

Abstract
Adult-onset neurodegenerative diseases (AONDs) comprise a heterogeneous group of neurological disorders characterized by a progressive, age-dependent decline in neuronal function and loss of selected neuronal populations. Alterations in synaptic function and axonal connectivity represent early and critical pathogenic events in AONDs, but molecular mechanisms underlying these defects remain elusive. The large size and complex subcellular architecture of neurons render them uniquely vulnerable to alterations in axonal transport (AT). Accordingly, deficits in AT have been documented in most AONDs, suggesting a common defect acquired through different pathogenic pathways. These observations suggest that many AONDs can be categorized as dysferopathies, diseases in which alterations in AT represent a critical component in pathogenesis. Topics here address various molecular mechanisms underlying alterations in AT in several AONDs. Illumination of such mechanisms provides a framework for the development of novel therapeutic strategies aimed to prevent axonal and synaptic dysfunction in several major AONDs.
Introduction
Adult-onset neurodegenerative diseases (AONDs) involve dysfunction and degeneration of different neuronal populations (Bossy-Wetzel et al., 2004; Mattson and Magnus, 2006). Despite their heterogeneous etiology, AONDs share several characteristics: they are progressive and their clinical phenotype results from an age-dependent decline in neuronal function that is initially associated with loss of synaptic activity rather than neuronal cell death, a late event in the disease process. Historically, the focus has been on mechanisms of cell death, but identification of mutant genes associated with familial, inheritable forms of AONDs provided new insights on pathogenesis (Coleman and Perry, 2002; Coleman, 2005; Wishart et al., 2006).
Familial and sporadic forms of AONDs often present with nearly indistinguishable clinical pathologies, suggesting common pathogenic mechanisms (Bossy-Wetzel et al., 2004). However, identification of AOND-related genes often did not illuminate mechanisms. In many cases, biological functions of AODN-associated gene products were unknown, and mutant gene products were ubiquitously expressed in both neuronal and non-neuronal cells. When the gene function was known, disease-causative mutations often did not affect normal biological function. Curiously, some familial forms of AONDs were associated with different mutations within a given protein, or with mutations in structurally unrelated proteins, all of which lead to phenotypically indistinguishable pathologies (Price et al., 1998; Dauer and Przedborski, 2003; Bruijn et al., 2004; Morfini et al., 2005; Fink, 2006). With few exceptions, pathogenic mutations failed to explain either the unique vulnerability of neurons in AONDs, or why affected neurons functioned normally for decades before manifestation of pathology. Development of effective therapeutic targets and strategies to slow or reverse degeneration of affected neurons requires an explanation that resolves these apparent paradoxes. Finding such explanations becomes increasingly urgent as significant increases in human lifespan predict a dramatic increase in the incidence of AONDs.
Early axonal and synaptic abnormalities in AONDs
Until recently, our understanding of the neurodegenerative process in AONDs was limited to study of postmortem brain tissue from patients. The marked cell loss observed in these tissues logically focused research efforts on neuronal cell death-related mechanisms (e.g., apoptosis). However, identification of AOND-related genes permitted development of animal models that recapitulated some of the cardinal features of specific human AONDs and facilitated study of early pathogenic events (Price et al., 2000; Wong et al., 2002). Significantly, behavioral and motor abnormalities were detectable before obvious signs of neuronal loss in most animal models. Pathological, behavioral and functional imaging studies in asymptomatic and early symptomatic familial AOND patients highlighted relevance of these findings, clearly demonstrating signs of neuronal dysfunction in the absence of overt neuronal cell death. Electrophysiological and ultrastructural studies further revealed early abnormalities in synaptic function and axonal connectivity, as well as degeneration of axons and synapses (Coleman, 2005; Wishart et al., 2006). Together, current evidence indicates that loss of neuronal connectivity constitutes a critical pathogenic event in most AONDs, and that neurons affected in AONDs follow a “dying back” pattern of degeneration (Fischer et al., 2004; Coleman, 2005; Conforti et al., 2007; Fischer and Glass, 2007). Supporting this idea, approaches based on inhibition of neuronal apoptosis failed to delay onset and progression of clinical symptoms in various AODNs significantly (Djaldetti et al., 2003; Gould et al., 2006; Waldmeier et al., 2006).
Axonal transport represents a unique challenge for neuronal cells
The ubiquitous tissue expression of most AOND-associated gene products contrasts with selective vulnerability of neurons observed in AONDs (Sieradzan and Mann, 2001; Dauer and Przedborski, 2003; Mattson and Magnus, 2006). What distinctive feature or component makes neuronal cells uniquely vulnerable to AOND-related mutations? Neurons are among the largest cells in humans, with >99.9% of their total cell volume corresponding to the axonal compartment. The lack of protein synthesis in axons and large distances separating neuronal cell bodies from axonal and synaptic domains represent unique challenges. Translocation and delivery of membrane proteins and lipid components along axons represents a process collectively referred to as fast axonal transport (FAT), which is essential for proper axonal and synaptic function (for review, see Morfini et al., 2006a). Materials necessary for maintenance and function of axons and synapses are anterogradely transported by microtubule-based molecular motors from sites of synthesis in the cell body. Conventional kinesins are the major species of plus-end directed molecular motors in brain (Wagner et al., 1989), being involved in anterograde FAT of various membrane-bounded organelles (MBOs), including mitochondria, synaptic vesicles and axolemmal precursors, among others (Leopold et al., 1992; Elluru et al., 1995). Conventional kinesin is a heterotetramer composed of two heavy chains (kinesin-1s, KHCs) and two light chains (KLCs) (DeBoer et al., 2008) (Fig. 1). Kinesin-1s are the mechanochemical subunits of conventional kinesin holoenzymes, containing both microtubule-binding and ATPase domains, whereas KLCs play roles in binding and targeting of conventional kinesin to selected MBOs (DeBoer et al., 2008). Neuronal maintenance and survival requires return of signaling complexes and MBOs carrying degradation products from synaptic terminals and axons to the neuronal cell body (Delcroix et al., 2004). Retrograde FAT is carried out by the multisubunit motor protein complex cytoplasmic dynein (CDyn) (Susalka and Pfister, 2000). Although the identity of major molecular motors translocating MBOs along microtubules is established, questions remained about how specific cargos are delivered to the right subcellular location, given the complex cellular architecture of neurons (Morfini et al., 2001).
Figure 1.
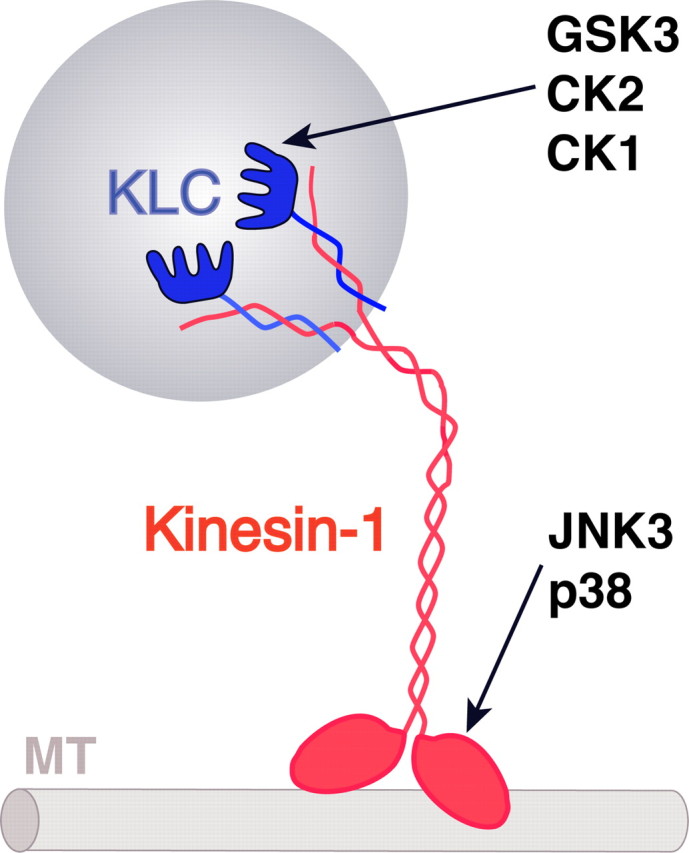
Protein kinases phosphorylate and regulate conventional kinesin. Conventional kinesin is composed of heavy chain (kinesin-1, in red) and light chain (in blue) homodimers (DeBoer et al., 2008). Kinesin-1s use energy derived from ATP hydrolysis to translocate along microtubules (MT). KLCs play a critical role in the binding of conventional kinesin to transported MBO cargoes (Stenoien and Brady, 1997). Recent studies identified various protein kinases which directly phosphorylate selected subunits of conventional kinesin. The functional consequence of each phosphorylation event is determined in part by the major function of the subunit targeted. For example, phosphorylation of KLCs by GSK3 (Morfini et al., 2002a) and CK2 (Morfini et al., 2001; Pigino et al., 2009) promotes the detachment of conventional kinesin from membranes, whereas phosphorylation of kinesin-1s by JNK inhibits the binding of conventional kinesin to microtubules (Morfini et al., 2006b, 2009). Similar regulatory mechanisms have been proposed for cytoplasmic dynein (Susalka and Pfister, 2000; Morfini et al., 2007c). These findings provide a molecular basis for the delivery of selected motor cargoes at specialized axonal compartments (Morfini et al., 2001). The heterogeneity of MBO cargoes suggests the involvement of additional kinases in the regulation of molecular motors.
Regulatory mechanisms of fast axonal transport
The need to deliver selected materials to specialized axonal subdomains implies the existence of targeting mechanisms for their delivery. For example, neurotransmitter-bearing synaptic vesicles need to be delivered to presynaptic terminals, whereas vesicles bearing sodium channels should be selectively delivered to nodes of Ranvier (Morfini et al., 2001; DeBoer et al., 2008). As a result, neurons rely heavily on phosphorylation-dependent intracellular signaling mechanisms, which help in the spatial and temporal coordination and regulation of many cellular processes within axons, including FAT (Morfini et al., 2001). Cumulative data indicates that phosphorylation of molecular motors represents a major mechanism for regulation of FAT in vivo (Hollenbeck, 1990; Morfini et al., 2001, 2002a, 2004; Donelan et al., 2002). Many of these findings stem from use of isolated squid axoplasm, a unique experimental model for study of axon-specific events (Brady et al., 1990). Consistent with heterogeneity of axonal MBOs and the need for delivering cargos to specific axonal compartments, biochemical, pharmacological and cell biological experiments identified multiple kinases that differentially regulate conventional kinesin (Morfini et al., 2001, 2002a, 2004, 2007a, 2009) and CDyn (Brill and Pfister, 2000). These kinases directly or indirectly modify specific subunits within molecular motor complexes, and differentially regulate enzymatic (i.e., binding to microtubules and ATPase activity) (Morfini et al., 2009) and nonenzymatic properties (i.e., attachment to transported cargoes) (Morfini et al., 2002a; Pigino et al., 2009) (Fig. 1). These regulatory mechanisms provided an explanation for delivery of selected motor cargoes to specialized axonal subcompartments, producing an unexpected insight: activity of many kinases regulating FAT are increased in AONDs, as reflected by aberrant patterns of protein phosphorylation (Wagey and Krieger, 1998).
Fast axonal transport deficits in AONDs
Early axonal and synaptic degeneration in AONDs are consistent with AT alterations. Evidence indicating that alterations in AT suffice to produced selective neuronal degeneration comes from genetic studies. Specifically, mutations in genes coding for microtubule-based molecular motor subunits can result in “dying back” neuropathologies (Hirokawa and Takemura, 2003; Morfini et al., 2005; Roy et al., 2005). For example, loss of function mutations in various subunits of conventional kinesin (Reid et al., 2002) and CDyn (Hafezparast et al., 2003; Puls et al., 2003, 2005; Farrer et al., 2009) result in selective degeneration of specific neuronal subtypes. Often the length of axons is assumed to correlate with increased vulnerability to AT defects. However, mutations affecting different subunits of the same motor protein may induce dramatically different pathologies, suggesting additional levels of complexity. For example, some mutations in CDyn subunits produce degeneration of motor neurons (MNs) (Hafezparast et al., 2003; Puls et al., 2005), while others lead to sensory neuron pathology (Chen et al., 2007) or degeneration of cortical and extrapyramidal neurons (Farrer et al., 2009). The basis for selective vulnerability of specific neuronal subtypes due to mutations in motor proteins is not well understood, but likely results from functional specializations of each neuronal subtype. Regardless, the unique reliance of neurons on FAT is well established. Together, these observations indicate that FAT deficits are sufficient to cause neurodegeneration. Accordingly, alterations in FAT have been documented in many AONDs (Morfini et al., 2005; Roy et al., 2005). However, most AONDs are not associated with mutations in molecular motors, so other mechanisms must be responsible for changes in FAT seen in AONDs (Morfini et al., 2005). Alterations in regulatory pathways for FAT provide a potential mechanism. Accordingly, abnormal activation of protein kinases and aberrant patterns of protein phosphorylation represents major AOND hallmarks (Wagey and Krieger, 1998). A review of how hallmark pathological changes in several AONDs can be related to abnormalities in AT suggest a common vulnerability in these diseases.
Pathogenic tau and β-amyloid inhibit FAT
Alzheimer's disease (AD) affects as many as 5.3 million patients in the United States. AD has two major pathological hallmarks: accumulation of filamentous tau and β amyloid (Aβ) in affected cell regions (Mattson, 2004). Significantly, pathological forms of both can affect FAT, albeit by different mechanisms (Morfini et al., 2002b).
Tau is a microtubule-associated protein thought to stabilize microtubules (Binder et al., 2005; Conde and Cáceres, 2009). Although its tissue distribution is widespread, larger quantities of this protein are expressed in the brain (Binder et al., 2005). Within brain, tau is highly concentrated in axons but is also present in other neuronal compartments and glia (Binder et al., 1985; Papasozomenos and Binder, 1987). In AD and other tauopathies, tau becomes highly phosphorylated and aggregated in polymers termed “straight” or “paired-helical” filaments. Side-to-side associations of these form neurofibrillary tangles (NFTs), neuropil threads, and are associated with dystrophic neurites in AD (Binder et al., 2005) (Fig. 2). In isolated axoplasm, t canonical tau monomers (made of bacterially expressed hTau40, the longest tau isoform in brain, 441aa) do not inhibit FAT (Morfini et al., 2007b) (Fig. 2A). However, polymeric hTau40 filaments selectively inhibited anterograde, but not retrograde FAT (Lapointe et al., 2009). This effect required an N-terminal amino acid stretch that is apparently hidden in the monomer (Fig. 2B). Pharmacological studies indicate that the underlying mechanism involves activation of protein phosphatase 1 (PP1). PP1 dephosphorylates and activates glycogen synthase kinase 3 (GSK3), which in turn phosphorylates KLCs and promotes detachment of conventional kinesin from its transported cargoes (Morfini et al., 2002a, 2004; Lapointe et al., 2009). Significantly, noncanonical tau isoforms 6P and 6D (Luo et al., 2004) lacking most of the proline-rich, microtubule binding and C-terminal regions of canonical tau isoforms also inhibit anterograde FAT, even though these cannot form polymers (Lapointe et al., 2009). Inhibitory effects of 6P and 6D on anterograde FAT suggests that the N-terminal region of tau is required and sufficient for tau toxicity (Lapointe et al., 2009). We hypothesize that aggregation of intact tau “presents” its N-terminal region to the cell—a region normally hidden in monomer due to folding events such as those suggested by the “paper clip” model of tau monomer structure (Jeganathan et al., 2006). This folded conformation requires MTBRs and the carboxy terminus, which are lacking in 6P/6D noncanonical isoforms (Jeganathan et al., 2006). Previous work indicated that both amino and carboxy-end truncation of tau occur as NFTs begin to form in affected AD neurons (Carmel et al., 1996; García-Sierra et al., 2003; Guillozet-Bongaarts et al., 2005). During NFT formation, tau undergoes C-terminal truncation (Gamblin et al., 2003) and may be truncated from the amino end as well (Horowitz et al., 2004), but until recently functional consequences of these proteolytic events were unknown. Current work suggests that truncation of tau from the amino end may remove a toxic region capable of activating PP1 and inhibiting FAT. We further posit that truncation of amino and carboxy termini in NFTs likely renders these structure relatively inert (Fig. 2C). That is not to say NFTs are harmless, but as they are thought to persist in neurons for >20 years in AD patients (Morsch et al., 1999), we suggest that they are less toxic than NFTs made from intact full-length canonical tau inhibiting anterograde FAT.
Figure 2.
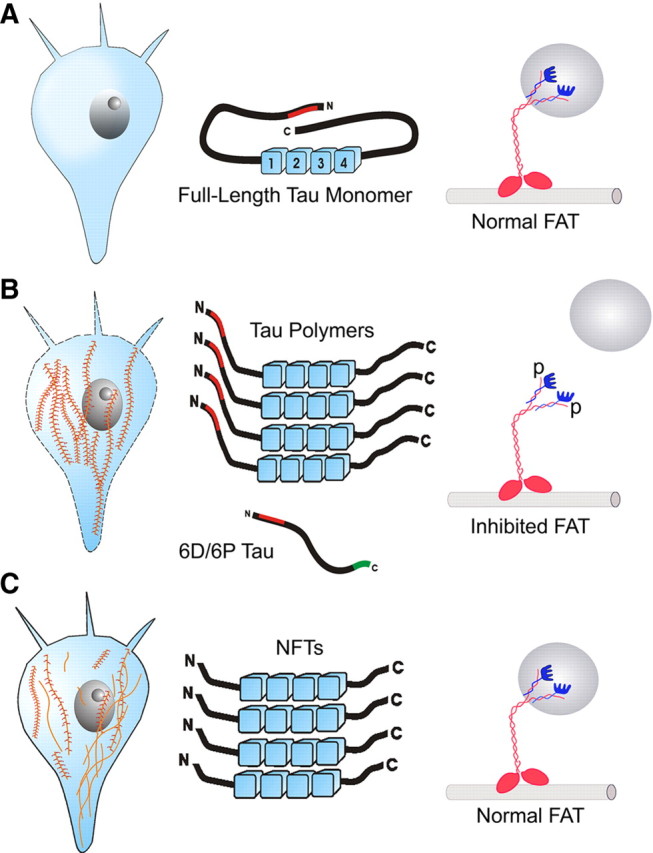
Schematic representation of tau-mediated toxicity to FAT. A, Healthy neurons: The canonical full-length isoform can fold into the “paper clip” conformation, which masks the toxic N-terminal region (red). FAT is unaffected by soluble monomeric full-length tau. B, Neuronal dysfunction: Polymers of canonical tau form, which leads to an unmaking of the toxic region. The truncated noncanonical 6D/6P tau monomers (also schematically depicted) cannot fold and presents an unmasked toxic region constitutively. Exposure of the toxic region in tau isoforms activates a PP1-GSK cascade. This results in phosphorylation of KLCs (blue), dissociation of the cargo (gray sphere), and FAT inhibition. C, Neuron survival: During NFT formation, modifications (e.g., truncation and phosphorylation) of tau in the polymers vie for preeminence in the neuron. If modified NFT formation predominates, FAT remains relatively unaffected and the cell likely survives for some years.
Accumulation of Aβ and its aggregates has long been considered instrumental in AD pathogenesis (Hardy and Selkoe, 2002). In recent years, this hypothesis was extended to include the concept that soluble oligomeric Aβ (oAβ) aggregates may play a major pathogenic role (Klein et al., 2004). FAT of several classes of MBOs, including mitochondria, endosomes and multivesicular bodies, as well as MBOs containing APP, synaptophysin, syntaxin and Trk receptors is inhibited in various AD mouse models (Pigino et al., 2003; Lazarov et al., 2007). Interestingly, the great majority of these mice accumulate soluble oAβ, but mechanisms linking oAβ species to FAT deficits remained to be defined. As a direct test of Aβ effects on FAT, various homogeneous Aβ species [unaggregated (uAβ), fibrillar (fAβ), and oAβ] were perfused into isolated squid axoplasm. Perfusion of oAβ dramatically inhibited both retrograde and anterograde FAT at physiological, low nanomolar concentrations (Pigino et al., 2009), whereas neither uAβ nor fAβ affected FAT. Inhibition of FAT by oAβ predicted failure of neurotransmission. Accordingly, presynaptic injection of oAβ in the squid giant synapse induced a profound inhibition of synaptic transmission, in association with a marked reduction in synaptic vesicle availability at active zones (Moreno et al., 2009).
Alterations in activity for protein kinases and abnormal phosphorylation of neuronal proteins are key features of AD (Pigino et al., 2001, 2003; Morfini et al., 2002b), and some kinases are abnormally deregulated in AD (Wang et al., 2007) can modulate FAT (see Fig. 6). Accordingly, we found that oAβ inhibits FAT by a mechanism involving CK2 activity. CK2 inhibits FAT by phosphorylation of KLCs and release of kinesin-1 from transport vesicles (Pigino et al., 2009), a mode of inhibition that parallels that of filamentous tau (Lapointe et al., 2009) and mutant PS1 (presenilin-1) (Pigino et al., 2003; Lazarov et al., 2007), both of which activate GSK3 (Morfini et al., 2002b).
Figure 6.

Relationships among microtubule-based molecular motors for FAT, protein kinases, and neuropathogenic polypeptides/compounds. Several protein kinases have been identified which phosphorylate and regulate specific functional activities of conventional kinesin and cytoplasmic dynein, the major microtubule-dependent motor proteins responsible for FAT. Significantly, various unrelated neuropathogenic proteins induce alterations in FAT through an independent mechanism involving the activation of specific kinases and phosphorylation of selected molecular motor subunits. The complexity of FAT regulation suggests the existence of additional pathogenic pathways. Inhibitory events are indicated by T-shaped lines; arrowheads indicate activation. Tau Fil, Filamentous tau; PS1, presenilin-1; AR, androgen receptor; Htt, Huntingtin; oAβ, oligomeric β-amyloid; MPP+, 1-methyl-4-phenylpyridinium; GSK3, glycogen synthase kinase 3; JNK3, cJun-amino terminal kinase 3; CK2, casein kinase 2; PKC, protein kinase C.
FAT defects in Huntington's disease
Huntington's disease (HD) is one of several polyglutamine (polyQ) expansion diseases characterized by expansion of unstable CAG repeats within the coding region of several unrelated proteins. A polymorphic polyQ tract in wild-type, nonpathogenic Htt (Huntingtin) (WT-Htt) normally comprises 6-35 glutamines, whereas Htt variants having >36Qs typically define an HD allele (polyQ-Htt) (Brinkman et al., 1997). At the cellular level, HD is characterized by marked degeneration of projection neurons within the striatum and cerebral cortex even though Htt protein is ubiquitously expressed in nearly all tissues (Sieradzan and Mann, 2001). Significantly, pathological studies in presymptomatic HD patients and HD mouse models documented “dying back” neuronal degeneration and loss of specific axonal tracts (Albin et al., 1992). As in other AONDs, synaptic dysfunction and axonal abnormalities long preceded neuronal cell death (Li and Li, 2004). Consistent with these observations, multiple lines of evidence showed FAT deficits in HD. Ultrastructural observations indicated a reduced number of synaptic vesicles, and abnormal MBO profiles within axons of affected neurons (Li et al., 2001). Subsequently, reductions in FAT in association with various polyQ-expanded pathogenic proteins were documented in various experimental systems (Gunawardena et al., 2003; Szebenyi et al., 2003; Lee et al., 2004; Morfini et al., 2006b; Her and Goldstein, 2008). Studies in isolated axoplasm showed that polyQ-Htt inhibits FAT in an axon-autonomous manner (Szebenyi et al., 2003), suggesting that inhibition of FAT might underlie axonal degeneration in HD (Morfini et al., 2005).
An explanation of how polyQ-Htt affects FAT emerged from experiments in isolated axoplasm. PolyQ-Htt, but not WT-Htt, inhibits FAT when presented at levels 100–1000-fold lower than molecular motors (Szebenyi et al., 2003), suggesting that molecular motor function was affected by an enzymatic activity, probably a kinase (Morfini et al., 2005). Consistent with this idea, abnormal phosphorylation of proteins and increased activation of various protein kinases represent well established features of HD (Liévens et al., 2002; DiProspero et al., 2004). Further, polyQ-expanded forms of androgen receptor inhibited FAT though a kinase-dependent pathway (Morfini et al., 2005). However, relevant pathogenic targets for these kinases were undefined, and the contribution of these kinases to HD pathogenesis uncertain (Morfini et al., 2005). Experimental evidence demonstrated that polyQ-Htt inhibits FAT by activation of cJun N-terminal kinase (JNK), a member of the mitogen activated protein kinase (MAPK) family (Morfini et al., 2009). Increased JNK activation was consistent with changes in gene transcription and activation of apoptotic pathways associated with HD (Sugars and Rubinsztein, 2003; Björkblom et al., 2008). Three JNK isoforms exist in mammals (JNK1, JNK2 and JNK3), and differential activation of JNKs was observed before disease onset in a well characterized HD mouse model (Morfini et al., 2009). Significantly, neuron-specific JNK3, but not ubiquitously expressed JNK1, mediated inhibition of FAT by polyQ-Htt, thereby providing a partial explanation for selective neuronal vulnerability in HD (Morfini et al., 2009). Providing a molecular basis for its effect on FAT, JNK3 (but not JNK1) was found to phosphorylate kinesin-1 subunits of conventional kinesin. Mass spectrometry studies mapped JNK3 phosphorylation of kinesin-1 to S176, a conserved residue in the motor domain of squid, mouse, and human kinesin-1s (Morfini et al., 2009). Collectively, these results demonstrated that polyQ-Htt activates the JNK pathway, and that JNK3 phosphorylates and inhibits kinesin-1 (Morfini et al., 2009) (Fig. 3). Activation of JNK3 by polyQ-Htt would promote reductions in kinesin-based transport, eventually leading to deficits in synaptic/axonal function and maintenance.
Figure 3.

Inhibition of FAT as a common mechanism for polyQ expansion disease pathogenesis. In this model, polyQ expansion of polypeptides directly or indirectly leads to activation of one or more MAPKKKs, which in turn activate one or more MAPKKs, finally activating JNK1 and JNK3. Increased JNK1/JNK3 activity represents a toxic gain of function common to several different pathogenic polyQ-expanded polypeptides (Morfini et al., 2006b, 2009). Altered folding of the pathogenic protein may lead to aggregate formation and/or altered association with binding partners could contribute to disease-specific characteristics for each polyQ disease (Morfini et al., 2005). The role of aggregates in this model remains unknown. Different consequences are associated with increased activation of each JNK isoform. Both ubiquitous JNK1 and neuron-specific JNK3 can alter gene expression in neurons (Sugars and Rubinsztein, 2003), but only JNK3 inhibits FAT (Morfini et al., 2009). Reductions in FAT lead to failure of neurotransmission and degeneration of the distal axon. When the extent of synaptic failure passes a critical threshold, cell death pathways are triggered in the neuronal cell body and apoptosis ensues.
Motor neuron diseases and FAT deficits
A number of AONDs primarily affect MNs, including Kennedy's disease, amyotrophic lateral sclerosis (ALS), hereditary spastic paraplegias (HSPs), and spinal muscular atrophies. The number of genes implicated in familial forms of motor neuron disease is remarkably large and diverse, comprising >50 genes or loci and ranging from molecular motors to cytoskeletal proteins, enzymes and proteins of unknown function (James and Talbot, 2006). Affected neurons may be upper or lower MNs, or both. The reasons for increased vulnerability of MNs are uncertain. As in other AONDs, they are projection neurons often with unusually long axons (Mattson and Magnus, 2006), which presumably increases their dependence on AT (Holzbaur, 2004).
Several components of the AT machinery are linked to MN diseases. Mutations in conventional kinesin and CDyn have been documented in humans (Hafezparast et al., 2003) and mice (Puls et al., 2003) with MN pathologies. A genome-wide single-nucleotide polymorphism (SNP) analysis of sporadic ALS (SALS) uncovered an SNP within the KIFAP3 gene, coding for an accessory subunit of kinesin-2 (Landers et al., 2009). SALS patients harboring a genotype with this SNP exhibit a 14 month survival advantage compared with individuals with other genotypes. Interestingly, the survival advantage of this allele correlates with decreased expression of KIFAP3 (Landers et al., 2009). Additionally, mutations in the nucleocytoplasmic, RNA/DNA binding proteins 43 kDa TAR DNA-binding protein (TDP43) (Rutherford et al., 2008) and fused in sarcoma/translated in liposarcoma (FUS/TLS) (Kwiatkowski et al., 2009) have been linked to ALS. Both are multifunctional proteins implicated in gene expression and RNA processing, including RNA export to cytoplasm and dendrites for local translation (Lagier-Tourenne and Cleveland, 2009).
ALS
ALS is characterized by relentless degeneration of upper and lower MNs, muscle atrophy, and eventual paralysis. Mean age of onset is 55, with an average survival of 3–5 years postdiagnosis (Bruijn et al., 2004). Over 90% of ALS cases have an unknown etiology and are classified as sporadic (SALS), whereas ≈10% of ALS cases are familial ALS (FALS) (Pasinelli and Brown, 2006). Clinically, SALS and FALS are nearly indistinguishable, suggesting that pathological events associated with each disease form eventually converge on a common pathway that leads to ALS (Pasinelli and Brown, 2006).
ALS animal models illuminated pathways and processes connected to ALS pathogenesis, including AT (Warita et al., 1999; Roy et al., 2005; De Vos et al., 2007). Mutations in Cu,Zn-superoxide dismutase (SOD1) collectively account for 20–25% of FALS cases (Pasinelli and Brown, 2006) and transgenic mice overexpressing FALS-linked human SOD1 (FALS mice) exhibit a phenotype that closely recapitulates human disease (Gurney et al., 1994; Bruijn et al., 1998). Detailed pathological analysis of these animals showed “dying back” degeneration of motor neurons (Fischer et al., 2004; Fischer and Glass, 2007). Impairments in AT of cytoskeletal components, mitochondria and selected MBOs have been demonstrated in FALS mice (Warita et al., 1999; Williamson and Cleveland, 1999). Deficits in AT were found early in the disease course, suggesting that alterations in AT play a causative role rather than representing an end-stage effect (Collard et al., 1995; Williamson and Cleveland, 1999). However, molecular mechanisms by which mutant SOD1 impairs AT remained unknown (De Vos et al., 2007).
Genetic evidence suggested that SOD1 mutations cause ALS through a toxic gain-of-function mechanism (Bruijn et al., 2004). Accordingly, vesicle motility assays in isolated squid axoplasm revealed a toxic gain of function in the context of AT. Specifically, recombinant FALS-linked SOD1 mutant proteins selectively inhibited anterograde FAT of MBOs, whereas wild-type SOD1 had no effect (G. Morfini, D. A. Bosco, R. H. Brown, Jr., and S. T. Brady, unpublished data). Effects of mutant SOD1 were not dependent on direct interactions with motors (Zhang et al., 2007) but instead involved selective activation of p38 MAP kinase. Significantly, hyperphosphorylation of neurofilaments represent a pathological hallmark of ALS, and p38 phosphorylates neurofilaments in vitro (Ackerley et al., 2004). Further, multiple independent reports documented p38 activation in ALS patients (Bendotti et al., 2004) and mutant SOD1 mice (Tortarolo et al., 2003; Bendotti et al., 2004). However, pathogenic p38 targets remained unknown, and thus the relationship of p38 activation to ALS pathogenesis was unclear. Significantly, biochemical studies revealed that KHCs are phosphorylated by p38, and this phosphorylation inhibited conventional kinesin-based motility. These data indicated that mutant SOD1 impairs anterograde FAT by activation of p38 MAPK.
Hereditary spastic paraplegias
HSPs comprise a group of neurodegenerative disorders characterized by progressive spasticity and weakness of lower limbs. Spasticity occurs in the absence (pure HSP) or presence (complicated HSP) of other neurological symptoms and is due to degeneration of distal portions of axons in the fasciculus gracilis and corticospinal tracts, which include the longest CNS axons. At least 41 spastic paraplegia (SPG) gene loci have been mapped and 17 genes with diverse functions and expression patterns identified (Salinas et al., 2008). HSPs represent classic examples of dying-back neuropathies. Accordingly, AT impairments have emerged as one potential convergent pathogenic mechanism (Crosby and Proukakis, 2002).
Several forms of HSP are due to mutations in genes involved in intracellular trafficking (Salinas et al., 2008), including a KHC gene, KIF5A, directly implicating AT impairments to HSP pathogenesis (Reid et al., 2002). Further, pathogenic spastin hampers FAT in squid axoplasm (Solowska et al., 2008). Mouse models of SPG7 and SPG4 provide in vivo evidence for AT impairment in HSPs. SPG7 is associated with mutations in paraplegin, a mitochondrial ATPase (Ferreirinha et al., 2004). SPG4 results from mutations in spastin, a microtubule-severing protein (Baas et al., 2005). SPG7 and SPG4 mice models develop axonal swellings filled with MBOs and cytoskeletal proteins (Tarrade et al., 2006). Injection of neurotracer into gastrocnemius muscle of SPG7 mice further demonstrated that retrograde AT is reduced in these mice (Ferreirinha et al., 2004).
AT defects were recently identified in an SPG2 mouse model. SPG2 and the allelic disorder Pelizaeus-Merzbacher disease (PMD) are caused by mutation in the X-linked proteolipid protein (PLP1) gene (Saugier-Veber et al., 1994) encoding proteolipid protein and DM20 (PLP/DM20), tetraspan membrane proteins expressed in the CNS by oligodendrocytes (Garbern, 2007). Plp1 gene knock-out mice synthesize normal amounts of myelin with subtle defects in compaction (Boison and Stoffel, 1994; Klugmann et al., 1997; Rosenbluth et al., 2006), but develop late onset axonal changes in the absence of primary demyelination or inflammation (Griffiths et al., 1998). The earliest morphological changes occur in small myelinated axons of optic nerve and fasciculus gracilis around postnatal day 40 (P40) and are characterized by accumulation of MBOs and swelling at distal juxtaparanodal regions. Axonal changes occur in proximal and distal axonal regions, but, as in PLP1-null patients, degeneration is restricted to distal portions of long axons (Fig. 4A) in spinal cord (Garbern et al., 2002). Interestingly, MBO-rich axonal swellings in Plp1 knock-out mice (Fig. 4B) resemble those seen in paraplegin and spastin mutants in association with impaired AT (Edgar et al., 2004). Mice deficient in another CNS myelin protein, 2′,3′-cyclic nucleotide-3′-phosphodiesterase (CNP) (Lappe-Siefke et al., 2003), develop morphologically similar axonal changes (Fig. 4C,D) and axonal degeneration. CNP is located in noncompact myelin, i.e., in the inner and outer tongue processes and paranodal loops, which form a channel linking myelin with the oligodendrocyte soma (Lappe-Siefke et al., 2003). Compact myelin appears normal in CNP-deficient mice, but abnormal enlargement of the inner tongue processes of myelin (Lappe-Siefke et al., 2003) (Fig. 4D,E) occurs concurrently with the earliest axonal defects, within days of myelination (Edgar et al., 2009).
Figure 4.

Axonal transport abnormalities in myelin protein-deficient mice. A, Schematic summarizing the changes that occur in axons in the Plp1-null mouse model of spastic paraplegia type 2. Despite almost normal myelination, axonal organelles accumulate at the distal juxtaparanodal regions of myelinated axons, eventually causing localized swelling of the axon. Axonal swellings are not preferentially localized to proximal or distal portions of an axon, and it is likely (though not confirmed) that a single axon contains multiple swellings. The distal portions of the long axons of the spinal cord eventually degenerate (represented by the dashed line). B–E, Electron micrographs of CNS fiber changes. Organelle accumulation and cytoskeletal disruption in a myelinated axon from a P120 Plp1-null mouse spinal cord (B). Morphologically similar organelle accumulations in the spinal cord (C) and optic nerve (D) of P120 Cnp1-null mice. Swelling of the inner tongue process of the oligodendrocyte (asterisks in D and E) is a feature of the Cnp1-null mouse. The swollen tongue appears not to compress the axon but seems to be accommodated by expansion of the compact myelin sheath, as illustrated in these P120 optic nerve fibers. Scale bars: B, C, 2 μm; D, E, 1 μm.
Pathological analysis of myelin mutants indicate that oligodendrocytes modulate various processes within their myelinated axon beneath, including AT (Witt and Brady, 2000). Providing a basis for regulation of AT by glia, localized changes in kinase and phosphatase activities occur at nodal/paranodal regions of myelinated axons, precisely the site at which axonal organelles accumulate in Plp1 knock-out mice This suggests that localized dysregulation of kinase/phosphatase activity and motor proteins might occur in the absence of various myelin-associated proteins. Such localized actions on motor proteins is supported by AT impairment independent of changes in motor protein transcription (Morfini et al., 2001, 2002a, 2004). Another explanation for impaired AT in myelin mutants is localized energy deficiencies. This is consistent with axonal organelle accumulations observed in paraplegin-deficient mice, which are morphologically similar to those in myelin mutants.
Parkinson's disease
The final example of AOND associated with altered AT is Parkinson's disease (PD). PD is the second most common neurodegenerative disease after AD, affecting >4 million people worldwide (Andersen et al., 2001). PD involves progressive dying back neuropathy of axons projecting to the striatum from the substantia nigra. When ∼80% of synapses from nigral dopaminergic neurons are dysfunctional, shortage of dopamine in the striatum causes movement defects that characterize PD (Dauer and Przedborski, 2003). Available treatments effectively delay clinical symptoms, but do not stop or slow progressive neuronal degeneration. Approximately 95% of PD cases are sporadic (sPD), with 5% due to mutations in functionally unrelated genes (fPD). An unknown fraction of sPD cases are due to toxic agents, including MPTP/MPP+, paraquat, and rotenone (Dauer and Przedborski, 2003). MPP+ is the best-characterized example of toxin-induced PD. These and other toxins are thought to target mitochondria and induce activation of caspases (Turmel et al., 2001; Viswanath et al., 2001). No common pathogenic mechanism emerges from a consideration of sPD, fPD and toxin-induced PD (Dauer and Przedborski, 2003; Cookson, 2005). Although there are links to mitochondrial function and ubiquitin/proteasome pathways in familial and toxin-induced PD forms, there is no evidence that these pathways are specifically activated in sPD. Intriguingly, fPD-associated mutant proteins are present in many neurons, yet nigrostriatal dopaminergic neurons are particularly affected. Further, many nondopaminergic neurons also degenerate in PD, and many dopaminergic neurons in other brain regions are largely spared. This diversity in PD etiology reflects the existence of multiple pathogenic pathways, perhaps converging on a common vulnerability in select neurons, but no consensus target has emerged.
Recent studies indicate MPP+ has a distinctive effect on AT. Perfusion of MPP+ into isolated axoplasm significantly increased retrograde FAT and reduced anterograde FAT of MBOs (Morfini et al., 2007c). This effect on AT was blocked by inhibitors of either PKCδ or caspase 3, which can activate PKCδ by proteolytic cleavage (Kaul et al., 2003). Increases in retrograde FAT contrast with the effects induced by other AOND-related pathogenic proteins examined, suggesting that proper balance between anterograde and retrograde AT is critical for neuronal function. A major consequence of such an imbalance was revealed by MPP+ treatment of the presynaptic terminal of the squid giant synapse (Serulle et al., 2007). Injection of MPP+ led to a failure of neurotransmission and a rapid block of the postsynaptic response. Failure of neurotransmission was prevented by coinjection of either caspase 3 or PKCδ inhibitors, which also prevented the effects MPP+ on FAT. Preliminary studies suggest that pathogenic forms of α-synuclein (the main component of Lewy bodies that are a pathological hallmark of PD (Duda et al., 2000; Spillantini and Goedert, 2000) may similarly affect AT (S. T. Brady, T. J. Chilcote, M. Sugimori, Y. Serulle, R. R. Llinas, and G. Morfini, unpublished data) (Fig. 5).
Figure 5.
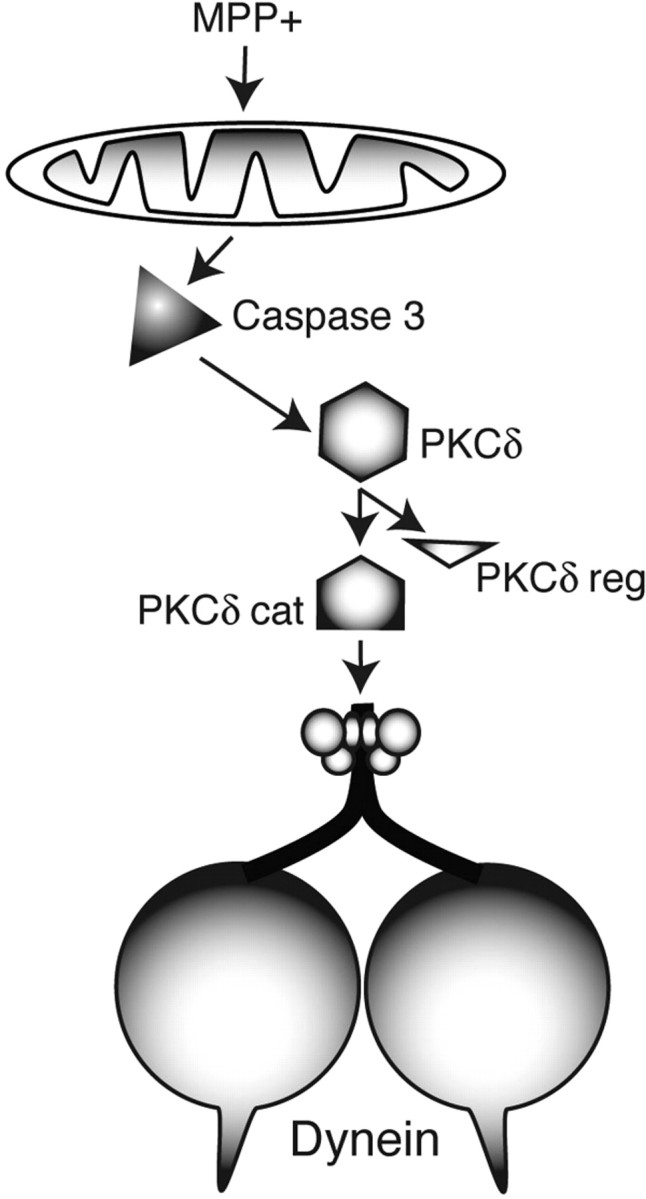
Model for alteration of FAT as a mechanism leading to MPTP/MPP+-induced parkinsonism. Certain toxic chemicals, like MPTP and its metabolite MPP+, can produce a severe parkinsonism, but the pathogenic process was poorly understood. Agents like MPP+, rotenone, and paraquat are thought to act at the level of mitochondria, but mitochondria are ubiquitous and the neurodegenerative parkinsonian phenotype is highly selective (Dauer and Przedborski, 2003). In isolated axons, MPP+ activates caspase 3, which cleaves and activates a specific isoform of protein kinase C, PKCδ. Activated PKCδ then phosphorylates dynein intermediate chains and activate retrograde FAT. Further, activation of PKCδ reduces anterograde FAT. These imbalances in FAT result in depletion of synaptic vesicles from synapses, failure of neurotransmission (Morfini et al., 2007c; Serulle et al., 2007), and altered trophic factor support (Delcroix et al., 2004). These events ultimately would lead to neuronal cell death. cat, Catalytic subunit; reg, regulatory subunit.
Conclusions
The accumulated evidence indicates that various unrelated neuropathogenic polypeptides induce alterations in AT by promoting activation of selected protein kinases, leading to abnormal phosphorylation and regulation of molecular motors (Morfini et al., 2002a, 2004, 2005, 2006b, 2007c; Pigino et al., 2003; Szebenyi et al., 2003) (Fig. 6). The emerging complexity and variety of regulatory pathways for AT thus appear consistent with a common pathogenic mechanism underlying different familial forms of AONDs and suggest that some AONDs represent a novel class of neurological disease that can be collectively characterized as “dysferopathies” (Morfini et al., 2007c; Pigino et al., 2009). Observations discussed here provide an explanation for similarities in pathological features of AD, HD, PD, ALS, and HSP patients and provide a mechanistic basis for abnormal phosphorylation of proteins, alterations in AT, and axonal degeneration characteristic of AONDs. Understanding pathogenic pathways and molecular components underlying AT defects in specific dysferopathies will provide a framework for development of novel therapeutic strategies to treat these diseases.
Footnotes
This work was supported by National Institutes of Health (NIH) Grants AG09466 to L.I.B. and T32 AG020506-07 to N.M.K.; ALS Association (ALSA) and ALS Therapy Alliance grants to D.A.B.; ALSA, Muscular Dystrophy Association, and NIH (NS23868, NS23320, NS41170) grants to S.T.B.; a Multiple Sclerosis Society (Scotland) grant to J.E.; and Huntington's Disease Society of America and American Parkinson Disease Association grants to G.M.
References
- Ackerley S, Grierson AJ, Banner S, Perkinton MS, Brownlees J, Byers HL, Ward M, Thornhill P, Hussain K, Waby JS, Anderton BH, Cooper JD, Dingwall C, Leigh PN, Shaw CE, Miller CC. p38alpha stress-activated protein kinase phosphorylates neurofilaments and is associated with neurofilament pathology in amyotrophic lateral sclerosis. Mol Cell Neurosci. 2004;26:354–364. doi: 10.1016/j.mcn.2004.02.009. [DOI] [PubMed] [Google Scholar]
- Albin RL, Reiner A, Anderson KD, Dure LS, 4th, Handelin B, Balfour R, Whetsell WO, Jr, Penney JB, Young AB. Preferential loss of striato-external pallidal projection neurons in presymptomatic Huntington's disease. Ann Neurol. 1992;31:425–430. doi: 10.1002/ana.410310412. [DOI] [PubMed] [Google Scholar]
- Andersen JK, Kumar J, Srinivas B, Kaur D, Hsu M, Rajagopalan S. The hunt for a cure for Parkinson's disease. Sci Aging Knowledge Environ. 2001;2001:re1. doi: 10.1126/sageke.2001.1.re1. [DOI] [PubMed] [Google Scholar]
- Baas PW, Karabay A, Qiang L. Microtubules cut and run. Trends Cell Biol. 2005;15:518–524. doi: 10.1016/j.tcb.2005.08.004. [DOI] [PubMed] [Google Scholar]
- Bendotti C, Atzori C, Piva R, Tortarolo M, Strong MJ, DeBiasi S, Migheli A. Activated p38MAPK is a novel component of the intracellular inclusions found in human amyotrophic lateral sclerosis and mutant SOD1 transgenic mice. J Neuropathol Exp Neurol. 2004;63:113–119. doi: 10.1093/jnen/63.2.113. [DOI] [PubMed] [Google Scholar]
- Binder LI, Frankfurter A, Rebhun LI. The distribution of tau in the mammalian central nervous system. J Cell Biol. 1985;101:1371–1378. doi: 10.1083/jcb.101.4.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder LI, Guillozet-Bongaarts AL, Garcia-Sierra F, Berry RW. Tau, tangles, and Alzheimer's disease. Biochim Biophys Acta. 2005;1739:216–223. doi: 10.1016/j.bbadis.2004.08.014. [DOI] [PubMed] [Google Scholar]
- Björkblom B, Vainio JC, Hongisto V, Herdegen T, Courtney MJ, Coffey ET. All JNKs can kill, but nuclear localization is critical for neuronal death. J Biol Chem. 2008;283:19704–19713. doi: 10.1074/jbc.M707744200. [DOI] [PubMed] [Google Scholar]
- Boison D, Stoffel W. Disruption of the compacted myelin sheath of axons of the central nervous system in proteolipid protein-deficient mice. Proc Natl Acad Sci U S A. 1994;91:11709–11713. doi: 10.1073/pnas.91.24.11709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossy-Wetzel E, Schwarzenbacher R, Lipton SA. Molecular pathways to neurodegeneration. Nat Med. 2004;10(Suppl):S2–S9. doi: 10.1038/nm1067. [DOI] [PubMed] [Google Scholar]
- Brady ST, Pfister KK, Leopold PL, Bloom GS. Fast axonal transport in isolated axoplasm. Cell Motil Cytoskeleton. 1990;17(Video Suppl 2):22. doi: 10.1002/cm.970150404. [DOI] [PubMed] [Google Scholar]
- Brill LB, 2nd, Pfister KK. Biochemical and molecular analysis of the mammalian cytoplasmic dynein intermediate chain. Methods. 2000;22:307–316. doi: 10.1006/meth.2000.1083. [DOI] [PubMed] [Google Scholar]
- Brinkman RR, Mezei MM, Theilmann J, Almqvist E, Hayden MR. The likelihood of being affected with Huntington disease by a particular age, for a specific CAG size. Am J Hum Genet. 1997;60:1202–1210. [PMC free article] [PubMed] [Google Scholar]
- Bruijn LI, Houseweart MK, Kato S, Anderson KL, Anderson SD, Ohama E, Reaume AG, Scott RW, Cleveland DW. Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science. 1998;281:1851–1854. doi: 10.1126/science.281.5384.1851. [DOI] [PubMed] [Google Scholar]
- Bruijn LI, Miller TM, Cleveland DW. Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu Rev Neurosci. 2004;27:723–749. doi: 10.1146/annurev.neuro.27.070203.144244. [DOI] [PubMed] [Google Scholar]
- Carmel G, Mager EM, Binder LI, Kuret J. The structural basis of monoclonal antibody Alz50's selectivity for Alzheimer's disease pathology. J Biol Chem. 1996;271:32789–32795. doi: 10.1074/jbc.271.51.32789. [DOI] [PubMed] [Google Scholar]
- Chen XJ, Levedakou EN, Millen KJ, Wollmann RL, Soliven B, Popko B. Proprioceptive sensory neuropathy in mice with a mutation in the cytoplasmic dynein heavy chain 1 gene. J Neurosci. 2007;27:14515–14524. doi: 10.1523/JNEUROSCI.4338-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman M. Axon degeneration mechanisms: commonality amid diversity. Nat Rev Neurosci. 2005;6:889–898. doi: 10.1038/nrn1788. [DOI] [PubMed] [Google Scholar]
- Coleman MP, Perry VH. Axon pathology in neurological disease: a neglected therapeutic target. Trends Neurosci. 2002;25:532–537. doi: 10.1016/s0166-2236(02)02255-5. [DOI] [PubMed] [Google Scholar]
- Collard JF, Côté F, Julien JP. Defective axonal transport in a transgenic mouse model of amyotrophic lateral sclerosis. Nature. 1995;375:61–64. doi: 10.1038/375061a0. [DOI] [PubMed] [Google Scholar]
- Conde C, Cáceres A. Microtubule assembly, organization and dynamics in axons and dendrites. Nat Rev Neurosci. 2009;10:319–332. doi: 10.1038/nrn2631. [DOI] [PubMed] [Google Scholar]
- Conforti L, Adalbert R, Coleman MP. Neuronal death: where does the end begin? Trends Neurosci. 2007;30:159–166. doi: 10.1016/j.tins.2007.02.004. [DOI] [PubMed] [Google Scholar]
- Cookson MR. The biochemistry of Parkinson's disease. Annu Rev Biochem. 2005;74:29–52. doi: 10.1146/annurev.biochem.74.082803.133400. [DOI] [PubMed] [Google Scholar]
- Crosby AH, Proukakis C. Is the transportation highway the right road for hereditary spastic paraplegia? Am J Hum Genet. 2002;71:1009–1016. doi: 10.1086/344206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauer W, Przedborski S. Parkinson's disease: mechanisms and models. Neuron. 2003;39:889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- DeBoer SR, You Y, Szodorai A, Kaminska A, Pigino G, Nwabuisi E, Wang B, Estrada-Hernandez T, Kins S, Brady ST, Morfini G. Conventional kinesin holoenzymes are composed of heavy and light chain homodimers. Biochemistry. 2008;47:4535–4543. doi: 10.1021/bi702445j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delcroix JD, Valletta J, Wu C, Howe CL, Lai CF, Cooper JD, Belichenko PV, Salehi A, Mobley WC. Trafficking the NGF signal: implications for normal and degenerating neurons. Prog Brain Res. 2004;146:3–23. doi: 10.1016/s0079-6123(03)46001-9. [DOI] [PubMed] [Google Scholar]
- De Vos KJ, Chapman AL, Tennant ME, Manser C, Tudor EL, Lau KF, Brownlees J, Ackerley S, Shaw PJ, McLoughlin DM, Shaw CE, Leigh PN, Miller CC, Grierson AJ. Familial amyotrophic lateral sclerosis-linked SOD1 mutants perturb fast axonal transport to reduce axonal mitochondria content. Hum Mol Genet. 2007;16:2720–2728. doi: 10.1093/hmg/ddm226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiProspero NA, Chen EY, Charles V, Plomann M, Kordower JH, Tagle DA. Early changes in Huntington's disease patient brains involve alterations in cytoskeletal and synaptic elements. J Neurocytol. 2004;33:517–533. doi: 10.1007/s11068-004-0514-8. [DOI] [PubMed] [Google Scholar]
- Djaldetti R, Lev N, Melamed E. Neuroprotection in progressive brain disorders. Isr Med Assoc J. 2003;5:576–580. [PubMed] [Google Scholar]
- Donelan MJ, Morfini G, Julyan R, Sommers S, Hays L, Kajio H, Briaud I, Easom RA, Molkentin JD, Brady ST, Rhodes CJ. Ca2+-dependent dephosphorylation of kinesin heavy chain on beta-granules in pancreatic beta-cells. Implications for regulated beta-granule transport and insulin exocytosis. J Biol Chem. 2002;277:24232–24242. doi: 10.1074/jbc.M203345200. [DOI] [PubMed] [Google Scholar]
- Duda JE, Lee VM, Trojanowski JQ. Neuropathology of synuclein aggregates. J Neurosci Res. 2000;61:121–127. doi: 10.1002/1097-4547(20000715)61:2<121::AID-JNR1>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Edgar JM, McLaughlin M, Yool D, Zhang SC, Fowler JH, Montague P, Barrie JA, McCulloch MC, Duncan ID, Garbern J, Nave KA, Griffiths IR. Oligodendroglial modulation of fast axonal transport in a mouse model of hereditary spastic paraplegia. J Cell Biol. 2004;166:121–131. doi: 10.1083/jcb.200312012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar JM, McLaughlin M, Werner HB, McCulloch MC, Barrie JA, Brown A, Faichney AB, Snaidero N, Nave KA, Griffiths IR. Early ultrastructural defects of axons and axon-glia junctions in mice lacking expression of Cnp1. Glia. 2009 doi: 10.1002/glia.20893. in press. [DOI] [PubMed] [Google Scholar]
- Elluru RG, Bloom GS, Brady ST. Fast axonal transport of kinesin in the rat visual system: functionality of the kinesin heavy chain isoforms. Mol Biol Cell. 1995;6:21–40. doi: 10.1091/mbc.6.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrer MJ, Hulihan MM, Kachergus JM, Dächsel JC, Stoessl AJ, Grantier LL, Calne S, Calne DB, Lechevalier B, Chapon F, Tsuboi Y, Yamada T, Gutmann L, Elibol B, Bhatia KP, Wider C, Vilariño-Güell C, Ross OA, Brown LA, Castanedes-Casey M, et al. DCTN1 mutations in Perry syndrome. Nat Genet. 2009;41:163–165. doi: 10.1038/ng.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreirinha F, Quattrini A, Pirozzi M, Valsecchi V, Dina G, Broccoli V, Auricchio A, Piemonte F, Tozzi G, Gaeta L, Casari G, Ballabio A, Rugarli EI. Axonal degeneration in paraplegin-deficient mice is associated with abnormal mitochondria and impairment of axonal transport. J Clin Invest. 2004;113:231–242. doi: 10.1172/JCI20138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink JK. Hereditary spastic paraplegia. Curr Neurol Neurosci Rep. 2006;6:65–76. doi: 10.1007/s11910-996-0011-1. [DOI] [PubMed] [Google Scholar]
- Fischer LR, Glass JD. Axonal degeneration in motor neuron disease. Neurodegener Dis. 2007;4:431–442. doi: 10.1159/000107704. [DOI] [PubMed] [Google Scholar]
- Fischer LR, Culver DG, Tennant P, Davis AA, Wang M, Castellano-Sanchez A, Khan J, Polak MA, Glass JD. Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp Neurol. 2004;185:232–240. doi: 10.1016/j.expneurol.2003.10.004. [DOI] [PubMed] [Google Scholar]
- Gamblin TC, Chen F, Zambrano A, Abraha A, Lagalwar S, Guillozet AL, Lu M, Fu Y, Garcia-Sierra F, LaPointe N, Miller R, Berry RW, Binder LI, Cryns VL. Caspase cleavage of tau: linking amyloid and neurofibrillary tangles in Alzheimer's disease. Proc Natl Acad Sci U S A. 2003;100:10032–10037. doi: 10.1073/pnas.1630428100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbern JY. Pelizaeus-Merzbacher disease: genetic and cellular pathogenesis. Cell Mol Life Sci. 2007;64:50–65. doi: 10.1007/s00018-006-6182-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbern JY, Yool DA, Moore GJ, Wilds IB, Faulk MW, Klugmann M, Nave KA, Sistermans EA, van der Knaap MS, Bird TD, Shy ME, Kamholz JA, Griffiths IR. Patients lacking the major CNS myelin protein, proteolipid protein 1, develop length-dependent axonal degeneration in the absence of demyelination and inflammation. Brain. 2002;125:551–561. doi: 10.1093/brain/awf043. [DOI] [PubMed] [Google Scholar]
- García-Sierra F, Ghoshal N, Quinn B, Berry RW, Binder LI. Conformational changes and truncation of tau protein during tangle evolution in Alzheimer's disease. J Alzheimers Dis. 2003;5:65–77. doi: 10.3233/jad-2003-5201. [DOI] [PubMed] [Google Scholar]
- Gould TW, Buss RR, Vinsant S, Prevette D, Sun W, Knudson CM, Milligan CE, Oppenheim RW. Complete dissociation of motor neuron death from motor dysfunction by Bax deletion in a mouse model of ALS. J Neurosci. 2006;26:8774–8786. doi: 10.1523/JNEUROSCI.2315-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths I, Klugmann M, Anderson T, Yool D, Thomson C, Schwab MH, Schneider A, Zimmermann F, McCulloch M, Nadon N, Nave KA. Axonal swellings and degeneration in mice lacking the major proteolipid of myelin. Science. 1998;280:1610–1613. doi: 10.1126/science.280.5369.1610. [DOI] [PubMed] [Google Scholar]
- Guillozet-Bongaarts AL, Garcia-Sierra F, Reynolds MR, Horowitz PM, Fu Y, Wang T, Cahill ME, Bigio EH, Berry RW, Binder LI. Tau truncation during neurofibrillary tangle evolution in Alzheimer's disease. Neurobiol Aging. 2005;26:1015–1022. doi: 10.1016/j.neurobiolaging.2004.09.019. [DOI] [PubMed] [Google Scholar]
- Gunawardena S, Her LS, Brusch RG, Laymon RA, Niesman IR, Gordesky-Gold B, Sintasath L, Bonini NM, Goldstein LS. Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in Drosophila. Neuron. 2003;40:25–40. doi: 10.1016/s0896-6273(03)00594-4. [DOI] [PubMed] [Google Scholar]
- Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX, Chen W, Zhai P, Sufit RL, Siddique T. Motor neuron degeneration in mice that express human Cu,Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- Hafezparast M, Klocke R, Ruhrberg C, Marquardt A, Ahmad-Annuar A, Bowen S, Lalli G, Witherden AS, Hummerich H, Nicholson S, Morgan PJ, Oozageer R, Priestley JV, Averill S, King VR, Ball S, Peters J, Toda T, Yamamoto A, Hiraoka Y, et al. Mutations in dynein link motor neuron degeneration to defects in retrograde transport. Science. 2003;300:808–812. doi: 10.1126/science.1083129. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Her LS, Goldstein LS. Enhanced sensitivity of striatal neurons to axonal transport defects induced by mutant huntingtin. J Neurosci. 2008;28:13662–13672. doi: 10.1523/JNEUROSCI.4144-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirokawa N, Takemura R. Biochemical and molecular characterization of diseases linked to motor proteins. Trends Biochem Sci. 2003;28:558–565. doi: 10.1016/j.tibs.2003.08.006. [DOI] [PubMed] [Google Scholar]
- Hollenbeck PJ. Kinesin heavy and light chains are phosphorylated in vivo in neurons. J Cell Biol. 1990;115:390a. [Google Scholar]
- Holzbaur EL. Motor neurons rely on motor proteins. Trends Cell Biol. 2004;14:233–240. doi: 10.1016/j.tcb.2004.03.009. [DOI] [PubMed] [Google Scholar]
- Horowitz PM, Patterson KR, Guillozet-Bongaarts AL, Reynolds MR, Carroll CA, Weintraub ST, Bennett DA, Cryns VL, Berry RW, Binder LI. Early N-terminal changes and caspase-6 cleavage of tau in Alzheimer's disease. J Neurosci. 2004;24:7895–7902. doi: 10.1523/JNEUROSCI.1988-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James PA, Talbot K. The molecular genetics of non-ALS motor neuron diseases. Biochim Biophys Acta. 2006;1762:986–1000. doi: 10.1016/j.bbadis.2006.04.003. [DOI] [PubMed] [Google Scholar]
- Jeganathan S, von Bergen M, Brutlach H, Steinhoff HJ, Mandelkow E. Global hairpin folding of tau in solution. Biochemistry. 2006;45:2283–2293. doi: 10.1021/bi0521543. [DOI] [PubMed] [Google Scholar]
- Kaul S, Kanthasamy A, Kitazawa M, Anantharam V, Kanthasamy AG. Caspase-3 dependent proteolytic activation of protein kinase C delta mediates and regulates 1-methyl-4-phenylpyridinium (MPP+)-induced apoptotic cell death in dopaminergic cells: relevance to oxidative stress in dopaminergic degeneration. Eur J Neurosci. 2003;18:1387–1401. doi: 10.1046/j.1460-9568.2003.02864.x. [DOI] [PubMed] [Google Scholar]
- Klein WL, Stine WB, Jr, Teplow DB. Small assemblies of unmodified amyloid beta-protein are the proximate neurotoxin in Alzheimer's disease. Neurobiol Aging. 2004;25:569–580. doi: 10.1016/j.neurobiolaging.2004.02.010. [DOI] [PubMed] [Google Scholar]
- Klugmann M, Schwab MH, Pühlhofer A, Schneider A, Zimmermann F, Griffiths IR, Nave KA. Assembly of CNS myelin in the absence of proteolipid protein. Neuron. 1997;18:59–70. doi: 10.1016/s0896-6273(01)80046-5. [DOI] [PubMed] [Google Scholar]
- Kwiatkowski TJ, Jr, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, Valdmanis P, Rouleau GA, Hosler BA, Cortelli P, de Jong PJ, Yoshinaga Y, Haines JL, Pericak-Vance MA, Yan J, Ticozzi N, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323:1205–1208. doi: 10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- Lagier-Tourenne C, Cleveland DW. Rethinking ALS: the FUS about TDP-43. Cell. 2009;136:1001–1004. doi: 10.1016/j.cell.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landers JE, Melki J, Meininger V, Glass JD, van den Berg LH, van Es MA, Sapp PC, van Vught PW, McKenna-Yasek DM, Blauw HM, Cho TJ, Polak M, Shi L, Wills AM, Broom WJ, Ticozzi N, Silani V, Ozoguz A, Rodriguez-Leyva I, Veldink JH, et al. Reduced expression of the kinesin-associated protein 3 (KIFAP3) gene increases survival in sporadic amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2009;106:9004–9009. doi: 10.1073/pnas.0812937106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapointe NE, Morfini G, Pigino G, Gaisina IN, Kozikowski AP, Binder LI, Brady ST. The amino terminus of tau inhibits kinesin-dependent axonal transport: implications for filament toxicity. J Neurosci Res. 2009;97:440–451. doi: 10.1002/jnr.21850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappe-Siefke C, Goebbels S, Gravel M, Nicksch E, Lee J, Braun PE, Griffiths IR, Nave KA. Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat Genet. 2003;33:366–374. doi: 10.1038/ng1095. [DOI] [PubMed] [Google Scholar]
- Lazarov O, Morfini GA, Pigino G, Gadadhar A, Chen X, Robinson J, Ho H, Brady ST, Sisodia SS. Impairments in fast axonal transport and motor neuron deficits in transgenic mice expressing familial Alzheimer's disease-linked mutant presenilin 1. J Neurosci. 2007;27:7011–7020. doi: 10.1523/JNEUROSCI.4272-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WC, Yoshihara M, Littleton JT. Cytoplasmic aggregates trap polyglutamine-containing proteins and block axonal transport in a Drosophila model of Huntington's disease. Proc Natl Acad Sci U S A. 2004;101:3224–3229. doi: 10.1073/pnas.0400243101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leopold PL, McDowall AW, Pfister KK, Bloom GS, Brady ST. Association of kinesin with characterized membrane-bounded organelles. Cell Motil Cytoskeleton. 1992;23:19–33. doi: 10.1002/cm.970230104. [DOI] [PubMed] [Google Scholar]
- Li H, Li SH, Yu ZX, Shelbourne P, Li XJ. Huntingtin aggregate-associated axonal degeneration is an early pathological event in Huntington's disease mice. J Neurosci. 2001;21:8473–8481. doi: 10.1523/JNEUROSCI.21-21-08473.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li SH, Li XJ. Huntingtin and its role in neuronal degeneration. Neuroscientist. 2004;10:467–475. doi: 10.1177/1073858404266777. [DOI] [PubMed] [Google Scholar]
- Liévens JC, Woodman B, Mahal A, Bates GP. Abnormal phosphorylation of synapsin I predicts a neuronal transmission impairment in the R6/2 Huntington's disease transgenic mice. Mol Cell Neurosci. 2002;20:638–648. doi: 10.1006/mcne.2002.1152. [DOI] [PubMed] [Google Scholar]
- Luo MH, Tse SW, Memmott J, Andreadis A. Novel isoforms of tau that lack the microtubule-binding domain. J Neurochem. 2004;90:340–351. doi: 10.1111/j.1471-4159.2004.02508.x. [DOI] [PubMed] [Google Scholar]
- Mattson MP. Pathways towards and away from Alzheimer's disease. Nature. 2004;430:631–639. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Magnus T. Ageing and neuronal vulnerability. Nat Rev Neurosci. 2006;7:278–294. doi: 10.1038/nrn1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno H, Yu E, Pigino G, Hernandez AI, Kim N, Moreira JE, Sugimori M, Llinás RR. Synaptic transmission block by presynaptic injection of oligomeric amyloid beta. Proc Natl Acad Sci U S A. 2009;106:5901–5906. doi: 10.1073/pnas.0900944106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morfini G, Szebenyi G, Richards B, Brady ST. Regulation of kinesin: implications for neuronal development. Dev Neurosci. 2001;23:364–376. doi: 10.1159/000048720. [DOI] [PubMed] [Google Scholar]
- Morfini G, Szebenyi G, Elluru R, Ratner N, Brady ST. Glycogen synthase kinase 3 phosphorylates kinesin light chains and negatively regulates kinesin-based motility. EMBO J. 2002a;23:281–293. doi: 10.1093/emboj/21.3.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morfini G, Pigino G, Beffert U, Busciglio J, Brady ST. Fast axonal transport misregulation and Alzheimer's disease. Neuromolecular Med. 2002b;2:89–99. doi: 10.1385/NMM:2:2:089. [DOI] [PubMed] [Google Scholar]
- Morfini G, Szebenyi G, Brown H, Pant HC, Pigino G, DeBoer S, Beffert U, Brady ST. A novel CDK5-dependent pathway for regulating GSK3 activity and kinesin-driven motility in neurons. EMBO J. 2004;23:2235–2245. doi: 10.1038/sj.emboj.7600237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morfini G, Pigino G, Brady ST. Polyglutamine expansion diseases: failing to deliver. Trends Mol Med. 2005;11:64–70. doi: 10.1016/j.molmed.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Morfini GA, Stenoien DL, Brady ST. Axonal transport. In: Siegel G, Albers RW, Brady S, Price D, editors. Basic neurochemistry. Ed 7. Burlington, MA: Elsevier Academic; 2006a. pp. 485–502. [Google Scholar]
- Morfini G, Pigino G, Szebenyi G, You Y, Pollema S, Brady ST. JNK mediates pathogenic effects of polyglutamine-expanded androgen receptor on fast axonal transport. Nat Neurosci. 2006b;9:907–916. doi: 10.1038/nn1717. [DOI] [PubMed] [Google Scholar]
- Morfini G, Pigino G, Brady ST. Approaches to kinesin-1 phosphorylation. Methods Mol Biol. 2007a;392:51–69. doi: 10.1007/978-1-59745-490-2_4. [DOI] [PubMed] [Google Scholar]
- Morfini G, Pigino G, Mizuno N, Kikkawa M, Brady ST. Tau binding to microtubules does not directly affect microtubule-based vesicle motility. J Neurosci Res. 2007b;85:2620–2630. doi: 10.1002/jnr.21154. [DOI] [PubMed] [Google Scholar]
- Morfini G, Pigino G, Opalach K, Serulle Y, Moreira JE, Sugimori M, Llinás RR, Brady ST. 1-Methyl-4-phenylpyridinium affects fast axonal transport by activation of caspase and protein kinase C. Proc Natl Acad Sci U S A. 2007c;104:2442–2447. doi: 10.1073/pnas.0611231104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morfini GA, You YM, Pollema SL, Kaminska A, Liu K, Yoshioka K, Björkblom B, Coffey ET, Bagnato C, Han D, Huang CF, Banker G, Pigino G, Brady ST. Pathogenic huntingtin inhibits fast axonal transport by activating JNK3 and phosphorylating kinesin. Nat Neurosci. 2009;12:864–871. doi: 10.1038/nn.2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morsch R, Simon W, Coleman PD. Neurons may live for decades with neurofibrillary tangles. J Neuropathol Exp Neurol. 1999;58:188–197. doi: 10.1097/00005072-199902000-00008. [DOI] [PubMed] [Google Scholar]
- Papasozomenos SC, Binder LI. Phosphorylation determined two distinct species of tau in the central nervous system. Cell Motil Cytoskeleton. 1987;8:210–226. doi: 10.1002/cm.970080303. [DOI] [PubMed] [Google Scholar]
- Pasinelli P, Brown RH. Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat Rev Neurosci. 2006;7:710–723. doi: 10.1038/nrn1971. [DOI] [PubMed] [Google Scholar]
- Pigino G, Pelsman A, Mori H, Busciglio J. Presenilin-1 mutations reduce cytoskeletal association, deregulate neurite growth, and potentiate neuronal dystrophy and tau phosphorylation. J Neurosci. 2001;21:834–842. doi: 10.1523/JNEUROSCI.21-03-00834.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pigino G, Morfini G, Pelsman A, Mattson MP, Brady ST, Busciglio J. Alzheimer's presenilin 1 mutations impair kinesin-based axonal transport. J Neurosci. 2003;23:4499–4508. doi: 10.1523/JNEUROSCI.23-11-04499.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pigino G, Morfini G, Atagi Y, Deshpande A, Yu C, Jungbauer L, LaDu M, Busciglio J, Brady S. Disruption of fast axonal transport is a pathogenic mechanism for intraneuronal amyloid beta. Proc Natl Acad Sci U S A. 2009;106:5907–5912. doi: 10.1073/pnas.0901229106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price DL, Tanzi RE, Borchelt DR, Sisodia SS. Alzheimer's disease: genetic studies and transgenic models. Annu Rev Genet. 1998;32:461–493. doi: 10.1146/annurev.genet.32.1.461. [DOI] [PubMed] [Google Scholar]
- Price DL, Wong PC, Markowska AL, Lee MK, Thinakaren G, Cleveland DW, Sisodia SS, Borchelt DR. The value of transgenic models for the study of neurodegenerative diseases. Ann N Y Acad Sci. 2000;920:179–191. doi: 10.1111/j.1749-6632.2000.tb06920.x. [DOI] [PubMed] [Google Scholar]
- Puls I, Jonnakuty C, LaMonte BH, Holzbaur EL, Tokito M, Mann E, Floeter MK, Bidus K, Drayna D, Oh SJ, Brown RH, Jr, Ludlow CL, Fischbeck KH. Mutant dynactin in motor neuron disease. Nat Genet. 2003;33:455–456. doi: 10.1038/ng1123. [DOI] [PubMed] [Google Scholar]
- Puls I, Oh SJ, Sumner CJ, Wallace KE, Floeter MK, Mann EA, Kennedy WR, Wendelschafer-Crabb G, Vortmeyer A, Powers R, Finnegan K, Holzbaur EL, Fischbeck KH, Ludlow CL. Distal spinal and bulbar muscular atrophy caused by dynactin mutation. Ann Neurol. 2005;57:687–694. doi: 10.1002/ana.20468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid E, Kloos M, Ashley-Koch A, Hughes L, Bevan S, Svenson IK, Graham FL, Gaskell PC, Dearlove A, Pericak-Vance MA, Rubinsztein DC, Marchuk DA. A kinesin heavy chain (KIF5A) mutation in hereditary spastic paraplegia (SPG10) Am J Hum Genet. 2002;71:1189–1194. doi: 10.1086/344210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbluth J, Nave KA, Mierzwa A, Schiff R. Subtle myelin defects in PLP-null mice. Glia. 2006;54:172–182. doi: 10.1002/glia.20370. [DOI] [PubMed] [Google Scholar]
- Roy S, Zhang B, Lee VM, Trojanowski JQ. Axonal transport defects: a common theme in neurodegenerative diseases. Acta Neuropathol. 2005;109:5–13. doi: 10.1007/s00401-004-0952-x. [DOI] [PubMed] [Google Scholar]
- Rutherford NJ, Zhang YJ, Baker M, Gass JM, Finch NA, Xu YF, Stewart H, Kelley BJ, Kuntz K, Crook RJ, Sreedharan J, Vance C, Sorenson E, Lippa C, Bigio EH, Geschwind DH, Knopman DS, Mitsumoto H, Petersen RC, Cashman NR, et al. Novel mutations in TARDBP (TDP-43) in patients with familial amyotrophic lateral sclerosis. PLoS Genet. 2008;4:e1000193. doi: 10.1371/journal.pgen.1000193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salinas S, Proukakis C, Crosby A, Warner TT. Hereditary spastic paraplegia: clinical features and pathogenetic mechanisms. Lancet Neurol. 2008;7:1127–1138. doi: 10.1016/S1474-4422(08)70258-8. [DOI] [PubMed] [Google Scholar]
- Saugier-Veber P, Munnich A, Bonneau D, Rozet JM, Le Merrer M, Gil R, Boespflug-Tanguy O. X-linked spastic paraplegia and Pelizaeus-Merzbacher disease are allelic disorders at the proteolipid protein locus. Nat Genet. 1994;6:257–262. doi: 10.1038/ng0394-257. [DOI] [PubMed] [Google Scholar]
- Serulle Y, Morfini G, Pigino G, Moreira JE, Sugimori M, Brady ST, Llinás RR. 1-Methyl-4-phenylpyridinium induces synaptic dysfunction through a pathway involving caspase and PKC{delta} enzymatic activities. Proc Natl Acad Sci U S A. 2007;104:2437–2441. doi: 10.1073/pnas.0611227104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieradzan KA, Mann DM. The selective vulnerability of nerve cells in Huntington's disease. Neuropathol Appl Neurobiol. 2001;27:1–21. doi: 10.1046/j.0305-1846.2001.00299.x. [DOI] [PubMed] [Google Scholar]
- Solowska JM, Morfini G, Falnikar A, Himes BT, Brady ST, Huang D, Baas PW. Quantitative and functional analyses of spastin in the nervous system: implications for hereditary spastic paraplegia. J Neurosci. 2008;28:2147–2157. doi: 10.1523/JNEUROSCI.3159-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini MG, Goedert M. The alpha-synucleinopathies: Parkinson's disease, dementia with Lewy bodies, and multiple system atrophy. Ann N Y Acad Sci. 2000;920:16–27. doi: 10.1111/j.1749-6632.2000.tb06900.x. [DOI] [PubMed] [Google Scholar]
- Stenoien DL, Brady ST. Immunochemical analysis of kinesin light chain function. Mol Biol Cell. 1997;8:675–689. doi: 10.1091/mbc.8.4.675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugars KL, Rubinsztein DC. Transcriptional abnormalities in Huntington disease. Trends Genet. 2003;19:233–238. doi: 10.1016/S0168-9525(03)00074-X. [DOI] [PubMed] [Google Scholar]
- Susalka SJ, Pfister KK. Cytoplasmic dynein subunit heterogeneity: implications for axonal transport. J Neurocytol. 2000;29:819–829. doi: 10.1023/a:1010995408343. [DOI] [PubMed] [Google Scholar]
- Szebenyi G, Morfini GA, Babcock A, Gould M, Selkoe K, Stenoien DL, Young M, Faber PW, MacDonald ME, McPhaul MJ, Brady ST. Neuropathogenic forms of huntingtin and androgen receptor inhibit fast axonal transport. Neuron. 2003;40:41–52. doi: 10.1016/s0896-6273(03)00569-5. [DOI] [PubMed] [Google Scholar]
- Tarrade A, Fassier C, Courageot S, Charvin D, Vitte J, Peris L, Thorel A, Mouisel E, Fonknechten N, Roblot N, Seilhean D, Diérich A, Hauw JJ, Melki J. A mutation of spastin is responsible for swellings and impairment of transport in a region of axon characterized by changes in microtubule composition. Hum Mol Genet. 2006;15:3544–3558. doi: 10.1093/hmg/ddl431. [DOI] [PubMed] [Google Scholar]
- Tortarolo M, Veglianese P, Calvaresi N, Botturi A, Rossi C, Giorgini A, Migheli A, Bendotti C. Persistent activation of p38 mitogen-activated protein kinase in a mouse model of familial amyotrophic lateral sclerosis correlates with disease progression. Mol Cell Neurosci. 2003;23:180–192. doi: 10.1016/s1044-7431(03)00022-8. [DOI] [PubMed] [Google Scholar]
- Turmel H, Hartmann A, Parain K, Douhou A, Srinivasan A, Agid Y, Hirsch EC. Caspase-3 activation in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-treated mice. Mov Disord. 2001;16:185–189. doi: 10.1002/mds.1037. [DOI] [PubMed] [Google Scholar]
- Viswanath V, Wu Y, Boonplueang R, Chen S, Stevenson FF, Yantiri F, Yang L, Beal MF, Andersen JK. Caspase-9 activation results in downstream caspase-8 activation and bid cleavage in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinson's disease. J Neurosci. 2001;21:9519–9528. doi: 10.1523/JNEUROSCI.21-24-09519.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagey RT, Krieger C. Abnormalities of protein kinases in neurodegenerative diseases. Prog Drug Res. 1998;51:133–183. doi: 10.1007/978-3-0348-8845-5_4. [DOI] [PubMed] [Google Scholar]
- Wagner MC, Pfister KK, Bloom GS, Brady ST. Copurification of kinesin polypeptides with microtubule-stimulated Mg-ATPase activity and kinetic analysis of enzymatic processes. Cell Motil Cytoskeleton. 1989;12:195–215. doi: 10.1002/cm.970120403. [DOI] [PubMed] [Google Scholar]
- Waldmeier P, Bozyczko-Coyne D, Williams M, Vaught JL. Recent clinical failures in Parkinson's disease with apoptosis inhibitors underline the need for a paradigm shift in drug discovery for neurodegenerative diseases. Biochem Pharmacol. 2006;72:1197–1206. doi: 10.1016/j.bcp.2006.06.031. [DOI] [PubMed] [Google Scholar]
- Wang JZ, Grundke-Iqbal I, Iqbal K. Kinases and phosphatases and tau sites involved in Alzheimer neurofibrillary degeneration. Eur J Neurosci. 2007;25:59–68. doi: 10.1111/j.1460-9568.2006.05226.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warita H, Itoyama Y, Abe K. Selective impairment of fast anterograde axonal transport in the peripheral nerves of asymptomatic transgenic mice with a G93A mutant SOD1 gene. Brain Res. 1999;819:120–131. doi: 10.1016/s0006-8993(98)01351-1. [DOI] [PubMed] [Google Scholar]
- Williamson TL, Cleveland DW. Slowing of axonal transport is a very early event in the toxicity of ALS-linked SOD1 mutants to motor neurons. Nat Neurosci. 1999;2:50–56. doi: 10.1038/4553. [DOI] [PubMed] [Google Scholar]
- Wishart TM, Parson SH, Gillingwater TH. Synaptic vulnerability in neurodegenerative disease. J Neuropathol Exp Neurol. 2006;65:733–739. doi: 10.1097/01.jnen.0000228202.35163.c4. [DOI] [PubMed] [Google Scholar]
- Witt A, Brady ST. Unwrapping new layers of complexity in axon/glial relationships. Glia. 2000;29:112–117. doi: 10.1002/(sici)1098-1136(20000115)29:2<112::aid-glia3>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Wong PC, Cai H, Borchelt DR, Price DL. Genetically engineered mouse models of neurodegenerative diseases. Nat Neurosci. 2002;5:633–639. doi: 10.1038/nn0702-633. [DOI] [PubMed] [Google Scholar]
- Zhang F, Ström AL, Fukada K, Lee S, Hayward LJ, Zhu H. Interaction between familial amyotrophic lateral sclerosis (ALS)-linked SOD1 mutants and the dynein complex. J Biol Chem. 2007;282:16691–16699. doi: 10.1074/jbc.M609743200. [DOI] [PubMed] [Google Scholar]