

Abstract
Primary immunodeficiencies (PIDs) are uncommon, chronic and severe disorders of the immune system in which patients cannot mount a sufficiently protective immune response, leading to an increased susceptibility to infections. The treatment of choice for PID patients with predominant antibody deficiency is intravenous immunoglobulin (Ig) replacement therapy. Despite major advances over the last 20 years in the molecular characterization of PIDs, many patients remain undiagnosed or are diagnosed too late, with severe consequences. Various strategies to ensure timely diagnosis of PIDs are in place, and novel approaches are being developed. In recent years, several patient registries have been established. Such registries shed light on the pathology and natural history of these varied disorders. Analyses of the registry data may also reveal which patients are likely to respond well to higher Ig infusion rates and may help to determine the optimal dosing of Ig products. Faster infusion rates may lead to improved convenience for patients and thus increase patient compliance, and may reduce nursing time and the need for hospital resources. Data from two recent studies suggest that Gamunex® and Privigen® are well tolerated at high infusion rates. Nevertheless, careful selection of patients for high infusion rates, based on co-morbid conditions and tolerance of the current infusion rate, is advisable. Based on the available data, intravenous Ig offers broad protection against encapsulated organisms. As vaccine trends change, careful monitoring of specific antibody levels in the general population, such as those against pneumococcal and meningococcal bacteria, should be implemented.
Keywords: antibody, common variable immunodeficiency, immunodeficiencies, registry, X-linked agammaglobulinaemia
Introduction
Primary immunodeficiencies (PIDs) are uncommon, chronic and severe disorders of the immune system in which patients cannot mount a sufficiently protective immune response, leading to an increased susceptibility to infections. Immunoglobulin (Ig) replacement therapy was first used in the 1950s for the treatment of PIDs characterized as predominant antibody deficiencies [1]. Since then, remarkable progress has been made in understanding the clinical improvements with Ig replacement, as well as in understanding the many genotypic and phenotypic variations that result in impairment or abrogation of cognate or innate immunity.
This session on immunodeficiencies, chaired by Drs Mark Ballow and José Luis Franco, opened with a presentation by Dr Luigi Notarangelo on the importance of timely diagnosis of PIDs. Drs Bodo Grimbacher and Charlotte Cunningham-Rundles then presented new data from large PID patient registries in Europe and the United States, respectively, and Dr Mark Stein reported on studies into the use of higher infusion rates for intravenous immunoglobulin (IVIg) administration in PID patients. The impact of differences in antibody concentrations for specific pathogens in Ig replacement therapies was discussed by Dr Matthew Helbert.
Improving diagnosis of primary immunodeficiencies
Despite major advances over the last 20 years in the molecular characterization of PIDs, many patients still go undiagnosed or are diagnosed late, with adverse clinical consequences. A collaborative Italian study has shown that delayed diagnosis of patients with X-linked agammaglobulinaemia (XLA) leads to an increased cumulative risk of chronic lung disease; patients who are not diagnosed until the age of 10 years have a 40% chance of developing chronic lung disease (Fig. 1) [2]. These patients are faced with lifelong disability as a result of late diagnosis and depend heavily upon costly treatment and health care resources. Therefore, timely diagnosis of PIDs is crucial.
Fig. 1.
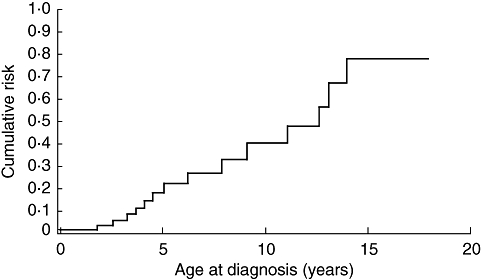
Cumulative risk of developing chronic lung disease in relation to age at diagnosis in patients with X-linked agammaglobulinaemia (XLA). Reproduced with permission from Plebani et al.[2].
Strategies to improve diagnosis of PID should include both global tools, that can be applied worldwide, and regional strategies that are particularly suited to the needs of the local geographical area. Various strategies are in place, and novel strategies and approaches are being developed.
One effective global tool for improving diagnosis of PIDs is the use of awareness campaigns, which may target the general population, primary care physicians and/or health authorities. Advocacy groups, such as the Jeffrey Modell Foundation and the Immune Deficiency Foundation, are particularly effective at raising awareness and can help to influence health authorities to implement policies that benefit patients. Modern multimedia technologies offer a broad range of opportunities for the distribution and sharing of awareness campaigns and educational programmes throughout the world.
Continuing education of physicians from different specialities is important, and as more and more PID patients are living into adulthood it becomes increasingly important to also educate those physicians who treat adolescents and adults. An analysis of the European Society for Immunodeficiencies (ESID) registry showed an increase in the percentage of patients older than 20 years and a decrease in those younger than 10 years from 2006 to 2009 [3].
The International Union of Immunological Societies (IUIS) Committee on PID updates the classifications of PIDs on a 2-yearly basis [4]. In addition, organizations such as the ESID and the Pan-American Group for Immunodeficiency (PAGID) have developed diagnostic guidelines [5]. Web-based algorithms are also being developed to assist the diagnosis of PID. Other online systems, such as PAGID's List Service, which allows physicians to interact with their peers, and PIDexpert, which aids physicians in the diagnosis of PID, encourage collaboration among primary care physicians, specialists and scientists. Such interactions provide diagnostic guidance and permit the sharing of ideas and knowledge among a wide range of health care providers.
Aside from these approaches that can be applied anywhere in the world, regional needs and opportunities exist. Some PIDs are particularly common in certain geographical areas because of, for example, founder effects, restricted genetic isolates or higher consanguinity rates. Along with disease-focused awareness campaigns, there is a need to support the development of appropriate diagnostic tools in these areas, including the use of immunological and molecular assays. Regional registries already exist in a number of countries, and may provide a way to expose regional trends, while the merger of regional registries into larger international databases may help to create a large pool of patient data that may reveal trends not apparent in smaller, regional registries.
In addition to these already available methods, novel diagnostic approaches are coming into practice. In particular, assessment of T cell receptor excision circles (TRECs) using dried blood spots collected at birth is being used currently in a pilot programme for newborn screening of severe combined immunodeficiency (SCID) in Wisconsin and Massachusetts [6,7]. Early identification of infants with SCID should lead to haematopoietic cell transplantation (HCT) in the very first months of life, when the chance of cure is >95%. Compared with other methods that may permit identification of babies with SCID (such as direct analysis of absolute lymphocyte count and subset distribution, or thymic ultrasound), assessment of TRECs on dried blood spots has the advantage of being quantitative, objective and highly reproducible. The assay is cheaper and can be automated and standardized easily. Customized PID gene chips represent another example of technological development that is expected to improve PID diagnosis in the future.
PID patient registries
In recent years, a number of groups have established patient registries in order to determine the incidence, complications, treatment and outcomes of patients with PIDs [8–17]. These initiatives have been based in various geographic regions, have highlighted different patient populations or concentrated on different facets of these conditions. The demand for these registries is driven by multiple reasons, including increased awareness of these diseases, the availability of potential new treatments, the extended life span of subjects with these disorders and the desire to uncover the pathogenesis and molecular causes of immune defects in humans.
The impact of these resources on clinical care in PIDs has been enormous. One of the first patient registries to be initiated in the United States by the Immune Deficiency Foundation was funded by the National Institutes of Health (NIH) to collect data on chronic granulomatous disease (CGD) [17]. The goal of this registry was to determine the health of patients with this rare granulocyte disorder, which had been brought to the forefront because interferon (IFN)-γ was an emerging therapy [18]. The database demonstrated that CGD was more common than perceived, and showed that significant morbidity and mortality were pervasive in CGD [17]. These facts served to accelerate bone marrow transplantation in CGD in the United States and in Europe [19,20]. In Europe, the seminal data collection on children with hyper-IgM syndrome revealed that severe liver diseases were more common than appreciated previously [21–23]. This led to work on the causes, microbiology and cellular mechanisms of hepatic disease [24] and to the realization that stem cell transplantation may be the optimal therapy for this immune defect [20,25,26]. For patients with XLA, registry data revealed that while two or three decades ago this condition was associated more commonly with lung failure and viral neurological disease, current patient survival is nearly the same as for age-matched controls. These data also showed that adults with XLA who are treated with the standard of care are doing well and have good quality of life [8,27,28]. In the past decade, several web-based registries have been established to collect data on selected immune deficiencies.
ESID online database
Centres from all over Europe specializing in PID are collaborating under the auspices of ESID to document their patients into a single database available online [3]. The ESID database contains information mainly from patients in European countries, but also from Iran, Egypt and Russia. The existence of national registries greatly facilitates the uploading of patient information. As of 14 January 2009, the database held data of more than 7500 patients.
The ESID registry encompassed all PIDs. Antibody disorders accounted for 54% of patients registered, of whom 37% were diagnosed with common variable immunodeficiency (CVID). Phagocytic disorders made up 13%, T cell defects 9%, complement deficiencies 2%, and other well-documented disorders, including Wiscott–Aldrich syndrome, made up 18%. Disorders of immune regulation, autoinflammatory syndromes and unclassified PIDs accounted for the rest of the patients.
A total of 2869 patients (42·1%) received Ig replacement therapy, and antibiotics were taken on a regular basis by 1537 patients (22·6%). Interestingly, Ig replacement was given for almost any PID (Table 1), although the rate of Ig replacement was the highest in the group of antibody-deficient patients, where 2317 (57·6%) of 4024 live patients received this form of therapy. There was a high rate of Ig replacement in both CVID and XLA patients, with 1285 of 1483 CVID patients (86·6%) and 390 of 415 XLA patients (93·1%) on Ig replacement (Table 1).
Table 1.
Percentage of live patients receiving immunoglobulin (Ig) replacement therapy within the largest primary immunodeficiency (PID) subgroups in the European Society for Immunodeficiencies (ESID) database.
| PID subcategory | Alive patients | Patients receiving Ig replacement (%) |
|---|---|---|
| Agammaglobulinaemias | 500 | 92·2 |
| Common variable immunodeficiency (CVID) | 1483 | 86·6 |
| Class switch recombination defects (CSR)/hyper-IgM syndromes | 218 | 57·8 |
| X-linked lymphoproliferative syndrome (XLP) | 36 | 50·0 |
| T-B- severe combined immunodeficiency (SCID) | 143 | 46·2 |
| Wiskott–Aldrich syndrome (WAS) | 235 | 43·8 |
| Other unclassified T cell disorders | 99 | 43·4 |
| Human leucocyte antigen class II deficiency | 31 | 41·9 |
| T-B+ severe combined immunodeficiency (SCID) | 155 | 41·3 |
| Chronic mucocutaneous candidiasis (CMC) | 27 | 29·6 |
| CD4 deficiency | 35 | 28·6 |
| Immunodeficiencies of unknown cause | 111 | 27·0 |
| Hypogammaglobulinaemias (without CVID) | 1823 | 24·4 |
| Hyper-IgE syndromes | 114 | 22·8 |
| DNA-breakage disorder | 353 | 22·1 |
| Familial haemophagocytic lymphohistiocytosis syndromes (FLH) | 52 | 17·3 |
| Autoimmune lymphoproliferative syndrome (ALPS) | 78 | 15·4 |
| Leucocyte adhesion deficiency (LAD) | 30 | 6·7 |
| DiGeorge syndrome | 230 | 4·3 |
| Severe congenital neutropenia and Kostmann syndrome | 258 | 3·9 |
| Chronic granulomatous disease (CGD) | 300 | 3·7 |
While the intravenous route is the most common method of administration for Ig therapy, the use of subcutaneous Ig (SCIg) is rising. Immunoglobulin was administered intravenously in 2168 patients (75·6%), while 694 patients (24·2%) made use of home therapy with self-administered subcutaneous infusions. One patient received intramuscular immunoglobulin, while for six patients the route of administration was not documented in the Registry. The use of SCIg as home therapy is available only in some European countries and its use varies between countries, being particularly high in Sweden. Among the countries currently represented in the ESID registry, Germany had the highest rate of SCIg use, with 53·4% of patients receiving immunoglobulin replacement via the SC route (Fig. 2). Between countries there were also differences in the interval of IVIg administration, with the mean administration frequency in many countries being every 3–4 weeks.
Fig. 2.
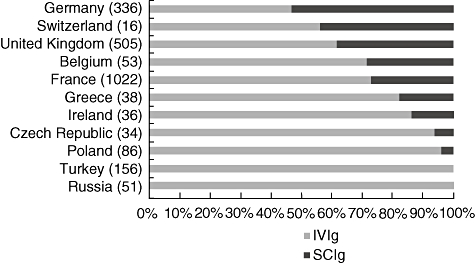
Proportion of patients treated with intravenous immunoglobulin (IVIg) and subcutaneous (SC)Ig in selected European countries, as documented in the European Society for Immunodeficiencies (ESID) database. The total number of documented patients receiving Ig replacement in each country is given in brackets.
A key target in Ig replacement is achievement of an adequate serum IgG trough level. The trough serum level measured immediately before the next administered dose of Ig should be at least 5 g/l, and ideally within the normal range of healthy adult individuals (7–17 g/l [29]). The mean IgG trough level in the ESID database was 6·86 g/l for CVID patients (731 patients receiving Ig replacement with information on the trough level available) and 7 g/l for XLA patients (180 patients).
The effect of IgG trough levels on patients' health was analysed. The analysis was restricted to the subgroup of antibody-deficient patients, because in this group antibody substitution plays a critical role, while in other PID groups, additional components of the immune system are defective and may be treated insufficiently by antibody replacement. The indicators used for measuring patients' health were days spent in hospital due to the antibody deficiency (not including visits to out-patient clinics), number of infectious episodes and number of serious bacterial infections (all per 365 days). Patients (for whom data were available) were divided into three groups according to the average IgG trough level: <5 g/l, ≥5 and <7 g/l and ≥7 g/l.
A higher trough level (>7 g/l) led to a reduction in the days spent in hospital and this trough level seems to have an advantage over a trough level of 5–7 g/l with regard to the patients' health, although the rate of infections does not seem to improve despite this increase in trough level. However, if the analysis is restricted to the subgroup of CVID patients, all indicators show an improvement with a higher trough level.
The CVID patients were analysed further according to the route of administration (IVIg versus SCIg). The results (Fig. 3) support the notion that a trough level below 5 g/l leads to infectious complications and that a level of >7 g/l produces the best results, independent of the route of administration.
Fig. 3.
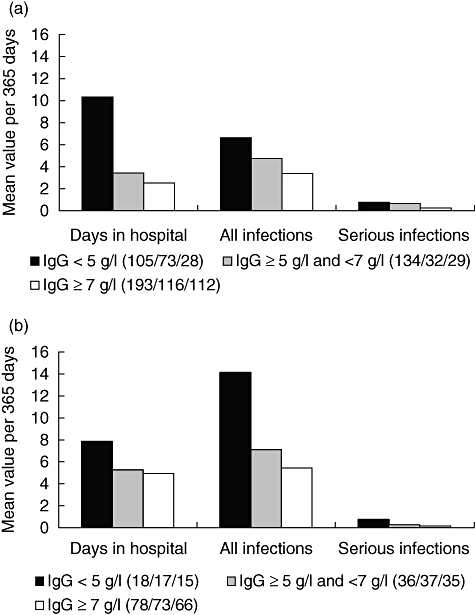
Health outcomes for patients receiving immunoglobulin (Ig) replacement therapy in relation to the mean IgG trough level. (a) Common variable immunodeficiency (CVID) patients on IVIg. (b) CVID patients on subcutaneous (SC)Ig. The number of evaluated patients per group and health indicator is given in brackets.
Although these data were acquired retrospectively and do not fulfil the requirements of a prospective and randomized clinical trial, they point to the need to investigate further the role of IgG trough levels in future prospective trials. In complex conditions such as immune deficiencies, there are certainly other factors besides the serum IgG level that influence the immune response. It will be necessary to identify these factors and to define subsets of patients who may need higher or lower IgG trough levels for protection from infection.
Update on CVID – data from a European CVID registry and the USIDNet registry
As seen from the ESID registry data, CVID is the most prevalent and clinically important PID, a heterogeneous immune deficiency considered to be genetic in origin [30]. Many of the medical complications were already noted in the first report of a large series in 1976 [31]. The most common clinical manifestation of CVID is pneumonia, which is present in 80% of patients, of whom 25% present with recurrent episodes [32]. Remarkably, earlier analyses showed that there is a lapse of approximately 6–8 years after the onset of known lung disease and the diagnosis of immune deficiency [33,34].
Similarly, a recent study of a CVID cohort conducted in seven European hospitals showed that the length of time between the first characteristic symptoms and diagnosis is still 8 years [35]. In addition to infections, many patients with CVID have other clinically challenging complications, including autoimmunity [36,37], granulomatous infiltrations [38–41], lymphadenopathy and/or splenomegaly or non-Hodgkin's lymphoma [35,42]. It appears that CVID subjects with only infectious complications tend to do well, but the presence of other complications leads to increased mortality [13]. Neither initial levels of serum IgG nor the length of diagnostic delay were predictive of earlier mortality [35]. Furthermore, although chronic lung disease continues to be of concern in CVID [43–45], initial serum IgG levels were not predictive of the presence of chronic lung disease in this cohort [35]. However, when present, bronchiectasis was also associated with reduced survival [35].
The USIDNet Registry for Primary Immune Deficiency is based on a Core Data form and a series of disease-specific forms [46]. The essential data fields map to the ESID data forms, so that harmonization can be achieved. As of April 2009, the data bank held clinical and laboratory information for 889 CVID subjects submitted by 122 physicians in 112 different medical institutions. Fifty-six per cent of the patients were female and 44% male. The majority of the patients (95%) were Caucasian non-Hispanic, 4·3% were Hispanic and 3·5% were African American. The median age at diagnosis of CVID was 32 years; however, as noted previously [47], diagnosis was made one decade earlier for males, at a median age of 25 years, compared with females, who were diagnosed at a median age of 36 years. The median Ig levels at the time of diagnosis were 210 mg/dl for IgG, 25 mg/dl for IgA and 8 mg/dl for IgM. Most patients (88%) had experienced at least one infectious complication, including pneumonia (62%) (Fig. 4), sinusitis (64%), bronchitis (50%) or otitis media (35%). Bronchiectasis was found in 7% of patients, and other forms of chronic lung disease in 11%. Malignancy occurred in 13% of patients, with lymphoma accounting for almost half of all cancers (6% of patients). Almost all patients were receiving IVIg at the time of the survey and many (45%) were also on prophylactic antibiotics, at least intermittently. Survival statistics for this group are not yet available, but a subanalysis of 71 deceased patients of another cohort of 338 subjects from Mount Sinai Medical Centre indicated a decreased life span for this cohort, as has been noted previously [47].
Fig. 4.
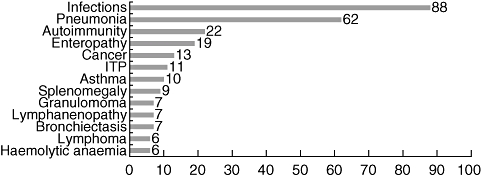
Percentage of clinical manifestations and complications in common variable immunodeficiency (CVID) subjects from the USIDnet registry.
Patient registries: summary
While the genetic causes of CVID are still unclear for the majority of subjects, clinically relevant methods of classification of subjects into diagnostic groups as, for example, by B cell phenotypes [48–50], have emerged as a means of better predicting outcomes. As for other PIDs, CVID is a rare condition, and only by the longitudinal collection of data from large patient populations can more accurate information be obtained. Most registry data have been collected from medical institutions that concentrate on immune deficiency diseases, and as a result are likely to see more complex cases; however, as shown in a number of other surveys on PIDs [8,17,51], a number of subjects with PID in the United States, presumably in a stable condition, receive care in medical practices where only a few patients are being followed. This may be especially true for antibody deficiencies such as CVID. Collecting data from these sources will be important to generate a more precise picture of the overall natural history of these immune defects.
Use of high infusion rates for IVIg in PID patients
As discussed above, patient registries for such complex syndromes as PIDs may help to reveal broad patterns related to these diseases and their management. According to the national survey of the Immune Deficiency Foundation, the average length of IVIg infusion in PID patients is 3·5 h. Shortening the length of infusion would provide increased convenience for the patient and may be more cost-effective, due to the shorter requirements for nursing time and hospital resources. However, the impact of higher infusion rates upon tolerability needs to be addressed. Currently, there are only two studies of more than 20 patients with PID in whom high infusion rates were used for IVIg therapy.
In a prospective, single-blind, randomized, cross-over trial involving 97 subjects, infusions of single doses of Gamunex® at a rate of 8 mg/kg/min or 14 mg/kg/min were compared[52]. All patients received a single dose at each infusion rate at their usual 3- or 4-week interval, and were randomized as to which rate they received first. In this study, the incidence of any adverse event was similar for the higher and lower infusion rates (37·1% and 32·0%, respectively), although the incidence of ‘predefined’ infusion-related adverse events was 10·3% at the higher infusion rate and 3·1% at the lower infusion rate.
A second trial was performed recently as an extension to the pivotal study of Privigen® in PID. In the pivotal study [53], Privigen® was shown to be well tolerated and effective for the treatment of PID. The extension study was a prospective, multi-centre, open-label, single-arm study designed to assess the tolerability and safety of Privigen® when used at higher infusion rates (i.e. up to 12 mg/kg/min) than those used in the initial pivotal study (i.e. up to 8 mg/kg/min). Of the 80 original subjects, 45 continued in the extension study and were eligible for the highest rate of infusion. The rate of infusion for each patient was determined by the on-site investigator. The main outcome measurement was the incidence of temporally associated (within 72 h) adverse events (TAAEs).
Forty-five patients from 10 centres in the United States participated in the trial and 33 patients completed the study. Of the 12 patients who withdrew from the study, only one withdrew due to a non-fatal adverse event. The patients were divided into two groups: previous maximum infusion rate (low) (LIR), i.e. ≤8 mg/kg/min; or high maximum infusion rate (HIR), i.e. 8–12 mg/kg/min. Proportion of infusions with TAAEs were compared between the two groups. Analysis of the study results indicates that the rate of TAAEs was not higher in the HIR group compared with the LIR group.
These two high-infusion-rate studies had major differences. In the Gamunex® study, all patients received a single dose at the higher infusion rate in a blinded fashion. The incidence of infusion-related adverse events was higher with the faster infusion rate, although the incidence was low at both rates [52]. The infusion rate used (14 mg/kg/min) was higher than the maximal rate (12 mg/kg/min) used in the Privigen® study. In the Privigen® study, 257 infusions were given at the maximal rate (12 mg/kg/min) over a 12-month period. Preliminary results indicate no positive correlation between infusion rate and the rate of TAAEs. However, there was probably some selection bias from investigators selecting patients who were perceived to be at lower risk for adverse reactions with high infusion rates. This selection of patients may account for the decreased rate of TAAEs, including headaches, in the HIR group.
These two studies suggest that Gamunex® and Privigen® are well tolerated at high infusion rates. Careful selection of patients for high infusion rates, based on co-morbid conditions and tolerance of the current infusion rate, is advisable. Patients who already are having TAAEs at lower infusion rates will probably not tolerate higher infusion rates.
Specific antibody responses: anti-polysaccharide antibodies in IVIg
Despite regular Ig therapy and the achievement of trough IgG levels thought to be protective (i.e. >7 g/l), PID patients continue to experience infections, particularly of the respiratory tract, as confirmed by the registry data presented above. The addition of prophylactic antibiotics does not prevent respiratory infection completely, which occurs predominantly with encapsulated organisms such as Haemophilus, Pneumococcus and Moraxella[54]. Rarely, patients on Ig replacement experience severe non-respiratory infection with organisms such as Neisseria meningitidis, even when trough IgG levels above 10 g/l have been achieved [55].
Polymorphisms in immune response genes and concomitant structural damage (for example, due to sinusitis or bronchiectasis) might confer ongoing risk for infection in patients on Ig therapy. Additionally there is a theoretical risk that Ig products do not always provide optimum levels of antibodies against encapsulated organisms. This could occur, for example, when plasma is sourced and used in different continents or different populations, either of which may differ in epidemiological characteristics. Alternatively, fractionation, purification or virus inactivation steps may reduce antibody levels quantitatively or affect their opsonic or complement-fixing function.
Most Haemophilus infections experienced by PID patients are non-typeable and the utility of antibody levels is difficult to determine. The measurement of antibodies against Moraxella species is in its infancy. Thus, most studies on anti-polysaccharide antibodies in IVIg have used Pneumococcus and Meningococcus as model bacterial infections. The technology of these assays has benefited from the drive to test commercial polysaccharide vaccines.
One study of antibody concentrations in IVIg showed considerable variation in the levels of antibodies against the eight strains of Pneumococcus investigated [56]. In this study, there were similar levels of antibodies in batches sourced in the United Kingdom and in the United States (Fig. 5). The highest levels of antibodies in serum investigated in the United Kingdom were against strains 6, 19 and 14, reflecting seroprevalence data in young adults [57]. Others have confirmed the finding that antibodies against serotypes 19 and 14 predominate in IVIg [58,59].
Fig. 5.
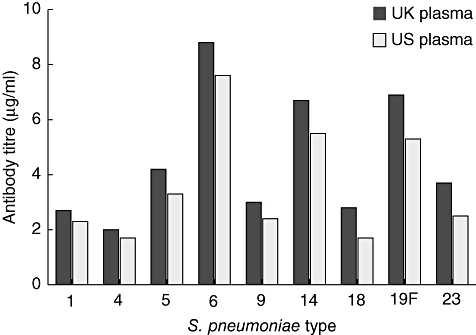
Levels of antibodies to different strains of Streptococcus pneumoniae in Vigam® liquid produced from UK or US plasma [56]. Three batches of Vigam® liquid from UK plasma and five from US plasma were analysed. Antibody titres to S. pneumoniae serotypes were determined by enzyme-linked immunosorbent assay using a standardized protocol.
These studies offer two explanations as to why patients on Ig therapy experience breakthrough pneumococcal infections. One explanation may be that these infections involve strains that otherwise cause infections in the elderly (predominantly serotypes 9 and 23). Ig products tend not to contain high levels of antibody against these pneumococcal strains. Serotyping the infecting strains isolated from PID patients could test this hypothesis readily. A more prosaic explanation may be that although IgG levels are corrected in manufacture, there may be batch-to-batch variation in the levels of anti-pneumococcal antibodies. Inter-batch variation of up to 25% in pneumococcal antibodies has indeed been observed [58].
Antibodies against N. meningitidis can also be measured using standard solid phase antibody capture assays. These have been used to show, for example, that IVIg batches sourced on either side of the Atlantic contain similar levels of antibody against Meningococcus[56]. However, the epidemiological studies used to determine levels of antibody that protect against Meningococcus used bacteriocidal assays, measuring the ability of the antibody to bind to the pathogen and activate complement. Reassuringly, in epidemiological studies on donated serum samples, bacteriocidal antibody levels correlate generally with geometric mean concentrations of IgG antibody against N. meningitides[60,61]. Comparable data showing both antibody levels and bacteriocidal activity in Ig replacement suggest there is a lack of concordance between in vivo bacteriocidal antibody and anti-meningococcal antibody levels measured by enzyme-linked immunosorbent assay (ELISA) in a PID patient on Ig replacement therapy [55]. The assumption remains that the rare patients developing meningococcal infection while on Ig therapy may have other problems contributing to infection, for example concomitant mannose-binding lectin (MBL) deficiency.
The major difficulty with these data may be the lack of understanding of how levels of specific antibodies in Ig products translate into antibody levels in the blood of recipients. The ideas explored here, namely absence of antibody specific to infecting strains of Pneumococcus, batch-to-batch variation and loss of bacteriocidal activity, may offer some explanation as to why patients on Ig replacement continue to experience infections. Therefore, it might be useful to evaluate how well in vitro antibody and bacteriocidal levels in Ig products correlate with the corresponding level in patients and to correlate these with the serotype of infecting organisms.
It will be of interest to see how, in the next few years, the impact of new vaccination schedules will influence these concerns. The first generations to receive routine meningococcal and pneumococcal vaccination will eventually begin to donate to the plasma pool, and it is not yet known whether high levels of antibody are sustained. It is known that following the introduction of conjugated meningococcal C vaccine in the United Kingdom in 1999, levels of bacteriocidal antibody levels have fallen in older individuals [61]. This may be a consequence of improving herd immunity against Meningococcus C in the young and reduced (boosting) exposure in the elderly. Further surveillance will be required to monitor these ramifications of changes in vaccine strategies. We can conclude that, based on the currently available data, Ig batches offer broad protection against encapsulated organisms.
Summary
A great deal of effort has been put forth to understand the immune defects in PIDs more clearly. Global and regional efforts towards improving collaboration among health care providers will be crucial for proper and timely diagnosis of patients with PIDs, which in turn will help to drive further improvements in patient outcomes. Excellent examples of successful and productive collaborations are patient database registries, such as those compiled by ESID in Europe and USIDNet in the United States. These patient registries have already and will continue to shed light on the pathology and natural history of these varied disorders. Analyses of the registry data may also reveal which patients are likely to respond well to higher Ig infusion rates and may help to determine the optimal dosing of Ig products. High infusion rates, in turn, may lead to improved convenience and thus increase patient compliance. Data from the registries show that infection remains the most common complication of PIDs. Careful monitoring of specific antibody levels in the general population, such as those against pneumococcal and meningococcal bacteria, should be implemented as vaccine trends change.
Overall, although numerous advances in our understanding of Ig treatment for immunodeficiencies have clearly been made, there is still much to be learned about these multi-faceted syndromes.
Acknowledgments
Dr Matthew Helbert would like to acknowledge Ray Borrow, Health Protection Agency, Manchester Royal Infirmary, UK and Jamie Findlow, Manchester Royal Infirmary, UK for their contributions. The authors would like to thank nspm ltd for providing medical writing services, with financial support through an unrestricted educational grant from CSL Behring.
Disclosures
BG has received funding of EUR 333 from CSL for preparing this publication. HO is a consultant for CSL Behring. MS was an investigator for CSL Behring in the study reported on rapid infusion rates. JF acted as a paid consultant for Baxter, but has received no funding for research carried out in this work. All other authors have declared that they have no conflicts of interest.
References
- 1.Bruton OC. Agammaglobulinemia. Pediatrics. 1952;9:722–8. [PubMed] [Google Scholar]
- 2.Plebani A, Soresina A, Rondelli R, et al. Clinical, immunological, and molecular analysis in a large cohort of patients with x-linked agammaglobulinemia: an Italian multicenter study. Clin Immunol. 2002;104:221–30. doi: 10.1006/clim.2002.5241. [DOI] [PubMed] [Google Scholar]
- 3.European Society for Immunodeficiencies Registry. Available at: http://www.esid.org/workingparty.php?party=2 (accessed 21 April 2009.
- 4.Geha RS, Notarangelo LD, Casanova JL, et al. Primary immunodeficiency diseases: an update from the International Union of Immunological Societies Primary Immunodeficiency Diseases Classification Committee. J Allergy Clin Immunol. 2007;120:776–94. doi: 10.1016/j.jaci.2007.08.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Conley ME, Notarangelo LD, Etzioni A. Diagnostic criteria for primary immunodeficiencies. Representing PAGID (Pan-American Group for Immunodeficiency) and ESID (European Society for Immunodeficiencies) Clin Immunol. 1999;93:190–7. doi: 10.1006/clim.1999.4799. [DOI] [PubMed] [Google Scholar]
- 6.Chan K, Puck JM. Development of population-based newborn screening for severe combined immunodeficiency. J Allergy Clin Immunol. 2005;115:391–8. doi: 10.1016/j.jaci.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 7.McGhee SA, Stiehm ER, Cowan M, Krogstad P, McCabe ER. Two-tiered universal newborn screening strategy for severe combined immunodeficiency. Mol Genet Metab. 2005;86:427–30. doi: 10.1016/j.ymgme.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 8.Winkelstein JA, Marino MC, Lederman HM, et al. X-linked agammaglobulinemia: report on a United States registry of 201 patients. Medicine. 2006;85:193–202. doi: 10.1097/01.md.0000229482.27398.ad. [DOI] [PubMed] [Google Scholar]
- 9.Leiva LE, Zelazco M, Oleastro M, et al. Primary immunodeficiency diseases in Latin America: the second report of the LAGID registry. J Clin Immunol. 2007;27:101–8. doi: 10.1007/s10875-006-9052-0. [DOI] [PubMed] [Google Scholar]
- 10.Eades-Perner AM, Gathmann B, Knerr V, et al. The European internet-based patient and research database for primary immunodeficiencies: results 2004–06. Clin Exp Immunol. 2007;147:306–12. doi: 10.1111/j.1365-2249.2006.03292.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Knerr V, Grimbacher B. Primary immunodeficiency registries. Curr Opin Allergy Clin Immunol. 2007;7:475–80. doi: 10.1097/ACI.0b013e3282f2162c. [DOI] [PubMed] [Google Scholar]
- 12.Kirkpatrick P, Riminton S. Primary immunodeficiency diseases in Australia and New Zealand. J Clin Immunol. 2007;27:517–24. doi: 10.1007/s10875-007-9105-z. [DOI] [PubMed] [Google Scholar]
- 13.Cunningham-Rundles C, Knight AK. Common variable immune deficiency: reviews, continued puzzles, and a new registry. Immunol Res. 2007;38:78–86. doi: 10.1007/s12026-007-0024-0. [DOI] [PubMed] [Google Scholar]
- 14.Al-Herz W. Primary immunodeficiency disorders in Kuwait: first report from Kuwait National Primary Immunodeficiency Registry (2004–2006) J Clin Immunol. 2008;28:186–93. doi: 10.1007/s10875-007-9144-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reda SM, Afifi HM, Amine MM. Primary immunodeficiency diseases in Egyptian children: a single-center study. J Clin Immunol. 2009;29:343–51. doi: 10.1007/s10875-008-9260-x. [DOI] [PubMed] [Google Scholar]
- 16.Razvi S, Schneider L, Jonas MM, Cunningham-Rundles C. Outcome of intravenous immunoglobulin-transmitted hepatitis C virus infection in primary immunodeficiency. Clin Immunol. 2001;101:284–8. doi: 10.1006/clim.2001.5132. [DOI] [PubMed] [Google Scholar]
- 17.Winkelstein JA, Marino MC, Johnston RB, Jr, et al. Chronic granulomatous disease. Report on a national registry of 368 patients. Medicine. 2000;79:155–69. doi: 10.1097/00005792-200005000-00003. [DOI] [PubMed] [Google Scholar]
- 18.Gallin JI. Interferon-gamma in the management of chronic granulomatous disease. Rev Infect Dis. 1991;13:973–8. doi: 10.1093/clinids/13.5.973. [DOI] [PubMed] [Google Scholar]
- 19.Schuetz C, Hoenig M, Schulz A, et al. Successful unrelated bone marrow transplantation in a child with chronic granulomatous disease complicated by pulmonary and cerebral granuloma formation. Eur J Pediatr. 2007;166:785–8. doi: 10.1007/s00431-006-0317-7. [DOI] [PubMed] [Google Scholar]
- 20.Porta F, Forino C, De Martiis D, et al. Stem cell transplantation for primary immunodeficiencies. Bone Marrow Transplant. 2008;41(Suppl. 2):S83–6. doi: 10.1038/bmt.2008.61. [DOI] [PubMed] [Google Scholar]
- 21.Levy J, Espanol-Boren T, Thomas C, et al. Clinical spectrum of X-linked hyper-IgM syndrome. J Pediatr. 1997;131:47–54. doi: 10.1016/s0022-3476(97)70123-9. [DOI] [PubMed] [Google Scholar]
- 22.Nagaraj N, Egwim C, Adler DG. X-linked hyper-IgM syndrome associated with poorly differentiated neuroendocrine tumor presenting as obstructive jaundice secondary to extensive adenopathy. Dig Dis Sci. 2007;52:2312–6. doi: 10.1007/s10620-006-9702-3. [DOI] [PubMed] [Google Scholar]
- 23.Malamut G, Ziol M, Suarez F, et al. Nodular regenerative hyperplasia: the main liver disease in patients with primary hypogammaglobulinemia and hepatic abnormalities. J Hepatol. 2008;48:74–82. doi: 10.1016/j.jhep.2007.08.011. [DOI] [PubMed] [Google Scholar]
- 24.Hayward AR, Levy J, Facchetti F, et al. Cholangiopathy and tumors of the pancreas, liver, and biliary tree in boys with X-linked immunodeficiency with hyper-IgM. J Immunol. 1997;158:977–83. [PubMed] [Google Scholar]
- 25.Horwitz ME, Barrett AJ, Brown MR, et al. Treatment of chronic granulomatous disease with nonmyeloablative conditioning and a T-cell-depleted hematopoietic allograft. N Engl J Med. 2001;344:881–8. doi: 10.1056/NEJM200103223441203. [DOI] [PubMed] [Google Scholar]
- 26.Seger RA, Gungor T, Belohradsky BH, et al. Treatment of chronic granulomatous disease with myeloablative conditioning and an unmodified hemopoietic allograft: a survey of the European experience, 1985–2000. Blood. 2002;100:4344–50. doi: 10.1182/blood-2002-02-0583. [DOI] [PubMed] [Google Scholar]
- 27.Howard V, Greene JM, Pahwa S, et al. The health status and quality of life of adults with X-linked agammaglobulinemia. Clin Immunol. 2006;118:201–8. doi: 10.1016/j.clim.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 28.Winkelstein JA, Conley ME, James C, Howard V, Boyle J. Adults with X-linked agammaglobulinemia: impact of disease on daily lives, quality of life, educational and socioeconomic status, knowledge of inheritance, and reproductive attitudes. Medicine. 2008;87:253–8. doi: 10.1097/MD.0b013e318187ed81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gonzalez-Quintela A, Alende R, Gude F, et al. Serum levels of immunoglobulins (IgG, IgA, IgM) in a general adult population and their relationship with alcohol consumption, smoking and common metabolic abnormalities. Clin Exp Immunol. 2008;151:42–50. doi: 10.1111/j.1365-2249.2007.03545.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chapel H, Cunningham-Rundles C. Update in understanding common variable immunodeficiency disorders (CVIDs) and the management of patients with these conditions. Br J Haematol. 2009;145:709–27. doi: 10.1111/j.1365-2141.2009.07669.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hermans PE, Diaz-Buxo JA, Stobo JD. Idiopathic late-onset immunoglobulin deficiency. Clinical observations in 50 patients. Am J Med. 1976;61:221–37. doi: 10.1016/0002-9343(76)90173-x. [DOI] [PubMed] [Google Scholar]
- 32.Busse PJ, Razvi S, Cunningham-Rundles C. Efficacy of intravenous immunoglobulin in the prevention of pneumonia in patients with common variable immunodeficiency. J Allergy Clin Immunol. 2002;109:1001–4. doi: 10.1067/mai.2002.124999. [DOI] [PubMed] [Google Scholar]
- 33.Cunningham-Rundles C. Common variable immunodeficiency. Curr Allergy Asthma Rep. 2001;1:421–9. doi: 10.1007/s11882-001-0027-1. [DOI] [PubMed] [Google Scholar]
- 34.Thickett KM, Kumararatne DS, Banerjee AK, Dudley R, Stableforth DE. Common variable immune deficiency: respiratory manifestations, pulmonary function and high-resolution CT scan findings. Q J Med. 2002;95:655–62. doi: 10.1093/qjmed/95.10.655. [DOI] [PubMed] [Google Scholar]
- 35.Chapel H, Lucas M, Lee M, et al. Common variable immunodeficiency disorders: division into distinct clinical phenotypes. Blood. 2008;112:277–86. doi: 10.1182/blood-2007-11-124545. [DOI] [PubMed] [Google Scholar]
- 36.Michel M, Chanet V, Galicier L, et al. Autoimmune thrombocytopenic purpura and common variable immunodeficiency: analysis of 21 cases and review of the literature. Medicine. 2004;83:254–63. doi: 10.1097/01.md.0000133624.65946.40. [DOI] [PubMed] [Google Scholar]
- 37.Wang J, Cunningham-Rundles C. Treatment and outcome of autoimmune hematologic disease in common variable immunodeficiency (CVID) J Autoimmun. 2005;25:57–62. doi: 10.1016/j.jaut.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 38.Mechanic LJ, Dikman S, Cunningham-Rundles C. Granulomatous disease in common variable immunodeficiency. Ann Intern Med. 1997;127:613–7. doi: 10.7326/0003-4819-127-8_part_1-199710150-00005. [DOI] [PubMed] [Google Scholar]
- 39.Fasano MB, Sullivan KE, Sarpong SB, et al. Sarcoidosis and common variable immunodeficiency. Report of 8 cases and review of the literature. Medicine. 1996;75:251–61. doi: 10.1097/00005792-199609000-00002. [DOI] [PubMed] [Google Scholar]
- 40.Morimoto Y, Routes JM. Granulomatous disease in common variable immunodeficiency. Curr Allergy Asthma Rep. 2005;5:370–5. doi: 10.1007/s11882-005-0008-x. [DOI] [PubMed] [Google Scholar]
- 41.Wheat WH, Cool CD, Morimoto Y, et al. Possible role of human herpesvirus 8 in the lymphoproliferative disorders in common variable immunodeficiency. J Exp Med. 2005;202:479–84. doi: 10.1084/jem.20050381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cunningham-Rundles C, Cooper DL, Duffy TP, Strauchen J. Lymphomas of mucosal-associated lymphoid tissue in common variable immunodeficiency. Am J Hematol. 2002;69:171–8. doi: 10.1002/ajh.10050. [DOI] [PubMed] [Google Scholar]
- 43.Busse PJ, Farzan S, Cunningham-Rundles C. Pulmonary complications of common variable immunodeficiency. Ann Allergy Asthma Immunol. 2007;98:1–8. doi: 10.1016/S1081-1206(10)60853-8. [DOI] [PubMed] [Google Scholar]
- 44.Quinti I, Soresina A, Spadaro G, et al. Long-term follow-up and outcome of a large cohort of patients with common variable immunodeficiency. J Clin Immunol. 2007;27:308–16. doi: 10.1007/s10875-007-9075-1. [DOI] [PubMed] [Google Scholar]
- 45.Gregersen S, Aalokken TM, Mynarek G, et al. High resolution computed tomography and pulmonary function in common variable immunodeficiency. Respir Med. 2009;103:873–80. doi: 10.1016/j.rmed.2008.12.015. [DOI] [PubMed] [Google Scholar]
- 46.United States Immunodeficiency Network. Available at: http://www.usidnet.org/ (accessed 23 April 2009.
- 47.Cunningham-Rundles C, Bodian C. Common variable immunodeficiency: clinical and immunological features of 248 patients. Clin Immunol. 1999;92:34–48. doi: 10.1006/clim.1999.4725. [DOI] [PubMed] [Google Scholar]
- 48.Wehr C, Kivioja T, Schmitt C, et al. The EUROclass trial: defining subgroups in common variable immunodeficiency. Blood. 2008;111:77–85. doi: 10.1182/blood-2007-06-091744. [DOI] [PubMed] [Google Scholar]
- 49.Warnatz K, Denz A, Drager R, et al. Severe deficiency of switched memory B cells (CD27+IgM-IgD-) in subgroups of patients with common variable immunodeficiency: a new approach to classify a heterogeneous disease. Blood. 2002;99:1544–51. doi: 10.1182/blood.v99.5.1544. [DOI] [PubMed] [Google Scholar]
- 50.Sanchez-Ramon S, Radigan L, Yu JE, Bard S, Cunningham-Rundles C. Memory B cells in common variable immunodeficiency: clinical associations and sex differences. Clin Immunol. 2008;128:314–21. doi: 10.1016/j.clim.2008.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Winkelstein JA, Marino MC, Ochs H, et al. The X-linked hyper-IgM syndrome: clinical and immunologic features of 79 patients. Medicine. 2003;82:373–84. doi: 10.1097/01.md.0000100046.06009.b0. [DOI] [PubMed] [Google Scholar]
- 52.Gelfand EW, Hanna K. Safety and tolerability of increased rate of infusion of intravenous immunoglobulin G, 10% in antibody-deficient patients. J Clin Immunol. 2006;26:284–90. doi: 10.1007/s10875-006-9014-6. [DOI] [PubMed] [Google Scholar]
- 53.Stein MR, Nelson RP, Church JA. Safety and efficacy of Privigen, a novel 10% liquid immunoglobulin preparation for intravenous use, in patients with primary immunodeficiencies. J Clin Immunol. 2009;29:137–44. doi: 10.1007/s10875-008-9231-2. [DOI] [PubMed] [Google Scholar]
- 54.Pettit SJ, Bourne H, Spickett GP. Survey of infection in patients receiving antibody replacement treatment for immune deficiency. J Clin Pathol. 2002;55:577–80. doi: 10.1136/jcp.55.8.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lear S, Eren E, Findlow J, Borrow R, Webster D, Jolles S. Meningococcal meningitis in two patients with primary antibody deficiency treated with replacement intravenous immunoglobulin. J Clin Pathol. 2006;59:1191–3. doi: 10.1136/jcp.2005.031054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Matejtschuk P, Chidwick K, Prince A, Moore JE, Goldblatt D. A direct comparison of the antigen-specific antibody profiles of intravenous immunoglobulins derived from US and UK donor plasma. Vox Sang. 2002;83:17–22. doi: 10.1046/j.1423-0410.2002.00186.x. [DOI] [PubMed] [Google Scholar]
- 57.Balmer P, Borrow R, Findlow J, et al. Age-stratified prevalences of pneumococcal-serotype-specific immunoglobulin G in England and their relationship to the serotype-specific incidence of invasive pneumococcal disease prior to the introduction of the pneumococcal 7-valent conjugate vaccine. Clin Vaccine Immunol. 2007;14:1442–50. doi: 10.1128/CVI.00264-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mikolajczyk MG, Concepcion NF, Wang T, et al. Characterization of antibodies to capsular polysaccharide antigens of Haemophilus influenzae type b and Streptococcus pneumoniae in human immune globulin intravenous preparations. Clin Diagn Lab Immunol. 2004;11:1158–64. doi: 10.1128/CDLI.11.6.1158-1164.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lejtenyi D, Mazer B. Consistency of protective antibody levels across lots of intravenous immunoglobulin preparations. J Allergy Clin Immunol. 2008;121:254–5. doi: 10.1016/j.jaci.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 60.Trotter C, Findlow J, Balmer P, et al. Seroprevalence of bactericidal and anti-outer membrane vesicle antibodies to Neisseria meningitidis group B in England. Clin Vaccine Immunol. 2007;14:863–8. doi: 10.1128/CVI.00102-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Trotter CL, Borrow R, Findlow J, et al. Seroprevalence of antibodies against serogroup C meningococci in England in the postvaccination era. Clin Vaccine Immunol. 2008;15:1694–8. doi: 10.1128/CVI.00279-08. [DOI] [PMC free article] [PubMed] [Google Scholar]