

Abstract
High-dose intravenous immunoglobulin (IVIg) preparations are used currently for the treatment of autoimmune or inflammatory diseases. Despite numerous studies demonstrating efficacy, the precise mode of action of IVIg remains unclear. Paradoxically, IgG can exert both pro- and anti-inflammatory activities, depending on its concentration. The proinflammatory activity of low-dose IVIg requires complement activation or binding of the Fc fragment of IgG to IgG-specific receptors (FcγR) on innate immune effector cells. In contrast, when administered in high concentrations, IVIg has anti-inflammatory properties. How this anti-inflammatory effect is mediated has not yet been elucidated fully, and several mutually non-exclusive mechanisms have been proposed. This paper represents the proceedings of a session entitled ‘IVIg – Understanding properties and mechanisms’ at the 6th International Immunoglobulin Symposium that was held in Interlaken on 26–28 March 2009. The presentations addressed how IgG may affect the cellular compartment, evidence for IVIg-mediated scavenging of complement fragments, the role of the dimeric fraction of IVIg, the anti-inflammatory properties of the minor fraction of sialylated IgG molecules, and the genetic organization and variation in FcγRs. These findings demonstrate the considerable progress that has been made in understanding the mechanisms of action of IVIgs, and may influence future perspectives in the field of Ig therapy.
Keywords: complement, dendritic cell, Fc receptor, immunoglobulin, sialylation
Introduction
Immunoglobulins and T cells are the key mediators of adaptive immunity. Deficiencies in either of these two arms can lead to a heightened susceptibility to bacterial, fungal or viral infections [1]. Primary immunodeficiency (PID) disorders, such as agammaglobulinaemias, hyperimmunoglobulin M (IgM) syndromes and common variable immunodeficiencies (CVID), are either caused by defined gene mutations or remain molecularly undefined [2]. In addition, hypogammaglobulinaemic phenotypes, termed secondary immunodeficiencies, can arise for example from viral infections, B cell malignancies, bone marrow transplantation or immunosuppressive therapy [1].
For most of the primary and secondary Ig deficiencies, Ig replacement therapy is the treatment of choice [3]. Therapeutic Ig preparations are comprised of normal, polyclonal, polyspecific Ig (consisting mainly of IgG) derived from plasma pools of thousands of healthy donors. The preparations contain antibodies to foreign (non-self) antigens, to self-antigens (natural autoantibodies) and to other antibodies (idiotypic antibodies). Traditionally, Ig replacement therapy has been administered via the intravenous route (IVIg), but in recent years subcutaneous administration has become increasingly popular. In addition to its use as substitutive therapy in primary and secondary immunodeficiencies, Ig therapy is used in a wide spectrum of autoimmune diseases believed to be mediated by autoantibodies or T cells and in systemic inflammatory conditions [4]. Currently licensed autoimmune applications for Ig therapy include Guillain–Barré syndrome, Kawasaki disease and chronic inflammatory demyelinating polyneuropathy (CIDP) [1].
The mechanism of activity of the substituted IgG is easily understood for immunodeficiency disorders. Antibodies with the intrinsic capacity to recognize foreign antigens or common pathogen-specific IgG antibodies are replaced by those from the donor pool. The immunomodulatory mechanisms of administered IgG, on the other hand, are more difficult to explain.
Paradoxically, IgG can exert both pro- and anti-inflammatory activities, depending upon its concentration. The proinflammatory activity of low-dose IVIg requires complement activation or binding of the Fc fragment of IgG to IgG-specific receptors (FcγR) on innate immune effector cells. This results in receptor clustering, recruitment of secondary effector functions and subsequent activation of signalling pathways, leading to an increase in intracellular calcium levels and cell activation. In contrast, when administered in high concentrations, IVIg has anti-inflammatory properties. How this anti-inflammatory effect is mediated has not yet been elucidated fully. Several mutually non-exclusive mechanisms have been proposed [1,5], including modulation of the expression and function of FcγRs, interference with activation of the complement cascade and the cytokine network, neutralization of autoantibodies and regulation of cell proliferation.
The genetic factors that predispose certain individuals to the development of autoimmune diseases are also poorly understood. The family of FcγRs consists of several activating members and one inhibitory member, FcγRIIb. The current paradigm in FcγR biology states that cell activation is balanced by the activating and inhibitory FcγRs. Alterations in the expression or function of these receptors may therefore result in unbalanced immunity and inflammation. Inter-individual differences in FcγR expression can arise from single nucleotide polymorphisms (SNP) or gene copy number variation (CNV). Such genetic mutations are now recognised increasingly as leading to differential responsiveness to infection and thus predisposition to autoimmune diseases [6,7].
Insights into the current understanding of the properties and mechanisms of action of IVIgs were provided in the session chaired by Drs Anne Durandy and Robert Rieben. How IgG may affect the cellular compartment was addressed by Dr Srini Kaveri, while Dr Milan Basta presented evidence for IVIg-mediated scavenging of complement fragments. Dr Sylvia Miescher discussed the role of the dimeric fraction of IVIg. The finding that a minor population of sialylated IgG molecules may be responsible for mediation of its anti-inflammatory effect was presented by Dr Jeffrey Ravetch. Finally, Dr Taco Kuijper's presentation focused upon the genetic organization of FcγRs and on the establishment of new techniques to unravel CNV in the FcγR family and the subsequent discovery of previously unknown gene alterations. These findings demonstrate the considerable progress that has been made in understanding the mechanisms of action of IVIgs, and may influence future perspectives in the field of Ig therapy.
Immune cell function and the impact of IVIg
Some of the beneficial effects of administered IgG extend beyond its half-life, suggesting that these effects are not due merely to passive clearance or competition with pathogenic autoantibodies. These observations raise the possibility that Ig therapy results in significant alterations in the cellular compartment of the immune system [1,5]. Several recent observations have emphasized the effects of Ig therapy on different cells of the innate and adaptive compartments of the immune system, including dendritic cells (DCs), the monocyte/macrophage system, granulocytes, natural killer (NK) cells, various subsets of T cells, in particular the regulatory T cell (Treg) subset, and B cells (Fig. 1). Collectively, these findings may help to explain the beneficial effects of administered IgG in disorders caused by dysregulated cellular immunity [5].
Fig. 1.
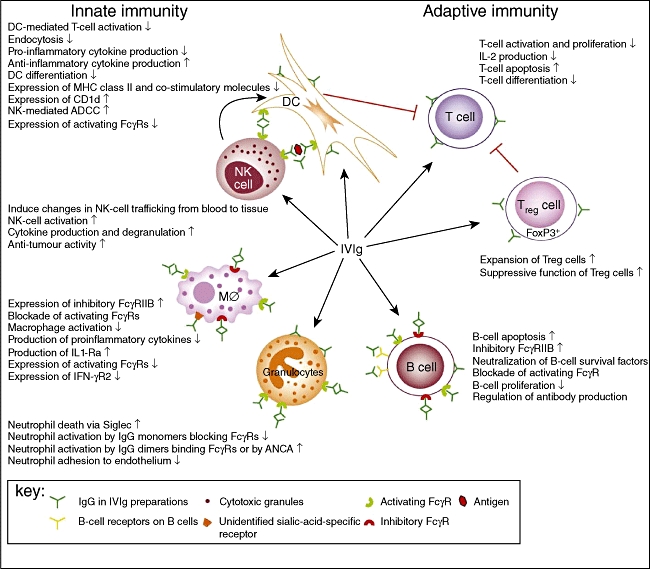
A schematic representation of the proposed mechanisms of action of intravenous immunoglobulin (IVIg) on cellular immunity. IVIg targets the cellular immune compartment at multiple levels, including innate and adaptive immune cells. IVIg interacts with dendritic cells (DCs), macrophages (MØ) and granulocytes, mainly via activating and inhibitory FC gamma receptors (FcγRs). Monomeric IgG in IVIg preparations can block the interaction of immune complexes with activating FcγRs, thereby inhibiting endocytosis and phagocytosis by DCs and macrophages and activation of granulocytes. IgG dimers in IVIg preparations binding to activating FcγRs on macrophages induce the expression of the inhibitory FcγRIIB and suppress expression of interferon (IFN)-γR2, thereby inhibiting macrophage functions. In addition, IgG dimers promote antibody-dependent cell-mediated cytotoxicity (ADCC) of DCs by natural killer (NK) cells, resulting in reduced T cell activation. IgG dimers suppress macrophage and B cell functions by ligating FcγRIIB. In addition, F(ab′)2-mediated effects of natural antibodies present in IVIg have been described for DCs, granulocytes and B cells. Interactions between IVIg and regulatory T cells (Tregs) lead to expansion and increased suppressive function of Tregs. The immunological effects depicted are not mutually exclusive and are likely to work in synergy. Reprinted from Tha-In et al.[28], copyright 2008, with permission from Elsevier.
The effect of Ig therapy on DCs was studied in view of their implicated role at several levels in the pathogenesis of autoimmune diseases, inflammatory disorders and allograft rejection. Ig therapy was shown to inhibit the differentiation and maturation of normal human DCs, to inhibit the up-regulation of the co-stimulatory molecules CD80 and CD86, and the ability of DCs to process and present self-antigens [8]. Both Fc and F(ab′)2 fragments of IgG were able to mediate the suppression of DCs. This suggests the involvement of FcγR and non-Fc-receptor-mediated signalling events in Ig-therapy-mediated modulation of DC function. At a dose used in the therapy of autoimmune and inflammatory conditions (25–35 mg/ml), IVIg also interfered with the differentiation of DCs from systemic lupus erythematosus (SLE) patients [9]. The IVIg-treated, immature DCs showed inhibited expression of human leucocyte antigen (HLA) and CD80/CD86 and displayed a reduced ability to ingest nucleosomes by up to 36%. Given the critical role of HLA molecules and co-stimulatory signals delivered by CD80 and CD86 for optimal antigen presentation and T cell activation, the inhibition of expression of these molecules by IVIg offers a plausible explanation for the efficacy of Ig therapy in SLE and other immunoinflammatory conditions. Similarly, IVIg was shown to suppress the expression of DCs in a model of autoimmune giant cell myocarditis [10] and to reduce the number of DCs in the cerebrospinal fluid of patients with Guillain–Barré syndrome and chronic inflammatory demyelinating polyradiculoneuropathy [11].
At a lower dose, administered generally to patients with immunodeficiencies, however, IVIg exerts a contrasting effect. DCs of patients with CVID differentiated in the presence of IVIg and presented with an up-regulated expression of CD1a and the co-stimulatory molecules CD80, CD86 and CD40 [12,13].
Defective functions of DCs have been associated with predisposition to several pathological conditions. CVID patients display high susceptibility to recurrent infections and autoimmune diseases that could be due in part to impaired DC functions [12,13]. Remission from pathological manifestations following infusion of IVIg, together with partially restored phenotypes of DC, points towards an active role of IVIg in maintaining immune homeostasis through interactions with the cellular compartment. Similarly, DCs of patients with X-linked agammaglobulinaemia (XLA), that were allowed to differentiate in the presence of autologous plasma reconstituted with IVIg to physiological levels, expressed increased levels of maturation markers compared with cells cultured without in vitro addition of immunoglobulins [14]. Together, the data suggest that the defective differentiation of DCs in CVID and XLA patients depends in part on low levels of circulating antibodies, and their substitution through IVIg ameliorates the defective functioning of the DC compartment. This process is mediated, at least to some extent, by anti-CD40 antibodies within IVIg, and is accompanied by increased interleukin (IL)-10 and decreased IL-12 production by DCs [8].
IgG also exerts anti-inflammatory effects through monocytes and macrophages. It alters transcription of various inflammatory genes [15], and lowers circulating levels of the proinflammatory cytokines tumour necrosis factor (TNF)-α and IL-1β[16]. Furthermore, IgG triggers monocyte production of IL-1 receptor antagonist (IL-1RA), a potent anti-inflammatory cytokine that counteracts IL-1. It blocks transiently the function of FcγR on splenic macrophages, as demonstrated by decreased clearance of anti-D-coated autologous erythrocytes in patients [17]. In addition, peripheral blood monocytes from idiopathic thrombocytopenic purpura (ITP) patients treated with IVIg exhibit a decreased ability to form rosettes with IgG-coated erythrocytes [18]. IVIg increases expression of FcγRIIB on the surface of effector macrophages in mice [1]. However, the IVIg-mediated beneficial effect was not observed in mice lacking colony-stimulating factor (CSF)-1-dependent macrophages. Thus, it has been proposed that CSF-1-dependent ‘regulatory’ macrophages in the marginal zone of the spleen regulate IVIg-mediated anti-inflammatory effects by enhancing the expression of FcγRIIB on the surface of effector macrophages. However, this concept is inconsistent with a recent report showing that overexpression of FcγRIIB on splenic macrophages does not influence the pathogenesis of autoimmune diseases in mice [19]. The effects of IVIg on FcγRIIB expression on human tissue macrophages have not been studied, but IVIg treatment does not alter FcγRIIB mRNA expression in circulating human monocytes [15]. Therefore, the role of FcγRIIB in the effects of IVIg treatment, especially in humans, remains unresolved [20].
Effects of IVIg on the adaptive cellular compartment include interaction with B and T cells. The therapeutic effects of IVIg in several antibody-mediated diseases reflect more than mere neutralization and FcγR blockade. They may include suppression of the expansion of autoreactive B lymphocytes through signalling via FcγRIIB, idiotype-mediated inhibition of B cell receptors and neutralization of cytokines such as the survival factors B cell activating factor (BAFF) and a proliferation-inducing ligand (APRIL) [21,22]. Interaction of the anti-idiotypic antibodies with membrane-bound IgG or IgM of B cells can transmit negative signals and result in down-modulation of pathogenic autoantibody production. IVIg has been shown to induce the secretion of IgG reacting against various self- and non-self antigens from a unique subset of human B lymphocytes. These de novo-induced antibodies might further help in controlling the reactivities of pathogenic autoantibodies either by idiotype-mediated mechanisms or by sequestering autoantigens [23,24]. Analogous to these in vitro findings, IVIg therapy in women with recurrent spontaneous abortion is accompanied by a decrease in the peripheral blood B cell numbers [25].
Although it is well established that suppressive effects of IVIg on T cells are significant, how IVIg affects the function of distinct T cell subsets is unclear. Initial findings, wherein IVIg renders a DC a tolerogenic potential, led to further examination of whether IVIg induces immune tolerance via modulation of T cell subsets, in particular Tregs. It was observed recently that IVIg can expand and enhance the functions of human and murine forkhead box P3 (FoxP3+) Tregs[26]. Accordingly, beneficial effects of IVIg in patients have been shown to be associated with an increase in Tregs both quantitatively and qualitatively. In animal studies of experimental autoimmune encephalomyelitis, IVIg failed to protect against inflammation in mice depleted of Tregs. The mechanisms underlying the IVIg-mediated expansion and enhanced suppressive properties of Tregs remain unclear and are subject to further investigation [26–28].
Thus, the ability of IVIg to interact with relevant immunoregulatory molecules and cells of the immune system, both innate and adaptive, provides the basis for the reestablishment of an immune equilibrium.
Scavenging of complement fragments by IVIg
Scavenging of active complement fragments by high doses of Igs has gained recognition as one of the beneficial mechanisms in autoimmune conditions. Binding of IgG molecules to potentially harmful complement fragments (C3b, C4b, C3a and C5a) blocks deposition of these fragments onto their targets and prevents subsequent immune damage that stems from cellular destruction and/or excessive inflammation [29].
The first series of experiments to investigate the impact of Ig on complement scavenging employed an in vitro complement uptake system [30]. In this assay, corpusculate immune complexes (red cells sensitized with specific anti-erythrocyte antibodies) were used to trigger complement activation and serve as targets for deposition of complement fragments. Supplementation of guinea pig serum with 50 mg/ml IVIg caused significant (50–60%) inhibition of C3b and C4b uptake compared with sera containing added human serum albumin [31,32]. Complement uptake inhibition was much more pronounced, and reached almost 100% in sera from IVIg-treated guinea pigs [33] and humans [32].
In subsequent studies, complement uptake was quantified in serum samples of patients subjected to IVIg therapy. In active dermatomyositis (DM), Kawasaki disease, autoimmune haemolytic anaemia (AIHA), Guillain–Barré syndrome and myasthenia gravis, diseases that are known or suspected to be mediated by complement, the baseline or pre-IVIg uptake was higher in comparison with healthy controls. Increased baseline uptake indicates a rapid turnover of complement fragments as a consequence of continuous complement activation and lends further support to the role of complement in pathogenesis of these conditions. In inclusion body myositis and chronic DM, conditions in which the participation of complement in pathogenesis has not been demonstrated, baseline uptake was not significantly different from values observed in normal controls. Post-IVIg uptake was inhibited markedly compared with the baseline values in active DM, Kawasaki and AIHA and correlated with the positive clinical response to IVIg infusions (Table 1). The uptake assays may have a role in clinical medicine based on the fact that they can provide evidence of complement activation in a given condition, and may be useful in terms of monitoring disease activity and measuring response to IVIg therapy [29].
Table 1.
C3b uptake in serum samples from patients with complement-mediated conditions, control diseases and healthy individuals. All values are means of triplicate measurements of C3 uptake in counts per minute.
| Disease | Pre-IVIg uptake | Post-IVIg uptake | Inhibition (%) |
|---|---|---|---|
| Active DM (n = 9) | 12 190 | 2 350 | 80·6 |
| Kawasaki (n = 6) | 19 120 | 12 120 | 36·6 |
| AIHA (n = 3) | 54 123 | 32 303 | 40·3 |
| Guillain–Barré (n = 9) | 11 590 | n.a. | n.a. |
| Myasthenia gravis (n = 10) | 10 570 | n.a. | n.a. |
| Chronic DM (n = 3) | 1 980 | 1 942 | 0·2 |
| IBM (n = 10) | 3 491 | 3 264 | 0·9 |
| Normal individuals (n = 10) | 3 459 | n.a. | n.a. |
AIHA: autoimmune haemolytic anaemia; DM: dermatomyositis; IBM: inclusion body myositis; IVIg: intravenous immunoglobulin; n.a.: not available.
In DM, terminal components of the classical complement cascade, assembled in the membrane attack complex (MAC), mediate the damage to endothelial cells in endomysial capillaries that leads subsequently to ischaemia of muscle fibres and muscle weakness. C3b fragments are observed in muscle biopsies taken before IVIg therapy. Immunohistochemical analysis of post-IVIg biopsies revealed significant reduction and even elimination of MAC complexes and C3b fragments [34].
Immortalized mast cells (HMC-1), a cell line that expresses both C3a and C5a receptors, were used to demonstrate that pre-incubation of C3a and C5a with Fab-containing IVIg prevented anaphylatoxin-mediated calcium signalling. Subsequent release of proinflammatory mediators, such as histamine and thromboxane, was also inhibited [35].
The ability of IgG to bind C3b and C4b is a function of the Fc region of IgG molecules, as shown by the uptake inhibition assays [29]. The acceptor site of C3b/C4b was mapped to residues 381–390 of the CH3 domain of the IgG Fc fragment. Surface plasmon resonance analysis indicated that the acceptor site for C3a and C5a resides within the constant domain of Fab. The Fc fragment of IgG did not interact with immobilized C3a and C5a, while a human immunodeficiency virus (HIV)-specific monoclonal antibody still bound to C3a even in the presence of specific antigen (gp120) that completely saturated its binding site [36].
Scavenging ability is not restricted to a particular Ig phenotype. All four IgG subclasses and different IgG allotypes exhibited potent inhibition of C3b/C4b uptake [29]. IgM was more effective than IgG, both in solid-phase immune complex essays [37,38] and in a xenogeneic system consisting of pig endothelial cells as targets for C3b and C4b deposition and human serum as a source of the activated fragments [39].
Scavenging of large complement fragments was investigated in two animal models of complement-mediated conditions. Clearance of chromate-labelled IgM-sensitized guinea pig erythrocytes is dependent upon their opsonization with C3b and subsequent removal from the circulation due to their adherence to C3b receptors on liver macrophages. In guinea pigs treated with IVIg, clearance of chromate-labelled, IgM-sensitized red cells was suppressed relative to albumin- and saline-treated animals, presumably because of decreased opsonization of target erythrocytes due to scavenging of C3b by IVIg [31]. In Forssman shock, complement-dependent destruction of pulmonary capillaries with ensuing rapidly fatal pulmonary oedema was prevented in up to 75% of IVIg-treated guinea pigs, while no control (non-treated or albumin-treated) animal survived this cataclysmic reaction [33]. In vivo scavenging of C5a was demonstrated in a porcine model of anaphylatoxin-induced cardiopulmonary distress. In this model, injection of recombinant human C5a (40 ng/kg) caused a maximum rise followed by complete loss of pulmonary arterial pressure, associated with decline and loss of systemic arterial pressure and death within minutes. In animals pretreated with 300 mg/kg Fab-containing IVIg the reaction was prevented even after injections of multiple (up to 12) lethal doses of C5a [35].
An asthma-like condition, characterized by cellular migration into bronchial and alveolar compartments, was induced in mice by systemic sensitization and airway challenge with ovalbumin. The role of C3a in pathogenesis of this condition was suggested by the reduced number of perivascular infiltrates (cuffs) in the lungs and a decrease of eosinophils in bronchoalveolar lavage fluid from mice homozygous for C3 gene deletion (C3−/−) and therefore lacking the parent C3 molecule. IVIg treatment of Balb/c mice resulted in a suppression of cellular migration similar in magnitude to that observed in C3−/− mice, supporting the hypothesis that IVIg exerts its beneficial effect by neutralizing C3a [35].
The complement-scavenging ability of Ig implies expansion of the use of IVIg to all diseases in which generation of complement fragments plays a crucial role in pathogenesis. Considering the large number of complement-mediated conditions and the limited supply of IVIg preparations, novel clinical applications should be limited to life-threatening diseases that represent a major global health problem. One condition that meets these criteria is stroke, the third leading cause of death and the most common reason for permanent neurological disability worldwide [40]. In 70–80% of cases, cerebral infarction is caused by arterial (most frequently middle cerebral) occlusion with subsequent neuronal ischaemia that triggers complement activation and in situ inflammation [41]. A mouse model of transient middle cerebral artery occlusion was employed to examine the effect of IVIg treatment on the outcome of experimental stroke. IVIg, administered at 2 g/kg either before or up to 3 h following ischaemia, practically eliminated mortality, reduced infarction size by 60% and diminished neurological deficit two- to threefold compared with control animals (non-treated or treated with albumin, IVIg vehicle and saline). The same protective effect was observed when the dose of IVIg was reduced to 0·5 g/kg [42].
Mice lacking the C5 gene (C5−/−) and subjected to ischaemia/reperfusion (I/R) brain injury showed improved functional outcome and less brain damage compared with their wild-type littermates. It was found that I/R brain injury caused an increase of C3b at the site of injury. IVIg treatment resulted in a highly significant suppression of binding of C3b, while vehicle and albumin had no such effect. Immunoprecipitation analysis of brain samples from the ischaemic hemisphere of IVIg-treated mice showed that human IgG bound mouse C3b (Fig. 2). Co-immunoprecipitation data support scavenging of complement fragments as the protective mechanism of action of IVIg in this model. Primary neuronal cell culture was subjected to in vitro oxygen and glucose deprivation, mimicking the in vivo situation caused by arterial obstruction. Under these conditions, neurones exhibited an increase of intrinsic C3 and caspase-3, suggesting that brain hypoxia may induce complement-mediated apoptosis. Both C3 and caspase-3 signals were suppressed by IVIg supplementation of the culture medium [42].
Fig. 2.
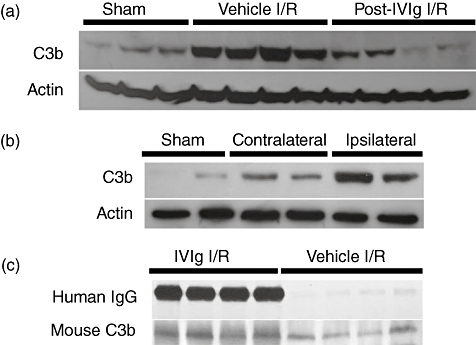
Attenuation of in situ C3b binding by intravenous immunoglobulin (IVIg). (a) Ischaemia/reperfusion (I/R) brain injury causes increased presence of the complement protein C3b in the affected tissue, and post-injury IVIg treatment reduces the I/R-mediated C3b binding. (b) C3b binding is strongest at the site of the lesion, i.e. in the ipsilateral region. (c) Immunoprecipitation analysis of brain samples from the ischaemic hemisphere of IVIg-treated mice, showing that human IgG binds mouse C3b. Shams: animals subjected to the same extent of anaesthesia and surgery as I/R animals, except for the middle artery occlusion; controls for the effect of surgical and anaesthesiological injury. Vehicle: animals infused with the stabilizer for the immunoglobulins in IVIg preparation; controls for the specificity of immunoglobulin effect in the IVIg preparation.
The results of the above studies are encouraging, and suggest strongly that clinical trials using low-dose IVIg infusions should be considered as a possible interventional therapy for stroke. In addition, IVIg should be tested in models of other serious human conditions in which ischaemia and reperfusion induce generation of pathogenic complement fragments.
Dimer formation in IVIgs
Therapeutic Ig formulations contain intact IgG molecules with an isotype distribution similar to that found in normal human serum. Besides the monomeric IgG, the formulations contain dimeric IgG molecules, first reported in 1982. At that time their clinical significance was not recognized, neither for their potential contribution to the mechanisms of action of supplemented IgG nor for their influence on infusion tolerability. Therapeutic Ig formulations contain variable amounts of monomeric and dimeric IgG existing in a dynamic equilibrium depending on the IgG concentration, the pH of the IgG solution, the storage temperature and the size of the donor pool for the starting material [43,44] (Fig. 3). Additionally, amphiphilic amino acids such as proline, which contain both hydrophilic and hydrophobic chemical groups, can interfere with dimer formation, probably by interacting with hydrophobic groups in the IgG molecule, preventing protein–protein association and stabilizing the IgG molecules in their monomeric form. Dimeric IgG has been observed by electron microscopy and can be separated by high performance lipid chromatography [45]. Previous reports suggest that IgG dimers are idiotype–anti-idiotype pairs, which contribute to the immunoregulatory actions of IVIg [46–48]. However, there is limited information on a potential selective partition of antibody specificities into the monomeric and dimeric fractions or on the immunological significance of the fractions.
Fig. 3.
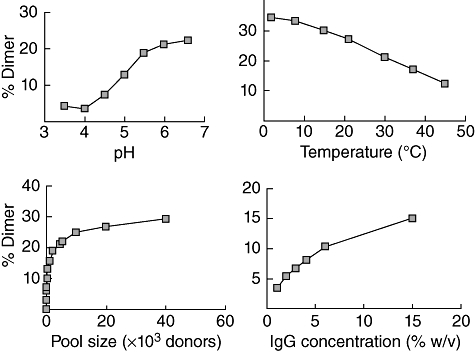
Factors affecting dimer formation. Larger donor pool size, low temperature, high pH and high immunoglobulin (Ig)G concentration increase the dimer content of therapeutic Ig formulations.
Highly purified fractions (> 95%) of monomeric and dimeric IgG were isolated by size-exclusion chromatography. The dimeric IgG fraction separated under physiological pH 7·0 conditions was labile and tended to dissociate, whereas the monomeric IgG fraction was stable at pH 7·0. In order to obtain a dynamically stable dimeric fraction for comparative analyses, all fractions were treated at pH 4·0, resulting in a stable dissociated dimer fraction (containing > 90% monomeric IgG), and ensuring that all fractions contained an equal number of F(ab)2 binding sites. Biochemical analysis by two-dimensional gel electrophoresis showed no significant differences between the fractions. In contrast, there was a segregation of the binding capacity in the different fractions according to the nature of the antigen. The dissociated dimeric fraction showed a markedly increased reactivity against certain self-antigens, in particular conserved intracellular self-antigens, as well as members of the Siglec family and some bacterial exotoxins [49–51].
Immunocytological studies on human epithelial type 2 (HEP-2) cells, which are used conventionally for the serological diagnosis of autoimmune diseases, revealed striking differences in staining depending on the Ig fraction used. Unseparated IVIg, monomeric and dimeric IgG clearly recognize self-antigens within the HEP-2 cells, demonstrating a perinuclear pattern with enhanced staining of both nuclear and cytoskeletal structures by the dimeric IgG fraction (Fig. 4b–d). No pathological autoimmune patterns were observed. For comparison, Fig. 4a shows a typical speckled anti-centromere staining using serum from an SLE patient. IgG purified from single donor serum or IgM from cord blood showed very similar perinuclear patterns as seen for the monomeric IgG (Fig. 4e,f).
Fig. 4.

Immunofluorescent staining patterns of the different immunoglobulin (Ig) preparations analysed by incubation on human epithelial type 2 (HEP-2) cells. (a) Positive anti-centromere control serum, showing typical speckled staining pattern. Inset shows May–Grünwald–Giemsa staining of HEP-2 cells and black inset is the conjugate control. Staining of perinuclear antigens characterizes the patterns for intravenous immunoglobulin (IVIg) (b) and the monomeric IgG fraction (c). The dimeric IgG fraction shows enhanced nuclear and cytoplasmic staining (d). IgG purified from adult single-donor serum (e) and IgM from cord-blood serum (f) showed similar staining on perinuclear structures comparable to monomeric IgG and IVIg.
A similar increase in activity of the dimeric fraction was observed against exotoxin A of Pseudomonas aeruginosa, an opportunistic pathogen of clinical relevance for immunocompromised patients. In particular, comparison of the dimer fraction versus the dissociated dimer fraction revealed substantially increased signals, when analysed by surface plasmon resonance. Taken together, these results support the hypothesis that the dimeric IgG fraction originating from large donor pools is enriched in antibodies of certain specificities but not others, which are unmasked in the dissociated dimer fraction, and this may represent an aspect of the idiotype network. This was investigated further using affinity-purified antibodies from IVIg against the Siglec-9 receptor. The Siglecs are a family of cell surface proteins found primarily on haematopoietic cells. They bind sialic-acid-containing glycan ligands, are linked to cell signalling pathways, and natural antibodies against Siglecs may contribute to the anti-inflammatory mechanisms of IVIg [51]. Immobilized affinity-purified anti-Siglec-9 antibodies showed no binding to the monomeric IgG fraction, whereas the dissociated dimer fraction clearly bound to its presumed anti-idiotypic partner, supporting the presence of the idiotype network in the dimer fraction.
The tolerability of liquid IVIgs during the infusion period is influenced by the level of dimeric IgG. In an investigational phase I study in healthy volunteers, blood samples were taken pre- and post-IVIg infusion over a period of 24 h. Dosage (0·4 g/kg body weight) and infusion rate were controlled and equal for all volunteers. Transient early adverse reactions, such as elevated temperature, chills, myalgias, malaise, headache, hypotension and nausea were monitored and a clinical score for well or poorly tolerated infusions was defined. Kinetic analysis showed elevated levels in the blood of proinflammatory cytokines (TNF-α, IL-6, IL-8 with maximal levels at 2·5, 3 and 4 h, respectively) followed by cytokine antagonists [IL-1Rα, soluble TNF receptor 1 (TNFRI) and TNFRII]. These changes in cytokine levels were accompanied by transient decreases in neutrophil, monocyte and lymphocyte counts, followed by an increase in neutrophil levels. All values returned to baseline 24 h after the infusions [52]. The cytokine levels and numbers of adverse events in individual volunteers were related directly to the tolerability of the infusions. Cytokines produced primarily by lymphocytes and not involved in early inflammatory reactions, e.g. IFN-γ, IL-2 and IL-4, were not raised during the infusions. Dimer levels measured in the products were related to the maximum proinflammatory cytokine levels measured after IVIg administration, and levels above approximately 12% were associated with poorly tolerated infusions, due to the induction of a transient inflammatory reaction. As there were no indications for complement activation, the probable mechanism for these transient inflammatory reactions is interaction of immune complexes, in the form of dimeric IgG, with cellular Fcγ receptors.
Despite their association with early adverse reactions, dimers most probably play an important immunoregulatory role in autoimmune diseases and therefore these specificities should not be removed from IVIg products. Instead, the dimer content must be controlled below a defined threshold level. As a consequence, the new liquid 10% IVIg product Privigen is formulated at pH 4·8 and stabilized with l-proline, an amphiphilic compound that restricts dimer formation, resulting in a stable and well-tolerated product.
Immunomodulation by sialylated IgG
Although the paradoxical pro- and anti-inflammatory properties of IgG were recognized, for a long time they were poorly understood. Despite numerous models being proposed, advances in the field were hampered by a lack of in vivo models that recapitulate the anti-inflammatory activity of high-dose IgG and an incomplete understanding of the biochemical composition of the active components within IVIg required for in vivo activity.
To address these shortcomings, murine models of inflammatory disease that are attenuated by the anti-inflammatory activity of high-dose IVIg or its Fc fragments, including immune thrombocytopenia [53], serum-induced arthritis [54] and nephrotoxic nephritis [55], were developed. The requirement for the IgG Fc fragment and the inhibitory receptor FcγRIIB was first demonstrated in a murine model for ITP. Administration of clinically protective doses of intact antibody or monomeric Fc fragments to wild-type or FcγR receptor-humanized mice prevented platelet consumption triggered by a pathogenic autoantibody. Disruption of FcγRIIB by either genetic deletion or with a monoclonal blocking antibody abolished the protective effect [53].
In an arthritis model, IVIg induced the expression of FcγRIIB on infiltrating macrophages, but not neutrophils, indicating a critical role for macrophage activation in antibody-induced inflammation. The protective effect was lost in mice deficient for CSF-1, highlighting the importance of a subset of CSF-1-dependent macrophages [54].
A novel γ-chain-dependent, activating Fc receptor, FcγRIV, was shown subsequently to be involved in the autoimmune disease process. Blocking FcγRIV binding to pathogenic anti-platelet antibodies was sufficient to protect mice from antibody-induced thrombocytopenia [55]. Co-expression of the activating FcγRIV and its inhibitory counterpart, FcγRIIB, on infiltrating macrophages sets a threshold for activation of these effector cells [56]. Blocking FcγRIV with a specific monoclonal antibody or high-dose IVIg to down-regulate FcγRIV and up-regulate FcγRIIB, protected mice from nephrotoxic nephritis.
As IgG glycosylation differs in patients with rheumatoid arthritis and several forms of autoimmune vasculitis, it had been suggested that individual IgG glycoforms may play a role in modulating antibody effector function in vivo. To define the potential role of the glycan structure on the IgG Fc in the anti-inflammatory activity of IVIg, these carbohydrates were removed and the resulting ability to inhibit inflammatory responses was assessed in a model of rheumatoid arthritis. Deglycosylated IVIg was unable to mediate anti-inflammatory activity [57]. Further characterization demonstrated that the anti-inflammatory activity of IVIg results from a minor population of the pooled IgG molecules that contains terminal α-2,6 sialic acid linkages on their Fc-linked glycans [58]. This fully processed glycan is found in 1–3% of IgG in IVIg, which may explain the requirement for a high dose of IVIg. Recent data demonstrate that the anti-inflammatory properties of IVIg can be recapitulated with a fully recombinant preparation of appropriately sialylated IgG Fc fragments (Fig. 5) [58]. The recombinant preparation had a 35-fold enhanced in vivo activity and could potentially be used at much lower doses than IVIg preparations.
Fig. 5.
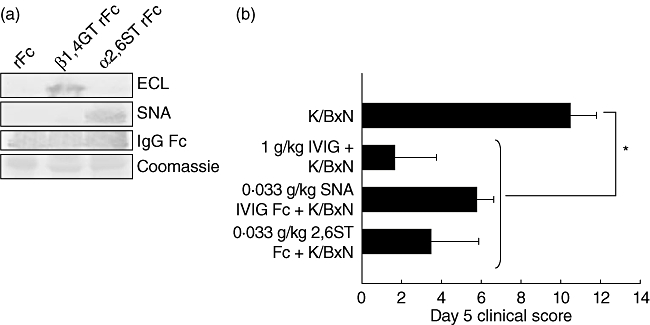
Recombinant, sialylated immunoglobulin (Ig)G Fc fragments are anti-inflammatory and comparable to native intravenous immunoglobulin (IVIg). Recombinant human IgG1 was digested with papain, and Fc fragments were purified. The recombinant Fc fragments were galactosylated and sialylated in vitro with α-2,6-sialyltransferase (α-2,6-ST). (a) Glycosylation was confirmed by lectin blotting for terminal galactose with enhanced chemiluminescence (ECL) (top) or α-2,6-sialic acid with Sambucus nigra agglutinin (SNA) (middle); Coomassie loading controls are shown (bottom). (b) Mice were administered 1 g/kg IVIg, 0·033 g/kg SNA+ IVIg Fc fragments, or 0·33 g/kg sialylated recombinant Fc (α-2,6-ST rFc) 1 h before K/BxN sera, and footpad swelling was monitored over the next several days. Means and standard deviations of clinical scores of four to five mice per group are plotted. *P < 0·05. From Anthony RM et al.[58]; reprinted with permission from the American Association for the Advancement of Science.
For mediating their anti-inflammatory effect, sialylated Fc fragments require a specific C-type lectin, SIGN-R1 (specific ICAM-3 grabbing non-integrin-related 1) to be expressed on macrophages in the splenic marginal zone [59]. Splenectomy, loss of SIGN-R1+ cells in the splenic marginal zone, blockade of the carbohydrate recognition domain (CRD) of SIGN-R1 or genetic deletion of SIGN-R1 abrogated the anti-inflammatory activity of IVIg or sialylated Fc fragments. Although SIGN-R1 has not been shown previously to bind to sialylated glycans, it has been demonstrated that SIGN-R1 binds preferentially to α2,6 sialylated Fc compared with similarly sialylated, biantennary glycoproteins, suggesting that a specific binding site is created by the sialylation of IgG Fc. A human orthologue of SIGN-R1, DC-SIGN, displays a binding specificity similar to that of SIGN-R1 but differs in its cellular distribution, accounting potentially for some of the species differences observed in IVIg protection.
With the discovery of FcγRIIB, sialylated IgG and SIGN-R1, some of the mechanistic steps accounting for the anti-inflammatory action of IVIg have been unravelled. In the future, it will be of particular interest to determine whether the reproduction of IVIgs anti-inflammatory properties by recombinant preparations can be translated into the treatment of human autoimmune diseases.
Immunomodulation via Fc receptor polymorphism
The genes for FcγRs are located on chromosome 1 as a series, resulting from gene duplication during evolution. Based on their affinity for monomeric IgG, the FcγRs can be subdivided into high-affinity receptors (type I) and low-affinity receptors (types II and III). The genes encoding the high-affinity receptors (FCGR1A, FCGR1B and FCGR1C) are located in the region 1q21, while the low-affinity FcγRs (FCGR2A, FCGR2B, FCGR2C, FCGR3A and FCGR3B) are located at 1q23–24 (Fig. 6) [60].
Fig. 6.
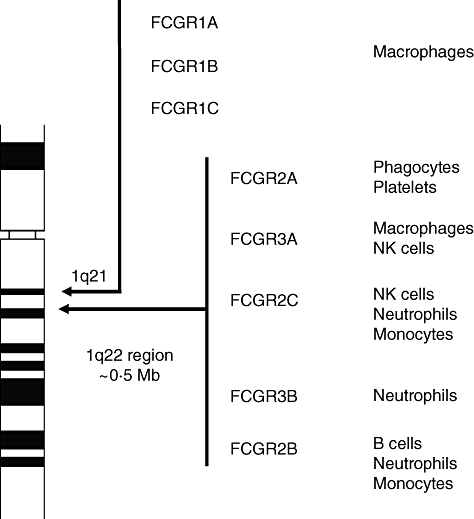
Human Fc gamma (Fcγ) receptors. In humans, the genes encoding for the Fcγ receptors are located on chromosome 1. The receptors are expressed on different cell types, as indicated on the right.
FcγRs are highly homologous, making them difficult to study. For example, FcγRIIA, FcγRIIB and FcγRIIC are encoded by three genes, which are 99% homologous. This hampers the use of monoclonal antibodies in this area, as they often recognize two or more isoforms. Of particular interest is FcγRIIC, which is a product of an unequal cross-over of the IIB and the IIIB cytoplasmic tail [61]. Due to a stop codon in exon 3, the gene was long considered to be a pseudogene [62].
SNPs have been detected in the second extracellular domain of the activating receptors and the transmembrane-spanning domain of the inactivating receptors and have been shown to alter receptor affinity. Such functional variations in human FcγR have been studied for some time and efforts have been undertaken to relate them to certain diseases and their outcome [63].
An additional layer of complexity was added by recent studies identifying CNV in large areas at chromosome 1q23–24 [64]. A linked deletion and duplication of the FCGR3B and FCGR2C genes [65–67] was described, and an association between a low copy number of FCGR3B and glomerulonephritis in SLE has been reported recently [68–70]. The low gene copy number correlates with reduced FcγRIIIB expression and is likely to contribute to the impaired clearance of immune complexes, a feature of SLE [68].
A new assay to detect simultaneously both SNPs and CNVs of the low-affinity FcγRs has been developed recently [71]. This multiplex ligation-dependent probe amplification (MLPA) assay allows for more accurate genotyping. It is able not only to detect the copy number of 50 DNA sequences in a simple, polymerase chain reaction (PCR)-based reaction, but also new unknown SNPs. Using MLPA, FCGR2C and FCGR3B CNV was confirmed, and a CNV for FCGR3A was identified. Similar to FCGR2C and FCGR3B, a gene dosage effect for FCGR3A was found which seemed to correlate with FCγRIIIa expression and function on NK cells [72]. Delineation of the approximate boundaries of the CNV at the FCGR locus showed that co-segregation of neighbouring FCGR genes was limited to five variants, with patterns of Mendelian inheritance. No CNV of the FCGR2A and FCGR2B genes was observed in more than 600 individuals [72].
In addition to the CNVs, a SNP in exon 3 of FCGR2C has been characterized recently [73]. This SNP changes the common stop codon into an open reading frame (ORF), turning the pseudogene FCGR2C-Stop into a formal gene FCGR2C-ORF. This change results in active transcription of the gene and expression of a novel FcγR isoform, FcγRIIc. Interestingly, in a cohort of ITP patients, the proportion of individuals carrying one or two alleles with this ORF was much higher than in a control group of healthy individuals (18% versus 34%) (Fig. 7) [73].
Fig. 7.
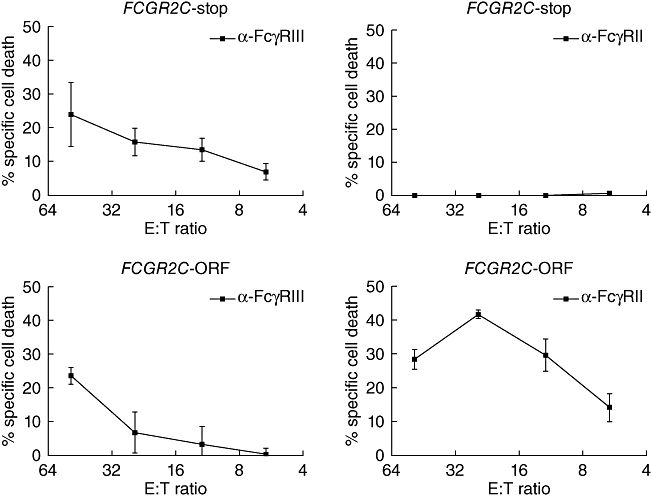
Redirected antibody-dependent cell-mediated cytotoxicity (rADCC) of effector natural killer (NK) cells against target cells. Peripheral blood lymphocytes were isolated and Fc gamma receptor IIC (FcγRIIC) functionality was assessed by rADCC. Cells from both FCGR2C-Stop and FCGR2C-open reading frame (ORF) genotyped donors killed anti-FcγRIII-coated targets with similar kinetics (left panels). In contrast, only cells from FCGR2C-ORF genotyped donors were capable of killing anti-FcγRII-coated targets (right panels) (n = 4). Data are expressed as mean ± standard error of the mean.
FcγRIIC is usually not expressed. In case of an ORF, however, it is expressed on NK cells, monocytes, neutrophils and DCs of ITP patients and has a distinct immunological function. For instance, in individuals with the common FCGR2C pseudogene and therefore without this receptor, NK cells did not show killing of target cells in an assay for redirected antibody-dependent cell-mediated cytotoxicity. Conversely, NK cells from individuals with an ORF were able show antibody-dependent killing of target cells via the activating IgG receptor FcγRIIc (Fig. 7). It was therefore proposed that the activating FCGR2C-ORF genotype predisposes to ITP by altering the balance of activating and inhibitory FcγRs on immune cells [73].
The effects of CNVs and SNPs on susceptibility to disease, severity and outcome are now being studied in cohorts of patients with Kawasaki disease, SLE and rheumatoid arthritis. The use of new analytical techniques, such as MLPA, facilitates the study of complex human genomic structures and the discovery of new SNPs and CNVs and may allow for new susceptibility loci for autoimmunity to be found [7].
Summary
The mechanisms by which IVIg preparations exert their immunomodulatory and anti-inflammatory effects are beginning to be understood, as are the genetic alterations that may predispose some individuals to the development of autoimmune disorders. Some of the findings presented in this session may have important implications for the future use of IVIg therapy. Should IVIg therapy become used routinely for the treatment of complement-mediated diseases, such as stroke, the adequate supply of therapeutic formulations may become critical. The modulation of the anti-inflammatory properties of IVIg by recombinant sialylated Fc fragments may be seen as a first step in the direction of the generation of recombinant therapeutics, but at present only mouse data are available and the road to clinical application is still very long.
Acknowledgments
Dr Miescher would like to thank the following researchers for their contributions: Alexander Schaub; Sandra Wymann; Stephan von Gunten; Martin Spycher; and Reinhard Bolli. Dr Kaveri expresses his gratitude towards Dr Jagadeesh Bayry. The authors also thank nspm ltd for providing medical writing services, with financial support through an unrestricted educational grant from CSL Behring.
Disclosures
RR receives financial support for his research on IVIg from CSL Behring AG and from Biotest Pharma AG. All other authors have declared that they have no conflicts of interest.
References
- 1.Nimmerjahn F, Ravetch JV. Anti-inflammatory actions of intravenous immunoglobulin. Annu Rev Immunol. 2008;26:513–33. doi: 10.1146/annurev.immunol.26.021607.090232. [DOI] [PubMed] [Google Scholar]
- 2.Conley ME, Dobbs AK, Farmer DM, et al. Primary B cell immunodeficiencies: comparisons and contrasts. Annu Rev Immunol. 2009;27:199–227. doi: 10.1146/annurev.immunol.021908.132649. [DOI] [PubMed] [Google Scholar]
- 3.Durandy A, Wahn V, Petteway S, Gelfand EW. Immunoglobulin replacement therapy in primary antibody deficiency diseases – maximizing success. Int Arch Allergy Immunol. 2005;136:217–29. doi: 10.1159/000083948. [DOI] [PubMed] [Google Scholar]
- 4.Gürcan HM, Ahmed AR. Efficacy of various intravenous immunoglobulin therapy protocols in autoimmune and chronic inflammatory disorders. Ann Pharmacother. 2007;41:812–23. doi: 10.1345/aph.1K037. [DOI] [PubMed] [Google Scholar]
- 5.Kazatchkine MD, Kaveri SV. Immunomodulation of autoimmune and inflammatory diseases with intravenous immune globulin. N Engl J Med. 2001;345:747–55. doi: 10.1056/NEJMra993360. [DOI] [PubMed] [Google Scholar]
- 6.Fanciulli M, Vyse TJ, Aitman TJ. Copy number variation of Fc gamma receptor genes and disease predisposition. Cytogenet Genome Res. 2008;123:161–8. doi: 10.1159/000184704. [DOI] [PubMed] [Google Scholar]
- 7.Schaschl H, Aitman TJ, Vyse TJ. Copy number variation in the human genome and its implication in autoimmunity. Clin Exp Immunol. 2009;156:12–6. doi: 10.1111/j.1365-2249.2008.03865.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bayry J, Lacroix-Desmazes S, Carbonneil C, et al. Inhibition of maturation and function of dendritic cells by intravenous immunoglobulin. Blood. 2003;101:758–65. doi: 10.1182/blood-2002-05-1447. [DOI] [PubMed] [Google Scholar]
- 9.Bayry J, Lacroix-Desmazes S, Delignat S, et al. Intravenous immunoglobulin abrogates dendritic cell differentiation induced by interferon-alpha present in serum from patients with systemic lupus erythematosus. Arthritis Rheum. 2003;48:3497–502. doi: 10.1002/art.11346. [DOI] [PubMed] [Google Scholar]
- 10.Shioji K, Kishimoto C, Sasayama S. Fc receptor-mediated inhibitory effect of immunoglobulin therapy on autoimmune giant cell myocarditis: concomitant suppression of the expression of dendritic cells. Circ Res. 2001;89:540–6. doi: 10.1161/hh1801.096263. [DOI] [PubMed] [Google Scholar]
- 11.Press R, Nennesmo I, Kouwenhoven M, et al. Dendritic cells in the cerebrospinal fluid and peripheral nerves in Guillain–Barré syndrome and chronic inflammatory demyelinating polyradiculoneuropathy. J Neuroimmunol. 2005;159:165–76. doi: 10.1016/j.jneuroim.2004.09.020. [DOI] [PubMed] [Google Scholar]
- 12.Bayry J, Lacroix-Desmazes S, Kazatchkine MD, et al. Common variable immunodeficiency is associated with defective functions of dendritic cells. Blood. 2004;104:2441–3. doi: 10.1182/blood-2004-04-1325. [DOI] [PubMed] [Google Scholar]
- 13.Scott-Taylor TH, Green MR, Eren E, Webster AD. Monocyte derived dendritic cell responses in common variable immunodeficiency. Clin Exp Immunol. 2004;138:484–90. doi: 10.1111/j.1365-2249.2004.02640.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bayry J, Lacroix-Desmazes S, Donkova-Petrini V, et al. Natural antibodies sustain differentiation and maturation of human dendritic cells. Proc Natl Acad Sci USA. 2004;101:14210–5. doi: 10.1073/pnas.0402183101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abe J, Jibiki T, Noma S, et al. Gene expression profiling of the effect of high-dose intravenous Ig in patients with Kawasaki disease. J Immunol. 2005;174:5837–45. doi: 10.4049/jimmunol.174.9.5837. [DOI] [PubMed] [Google Scholar]
- 16.Rhoades CJ, Williams MA, Kelsey SM, Newland AC. Monocyte–macrophage system as targets for immunomodulation by intravenous immunoglobulin. Blood Rev. 2000;14:14–30. doi: 10.1054/blre.1999.0121. [DOI] [PubMed] [Google Scholar]
- 17.Fehr J, Hofmann V, Kappeler U. Transient reversal of thrombocytopenia in idiopathic thrombocytopenic purpura by high-dose intravenous gamma globulin. N Engl J Med. 1982;306:1254–8. doi: 10.1056/NEJM198205273062102. [DOI] [PubMed] [Google Scholar]
- 18.Kimberly RP, Salmon JE, Bussel JB, et al. Modulation of mononuclear phagocyte function by intravenous gamma-globulin. J Immunol. 1984;132:745–50. [PubMed] [Google Scholar]
- 19.Brownlie RJ, Lawlor KE, Niederer HA, et al. Distinct cell-specific control of autoimmunity and infection by FcgammaRIIb. J Exp Med. 2008;205:883–95. doi: 10.1084/jem.20072565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaveri SV, Lacroix-Desmazes S, Bayry J. The antiinflammatory IgG. N Engl J Med. 2008;359:307–9. doi: 10.1056/NEJMcibr0803649. [DOI] [PubMed] [Google Scholar]
- 21.Pottier L, Sapir T, Bendaoud B, et al. Intravenous immunoglobulin and cytokines: focus on tumor necrosis factor family members BAFF and APRIL. Ann NY Acad Sci. 2007;1110:426–32. doi: 10.1196/annals.1423.044. [DOI] [PubMed] [Google Scholar]
- 22.Seite JF, Shoenfeld Y, Youinou P, Hillion S. What are the contents of the magic draft IVIg? Autoimmun Rev. 2008;7:435–9. doi: 10.1016/j.autrev.2008.04.012. [DOI] [PubMed] [Google Scholar]
- 23.de Grandmont MJ, Racine C, Roy A, et al. Intravenous immunoglobulins induce the in vitro differentiation of human B lymphocytes and the secretion of IgG. Blood. 2003;101:3065–73. doi: 10.1182/blood-2002-06-1684. [DOI] [PubMed] [Google Scholar]
- 24.Lemieux R, Bazin R, Néron S. Therapeutic intravenous immunoglobulins. Mol Immunol. 2005;42:839–48. doi: 10.1016/j.molimm.2004.07.046. [DOI] [PubMed] [Google Scholar]
- 25.Rigal D, Vermot-Desroches C, Heitz S, et al. Effects of intravenous immunoglobulins (IVIG) on peripheral blood B, NK, and T cell subpopulations in women with recurrent spontaneous abortions: specific effects on LFA-1 and CD56 molecules. Clin Immunol Immunopathol. 1994;71:309–14. doi: 10.1006/clin.1994.1091. [DOI] [PubMed] [Google Scholar]
- 26.Ephrem A, Chamat S, Miquel C, et al. Expansion of CD4+CD25+ regulatory T cells by intravenous immunoglobulin: a critical factor in controlling experimental autoimmune encephalomyelitis. Blood. 2008;111:715–22. doi: 10.1182/blood-2007-03-079947. [DOI] [PubMed] [Google Scholar]
- 27.Kessel A, Ammuri H, Peri R, et al. Intravenous immunoglobulin therapy affects T regulatory cells by increasing their suppressive function. J Immunol. 2007;179:5571–5. doi: 10.4049/jimmunol.179.8.5571. [DOI] [PubMed] [Google Scholar]
- 28.Tha-In T, Bayry J, Metselaar HJ, et al. Modulation of the cellular immune system by intravenous immunoglobulin. Trends Immunol. 2008;29:608–15. doi: 10.1016/j.it.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 29.Basta M. Ambivalent effect of immunoglobulins on the complement system: activation versus inhibition. Mol Immunol. 2008;45:4073–9. doi: 10.1016/j.molimm.2008.07.012. [DOI] [PubMed] [Google Scholar]
- 30.Basta M. Modulation of complement-mediated immune damage by intravenous immune globulin. Clin Exp Immunol. 1996;104(Suppl 1):21–5. [PubMed] [Google Scholar]
- 31.Basta M, Langlois PF, Marques M, et al. High-dose intravenous immunoglobulin modifies complement-mediated in-vivo clearance. Blood. 1989;74:326–33. [PubMed] [Google Scholar]
- 32.Basta M, Fries LF, Frank MM. High dose of intravenous Ig inhibit in vitro uptake of C4 fragments onto sensitized erythrocytes. Blood. 1991;77:376–80. [PubMed] [Google Scholar]
- 33.Basta M, Kirshbom P, Frank MM, Fries LF. Mechanism of therapeutic effect of high-dose intravenous immunoglobulin. Attenuation of acute, complement-dependent immune damage in a guinea pig model. J Clin Invest. 1989;84:1974–8. doi: 10.1172/JCI114387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Basta M, Dalakas MC. High-dose intravenous immunoglobulin exerts its beneficial effect in patients with dermatomyositis by blocking endomysial deposition of activated complement fragments. J Clin Invest. 1994;94:1729–35. doi: 10.1172/JCI117520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Basta M, Van Goor F, Luccioli S, et al. F(ab)′2-mediated neutralization of C3a and C5a anaphylatoxins: a novel effector function of immunoglobulins. Nat Med. 2003;9:431–8. doi: 10.1038/nm836. [DOI] [PubMed] [Google Scholar]
- 36.Frank MM, Miletic VD, Jiang H. Immunoglobulin in the control of complement action. Immunol Res. 2000;22:137–46. doi: 10.1385/IR:22:2-3:137. [DOI] [PubMed] [Google Scholar]
- 37.Miletic VD, Hester CG, Frank MM. Regulation of complement activity by immunoglobulin. I. Effect of immunoglobulin isotype on C4 uptake on antibody sensitized sheep erythrocytes and solid phase immune complexes. J Immunol. 1996;156:749–57. [PubMed] [Google Scholar]
- 38.Rieben R, Roos A, Muizert Y, et al. Immunoglobulin M-enriched human intravenous immunoglobulin prevents complement activation in-vitro and in-vivo in a rat model of acute inflammation. Blood. 1999;93:942–51. [PubMed] [Google Scholar]
- 39.Roos A, Rieben R, Faber-Krol MC, Daha MR. IgM-enriched human intravenous immunoglobulin strongly inhibits complement-dependent porcine cell cytotoxicity mediated by human xenoreactive antibodies. Xenotransplantation. 2003;10:596–605. doi: 10.1034/j.1399-3089.2003.00063.x. [DOI] [PubMed] [Google Scholar]
- 40.Adams HP. Stroke: a vascular pathology with inadequate management. J Hypertens Suppl. 2003;21:S3–7. doi: 10.1097/00004872-200306005-00002. [DOI] [PubMed] [Google Scholar]
- 41.D'Ambrosio AL, Pinsky DJ, Connolly ES. The role of the complement cascade in ischemia/reperfusion injury: implications for neuroprotection. Mol Med. 2001;7:367–82. [PMC free article] [PubMed] [Google Scholar]
- 42.Arumugam TV, Tang SC, Lathia JD, et al. Intravenous immunoglobulin (IVIG) protects the brain against experimental stroke by preventing complement-mediated neuronal cell death. Proc Natl Acad Sci USA. 2007;104:14104–9. doi: 10.1073/pnas.0700506104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gronski P, Bauer R, Bodenbender L, et al. On the nature of IgG dimers. I. Dimers in human polyclonal IgG preparations: kinetic studies. Behring Inst Mitt. 1988;82:127–43. [PubMed] [Google Scholar]
- 44.Gronski P, Bauer R, Bodenbender L, et al. On the nature of IgG dimers. II. Idiotype-anti-idiotype complexes of polyclonal and monoclonal origin: size distribution patterns and molecular geometries. Dimers in human polyclonal IgG preparations: kinetic studies. Behring Inst Mitt. 1988;82:144–53. [PubMed] [Google Scholar]
- 45.Roux KH, Tankersley DL. A view of the human idiotypic repertoire. Electron microscopic and immunologic analyses of spontaneous idiotype–anti-idiotype dimers in pooled human IgG. J Immunol. 1990;144:1387–95. [PubMed] [Google Scholar]
- 46.Tankersley DL. Dimer formation in immunoglobulin preparations and speculations on the mechanism of action of intravenous immune globulin in autoimmune diseases. Immunol Rev. 1994;139:159–72. doi: 10.1111/j.1600-065x.1994.tb00861.x. [DOI] [PubMed] [Google Scholar]
- 47.Vassilev TL. Variable region-connected, dimeric fraction of intravenous immunoglobulin enriched in natural autoantibodies. J Autoimmun. 1995;8:405–13. doi: 10.1006/jaut.1995.0032. [DOI] [PubMed] [Google Scholar]
- 48.Sultan Y, Rossi F, Kazatchkine MD. Recovery from anti-VIII:C (antihemophilic factor) autoimmune disease is dependent on generation of antiidiotypes against anti-VIII:C autoantibodies. Proc Natl Acad Sci USA. 1987;84:828–31. doi: 10.1073/pnas.84.3.828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schaub A, Wymann S, Heller M, et al. Self-reactivity in the dimeric intravenous immunoglobulin fraction. Ann NY Acad Sci. 2007;1110:681–93. doi: 10.1196/annals.1423.071. [DOI] [PubMed] [Google Scholar]
- 50.Wymann S, Ghielmetti M, Schaub A, et al. Monomerization of dimeric IgG of intravenous immunoglobulin (IVIg) increases the antibody reactivity against intracellular antigens. Mol Immunol. 2008;45:2621–8. doi: 10.1016/j.molimm.2007.12.020. [DOI] [PubMed] [Google Scholar]
- 51.VonGunten S, Simon HU. Natural anti-Siglec autoantibodies mediate potential immunoregulatory mechanisms: implications for the clinical use of IVIG. Autoimmun Rev. 2008;7:453–6. doi: 10.1016/j.autrev.2008.03.015. [DOI] [PubMed] [Google Scholar]
- 52.Spycher MO, Bolli R, Hodler G, et al. Well-tolerated liquid intravenous immunoglobulin G preparations (IVIG) have a low immunoglobulin G dimer (IgG-dimer) content. J Autoimmun. 1996;96(Suppl 1):96. [Google Scholar]
- 53.Samuelsson A, Towers TL, Ravetch JV. Anti-inflammatory activity of IVIG mediated through the inhibitory Fc receptor. Science. 2001;291:484–6. doi: 10.1126/science.291.5503.484. [DOI] [PubMed] [Google Scholar]
- 54.Bruhns P, Samuelsson A, Pollard JW, Ravetch JV. Colony-stimulating factor-1-dependent macrophages are responsible for IVIG protection in antibody-induced autoimmune disease. Immunity. 2003;18:573–81. doi: 10.1016/s1074-7613(03)00080-3. [DOI] [PubMed] [Google Scholar]
- 55.Nimmerjahn F, Ravetch JV. Divergent immunoglobulin g subclass activity through selective Fc receptor binding. Science. 2005;310:1510–2. doi: 10.1126/science.1118948. [DOI] [PubMed] [Google Scholar]
- 56.Kaneko Y, Nimmerjahn F, Madaio MP, Ravetch JV. Pathology and protection in nephrotoxic nephritis is determined by selective engagement of specific Fc receptors. J Exp Med. 2006;203:789–97. doi: 10.1084/jem.20051900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kaneko Y, Nimmerjahn F, Ravetch JV. Anti-inflammatory activity of immunoglobulin G resulting from Fc sialylation. Science. 2006;313:670–3. doi: 10.1126/science.1129594. [DOI] [PubMed] [Google Scholar]
- 58.Anthony RM, Nimmerjahn F, Ashline DJ, et al. Recapitulation of IVIG anti-inflammatory activity with a recombinant IgG Fc. Science. 2008;320:373–6. doi: 10.1126/science.1154315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Anthony RM, Wermeling F, Karlsson MC, Ravetch JV. Identification of a receptor required for the anti-inflammatory activity of IVIG. Proc Natl Acad Sci USA. 2008;105:19571–8. doi: 10.1073/pnas.0810163105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nimmerjahn F, Ravetch JV. Fcgamma receptors: old friends and new family members. Immunity. 2006;24:19–28. doi: 10.1016/j.immuni.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 61.Warmerdam PA, Nabben NM, van de Graaf SA, et al. The human low affinity immunoglobulin G Fc receptor IIC gene is a result of an unequal crossover event. J Biol Chem. 1993;268:7346–9. [PubMed] [Google Scholar]
- 62.Qiu WQ, de Bruin D, Brownstein BH, et al. Organization of the human and mouse low-affinity Fc gamma R genes: duplication and recombination. Science. 1990;248:732–5. doi: 10.1126/science.2139735. [DOI] [PubMed] [Google Scholar]
- 63.Gutierrez-Roelens I, Lauwerys BR. Genetic susceptibility to autoimmune disorders: clues from gene association and gene expression studies. Curr Mol Med. 2008;8:551–61. doi: 10.2174/156652408785747906. [DOI] [PubMed] [Google Scholar]
- 64.Redon R, Ishikawa S, Fitch KR, et al. Global variation in copy number in the human genome. Nature. 2006;444:444–54. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Koene HR, Kleijer M, Roos D, et al. Fc gamma RIIIB gene duplication: evidence for presence and expression of three distinct Fc gamma RIIIB genes in NA(1+,2+)SH(+) individuals. Blood. 1998;91:673–9. [PubMed] [Google Scholar]
- 66.De Haas M, Kleijer M, van Zwieten R, et al. Neutrophil Fc gamma RIIIb deficiency, nature, and clinical consequences: a study of 21 individuals from 14 families. Blood. 1995;86:2403–13. [PubMed] [Google Scholar]
- 67.Huizinga TW, Kuijpers RW, Kleijer M, et al. Maternal genomic neutrophil FcRIII deficiency leading to neonatal isoimmune neutropenia. Blood. 1990;76:1927–32. [PubMed] [Google Scholar]
- 68.Aitman TJ, Dong R, Vyse TJ, et al. Copy number polymorphism in Fcgr3 predisposes to glomerulonephritis in rats and humans. Nature. 2006;439:851–5. doi: 10.1038/nature04489. [DOI] [PubMed] [Google Scholar]
- 69.Willcocks LC, Lyons PA, Clatworthy MR, et al. Copy number of FCGR3B, which is associated with systemic lupus erythematosus, correlates with protein expression and immune complex uptake. J Exp Med. 2008;205:1573–82. doi: 10.1084/jem.20072413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fanciulli M, Norsworthy PJ, Petretto E, et al. FCGR3B copy number variation is associated with susceptibility to systemic, but not organ-specific, autoimmunity. Nat Genet. 2007;39:721–3. doi: 10.1038/ng2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schouten JP, McElgunn CJ, Waaijer R, et al. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 2002;30:e57. doi: 10.1093/nar/gnf056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Breunis WB, van Mirre E, Geissler J, et al. Copy number variation at the FCGR locus includes FCGR3A, FCGR2C and FCGR3B but not FCGR2A and FCGR2B. Hum Mutat. 2009;30:E640–50. doi: 10.1002/humu.20997. [DOI] [PubMed] [Google Scholar]
- 73.Breunis WB, van Mirre E, Bruin M, et al. Copy number variation of the activating FCGR2C gene predisposes to idiopathic thrombocytopenic purpura. Blood. 2008;111:1029–38. doi: 10.1182/blood-2007-03-079913. [DOI] [PubMed] [Google Scholar]