Rev. 2006;1:CD002063. doi: 10.1002/14651858.CD002063.pub3. [DOI] [PubMed] [Google Scholar]
- 45.Gürses N, Uysal S, Cetinkaya F, et al. Intravenous immunoglobulin treatment in children with Guillain–Barré syndrome. Scand J Infect Dis. 1995;27:241–3. doi: 10.3109/00365549509019016. [DOI] [PubMed] [Google Scholar]
- 46.Wang T, Feng A, Sun W, Wen Z. Intravenous immunoglobulin in children with Guillain–Barré syndrome. J Appl Clin Pediatr. 2001;16:223–4. [Google Scholar]
- 47.Korinthenberg R, Schessl J, Kirschner J, Mönting JS. Intravenously administered immunoglobulin in the treatment of childhood Guillain–Barré syndrome: a randomized trial. Pediatrics. 2005;116:8–14. doi: 10.1542/peds.2004-1324. [DOI] [PubMed] [Google Scholar]
- 48.Dalakas MC. Advances in the immunobiology and treatment of inflammatory myopathies. Curr Rheumatol Rep. 2007;9:291–7. doi: 10.1007/s11926-007-0047-5. [DOI] [PubMed] [Google Scholar]
- 49.Dalakas M. Intravenous immunoglobulin in autoimmune neuromuscular diseases. JAMA. 2004;29:2367–75. doi: 10.1001/jama.291.19.2367. [DOI] [PubMed] [Google Scholar]
- 50.Dalakas MC. Mechanisms of disease: signaling pathways and immunobiology of inflammatory myopathies. Nat Clin Pract Rheumatol. 2006;2:219–27. doi: 10.1038/ncprheum0140. [DOI] [PubMed] [Google Scholar]
- 51.Dalakas MC. Clinical benefits and immunopathological correlates of intravenous immune globulin in the treatment of inflammatory myopathies. Clin Exp Immunol. 1996;104:55–60. [PubMed] [Google Scholar]
- 52.Amemiya K, Semino-Mora C, Granger RP, Dalakas MC. Downregulation of TGF-beta1 mRNA and protein in the muscles of patients with inflammatory myopathies after treatment with high-dose intravenous immunoglobulin. Clin Immunol. 2000;94:99–104. doi: 10.1006/clim.1999.4823. [DOI] [PubMed] [Google Scholar]
- 53.Cherin P, Pelletier S, Teixeira A, et al. Results and long-term followup of intravenous immunoglobulin infusions in chronic, refractory polymyositis: an open study with thirty-five adult patients. Arthritis Rheum. 2002;46:467–74. doi: 10.1002/art.10053. [DOI] [PubMed] [Google Scholar]
- 54.Greenberg SA. Inclusion body myositis: review of recent literature. Curr Neurol Neurosci Rep. 2009;9:83–9. doi: 10.1007/s11910-009-0013-x. [DOI] [PubMed] [Google Scholar]
- 55.Dalakas MC, Sonies B, Dambrosia J, et al. Treatment of inclusion-body myositis with IVIg: a double-blind, placebo-controlled study. Neurology. 1997;48:712–6. doi: 10.1212/wnl.48.3.712. [DOI] [PubMed] [Google Scholar]
- 56.Dalakas MC, Koffman B, Fujii M, et al. A controlled study of intravenous immunoglobulin combined with prednisone in the treatment of IBM. Neurology. 2001;56:323–7. doi: 10.1212/wnl.56.3.323. [DOI] [PubMed] [Google Scholar]
- 57.Raju R, Dalakas MC. Gene expression profile in the muscles of patients with inflammatory myopathies: effect of therapy with IVIg and biological validation of clinically relevant genes. Brain. 2005;128:1887–96. doi: 10.1093/brain/awh518. [DOI] [PubMed] [Google Scholar]
- 58.Games D, Adams D, Alessandrini R, et al. Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature. 1995;373:523–7. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- 59.Schenk D, Barbour R, Dunn W, et al. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400:173–7. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- 60.Janus C, Pearson J, McLaurin J, et al. A beta peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer's disease. Nature. 2000;408:979–82. doi: 10.1038/35050110. [DOI] [PubMed] [Google Scholar]
- 61.Morgan D, Diamond DM, Gottschall PE, et al. A beta peptide vaccination prevents memory loss in an animal model of Alzheimer's disease. Nature. 2000;408:982–5. doi: 10.1038/35050116. [DOI] [PubMed] [Google Scholar]
- 62.Orgogozo JM, Gilman S, Dartigues JF, et al. Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology. 2003;61:46–54. doi: 10.1212/01.wnl.0000073623.84147.a8. [DOI] [PubMed] [Google Scholar]
- 63.Gilman S, Koller M, Black RS, et al. Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology. 2005;64:1553–62. doi: 10.1212/01.WNL.0000159740.16984.3C. [DOI] [PubMed] [Google Scholar]
- 64.Bard F, Cannon C, Barbour R, et al. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6:916–9. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- 65.Sohn JH, So JO, Kim H, et al. Reduced serum level of antibodies against amyloid beta peptide is associated with aging in Tg2576 mice. Biochem Biophys Res Commun. 2007;361:800–4. doi: 10.1016/j.bbrc.2007.07.107. [DOI] [PubMed] [Google Scholar]
- 66.Kellner A, Matschke J, Bernreuther C, et al. Autoantibodies against β-amyloid are common in Alzheimer's disease and help control plaque burden. Ann Neurol. 2009;65:24–31. doi: 10.1002/ana.21475. [DOI] [PubMed] [Google Scholar]
- 67.Dodel R, Hampel H, Depboylu C, et al. Human antibodies against amyloid beta peptide: a potential treatment for Alzheimer's disease. Ann Neurol. 2002;52:253–6. doi: 10.1002/ana.10253. [DOI] [PubMed] [Google Scholar]
- 68.Dodel R, Du Y, Depboylu C, et al. Intravenous immunoglobulins containing antibodies against beta-amyloid for the treatment of Alzheimer's disease. J Neurol Neurosurg Psychiatry. 2004;75:1472–4. doi: 10.1136/jnnp.2003.033399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tsakanikas D, Shah K, Flores C, et al. Effects of uninterrupted intravenous immunoglobulin treatment of Alzheimer's disease for 9 months. pp. P4–351. International Conference on Alzheimer's Disease Abstract.