

Abstract
Presenilins (PS) play a central role in γ-secretase-mediated processing of β-amyloid precursor protein (APP) and numerous type I transmembrane proteins. Expression of mutant PS1 variants causes familial forms of Alzheimer's disease (FAD). In cultured mammalian cells that express FAD-linked PS1 variants, the intracellular trafficking of several type 1 membrane proteins is altered. We now report that the anterograde fast axonal transport (FAT) of APP and Trk receptors is impaired in the sciatic nerves of transgenic mice expressing two independent FAD-linked PS1 variants. Furthermore, FAD-linked PS1 mice exhibit a significant increase in phosphorylation of the cytoskeletal proteins tau and neurofilaments in the spinal cord. Reductions in FAT and phosphorylation abnormalities correlated with motor neuron functional deficits. Together, our data suggests that defects in anterograde FAT may underlie FAD-linked PS1-mediated neurodegeneration through a mechanism involving impairments in neurotrophin signaling and synaptic dysfunction.
Keywords: Alzheimer's disease, presenilin, kinesin, protein trafficking, axonal transport, motor neurons, tau, neurofilament, kinase, GSK-3
Introduction
Presenilins (PS1 and PS2) are multipass membrane proteins that play a central role in intramembranous, “γ-secretase” processing of the β-amyloid precursor protein (APP) to generate β-amyloid (Aβ) peptides (Sisodia and St George-Hyslop, 2002). Inheritance of mutations in genes encoding PS1, PS2, and APP cause familial Alzheimer's disease (FAD), which correlates with increased production of highly fibrillogenic Aβ42 peptides that aggregate and subsequently deposit in the brains of human subjects affected with the disease (for review, see Price and Sisodia, 1998). The brains of affected individuals also exhibit intraneuronal accumulations of paired helical filaments (PHFs) composed of hyperphosphorylated forms of the microtubule-associated protein tau (Trojanowski and Lee, 1994).
A large body of evidence supports the notion that, in addition to their role in processing of numerous type I membrane proteins, PSs might play additional physiological roles. For example, studies using cultured primary neurons suggest that PS1 plays a role in regulation of intracellular trafficking of selected proteins, including APP (Naruse et al., 1998; Capell et al., 2000; Kim et al., 2001; Cai et al., 2003), the neurotrophin receptor TrkB (Naruse et al., 1998), N-cadherin (Uemura et al., 2004), the PS1-interacting proteins intercellular adhesion molecule-5 (ICAM-5)/telencephalin (Annaert et al., 2001), and nicastrin (Edbauer et al., 2002; Leem et al., 2002), whereas PS2 has been shown to regulate the trafficking of cystatin C (Ghidoni et al., 2006). More recently, Wang et al. (2006) have reported that, in the absence of PS1, the retinal pigment epithelium fails to undergo pigmentation because of mislocalization of 50 nm post-Golgi vesicles containing tyrosinase that would normally be transported to melanosomes. Finally, there is evidence that PS1 may play a role in the modulation of some protein kinase activities (Takashima et al., 1998; Pigino et al., 2003).
In vitro reconstitution studies have revealed that the budding of endoplasmic reticulum (ER)- and trans-Golgi network (TGN)-derived APP-containing vesicles is dramatically increased in PS1-deficient cells, or cells expressing a dominant-negative PS1 D385A variant (Cai et al., 2003). Supporting these observations, cell-surface labeling studies have shown increased steady-state levels of APP on the surface of cells lacking PS1 or expressing PS1 D385A. Conversely, vesicle biogenesis and the extent of cell-surface APP is reduced in membrane preparations from cells expressing FAD-linked PS1 variants (Cai et al., 2003). Extending these observations, recent studies revealed that the PS1-interacting protein phospholipase D1 plays a role in PS1-regulated APP trafficking in neuronal cultures (Cai et al., 2006a,b). Although these latter studies in cultured cells strongly suggest a role for PS1 in protein trafficking, no correlate had been documented for FAD using in vivo animal models.
Previously, we demonstrated that APP is axonally transported by anterograde fast axonal transport (FAT) in both peripheral and central nerves (Koo et al., 1990; Sisodia et al., 1993; Buxbaum et al., 1998; Lazarov et al., 2005). Using the sciatic nerve ligation paradigm, we addressed the possibility that PS1 might play a role in FAT of APP and other membrane proteins in vivo. Unfortunately, mice with genomic deletions of PSEN1 die in late embryogenesis and preclude a direct evaluation of physiological PS1 function contributions to FAT in vivo. However, we generated transgenic mice that express human wild-type or FAD-linked PS1 variants and exploited these animals to assess FAT of APP and neurotrophin receptors (Trks) in the sciatic nerve. Here we show that FAT of both APP and Trk receptors, but not of the glycosylphosphatidylinositol (GPI)-linked prion protein (PrP), is markedly impaired in the sciatic nerves of transgenic mice expressing FAD-linked PS1 mutants. We further show reductions in both steady-state levels and accumulation of kinesin-1 in nerves of mice expressing mutant PS1 relative to nerves of mice expressing human wild-type PS1 (PS1HWT). These findings are consistent with and extend our recent observations that expression of FAD-linked PS1 mutants in embryonic cultured neurons leads to increased glycogen synthase kinase 3 (GSK-3) activity and reductions in kinesin-1-based transport of selected membrane proteins (Pigino et al., 2003).
To assess functional consequences of FAD-linked PS1 variant-induced inhibition of FAT in vivo, we evaluated both morphological and behavioral parameters. We report that anterior horn motor neurons in the lumbar spinal cord of FAD-linked mutant PS1 mice exhibit AD-like increases in the phosphorylation of tau and neurofilaments, two major cytoskeletal proteins. Significantly, reductions in FAT and alterations in cytoskeletal protein phosphorylation observed in the spinal cord of FAD-linked PS1 mice were paralleled by functional impairments in motor activity. Our data support the notion that expression of FAD-linked PS1 leads to reductions in the supply of both neurotrophin receptors and presynaptic membrane components at nerve terminals, resulting in compromised motor neuron function.
Materials and Methods
Transgenic mice.
Mice expressing FAD mutant human PS1 variants (PS1HWT, PS1M146L, and PS1ΔE9) and/or a chimeric mouse/human APP695 harboring a human Aβ domain and mutations (K595N and M596L) linked to Swedish FAD pedigrees (APPswe) have been described previously (Borchelt et al., 1996; Lee et al., 1997). The background strains for APPswe are [C3H/HeJ × C57BL/6J F3] × C57BL/6J n1 and for PS1ΔE9 are C3H/HeJ × C57BL/6J F3 mice. Mice used in this study were 5–12 months old.
Antibodies and Western blot analysis.
For Western blot, the following antibodies were used: CT15 (Sisodia et al., 1993), mAB22C11 (Weidemann et al., 1989; Hilbich et al., 1993), mouse anti-α tubulin (1:10,000; Sigma, St. Louis, MO), rabbit anti-mouse PS1NTF (Thinakaran et al., 1997), 63–90 (Stenoien and Brady, 1997), H2 (Brady et al., 1990), anti-Trk (C-20, 1:1000; Santa Cruz Biotechnology, Santa Cruz, CA), anti-PrP antibodies (kindly provided by Dr. James Mastrianni, The University of Chicago, Chicago, IL), tau-5 (Biosource International, Camarillo, CA) tau-1 (1:2000; Roche, Indianapolis, IN), PHF-1 monoclonal antibody (mAb) (1:2000; Abnova, Heidelberg, Germany). Phosphorylation-sensitive neurofilament antibodies SMI31, SMI32 (Sternberger Monoclonals, Lutherville, MD), and RM055 (Black and Lee, 1988). Western blots were performed as described previously (Morfini et al., 2002b, 2004; Lazarov et al., 2005).
Sciatic nerve ligation.
Mice were anesthetized using a mixture of ketamine (75 mg/kg) and xylazine (4 mg/kg). The skin on the lateral surface of the right thigh was incised, and a section was made directly through the biceps femoris muscle. The sciatic nerve was exposed and doubly ligated using 4-0 silk sutures (Ethicon, New Brunswick, NJ). The contralateral nerve was left intact. Six or 24 h later, nerve segments (up to 0.5 cm) proximal and distal to the ligature were collected and frozen in liquid nitrogen. Corresponding contralateral nerve and cortex tissues were collected as well.
Protein extraction from nerve.
Sciatic nerve segments were homogenized in extraction buffer [50 mm Tris, pH 7.2, 150 mm NaCl, 5 mm EDTA, protease inhibitor mixture (Sigma), and 100 mm PMSF] using a glass-made micro homogenizer. After addition of 1% SDS, the homogenate was centrifuged for 10 min. Protein concentration of clarified supernatants was determined using BCA protein assay kit (Pierce, Rockford, IL). Equal amount of proteins were analyzed by Western blot.
Spinal cord lysate preparation.
Spinal cord samples were homogenized in 1 ml of ROLB buffer (10 mm HEPES, pH 7.4, 0.5% Triton X-100, 80 mm β-glycerophosphate, 50 mm sodium fluoride, 2 mm sodium orthovanadate, 100 nm staurosporine, 100 nm K252a, 50 nm okadaic acid, 50 nm microcystin, 100 mm potassium phosphate, and mammalian protease inhibitor cocktail (Sigma). Lysates were clarified by centrifugation using a TL100.3 rotor at 21,000 × g for 5 min at 4°C. This centrifugation step was repeated once, and protein concentration was determined using BCA kit (Pierce).
Rotorod test.
Transgenic mice harboring FAD-linked PS1 variants at age of 5 or 10 months were subject to habituation 4 d before the experiment, three times for 3 min a day. Mice were placed on the rotorod (Rotomex; Columbus Instruments, Columbus, OH), and rotation speed was accelerated from 4 to 40 m/s. Time on the rotorod (in seconds) has been recorded for each mouse. The first set of experiments took place at 5 months of age and the second trial at 10 months of age.
Electrophysiological examination of compound muscle action potentials.
Mice were anesthetized with Avertin (0.5 mg/g, i.p.) and placed on a warm pad at a temperature of ∼32–34°C. Recording needle electrodes were placed subcutaneously in the footpad. Supramaximal stimulation of sciatic nerves was performed with a 0.1–0.2 ms rectangular pulse, stimulating distally at the ankle and proximally at the sciatic notch with needle electrodes. Recordings were obtained on a Nicolet ViaSys (Madison, WI) VikingQuest EMG machine with a filter setting of 2 Hz to 10 kHz. Latencies correspond to the time lapse between the stimulus and the onset of compound muscle action potentials (CMAPs). Conduction velocities were calculated as follows: conduction velocities = distance/(proximal latency − distal latency). The peak-to-peak amplitudes of CMAPs were measured, and the ratio of proximal versus distal amplitude was used to determine the presence or absence of partial conduction block.
Immunohistochemical analysis of spinal cord sections.
Spinal cords were obtained from transgenic mice harboring FAD-linked PS1ΔE9 or PS1HWT at 8–9 months of age. Dissected spinal cords were frozen in dry ice-cold isopropanol and embedded in Tissue-Tek (Sakura, Tokyo, Japan). Transverse spinal cord cryosections (20 μm) were fixed in 3.7% paraformaldehyde solution for 30 min. Sections were then washed in TBS and simultaneously blocked and permeabilized using 5% donkey serum and 0.5% Triton X-100 for 2 h at room temperature. Sections were incubated with PHF-1 mAb (1:2000; Abnova) overnight at 4°C, washed, and incubated with fluorophore-conjugated secondary antibodies for 1 h at room temperature. Sections were washed and mounted with glycerol-based mounting medium (polyvinyl alcohol mounting medium with antifading 1,4-diazabicyclo-[2.2.2]octane; Sigma).
Results
Expression of FAD-linked PS1 mutants impairs anterograde fast axonal transport of APP
A series of studies have documented deficits in the trafficking and/or maturation of APP in PS1-deficient cells or cells that express several FAD-linked PS1 variants (Naruse et al., 1998; Capell et al., 2000; Kim et al., 2001; Cai et al., 2003). Studies in cultured neuroblastoma cells have revealed that expression of FAD-linked mutant PS1 impairs biogenesis of APP-containing vesicles derived from the ER and Golgi (Cai et al., 2003, 2006a,b). Furthermore, studies in cultured embryonic primary hippocampal neurons expressing FAD-linked mutant PS1 revealed reductions in kinesin-1-mediated delivery of selected synaptic markers and mitochondria to growing neurites (Pigino et al., 2003).
To evaluate whether these latter observations could be extended to a relevant protein trafficking paradigm in vivo, we examined FAT of APP in the sciatic nerve of adult transgenic mice. We placed double ligatures in the sciatic nerves of transgenic mice harboring FAD-linked PS1HWT or the FAD-linked PS1ΔE9 mutant (Borchelt et al., 1996; Lee et al., 1997). Ligatures were introduced immediately distal to the point at which sensory fibers emanating from the L4/L5 dorsal root ganglia merge with fibers from lumbar motor neurons. At 6 h after ligation, we prepared detergent-soluble extracts from nerve segments proximal and distal to the ligatures. Western blot analysis using the APP C-terminal-specific antibody CT-15 (Koo et al., 1990; Sisodia et al., 1993) revealed that highly glycosylated forms of full-length APP accumulate in the segment proximal to the ligature in sciatic nerves of PS1HWT mice (Fig. 1A, Fl-APP panel, lane 2), as described previously (Koo et al., 1990; Sisodia et al., 1993). In contrast, accumulation of APP at the proximal segment of the ligature was markedly reduced in PS1ΔE9 mice (Fig. 1A, Fl-APP panel, lane 8). We then crossed the PS1ΔE9 mice with FAD-linked APPswe mice (Borchelt et al., 1996, 1997) to examine the effects of the mutant PS1 transgene on trafficking of a transgene-encoded APP. Double ligatures were placed in sciatic nerves for 6 h, and detergent extracts were prepared from segments proximal and distal to the ligature; detergent extracts from a segment of the contralateral, unligated nerve served as a control. We observed elevated levels of mature APPswe that accumulated at the segment proximal to the ligature in wild-type mice (Fig. 1A, Fl-APP panel, lane 5). However, levels of full-length APP that accumulated at the proximal segment of the ligature in APPswe/PS1ΔE9 mice were markedly diminished (Fig. 1A, Fl-APP panel, lane 11). In contrast to nontransgenic mice, immature nonglycosylated APP was present in all nerve segments of mice expressing APPswe. This was likely a consequence of expressing the APPswe transgene using the mouse PrP promoter, which leads to APP expression in non-neuronal cells of the sciatic nerve (i.e., Schwann cells, fibroblasts, and microglia). The fact that these immature APPswe polypeptides do not accumulate at the proximal ligature and hence, were not transported is consistent with this idea. Notably, mice expressing FAD-linked APPswe variant (Borchelt et al., 1996, 1997) do not show any impairment of APP accumulation (Fig. 1A, Fl-APP panel, compare lanes 2 and 5).
Figure 1.

Reduction in anterograde FAT of full-length APP in ligated sciatic nerves of FAD-linked PS1 variants. A, Fl-APP, APP is accumulated at the proximal stump of ligated nerves 6 h after ligation in mice harboring PS1HWT. Although steady-state levels of APP are comparable between intact sciatic nerve segments of PS1HWT and PS1ΔE9 mice, accumulation of APP at the proximal stump of ligated nerves is significantly reduced in PS1ΔE9 mice. Similarly, transgenic mice harboring APPswe × PS1ΔE9 exhibit reduced accumulation of APP at the proximal stump of the ligated sciatic nerve compared with APPswe mice. Levels of APP accumulated at the distal site of the ligation remained unchanged in all cases. Expression of full-length PS1, PS1NTF, or full-length PS1ΔE9 is unchanged in the intact and ligated sciatic nerve of FAD-linked PS1 and APP variants. Immunoblot using anti-α-tubulin antibodies was used to control for equal amounts of protein loading. B, Densitometric analysis of APP accumulation in nerves of FAD-linked APP and PS1 variants 6 h after ligation as shown in A. Graphs show APP levels normalized to α-tubulin levels. C, APP accumulation at the proximal stump of the ligature is also reduced in the sciatic nerve of mice harboring PS1M146L compared with mice harboring PS1HWT. D, Densitometric analysis of APP accumulation in nerves of FAD-linked PS1HWT and PS1M146L variants 6 h after ligation as shown in D, Fl-APP panel. Fl-APP, Full-length APP; I, intact nerve segment; P, proximal nerve segment; D, distal nerve segment. Molecular weight markers are indicated in kilodaltons.
Densitometric analysis of immunoblot bands confirmed Western blot analysis and revealed a marked reduction (∼90%) in accumulation of APP in the proximal segment of PS1ΔE9 mice compared with PS1HWT. Also, a significant reduction (∼35%) in accumulation of transgenic and endogenous APP was seen in mice harboring APPswe/PS1ΔE9 compared with mice harboring APPswe (Fig. 1B). Notably, and consistent with our previous report (Lazarov et al., 2005), there is neither an accumulation of ∼43 kDa full-length human PS1 and ∼29 kDa PS1 derivatives (Fig. 1A, PS1 panel, lane 2) nor of the ∼42 kDa PS1ΔE9 variant, a polypeptide that is not subject to endoproteolysis (Thinakaran et al., 1996) (Fig. 1A, PS1 panel, lanes 8, 11). Thus, neither endogenous PS1 nor transgene-encoded PS1 displayed the rates of accumulation pattern observed for APP and other vesicular proteins (Lazarov et al., 2005).
To establish that the reduction of APP transport in nerves of PS1ΔE9 animals is not simply attributable to perturbations induced by transgene integration effects or the result of expressing the unprocessed PS1ΔE9 mutant, we compared the accumulation of APP in the proximal segment of nerves obtained from mice expressing PS1HWT and PS1M146L variants (Lee et al., 1997). We show that the levels of endogenous APP that accumulated at the proximal segment of ligated nerves from PS1M146L mice were markedly diminished compared with levels accumulated in mice expressing PS1HWT (Fig. 1C, compare lanes 5 and 2, respectively). Quantitative analysis confirmed these reductions (∼28%) in accumulation of APP at the proximal segment of PS1M146L compared with PS1HWT mice (Fig. 1D). Thus, in vivo expression of two independent FAD-linked PS1 mutants impairs the anterograde FAT of APP in sciatic nerve.
FAD-linked PS1ΔE9 impairs anterograde fast axonal transport of selected membrane proteins
Previous studies have shown that the trafficking of several integral membrane proteins, including the neurotrophin receptor TrkB (Naruse et al., 1998; Hamano et al., 2005), the PS1-interacting protein telencephalin [ICAM-5 (Annaert et al., 2001)], and the PS1-associated protein nicastrin (Edbauer et al., 2002; Leem et al., 2002), are altered in mammalian cells that either lack PS1 or express FAD-linked PS1 variants. In addition, our previous studies on the delivery of membrane proteins into neurites of cultured hippocampal neurons revealed a selective reduction in the levels of synaptophysin and syntaxin-I-containing vesicles but not of synaptosome-associated protein of 25 kDa-containing vesicles in neurons expressing FAD-linked PS1 compared with PS1HWT-expressing ones (Pigino et al., 2003).
Extending these latter studies to an in vivo setting in mature neurons, we assessed the effects of expressing FAD-linked PS1 variants to several polypeptides that are subject to anterograde FAT, including neurotrophin receptors (Ehlers et al., 1995; Kamal et al., 2001) and the GPI-linked PrP (Borchelt et al., 1994). We placed double ligatures in sciatic nerves of transgenic mice expressing either PS1HWT or the FAD-linked PS1ΔE9 variant for 6 h and prepared detergent extracts from segments proximal and distal to the ligature. An equivalent segment obtained from the intact contralateral, unligated sciatic nerve served as a control (Fig. 2A). As expected, levels of accumulated APP were markedly reduced in the proximal segment of sciatic nerves obtained from PS1ΔE9 mice (Fig. 2A, APP panel, lane 4). Reprobing the membrane with the C14 antibody, raised against a peptide sequence shared by all neurotrophin receptors, revealed a marked decrease in levels of accumulated Trks in the proximal segment of ligated nerves from PS1ΔE9 mice compared with corresponding segments from PS1HWT mice (Fig. 2A, Trk panel, compare lanes 4 and 2, respectively). In contrast, similar levels of PrP accumulated in the proximal segments of mice expressing PS1HWT or PS1ΔE9 (Fig. 2A, PrP panel, compare lanes 2 and 4, respectively). Densitometric analysis of immunoblot bands confirmed these observations above (Fig. 2B, 16 and 37% reductions in the levels of accumulated APP and Trk, respectively, at the proximal segments of ligated nerves from PS1ΔE9 mice compared with PS1HWT mice). Levels of accumulated PrP remained unchanged (Fig. 2B). Thus, expression of an FAD-linked PS1 variant selectively impairs FAT of membrane cargo vesicles containing APP and Trk, without affecting FAT of cargo vesicles containing PrP.
Figure 2.

Reduced rate of anterograde FAT of selected membrane proteins in ligated nerves of FAD-linked PS1ΔE9 mutant compared with PS1HWT. The levels of APP, Trk receptor, KHC, and KLC accumulated at the proximal stump of the ligation site in sciatic nerve of PS1ΔE9 mice are reduced compared with levels observed in nerves of PS1HWT mice. However, accumulation of PrP molecules in the proximal stump is comparable in sciatic nerves of PS1HWT and PS1ΔE9. α-Tubulin was used as loading control. I, Intact nerve segment; P, proximal nerve segment. Molecular weight markers are in kilodaltons. B, Densitometric analysis of data in A. Graphs show APP, Trk, PrP, KHC, and KLC levels normalized to α-tubulin levels. A.U., Arbitrary units.
Reduced FAT and enhanced degradation of kinesin-1 in sciatic nerve of mice expressing FAD-linked PS mutant
Previous studies revealed that expression of FAD-linked PS1 variants leads to an elevation in GSK-3 kinase activity (Takashima et al., 1998). Furthermore, increased GSK-3 activity and reduced levels of kinesin-1 in membrane fractions was reported in mice that express the FAD-linked PS1M146V variant (Pigino et al., 2003). These findings, together with the demonstration that GSK-3-mediated phosphorylation of kinesin-1 light chains (KLCs) leads to dissociation of kinesin-1 from vesicles (Morfini et al., 2002b), suggested that mutant PS1 might impair FAT of selected cargos by affecting the association of kinesin-1 with membranous organelles (Pigino et al., 2003). To examine this issue, we probed the samples shown in Figure 2A with H2 antibody that recognize all kinesin-1 heavy chain gene products (KHCs) and 63–90 antibodies that recognize KLCs. Remarkably, steady-state levels of full-length KHC and KLC in unligated nerves from PS1ΔE9 mice were lower than levels detected in PS1HWT nerves (Fig. 2A, KHC and KLC panels, lanes 3 and 1, respectively), as described previously for PS1M146V mutant mice (Pigino et al., 2003). Moreover, the levels of KHC and KLC that accumulated at the proximal segment of the ligature of PS1ΔE9 nerves were lower than levels seen in an equivalent segment from PS1HWT nerves (Fig. 2A, KHC and KLC panels, lanes 4 and 2, respectively). Thus, expression of FAD-linked mutant PS1 impairs FAT of kinesin-1. Significantly, we detected multiple H2-immunoreactive polypeptides of lower molecular weight in both unligated and proximal nerve segments of mice expressing PS1ΔE9 (Fig. 2A, KHC panels, lanes 3 and 4). These polypeptides may represent proteolytic fragments of kinesin-1, consistent with previous suggestions of kinesin-1 degradation after removal from vesicles (Morfini et al., 2002b).
Transgenic mice harboring FAD-linked PS1ΔE9 exhibit impaired motor activity
Recent studies have identified impairments of FAT as a mechanism that underlies pathogenesis of numerous neurodegenerative diseases, and, in many cases, motor deficits are part of the clinical phenotype (Morfini et al., 2002a, 2005, 2006; Reid et al., 2002; Szebenyi et al., 2003; Gould and Brady, 2004). Although FAD-linked PS1 variant mice do not exhibit gross motor behavior in the home cage, motor function has not been formally tested in these animals. Our observations of FAT impairments in the sciatic nerve of FAD-linked PS1ΔE9 mice prompted us to assess motor activity of PS1ΔE9 mice by examining their performance on the rotorod (Jones and Roberts, 1968a,b). For these studies, we compared the rotorod activity of a cohort of eight mice harboring FAD-linked PS1HWT with a cohort of eight mice harboring PS1ΔE9 at the ages of 5 and 10 months. Significantly, transgenic mice harboring the FAD-linked PS1ΔE9 variant exhibited reduced rotorod activity compared with PS1HWT mice at both time points (Fig. 3). The average ± SE running time of the PS1HWT mice was 116 ± 7 min at age of 5 months and 113 ± 6 min at age of 10 months. The average running time of the PS1ΔE9 mice was 92 ± 6 and 76 ± 3 min at age of 5 and 10 months, respectively (Fig. 3). These animals will be referred to as “rotorod mice” throughout the text.
Figure 3.

FAD-linked mutant PS1ΔE9 mice exhibit impaired motor performance. Average running time (minutes) on the rotorod for transgenic mice harboring PS1HWT (gray bar) or PS1ΔE9 (black bar) at 5 and 10 months of age. FAD-linked PS1ΔE9 mice exhibit impaired performance on the rotorod with shorter time periods spent on the cylinder (*p ≤ 0.005, ANOVA).
Morphological and electrophysiological parameters are unaffected in the sciatic nerve of mice expressing FAD-linked PS1ΔE9
We evaluated the possibility that impaired motor performance of the FAD-linked PS1ΔE9 on the rotorod might be the result of impairments unrelated to deficits in anterograde FAT. To this end, we first examined the possibility that FAD-linked PS1 transgenic mice might exhibit functional deficits in nerve conduction velocities, shown previously to be an early indication of neuropathology in a mutant mouse animal model of amyotrophic lateral sclerosis, a human motoneuron disease (Holtmann et al., 1999). To examine this issue, mice that were subject to the rotorod test (Fig. 3, age of ∼11 months) were reassessed by electromyography. CMAPs were recorded after distal and proximal stimulation of the sciatic nerve. Our examination failed to reveal any differences in CMAP amplitudes, distal latencies, or conduction velocities between the two groups of transgenic mice (Fig. 4A). In addition, there was no evidence of partial conduction block or temporal dispersion that would indicate the presence of a demyelination neuropathy.
Figure 4.

Conduction velocity, muscle tissue morphology, and myelination are intact in sciatic nerve of FAD-linked PS1 variants. A, CMAPs were recorded after distal and proximal stimulation of sciatic nerves of transgenic mice harboring FAD-linked PS1HWT and PS1ΔE9. Waveform, latencies, and amplitudes of CMAPs were found to be similar in both groups. DL, Distal latency; PL, proximal latency; CV, conduction velocity; AMP (D), distal amplitude (DA); AMP (P), proximal amplitude (PA). Ba, Bc, Muscle sections of PS1HWT (a) and FAD-linked PS1ΔE9 (c) mice. Immunostaining with antibodies raised against slow-tonic myosin heavy chain isoforms, indicating muscle morphology (green), and with antibodies raised against collagen IV (red) revealed no signs of muscular atrophy. Scale bar, 10 μm. Bb, Bd, Nodal and paranodal structures of sciatic nerves of PS1HWT (b) and FAD-linked PS1ΔE9 (d) mice were visualized using antibodies raised against pan VGSC (red) and antibodies raised against paranodin/Caspr (green), respectively. No morphological abnormalities could be detected in PS1ΔE9 mice. Scale bar, 50 μm.
We next examined the morphology of nodal and paranodal structures in the sciatic nerves of the wild-type or FAD-linked PS1ΔE9 rotorod mice (Scherer and Arroyo, 2002), because changes in these structures might account for the observed motor deficits (Samsam et al., 2002; Court et al., 2004; Taylor et al., 2004). We prepared single fibers from teased sciatic nerves (see Materials and Methods) (Krinke et al., 2000) and performed immunostaining with antibodies raised against Pan voltage-gated sodium channel (VGSC) or antibodies raised against paranodin/contactin-associated protein (Caspr). We did not observe alterations in the distributions of VGSC or paranodin/Caspr in the sciatic nerves of the FAD-linked PS1ΔE9 rotorod mice compared with the wild-type mice exposed to the rotorod (Fig. 4Bb,Bd). This immunohistochemical analysis also suggested that VGSC and paranodin/Caspr, along with PrP, do not show defects in their axonal compartmentalization and anterograde FAT. Collectively, these findings argue that the motor impairments of PS1ΔE9 transgenic mice are not attributable to major morphological or functional deficits in their sciatic nerves.
We then evaluated whether motor deficits in the PS1ΔE9 mice might result from primary muscle atrophy (Vallat, 2003). Muscle morphology was examined in serial sections of the quadriceps dissected from the rotorod mice. Muscle stained with antibodies raised against slow-tonic myosin heavy chain isoforms appeared intact in both PS1HWT and PS1ΔE9 mice (Fig. 4Ba,Bc), suggesting that impaired motor activity in the PS1ΔE9 mice is not attributable to pathology in the muscle itself.
Altered cytoskeletal protein phosphorylation in FAD-linked PS1ΔE9 transgenic mice lumbar spinal cord
Hyperphosphorylation of tau is a hallmark of AD. Significantly, alterations in phosphorylation-dependent regulatory pathways for FAT and misregulation of kinase activities have been shown to underlie FAT defects in various experimental models of human neurodegenerative diseases (Morfini et al., 2002a,b, 2006, 2007). Intriguingly, recent studies have reported axonal pathology in the spinal cord of mice that coexpress the FAD-linked APPswe and PS1ΔE9 variants (Wirths et al., 2006). These observations led us to explore alterations in kinase activities in the spinal cord of FAD-linked mutant PS1ΔE9 mice. The phosphorylation status of both tau and neurofilament protein was analyzed in lumbar spinal cords of 5-month-old PS1HWT and PS1ΔE9 transgenic mice using phospho-specific mAbs. Using tau-5, an antibody that recognizes all tau isoforms, we observed no changes in steady-state levels of tau isoforms in spinal cord samples from PS1HWT and PS1ΔE9 transgenic mice (Fig. 5A, Tau5 panel, arrows). Reprobing the membrane with PHF-1 mAb, which recognizes a phosphorylated epitope at Ser-396/Ser-404 of tau (Seubert et al., 1995; Hernandez et al., 2003), revealed increased levels of phosphorylation for medium-molecular-weight (arrows) and high-molecular-weight (arrowheads) tau isoforms in PS1ΔE9 transgenic mice compared with mice expressing PS1HWT (Fig. 5A, PHF-1 panel). High-molecular-weight tau isoforms (approximate molecular weight of 110 kDa) are predominantly expressed in the peripheral nervous system and in spinal cord motor neurons that project to the peripheral nervous system (Georgieff et al., 1991, 1993). In support of our observation, high-molecular-weight tau isoforms could not be detected in protein extracts of cortex and hippocampus of these mice (data not shown). To establish immunolocalization of PHF-1 positive spinal structures, spinal cord sections of mice harboring FAD-linked PS1HWT or PS1ΔE9 were immunolabeled with PHF-1 mAb. Increased immunostaining was detected in all areas of white matter (i.e., anterior, lateral, and posterior funiculus and anterior commissure) and in cell bodies in the ventral horn (Fig. 5B; indicated with arrows in Fig. 5).
Figure 5.

Neuropathology in lumbar spinal cord of FAD-linked PS1ΔE9 mice. A, Western blot analysis of tau phosphorylation in detergent-soluble protein extracts of lumbar spinal cord of PS1HWT and PS1ΔE9 mice. Note the increased immunoreactivity for PHF-1 in the spinal cord of PS1ΔE9 compared with PS1HWT mice. No significant change in the expression of total tau (Tau5) was observed between PS1ΔE9 and PS1HWT mice samples. Tau isoforms of medium (arrows) and high (arrows) molecular weight are indicated. B, Distribution and localization of PHF-1 expression in lumbar spinal cord sections of PS1ΔE9 and PS1HWT mice. Low-power (a, b) and high-power (c–f) images of PHF-1 immunoreactivity in spinal cord sections of PS1HWT (b, d, f) and PS1ΔE9 (a, c, e) show increased PHF-1 immunoreactivity in spinal cord sections of PS1ΔE9 mice. Higher levels of PHF-1 immunoreactivity were detected in white matter and in cell bodies in the ventral horn (arrowheads). Scale bars: a, b, 750 μm; c, d, 110 μm; e, f, 65 μm. C, Phosphorylation of NF-M is markedly increased in detergent-soluble protein extracts of lumbar spinal cord of PS1ΔE9 mice compared with PS1HWT mice, as shown by SMI34 and SMI31immunoreactivity. These antibodies recognize a phosphorylated epitope on both NF-H and NF-M subunits. Note that not all phospho-epitopes on NF-M are altered in PS1ΔE9, as shown by RM055, a monoclonal phospho-specific antibody that binds to a phosphorylated epitope in the tail domain of NF-M (Virginia Lee, personal communication).
Neurofilaments are major cytoskeletal phosphoproteins in axonal compartments, and these are composed of three polypeptide subunits of diverse molecular weight: NF-H (heavy, ∼200 kDa), NF-M (medium, ∼150 kDa), and NF-L (light, ∼70 kDa). In vivo, both NF-Hs and NF-Ms are heavily phosphorylated by various protein kinases, most notably cyclin-dependent protein kinase 5 and GSK-3 (Guidato et al., 1996; Bajaj and Miller, 1997; Julien and Mushynski, 1998; Hallows et al., 2003). Immunoblotting with phospho-specific mAbs SMI31 and SMI34 raised against KSP residues on both NF-H and NF-M show increased immunoreactivity for NF-M on spinal cord samples from PS1ΔE9 compared with control PS1HWT cortical samples (Fig. 5C). Remarkably, other NF-M phosphoepitopes remained unchanged, as revealed by RMO55, a phospho-specific anti-NF-M mAb (Fig. 5C). SMI31 or SMI34 immunoreactivity for NF-H showed no consistent differences between PS1ΔE9 or PS1HWT samples for either (data not shown). Interestingly, PHF-1 mAb recognizes phosphorylated tau epitopes preferentially phosphorylated by GSK-3 (Godemann et al., 1999; Lee et al., 2003), and SMI31 and SMI34 phosphoepitopes are altered in mice displaying increased GSK-3 activity (Hallows et al., 2003). Increased tau and neurofilament protein phosphorylation in spinal cords of FAD-linked PS1ΔE9 mice strongly suggest deregulation of GSK-3 and perhaps other kinase activities in the spinal cords of PS1 mutant mice. These alterations in kinase activity correlated with deficits in FAT and preceded motor neuron dysfunction in spinal cord.
Discussion
Compelling evidence has emerged suggesting that PS1 plays a central role in γ-secretase-mediated intramembranous processing of APP and a variety of additional type I integral membrane proteins (for review, see De Strooper, 2003). In addition, it is becoming increasingly clear that PS1 regulates the intracellular trafficking of several membrane proteins in neuronal and non-neuronal cells (Naruse et al., 1998; Capell et al., 2000; Annaert et al., 2001; Kim et al., 2001; Edbauer et al., 2002; Leem et al., 2002; Cai et al., 2003; Pigino et al., 2003; Wang et al., 2006). For example, expression of FAD-linked PS1 variants markedly impairs intracellular trafficking of membrane proteins in cell-free reconstitution systems (Cai et al., 2003). The evidence also indicates that mutations in PS1 may compromise neuronal function by increasing GSK-3 activity and kinesin-1-based transport of selected membrane proteins (Pigino et al., 2003). In the present report, we extended these latter observations by examining the effects of FAD-linked PS1 expression on anterograde FAT of kinesin-1 and various membrane proteins in vivo and now provide several novel insights. First, nerve ligation experiments demonstrated that transgenic mice harboring FAD-linked mutant PS1 exhibit a marked impairment in the anterograde FAT of several membrane proteins, including APP and Trk receptors. In these settings, FAT of PrP, a GPI-linked cargo, seems to be unaffected, suggesting a selective effect of pathogenic PS1 on a subset of kinesin-1-transported cargos. Our observation that expression of FAD-linked mutant PS1 impairs FAT of Trk receptors has significant implications regarding AD pathogenesis, particularly in view of the crucial roles of Trk receptors in synaptic transmission, synaptic plasticity (Kang and Schuman, 1995; Kang et al., 1996, 1997; Rosch et al., 2005), and neuronal survival through neurotrophin signaling (for review, see Huang and Reichardt, 2003).
Second, we demonstrate that the steady-state levels and transport of kinesin-1, a major motor protein responsible for the actual execution of anterograde FAT, are markedly reduced in axons of mice expressing the FAD-linked PS1ΔE9 variant. The reduction in axonal levels of kinesin-1 was accompanied by the appearance of several kinesin-1 antibody-immunoreactive polypeptides of lower molecular weight, which may represent degradative intermediates (Morfini et al., 2002b). Interestingly, GSK-3-mediated phosphorylation of kinesin-1 promotes its detachment from the transported membrane cargoes (Morfini et al., 2002b), and the lack of kinesin-1 accumulation at synaptic terminals suggested that most kinesin-1 is degraded shortly after cargo delivery (Li et al., 1999).
GSK-3-mediated phosphorylation of kinesin-1 represents one of multiple regulatory pathways for kinesin-1-based motility (Morfini et al., 2002b). Interestingly, PS1 associates with GSK-3 (Takashima et al., 1998; Tesco and Tanzi, 2000; Pigino et al., 2003), and expression of FAD-linked PS1 in mammalian cells has been reported to increase GSK-3 activity (Takashima et al., 1998; Weihl et al., 1999; Pigino et al., 2003; Baki et al., 2004). Furthermore, PS1M146V knock-in mice show increased GSK-3 activity and deregulated FAT of selected synaptic markers and mitochondria along neuritic processes of cultured primary hippocampal neurons (Pigino et al., 2003). Thus, mice transgenic for different human FAD-associated PS1 mutations show in vivo deficiencies in FAT of selected membrane cargos, which correlates with activation of GSK-3.
Third, we report that expression of the FAD-linked PS1ΔE9 variant leads to increased phosphorylation of NF-M as well as low- and high-molecular-weight tau isoforms in the lumbar spinal cord, as shown by increased SMI32, SMI34, and PHF-1 immunoreactivities. The molecular mechanisms by which PS1 mutations lead to increased GSK-3 activation are not explicitly addressed in this work, but a possibility is that mutant PS1 induced alterations in the activities of regulatory pathways for GSK-3 (Baki et al., 2004; Morfini et al., 2004). Direct or indirect alterations in GSK-3 activity would lead to impaired FAT by deregulating kinesin-1 function (i.e., kinesin-1 association to transported cargoes).
Fourth, we documented that expression of FAD-linked mutant PS1ΔE9 leads to functional motor deficits. Rotorod tests at additional time points would enable an accurate assessment of the dynamic nature of motor deficits induced by FAD-linked PS1 mutations. It should be emphasized that, in our study, motor deficits were not examined in other transgenic variants. It is not presently known whether these behavioral manifestations are a direct result of impairments in FAT and/or tau pathology, but it is significant that loss-of-function mutations in a single kinesin-1 heavy chain gene (KIF5A) suffice to produce motor neuron degeneration in some forms of hereditary spastic paraplegia (Reid et al., 2002; Fichera et al., 2004). Regardless, we considered the possibility that impaired motor function may result from pathological mechanisms unrelated to impaired FAT, but we did not observe altered myelination in the sciatic nerve, abnormal CMAP, or neurogenic muscular atrophy.
We now propose a model that accommodates our principal findings (Fig. 6). According to this model, expression of FAD-linked mutant PS1 induces an increase in GSK-3 activity, possibly through alterations in activity levels of enzymes that play a role in GSK-3 phosphorylation (Beffert et al., 2002; Morfini et al., 2004), leading to enhanced phosphorylation of kinesin-1 and its dissociation from cargoes. Consistent with this model, tau and NF-M exhibit increased phosphorylation at GSK-3-preferred sites. As a result, FAT of membrane proteins from the cell body to axonal, dendritic, and synaptic destinations would be compromised, leading to impairments in both synaptic and neurotrophic function. These changes would ultimately lead to neurodegeneration and motor neuron dysfunction.
Figure 6.
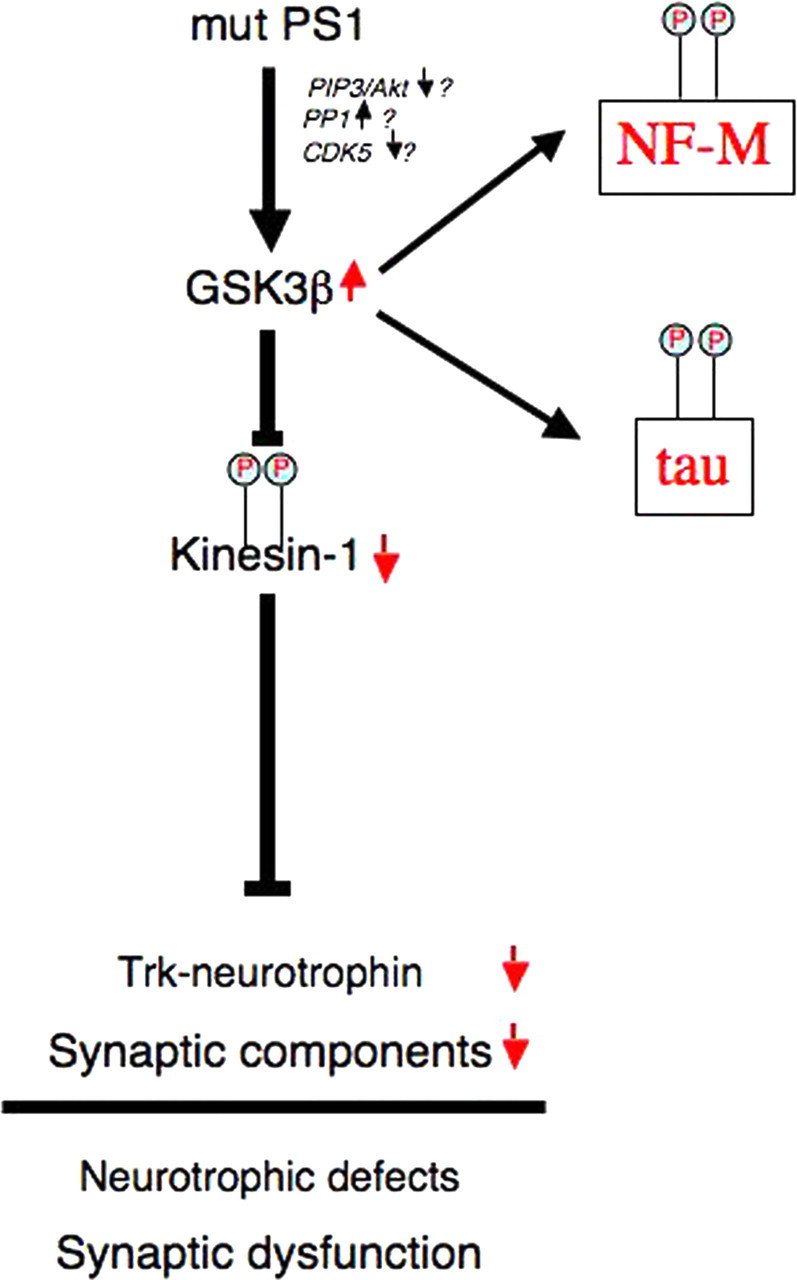
FAD-linked mutant PS1-induced neuropathology. Appropriate neurotrophic support and synaptic function depends on kinesin 1-mediated transport of neurotrophin receptors (Trk) and synaptic cargoes to their final sites of utilization. In a model encompassing our data, expression of FAD-linked PS1 mutants (mut PS1) results in increased GSK-3 activation (Pigino et al., 2003), possibly by altering the activity of phospho-transferases involved in GSK-3 regulation (Beffert et al., 2002; Morfini et al., 2004). Activated GSK-3 phosphorylates KLC (P), which in turn promotes dissociation of kinesin-1 from its transported cargo (Morfini et al., 2002b), resulting in impaired anterograde FAT of critical axonal cargoes (Pigino et al., 2003). Additionally, increased GSK-3 activation leads to the characteristic increases in tau and neurofilament (NF) phosphorylation, widely reported in AD. FAD-linked mutant PS1-induced reductions in FAT would result in impaired neurotrophin signaling and synaptic dysfunction. PIP3, Phosphatidylinositol 3,4,5-triphosphate; PP1, protein phosphatase-1; CDK5, cyclin-dependent kinase-5.
The demonstration that expression of FAD-linked mutant PS1 impairs FAT of a subset of membrane proteins in peripheral nerves might also have mechanistic implications for understanding a variant clinical phenotype, termed spastic paraparesis, described in several pedigrees exhibiting early-onset AD caused by mutations in PSEN1, including a PSEN1 variant that lacks exon 9 (PS1ΔE9) (Kwok et al., 1997; Crook et al., 1998; Hiltunen et al., 2000; O'Riordan et al., 2002; Orlacchio et al., 2002; Tabira et al., 2002; Assini et al., 2003; Brooks et al., 2003; Rogaeva et al., 2003; Moretti et al., 2004; Munhoz et al., 2006). Patients with spastic paraparesis exhibit lower limb weakness and hyperreflexia associated with severe degeneration of corticospinal tracts that are among the longest motor and sensory axons in the body (Bruyn, 1992). It is not certain whether corticospinal degeneration is the result of impaired FAT, but it seems noteworthy that a family with autosomal dominant hereditary spastic paraplegia has been described recently in association with loss-of-function mutations in KIF5A (Reid et al., 2002). In addition, a missense mutation in the microtubule-binding domain of dynactin that causes human motor neuron disease (Puls et al., 2003), and dominant mutations in cytoplasmic dynein heavy chain cause motor neuron degeneration in mice (Hafezparast et al., 2003). All of these observations strongly suggest that alterations in regulatory mechanisms for either anterograde or retrograde FAT might play critical roles in the pathogenesis of neurodegenerative diseases (Morfini et al., 2005, 2007).
Footnotes
This work was supported by National Institutes of Health Grant AG021494 (S.S.S.), National Institute of Neurological Disorders and Stroke Grants NS23868, NS23320, NS41170, and NS43408 (S.T.B.), the Huntington's Disease Society of America (G.A.M.), Eisai Labs Inc., and Torrey Pines Therapeutics Inc. We thank Evelyn Nwabuisi and Bin Wang for their excellent technical assistance. S.S.S. is a paid consultant of Neuropharma Inc.
References
- Annaert WG, Esselens C, Baert V, Boeve C, Snellings G, Cupers P, Craessaerts K, De Strooper B. Interaction with telencephalin and the amyloid precursor protein predicts a ring structure for presenilins. Neuron. 2001;32:579–589. doi: 10.1016/s0896-6273(01)00512-8. [DOI] [PubMed] [Google Scholar]
- Assini A, Terreni L, Borghi R, Giliberto L, Piccini A, Loqui D, Fogliarino S, Forloni G, Tabaton M. Pure spastic paraparesis associated with a novel presenilin 1 R278K mutation. Neurology. 2003;60:150. doi: 10.1212/01.wnl.0000040252.43269.83. [DOI] [PubMed] [Google Scholar]
- Bajaj NP, Miller CC. Phosphorylation of neurofilament heavy-chain side-arm fragments by cyclin-dependent kinase-5 and glycogen synthase kinase-3alpha in transfected cells. J Neurochem. 1997;69:737–743. doi: 10.1046/j.1471-4159.1997.69020737.x. [DOI] [PubMed] [Google Scholar]
- Baki L, Shioi J, Wen P, Shao Z, Schwarzman A, Gama-Sosa M, Neve R, Robakis NK. PS1 activates PI3K thus inhibiting GSK-3 activity and tau overphosphorylation: effects of FAD mutations. EMBO J. 2004;23:2586–2596. doi: 10.1038/sj.emboj.7600251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beffert U, Morfini G, Bock HH, Reyna H, Brady ST, Herz J. Reelin-mediated signaling locally regulates protein kinase B/Akt and glycogen synthase kinase 3beta. J Biol Chem. 2002;277:49958–49964. doi: 10.1074/jbc.M209205200. [DOI] [PubMed] [Google Scholar]
- Black MM, Lee VM. Phosphorylation of neurofilament proteins in intact neurons: demonstration of phosphorylation in cell bodies and axons. J Neurosci. 1988;8:3296–3305. doi: 10.1523/JNEUROSCI.08-09-03296.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borchelt DR, Koliatsos VE, Guarnieri M, Pardo CA, Sisodia SS, Price DL. Rapid anterograde axonal transport of the cellular prion glycoprotein in the peripheral and central nervous systems. J Biol Chem. 1994;269:14711–14714. [PubMed] [Google Scholar]
- Borchelt DR, Thinakaran G, Eckman CB, Lee MK, Davenport F, Ratovitsky T, Prada CM, Kim G, Seekins S, Yager D, Slunt HH, Wang R, Seeger M, Levey AI, Gandy SE, Copeland NG, Jenkins NA, Price DL, Younkin SG, Sisodia SS. Familial Alzheimer's disease-linked presenilin 1 variants elevate Abeta1–42/1–40 ratio in vitro and in vivo. Neuron. 1996;17:1005–1013. doi: 10.1016/s0896-6273(00)80230-5. [DOI] [PubMed] [Google Scholar]
- Borchelt DR, Ratovitski T, van Lare J, Lee MK, Gonzales V, Jenkins NA, Copeland NG, Price DL, Sisodia SS. Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron. 1997;19:939–945. doi: 10.1016/s0896-6273(00)80974-5. [DOI] [PubMed] [Google Scholar]
- Brady ST, Pfister KK, Bloom GS. A monoclonal antibody against kinesin inhibits both anterograde and retrograde fast axonal transport in squid axoplasm. Proc Natl Acad Sci USA. 1990;87:1061–1065. doi: 10.1073/pnas.87.3.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks WS, Kwok JB, Kril JJ, Broe GA, Blumbergs PC, Tannenberg AE, Lamont PJ, Hedges P, Schofield PR. Alzheimer's disease with spastic paraparesis and “cotton wool” plaques: two pedigrees with PS-1 exon 9 deletions. Brain. 2003;126:783–791. doi: 10.1093/brain/awg084. [DOI] [PubMed] [Google Scholar]
- Bruyn RP. The neuropathology of hereditary spastic paraparesis. Clin Neurol Neurosurg. 1992;94([Suppl]):S16–S18. doi: 10.1016/0303-8467(92)90010-z. [DOI] [PubMed] [Google Scholar]
- Buxbaum JD, Thinakaran G, Koliatsos V, O'Callahan J, Slunt HH, Price DL, Sisodia SS. Alzheimer amyloid protein precursor in the rat hippocampus: transport and processing through the perforant path. J Neurosci. 1998;18:9629–9637. doi: 10.1523/JNEUROSCI.18-23-09629.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai D, Leem JY, Greenfield JP, Wang P, Kim BS, Wang R, Lopes KO, Kim SH, Zheng H, Greengard P, Sisodia SS, Thinakaran G, Xu H. Presenilin-1 regulates intracellular trafficking and cell surface delivery of beta-amyloid precursor protein. J Biol Chem. 2003;278:3446–3454. doi: 10.1074/jbc.M209065200. [DOI] [PubMed] [Google Scholar]
- Cai D, Zhong M, Wang R, Netzer WJ, Shields D, Zheng H, Sisodia SS, Foster DA, Gorelick FS, Xu H, Greengard P. Phospholipase D1 corrects impaired betaAPP trafficking and neurite outgrowth in familial Alzheimer's disease-linked presenilin-1 mutant neurons. Proc Natl Acad Sci USA. 2006a;103:1936–1940. doi: 10.1073/pnas.0510710103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai D, Netzer WJ, Zhong M, Lin Y, Du G, Frohman M, Foster DA, Sisodia SS, Xu H, Gorelick FS, Greengard P. Presenilin-1 uses phospholipase D1 as a negative regulator of beta-amyloid formation. Proc Natl Acad Sci USA. 2006b;103:1941–1946. doi: 10.1073/pnas.0510708103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capell A, Steiner H, Romig H, Keck S, Baader M, Grim MG, Baumeister R, Haass C. Presenilin-1 differentially facilitates endoproteolysis of the beta-amyloid precursor protein and Notch. Nat Cell Biol. 2000;2:205–211. doi: 10.1038/35008626. [DOI] [PubMed] [Google Scholar]
- Court FA, Sherman DL, Pratt T, Garry EM, Ribchester RR, Cottrell DF, Fleetwood-Walker SM, Brophy PJ. Restricted growth of Schwann cells lacking Cajal bands slows conduction in myelinated nerves. Nature. 2004;431:191–195. doi: 10.1038/nature02841. [DOI] [PubMed] [Google Scholar]
- Crook R, Verkkoniemi A, Perez-Tur J, Mehta N, Baker M, Houlden H, Farrer M, Hutton M, Lincoln S, Hardy J, Gwinn K, Somer M, Paetau A, Kalimo H, Ylikoski R, Poyhonen M, Kucera S, Haltia M. A variant of Alzheimer's disease with spastic paraparesis and unusual plaques due to deletion of exon 9 of presenilin 1. Nat Med. 1998;4:452–455. doi: 10.1038/nm0498-452. [DOI] [PubMed] [Google Scholar]
- De Strooper B. Aph-1, Pen-2, and nicastrin with presenilin generate an active gamma-secretase complex. Neuron. 2003;38:9–12. doi: 10.1016/s0896-6273(03)00205-8. [DOI] [PubMed] [Google Scholar]
- Edbauer D, Winkler E, Haass C, Steiner H. Presenilin and nicastrin regulate each other and determine amyloid beta-peptide production via complex formation. Proc Natl Acad Sci USA. 2002;99:8666–8671. doi: 10.1073/pnas.132277899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers MD, Kaplan DR, Price DL, Koliatsos VE. NGF-stimulated retrograde transport of trkA in the mammalian nervous system. J Cell Biol. 1995;130:149–156. doi: 10.1083/jcb.130.1.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fichera M, Lo Giudice M, Falco M, Sturnio M, Amata S, Calabrese O, Bigoni S, Calzolari E, Neri M. Evidence of kinesin heavy chain (KIF5A) involvement in pure hereditary spastic paraplegia. Neurology. 2004;63:1108–1110. doi: 10.1212/01.wnl.0000138731.60693.d2. [DOI] [PubMed] [Google Scholar]
- Georgieff IS, Liem RK, Mellado W, Nunez J, Shelanski ML. High molecular weight tau: preferential localization in the peripheral nervous system. J Cell Sci. 1991;100:55–60. doi: 10.1242/jcs.100.1.55. [DOI] [PubMed] [Google Scholar]
- Georgieff IS, Liem RK, Couchie D, Mavilia C, Nunez J, Shelanski ML. Expression of high molecular weight tau in the central and peripheral nervous systems. J Cell Sci. 1993;105:729–737. doi: 10.1242/jcs.105.3.729. [DOI] [PubMed] [Google Scholar]
- Ghidoni R, Benussi L, Paterlini A, Missale C, Usardi A, Rossi R, Barbiero L, Spano P, Binetti G. Presenilin 2 mutations alter cystatin C trafficking in mouse primary neurons. Neurobiol Aging. 2006;28:371–376. doi: 10.1016/j.neurobiolaging.2006.01.007. [DOI] [PubMed] [Google Scholar]
- Godemann R, Biernat J, Mandelkow E, Mandelkow EM. Phosphorylation of tau protein by recombinant GSK-3beta: pronounced phosphorylation at select Ser/Thr-Pro motifs but no phosphorylation at Ser262 in the repeat domain. FEBS Lett. 1999;454:157–164. doi: 10.1016/s0014-5793(99)00741-3. [DOI] [PubMed] [Google Scholar]
- Gould RM, Brady ST. Neuropathology: many paths lead to hereditary spastic paraplegia. Curr Biol. 2004;14:R903–R904. doi: 10.1016/j.cub.2004.09.076. [DOI] [PubMed] [Google Scholar]
- Guidato S, Tsai LH, Woodgett J, Miller CC. Differential cellular phosphorylation of neurofilament heavy side-arms by glycogen synthase kinase-3 and cyclin-dependent kinase-5. J Neurochem. 1996;66:1698–1706. doi: 10.1046/j.1471-4159.1996.66041698.x. [DOI] [PubMed] [Google Scholar]
- Hafezparast M, Klocke R, Ruhrberg C, Marquardt A, Ahmad-Annuar A, Bowen S, Lalli G, Witherden AS, Hummerich H, Nicholson S, Morgan PJ, Oozageer R, Priestley JV, Averill S, King VR, Ball S, Peters J, Toda T, Yamamoto A, Hiraoka Y, et al. Mutations in dynein link motor neuron degeneration to defects in retrograde transport. Science. 2003;300:808–812. doi: 10.1126/science.1083129. [DOI] [PubMed] [Google Scholar]
- Hallows JL, Chen K, DePinho RA, Vincent I. Decreased cyclin-dependent kinase 5 (cdk5) activity is accompanied by redistribution of cdk5 and cytoskeletal proteins and increased cytoskeletal protein phosphorylation in p35 null mice. J Neurosci. 2003;23:10633–10644. doi: 10.1523/JNEUROSCI.23-33-10633.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamano T, Mutoh T, Tabira T, Araki W, Kuriyama M, Mihara T, Yano S, Yamamoto H. Abnormal intracellular trafficking of high affinity nerve growth factor receptor, Trk, in stable transfectants expressing presenilin 1 protein. Brain Res Mol Brain Res. 2005;137:70–76. doi: 10.1016/j.molbrainres.2005.02.018. [DOI] [PubMed] [Google Scholar]
- Hernandez F, Lucas JJ, Cuadros R, Avila J. GSK-3 dependent phosphoepitopes recognized by PHF-1 and AT-8 antibodies are present in different tau isoforms. Neurobiol Aging. 2003;24:1087–1094. doi: 10.1016/j.neurobiolaging.2003.04.002. [DOI] [PubMed] [Google Scholar]
- Hilbich C, Monning U, Grund C, Masters CL, Beyreuther K. Amyloid-like properties of peptides flanking the epitope of amyloid precursor protein-specific monoclonal antibody 22C11. J Biol Chem. 1993;268:26571–26577. [PubMed] [Google Scholar]
- Hiltunen M, Helisalmi S, Mannermaa A, Alafuzoff I, Koivisto AM, Lehtovirta M, Pirskanen M, Sulkava R, Verkkoniemi A, Soininen H. Identification of a novel 4.6-kb genomic deletion in presenilin-1 gene which results in exclusion of exon 9 in a Finnish early onset Alzheimer's disease family: an Alu core sequence-stimulated recombination? Eur J Hum Genet. 2000;8:259–266. doi: 10.1038/sj.ejhg.5200423. [DOI] [PubMed] [Google Scholar]
- Holtmann B, Zielasek J, Toyka KV, Sendtner M. Comparative analysis of motoneuron loss and functional deficits in PMN mice: implications for human motoneuron disease. J Neurol Sci. 1999;169:140–147. doi: 10.1016/s0022-510x(99)00237-3. [DOI] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. TRK receptors: roles in neuronal signal transduction. Annu Rev Biochem. 2003;72:609–642. doi: 10.1146/annurev.biochem.72.121801.161629. [DOI] [PubMed] [Google Scholar]
- Jones BJ, Roberts DJ. The quantiative measurement of motor inco-ordination in naive mice using an acelerating rotarod. J Pharm Pharmacol. 1968a;20:302–304. doi: 10.1111/j.2042-7158.1968.tb09743.x. [DOI] [PubMed] [Google Scholar]
- Jones BJ, Roberts DJ. A rotarod suitable for quantitative measurements of motor incoordination in naive mice. Naunyn Schmiedebergs Arch Exp Pathol Pharmakol. 1968b;259:211. doi: 10.1007/BF00537801. [DOI] [PubMed] [Google Scholar]
- Julien JP, Mushynski WE. Neurofilaments in health and disease. Prog Nucleic Acid Res Mol Biol. 1998;61:1–23. doi: 10.1016/s0079-6603(08)60823-5. [DOI] [PubMed] [Google Scholar]
- Kamal A, Almenar-Queralt A, LeBlanc JF, Roberts EA, Goldstein LS. Kinesin-mediated axonal transport of a membrane compartment containing beta-secretase and presenilin-1 requires APP. Nature. 2001;414:643–648. doi: 10.1038/414643a. [DOI] [PubMed] [Google Scholar]
- Kang H, Schuman EM. Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science. 1995;267:1658–1662. doi: 10.1126/science.7886457. [DOI] [PubMed] [Google Scholar]
- Kang H, Jia LZ, Suh KY, Tang L, Schuman EM. Determinants of BDNF-induced hippocampal synaptic plasticity: role of the Trk B receptor and the kinetics of neurotrophin delivery. Learn Mem. 1996;3:188–196. doi: 10.1101/lm.3.2-3.188. [DOI] [PubMed] [Google Scholar]
- Kang H, Welcher AA, Shelton D, Schuman EM. Neurotrophins and time: different roles for TrkB signaling in hippocampal long-term potentiation. Neuron. 1997;19:653–664. doi: 10.1016/s0896-6273(00)80378-5. [DOI] [PubMed] [Google Scholar]
- Kim SH, Leem JY, Lah JJ, Slunt HH, Levey AI, Thinakaran G, Sisodia SS. Multiple effects of aspartate mutant presenilin 1 on the processing and trafficking of amyloid precursor protein. J Biol Chem. 2001;276:43343–43350. doi: 10.1074/jbc.M108245200. [DOI] [PubMed] [Google Scholar]
- Koo EH, Sisodia SS, Archer DR, Martin LJ, Weidemann A, Beyreuther K, Fischer P, Masters CL, Price DL. Precursor of amyloid protein in Alzheimer disease undergoes fast anterograde axonal transport. Proc Natl Acad Sci USA. 1990;87:1561–1565. doi: 10.1073/pnas.87.4.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krinke GJ, Vidotto N, Weber E. Teased-fiber technique for peripheral myelinated nerves: methodology and interpretation. Toxicol Pathol. 2000;28:113–121. doi: 10.1177/019262330002800114. [DOI] [PubMed] [Google Scholar]
- Kwok JB, Taddei K, Hallupp M, Fisher C, Brooks WS, Broe GA, Hardy J, Fulham MJ, Nicholson GA, Stell R, St George Hyslop PH, Fraser PE, Kakulas B, Clarnette R, Relkin N, Gandy SE, Schofield PR, Martins RN. Two novel (M233T and R278T) presenilin-1 mutations in early-onset Alzheimer's disease pedigrees and preliminary evidence for association of presenilin-1 mutations with a novel phenotype. NeuroReport. 1997;8:1537–1542. doi: 10.1097/00001756-199704140-00043. [DOI] [PubMed] [Google Scholar]
- Lazarov O, Morfini GA, Lee EB, Farah MH, Szodorai A, DeBoer SR, Koliatsos VE, Kins S, Lee VM, Wong PC, Price DL, Brady ST, Sisodia SS. Axonal transport, amyloid precursor protein, kinesin-1, and the processing apparatus: revisited. J Neurosci. 2005;25:2386–2395. doi: 10.1523/JNEUROSCI.3089-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CW, Lau KF, Miller CC, Shaw PC. Glycogen synthase kinase-3 beta-mediated tau phosphorylation in cultured cell lines. NeuroReport. 2003;14:257–260. doi: 10.1097/00001756-200302100-00020. [DOI] [PubMed] [Google Scholar]
- Lee MK, Borchelt DR, Kim G, Thinakaran G, Slunt HH, Ratovitski T, Martin LJ, Kittur A, Gandy S, Levey AI, Jenkins N, Copeland N, Price DL, Sisodia SS. Hyperaccumulation of FAD-linked presenilin 1 variants in vivo. Nat Med. 1997;3:756–760. doi: 10.1038/nm0797-756. [DOI] [PubMed] [Google Scholar]
- Leem JY, Vijayan S, Han P, Cai D, Machura M, Lopes KO, Veselits ML, Xu H, Thinakaran G. Presenilin 1 is required for maturation and cell surface accumulation of nicastrin. J Biol Chem. 2002;277:19236–19240. doi: 10.1074/jbc.C200148200. [DOI] [PubMed] [Google Scholar]
- Li JY, Pfister KK, Brady S, Dahlstrom A. Axonal transport and distribution of immunologically distinct kinesin heavy chains in rat neurons. J Neurosci Res. 1999;58:226–241. [PubMed] [Google Scholar]
- Moretti P, Lieberman AP, Wilde EA, Giordani BI, Kluin KJ, Koeppe RA, Minoshima S, Kuhl DE, Seltzer WK, Foster NL. Novel insertional presenilin 1 mutation causing Alzheimer disease with spastic paraparesis. Neurology. 2004;62:1865–1868. doi: 10.1212/01.wnl.0000126447.91111.a1. [DOI] [PubMed] [Google Scholar]
- Morfini G, Pigino G, Beffert U, Busciglio J, Brady ST. Fast axonal transport misregulation and Alzheimer's disease. Neuromolecular Med. 2002a;2:89–99. doi: 10.1385/NMM:2:2:089. [DOI] [PubMed] [Google Scholar]
- Morfini G, Szebenyi G, Elluru R, Ratner N, Brady ST. Glycogen synthase kinase 3 phosphorylates kinesin light chains and negatively regulates kinesin-based motility. EMBO J. 2002b;21:281–293. doi: 10.1093/emboj/21.3.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morfini G, Szebenyi G, Brown H, Pant HC, Pigino G, DeBoer S, Beffert U, Brady ST. A novel CDK5-dependent pathway for regulating GSK3 activity and kinesin-driven motility in neurons. EMBO J. 2004;23:2235–2245. doi: 10.1038/sj.emboj.7600237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morfini G, Pigino G, Brady ST. Polyglutamine expansion diseases: failing to deliver. Trends Mol Med. 2005;11:64–70. doi: 10.1016/j.molmed.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Morfini G, Pigino G, Szebenyi G, You Y, Pollema S, Brady ST. JNK mediates pathogenic effects of polyglutamine-expanded androgen receptor on fast axonal transport. Nat Neurosci. 2006;9:907–916. doi: 10.1038/nn1717. [DOI] [PubMed] [Google Scholar]
- Morfini G, Pigino G, Opalach K, Serulle Y, Moreira JE, Sugimori M, Llinas RR, Brady ST. 1-Methyl-4-phenylpyridinium affects fast axonal transport by activation of caspase and protein kinase C. Proc Natl Acad Sci USA. 2007;104:2442–2447. doi: 10.1073/pnas.0611231104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munhoz RP, Kawarai T, Teive HA, Raskin S, Sato C, Liang Y, St George-Hyslop PH, Rogaeva E. Clinical and genetic study of a Brazilian family with spastic paraplegia (SPG6 locus) Mov Disord. 2006;21:279–281. doi: 10.1002/mds.20775. [DOI] [PubMed] [Google Scholar]
- Naruse S, Thinakaran G, Luo JJ, Kusiak JW, Tomita T, Iwatsubo T, Qian X, Ginty DD, Price DL, Borchelt DR, Wong PC, Sisodia SS. Effects of PS1 deficiency on membrane protein trafficking in neurons. Neuron. 1998;21:1213–1221. doi: 10.1016/s0896-6273(00)80637-6. [DOI] [PubMed] [Google Scholar]
- O'Riordan S, McMonagle P, Janssen JC, Fox NC, Farrell M, Collinge J, Rossor MN, Hutchinson M. Presenilin-1 mutation (E280G), spastic paraparesis, and cranial MRI white-matter abnormalities. Neurology. 2002;59:1108–1110. doi: 10.1212/wnl.59.7.1108. [DOI] [PubMed] [Google Scholar]
- Orlacchio A, Kawarai T, Rogaeva E, Song YQ, Paterson AD, Bernardi G, St George-Hyslop PH. Clinical and genetic study of a large Italian family linked to SPG12 locus. Neurology. 2002;59:1395–1401. doi: 10.1212/01.wnl.0000031423.43482.19. [DOI] [PubMed] [Google Scholar]
- Pigino G, Morfini G, Pelsman A, Mattson MP, Brady ST, Busciglio J. Alzheimer's presenilin 1 mutations impair kinesin-based axonal transport. J Neurosci. 2003;23:4499–4508. doi: 10.1523/JNEUROSCI.23-11-04499.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price DL, Sisodia SS. Mutant genes in familial Alzheimer's disease and transgenic models. Annu Rev Neurosci. 1998;21:479–505. doi: 10.1146/annurev.neuro.21.1.479. [DOI] [PubMed] [Google Scholar]
- Puls I, Jonnakuty C, LaMonte BH, Holzbaur EL, Tokito M, Mann E, Floeter MK, Bidus K, Drayna D, Oh SJ, Brown RH, Jr, Ludlow CL, Fischbeck KH. Mutant dynactin in motor neuron disease. Nat Genet. 2003;33:455–456. doi: 10.1038/ng1123. [DOI] [PubMed] [Google Scholar]
- Reid E, Kloos M, Ashley-Koch A, Hughes L, Bevan S, Svenson IK, Graham FL, Gaskell PC, Dearlove A, Pericak-Vance MA, Rubinsztein DC, Marchuk DA. A kinesin heavy chain (KIF5A) mutation in hereditary spastic paraplegia (SPG10) Am J Hum Genet. 2002;71:1189–1194. doi: 10.1086/344210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogaeva E, Bergeron C, Sato C, Moliaka I, Kawarai T, Toulina A, Song YQ, Kolesnikova T, Orlacchio A, Bernardi G, St George-Hyslop PH. PS1 Alzheimer's disease family with spastic paraplegia: the search for a gene modifier. Neurology. 2003;61:1005–1007. doi: 10.1212/wnl.61.7.1005. [DOI] [PubMed] [Google Scholar]
- Rosch H, Schweigreiter R, Bonhoeffer T, Barde YA, Korte M. The neurotrophin receptor p75NTR modulates long-term depression and regulates the expression of AMPA receptor subunits in the hippocampus. Proc Natl Acad Sci USA. 2005;102:7362–7367. doi: 10.1073/pnas.0502460102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samsam M, Frei R, Marziniak M, Martini R, Sommer C. Impaired sensory function in heterozygous P0 knockout mice is associated with nodal changes in sensory nerves. J Neurosci Res. 2002;67:167–173. doi: 10.1002/jnr.10115. [DOI] [PubMed] [Google Scholar]
- Scherer SS, Arroyo EJ. Recent progress on the molecular organization of myelinated axons. J Peripher Nerv Syst. 2002;7:1–12. doi: 10.1046/j.1529-8027.2002.02001.x. [DOI] [PubMed] [Google Scholar]
- Seubert P, Mawal-Dewan M, Barbour R, Jakes R, Goedert M, Johnson GV, Litersky JM, Schenk D, Lieberburg I, Trojanowski JQ, Lee VM-Y. Detection of phosphorylated Ser262 in fetal tau, adult tau, and paired helical filament tau. J Biol Chem. 1995;270:18917–18922. doi: 10.1074/jbc.270.32.18917. [DOI] [PubMed] [Google Scholar]
- Sisodia SS, St George-Hyslop PH. gamma-Secretase, Notch, Abeta and Alzheimer's disease: where do the presenilins fit in? Nat Rev Neurosci. 2002;3:281–290. doi: 10.1038/nrn785. [DOI] [PubMed] [Google Scholar]
- Sisodia SS, Koo EH, Hoffman PN, Perry G, Price DL. Identification and transport of full-length amyloid precursor proteins in rat peripheral nervous system. J Neurosci. 1993;13:3136–3142. doi: 10.1523/JNEUROSCI.13-07-03136.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenoien DL, Brady ST. Immunochemical analysis of kinesin light chain function. Mol Biol Cell. 1997;8:675–689. doi: 10.1091/mbc.8.4.675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szebenyi G, Morfini GA, Babcock A, Gould M, Selkoe K, Stenoien DL, Young M, Faber PW, MacDonald ME, McPhaul MJ, Brady ST. Neuropathogenic forms of huntingtin and androgen receptor inhibit fast axonal transport. Neuron. 2003;40:41–52. doi: 10.1016/s0896-6273(03)00569-5. [DOI] [PubMed] [Google Scholar]
- Tabira T, Chui de H, Nakayama H, Kuroda S, Shibuya M. Alzheimer's disease with spastic paresis and cotton wool type plaques. J Neurosci Res. 2002;70:367–372. doi: 10.1002/jnr.10392. [DOI] [PubMed] [Google Scholar]
- Takashima A, Murayama M, Murayama O, Kohno T, Honda T, Yasutake K, Nihonmatsu N, Mercken M, Yamaguchi H, Sugihara S, Wolozin B. Presenilin 1 associates with glycogen synthase kinase-3beta and its substrate tau. Proc Natl Acad Sci USA. 1998;95:9637–9641. doi: 10.1073/pnas.95.16.9637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor BV, Dyck PJ, Engelstad J, Gruener G, Grant I. Multifocal motor neuropathy: pathologic alterations at the site of conduction block. J Neuropathol Exp Neurol. 2004;63:129–137. doi: 10.1093/jnen/63.2.129. [DOI] [PubMed] [Google Scholar]
- Tesco G, Tanzi RE. GSK3 beta forms a tetrameric complex with endogenous PS1-CTF/NTF and beta-catenin. Effects of the D257/D385A and FAD-linked mutations. Ann NY Acad Sci. 2000;920:227–232. doi: 10.1111/j.1749-6632.2000.tb06927.x. [DOI] [PubMed] [Google Scholar]
- Thinakaran G, Borchelt DR, Lee MK, Slunt HH, Spitzer L, Kim G, Ratovitsky T, Davenport F, Nordstedt C, Seeger M, Hardy J, Levey AI, Gandy SE, Jenkins NA, Copeland NG, Price DL, Sisodia SS. Endoproteolysis of presenilin 1 and accumulation of processed derivatives in vivo. Neuron. 1996;17:181–190. doi: 10.1016/s0896-6273(00)80291-3. [DOI] [PubMed] [Google Scholar]
- Thinakaran G, Harris CL, Ratovitski T, Davenport F, Slunt HH, Price DL, Borchelt DR, Sisodia SS. Evidence that levels of presenilins (PS1 and PS2) are coordinately regulated by competition for limiting cellular factors. J Biol Chem. 1997;272:28415–28422. doi: 10.1074/jbc.272.45.28415. [DOI] [PubMed] [Google Scholar]
- Trojanowski JQ, Lee VM. Paired helical filament tau in Alzheimer's disease. The kinase connection. Am J Pathol. 1994;144:449–453. [PMC free article] [PubMed] [Google Scholar]
- Uemura K, Kuzuya A, Shimohama S. Protein trafficking and Alzheimer's disease. Curr Alzheimer Res. 2004;1:1–10. doi: 10.2174/1567205043480528. [DOI] [PubMed] [Google Scholar]
- Vallat JM. Dominantly inherited peripheral neuropathies. J Neuropathol Exp Neurol. 2003;62:699–714. doi: 10.1093/jnen/62.7.699. [DOI] [PubMed] [Google Scholar]
- Wang R, Tang P, Wang P, Boissy RE, Zheng H. Regulation of tyrosinase trafficking and processing by presenilins: partial loss of function by familial Alzheimer's disease mutation. Proc Natl Acad Sci USA. 2006;103:353–358. doi: 10.1073/pnas.0509822102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidemann A, Konig G, Bunke D, Fischer P, Salbaum JM, Masters CL, Beyreuther K. Identification, biogenesis, and localization of precursors of Alzheimer's disease A4 amyloid protein. Cell. 1989;57:115–126. doi: 10.1016/0092-8674(89)90177-3. [DOI] [PubMed] [Google Scholar]
- Weihl CC, Ghadge GD, Kennedy SG, Hay N, Miller RJ, Roos RP. Mutant presenilin-1 induces apoptosis and downregulates Akt/PKB. J Neurosci. 1999;19:5360–5369. doi: 10.1523/JNEUROSCI.19-13-05360.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirths O, Weis J, Szczygielski J, Multhaup G, Bayer TA. Axonopathy in an APP/PS1 transgenic mouse model of Alzheimer's disease. Acta Neuropathol (Berl) 2006;111:312–319. doi: 10.1007/s00401-006-0041-4. [DOI] [PubMed] [Google Scholar]