

Abstract
The ability to simultaneously quantify multiple signaling molecule protein levels from microscopic neural tissue samples would be of great benefit to deciphering how they affect brain function. This follows from evidence that indicates signaling molecules can be pleiotropic and can have complex interactive behavior that is regionally and cellularly heterogeneous. Multiplexed examination of tissue proteins has been exceedingly difficult because of the absence of available techniques. This void now has been removed by the commercial availability of bead-based immunoassays for targeted proteins that allow analyses of up to 100 (6–150 kDa) proteins from as little as 12 μl. Thus far used only for sera (human and mouse) and culture media, we demonstrate here that sensitive (as low as 2 pg/ml), wide-ranging (up to 2–32 000 pg/ml), accurate (8% intra-assay covariance) and reliable (4–7% inter-assay covariance) measurements can be made of nine exemplary cytokines (e.g., IL-1α, IL-1β, IL-2, IL-4, IL-6, IL-10, GM-CSF, IFN-γ, TNF-α) simultaneously not only from rat serum but, for the first time, also brain tissue. Furthermore, we describe animal handling procedures that minimize stress as determined by serum glucocorticoid levels since they can influence cytokine expression.
Keywords: Cytokines, Protein, Corticosterone, Kainic acid, Seizure, Epilepsy, Luminex™, Bio-Plex™
1. Introduction
Simultaneous measurement of multiple proteins can provide improved insight to brain function because of the inherent capacity for this experimental strategy to resolve complex interactions among signaling molecules (del Zoppo et al., 2000). Furthermore, application of such experimental strategies to brain should ideally be feasible on samples of exceedingly small size. This conclusion follows from the notion that brain shows marked structural and functional heterogeneity that in part may result from cell-specific (orregion-specific) production, expression, and subsequent effects of various proteins including ligands and ligand receptors. Interleukin-1 (IL-1) function within brain is a prime example of such complex interaction and potential local specificity.
IL-1 is a prototypic inflammatory cytokine with widespread impact on neural function in health (Lynch, 2002; Schneider et al., 1998) and disease (Patel et al., 2003; Rothwell and Luheshi, 2000) that is produced both systemically (Dinarello, 1996) and centrally (Breder et al., 1988). The effects of IL-1 (including those in brain) are sometimes inferred from the behavior of related mRNA species (Dinarello, 1996) since cytokines are typically produced upon need (Oppenheim and Feldmann, 2001). However, newly synthesized IL-1β mRNA is not always transcribed to protein, making measurement of protein change more critical for deciphering function of this inflammatory mediator (Dinarello, 1996). Furthermore, IL-1 family cytokines consist of six principle, related family members, i.e., three receptor ligands (IL-1α, IL-1β and IL-1 receptor antagonist (IL-1Ra)), two receptor subtypes (IL-1RI and IL-1RII) and an accessory protein (IL-1AcP) (Dinarello, 1996). These IL-1 family inflammatory mediators show cell-specific patterns of production, expression and release since ligands are made principally by glia (and some neurons) while astrocytes and neurons express the signal transducing IL-1 receptor, IL-1RI (Ban et al., 1991, 1993; Fontana et al., 1982; Giulian et al., 1986; Lechan et al., 1990). In addition, neurons (and glia) can be differentially distributed between and within various brain regions (Dombrowski et al., 2001). Thus to most accurately characterize the net effects of IL-1 on brain function, not only should simultaneous measurements be made of six key IL-1 family cytokine proteins, but they should be made from sample volumes small enough to account for potential regional variations of the related variables.
Other evidences of the potential for complex interactions among signaling molecules in brain exist. For example, injury from brain disease can result in temporally and spatially distinct expression patterns of specific cytokines (Jankowsky and Patterson, 1999; Jean Harry et al., 2003), which can influence their own expression as well as that of counterparts (Oppenheim and Feldmann, 2001). Additionally, other classes of signaling molecules such as glucocorticoids (Dinkel et al., 2003; Goujon et al., 1997; John and Buckingham, 2003; Nadeau and Rivest, 2003) and prostaglandins (del Zoppo et al., 2000; Hori et al., 2000; Vane et al., 1998) can either reduce or promote inflammatory cytokine responses, respectively. Finally, similar scenarios of spatially and temporally distinct, yet interactive and thus complex, behavior can be expected for other intracellular, autocrine, paracrine and endocrine signaling molecule systems. However, because of the prior absence of easily available experimental techniques, few studies have centered on experimental strategies that included simultaneous measurements of multiple signaling molecules.
Recent availability of commercial instruments based on bead-based immunoassay technology (Dasso et al., 2002; de Jager et al., 2003; Dunbar et al., 2003; Earley et al., 2002; Kellar and Iannone, 2002; Kellar et al., 2001; Prabhakar et al., 2002; Vignali, 2000) is likely to help remove this void. For example, the Bio-Plex™ Suspension Array system (Bio-Rad; Hercules, CA) is an easy-to-use and flexible unit capable of simultaneously analyzing up to 100 (6–150 kDa) proteins from as little as 12 μl of sample, which has been done for analyses of serum (de Jager et al., 2003). The system is based on using spectrally addressed 5.5 μm diameter polystyrene beads that serve as the solid phase for this capture sandwich assay. For protein analyses, a monoclonal antibody directed against a desired analyte is covalently coupled to the dyed beads. The conjugated beads are then allowed to react with the sample containing an unknown amount of targeted protein (or a standard solution containing a known amount of the same protein—for calibration as well as positive controls). Next, a biotinylated antibody that is specific for the protein of interest is added to the reaction. This results in the formation of a sandwich of antibodies around the targeted protein. Then, the reaction mixture is detected by the addition of streptavidin–phycoerythrin, which binds to the biotinylated detection antibodies. Finally, the reaction mixture is read using specially designed hardware and software. To date however, measurements have only been reported for sera and culture media using bead-based immunoassays.
Here we present experimental strategies for animal preparation that minimize spurious activation of inflammatory cytokine expression from stress. Furthermore, we detail protocols for easy-to-use methods that confirm the utility of bead-based immunoassays to provide sensitive, accurate and reproducible measurement of nine exemplary cytokine proteins from rat serum. Importantly these methods, for the first time, are extended to similar measurements from brain tissue.
2. Materials and methods
Catalog numbers are given where specific products are essential.
2.1. Animals
Adult (250–300 g) male Wistar rats (Charles River Laboratories, Portage, MI) were housed two animals per cage with a 7.6 cm diameter PVC curved tube (38 cm long) for nesting in a 12:12 light/dark cycle with ad libitum access to food and water. Forty animals were used for studies here (20 for corticosterone measurements (two groups of 10 animals); 10 for cytokine assays (two groups of 5 animals); and 10 for seizure induced cytokine measurements (five sham controls and five experimental animals). All experiments were performed within the guidelines described in the US Public Health Service Policy on Humane Care and Use of Laboratory Animals.
2.2. Serum harvest and preparation
Initially, blood was obtained from rats that were individually transferred from their housing quarters to the harvesting lab. There they were weighed and subsequently anesthetized, initially with inhalational halothane (5% mixed with air), then anesthesia was maintained with intraperitoneal pentobarbital (50 mg/kg) before decapitation and collection of blood and brain samples. However, this produced an unacceptably wide variance in serum corticosterone levels (see Section 3) probably due to the variable time and stress associated with sample collection. Thus, we developed a group-collection paradigm wherein all animals from a given cage were simultaneously anesthetized and then held under anesthetized conditions before subsequent, rapid tissue harvest. We anticipated that such a collection paradigm would be particularly useful when harvesting serum and tissue from animals exposed to environmental enrichment paradigms (Kraig et al., 2003) where groups of animals are housed together.
The group-anesthesia and collection strategy was performed as follows. First, a single animal cage (containing two animals and measuring (l, w, h) 45cm×25 cm×20 cm; i.e., 171 volume) was covered with an opaque drop cloth and hand-carried to the harvesting lab (which was separate from the room used for housing). There, animals were anesthetized with inhalational halothane (~10%; Linde, 1971). The latter was accomplished by placing 10 ml of halothane in a wire-covered pan (10 cm × 8cm × 6 cm) containing two paper towels to enhance the rate of halothane evaporation and quickly placing this assembly into the original housing cage. Next, a solid cover was placed over the cage to prevent vapor leakage. After animals became motionless (within 2 min), they were quickly transferred to a holding chamber maintained at 3% halothane mixed with air using a vaporizer. Individual animals were withdrawn from the holding chamber, decapitated, and truncal blood collected. Heads were placed on ice for subsequent harvesting of brains after both animals per cage were sacrificed. Collection of blood took less than 5 min and collection of brain tissue samples less than 10 min per cage. All blood and tissue harvesting started at 10 a.m. to account for the diurnal nature of corticosterone expression.
Serum used for corticosterone and cytokine analyses was prepared by collecting blood into a Microtainer® Brand Serum Seperator Tube (#365956; Beckton Dickinson; Franklin Lakes, NJ) and allowing it to clot for 10 min at room temperature. Then, it was centrifuged (12 000 rpm) for 5 min at room temperature. Serum was collected and stored at −80 °C. For filtered serum samples, serum was first diluted (e.g., at 1:4, 1:8; 1:16) in sample diluent (Bio-Plex™ Diluent Kit #171-305-008; Bio-Rad) and placed in low protein binding Ultrafree® MC-centrifugal filter device (#UFC30GVNB; Millipore Corporation; Billerica, MA) and centrifuged for 5 min (3000 rpm) at room temperature.
2.3. Hippocampal tissue harvest and preparation
Brains were removed and placed in cold (2–4 °C) phosphate (10 mM) buffered (pH 7.4) saline (PBS). There, left and right hippocampal were isolated using a stereomicroscope. Hippocampal samples were frozen on dry ice and stored at −80 °C.
For analyses, whole hippocampal samples were thawed in 1.5 ml of cell lysis buffer (from The Cell Lysis Kit (#171-304012) Bio-Rad; Hercules, CA; see Table 1) containing protease inhibitor cocktail (#171-304012; Bio-Rad) and 3 μl of a stock solution containing 500 mM phenylmethylsulfonyl fluoride (#P-7626) in dimethyl sulphoxide (#D2650, both from Sigma, St. Louis, MO). Then, samples were processed by either of two procedures. Tissue was disrupted by drawing samples up and down through a 1 ml pipette tip (cut back to a 2 mm opening) 20 times followed by orbital agitation for 20 min at 300 rpm and 4 °C. Alternatively, tissue samples were homogenized with an ultrasonic dismembrator (model 100; Fisher Scientific, Pittsburgh, PA) at 4 °C using three rapid pulses (<1 s each). Samples derived from both methods of homogenation were then centrifuged at 4500×g for 15 min at 4 °C, supernatants collected and stored at −80 °C.
Table 1.
Bio-Plex™ Protein Array System kit components
| Cytokine assay kit | Cytokine reagent kit | Cell lysis kit | Diluent kit (Rat) |
|---|---|---|---|
| Anti-cytokine conjugated beads | Assay buffer | Cell lysis buffer | Sample diluent |
| Cytokine detection antibody | Cell wash buffer | Factor 1 | Standard diluent |
| Cytokine standards | Detection antibody diluent | Factor 2 | |
| Streptavidin-PE | |||
| Sterile 96-well filter plate | |||
| Sealing tape |
Total protein concentration was determined using a DC Protein Assay Kit (#500-0113; Bio-Rad) and spectrophotometer (SmartSpec 3000™; #171-2501; Bio-Rad). All tissue samples were diluted with cell lysis buffer as needed to a final total protein concentration of 500 μg/ml.
2.4. Corticosterone measurements
Serum corticosterone concentrations were measured by ELISA (catalog #026-AF-14F1; American Laboratory Products; Windhamn, NH) following the manufacturer’s instructions. Samples were read using a Bio-Tek EL800 microplate reader that utilized a four-parameter logistic regression technique (Bio-Tek Instruments, Winooski, VT).
2.5. Cytokine measurements
Sera and tissue homogenates were assayed for cytokines using single (IL-1β; #X80000003N) and multiplexed (IL-1α, IL-1β, IL-2, IL-4, IL-6, IL-10, GM-CSF, IFN-γ, and TNF-α; #171-K11070) bead-based immunoassay kits (Bio-Rad) combined with a Cytokine Reagent Kit (#171-304000; Bio-Rad) and a Bio-Plex™ Diluent Kit for serum or a Cell Lysis Kit for tissue. Specific components of each kit are listed in Table 1. Cytokine assay plate layout was stereotypic and consisted of: (1) eight standards in duplicate; (2) three positive controls in triplicate (see below); (3) two blank wells (for background fluorescence subtraction); and (4) up to 69 wells for 23 samples in triplicate.
Cytokine assays were performed as follows. First, lyophilized cytokine standards (Cytokine Assay Kit) were reconstituted to a stock concentration of 500 000 pg/ml in cell lysis buffer (Cell Lysis Kit) for tissue assays or sample diluent (Diluent Kit) for serum assays. Furthermore, standards and positive controls (see below) were diluted to chosen concentrations in these same solutions, respectively. Second, 5.5 μm polystyrene beads (conjugated to specific primary capture antibodies (Cytokine Assay Kit)) were pelleted on a desktop centrifuge for 1 min at 300 rpm; sonicated in a water bath for 1 min; and then vortexed vigorously for 30 s to ensure mono dispersion. Beads were then diluted from a 100× stock to 1× working solution using assay buffer (Cytokine Reagent Kit). Then, the latter was immediately added (50 μl per well) to wells of a 96-well multiscreen plate (provided with Cytokine Reagent Kit; or #MABVN1210; MultiScreen®-BV Clear Plates; Millipore Corporation, Billerica, MA). Fluid was uniformly removed (after incubation) from each well using a MultiScreen Resist Vacuum Manifold (#MAVM0960R: Millipore) at constant pressure (~63 mmHg). Wells were washed twice with cell wash buffer (Cytokine Reagent Kit; 100 μl/ea.). Third, 50 μl each of standards (Cytokine Assay Kit), positive controls (Cytokine Assay Kit) and samples were then incubated with beads for 90 min (tissue samples) or 30 min (serum samples) at room temperature with orbital shaking (model MTS-4 shaker; IKA-Works, Inc., Wilmington, NC) at 1100 rpm for 1 min and then 300 rpm for the remainder of the incubation period. Blank wells consisted of 50 μl of either sample diluent (Diluent Kit) or cell lysis buffer (Cell Lysis Kit) for serum and tissue, respectively. A series of three predefined standard concentrations (i.e., positive control samples) were incorporated as measures of demonstrating assay standard curve accuracy. Materials for the positive controls were obtained from the Cytokine Assay Kit. The latter consisted of cytokines at 4000, 800 and 100 pg/ml that were created by either diluting in sample diluent (serum assay) or Cell Lysis Buffer (brain tissue assay)—a range that generally encompassed the concentrations of all nine analytes expected for serum and brain tissue. After incubation, fluid was removed again as defined above. Fourth, wells were washed (100 μl/ea.) three times with cell wash buffer using a multi-channel pipette (#EDP3-Plus™; Rainin, Oakland, CA). Such multiple wash steps were included at appropriate assay steps (see below) to reduce background noise (Prabhakar et al., 2002). Fifth, secondary reporter antibodies (Cytokine Assay Kit) were then added to all wells. Here, a 25× stock solution was diluted to 1× using detection antibody diluent (Cytokine Reagent Kit) and 25 μl added to each well followed by incubation for 30 min (tissue and serum) at room temperature. Again as previously done, shaking began at 1100 rpm for 1 min followed by constant shaking at 300 rpm. Fluid again was removed at constant, low pressure, and wells washed (100 μl/ea.) three times with cell wash buffer. Sixth, the fluorophore containing Streptavidin-PE complex (Cytokine Reagent Kit) was prepared by diluting a 100× stock to 1× with assay buffer and 50 μl added to each well for 10 min at room temperature and shaking as noted above. Seventh, the 96-well plate was washed (100 μl per well) three times with cell wash buffer using vacuum removal after each wash. Finally, assay buffer was added (125 μl per well) to resuspend beads.
At all shaking points noted above, 96-well assay plates were covered with sealing tape (Cytokine Reagent Kit) and aluminum foil (to retard photobleaching). In addition after each wash step, the underside of the plate was blotted with a paper towel.
After the above procedures, standards and samples were immediately processed using the Bio-Plex™ Protein Array System and related Bio-Plex Manager™ (Version 3) software (Bio-Rad). Samples and positive controls exhibiting a coefficient of variation (CV) >10% were omitted from final data analyses (Kellar and Iannone, 2002). Furthermore, all signals were normalized by inclusion of fluorescence from duplicate blank wells that consisted initially of only Cell Lysis Buffer (50 μl per well) for tissue and sample diluent (50 μl per well) for serum. Each well was sampled using a volume of 50 μl and a bead count of 100 per analyte before advancing to the next well. Both capture and reporter antibodies were chosen by the manufacturer to minimize potential cross-reactivity (Song et al., 2000). In addition to the normal start-up procedures (i.e., a series of fluidic functions) suggested by the manufacturer, we completed three wash cycles (using deionized water to clean tubing) and one priming cycle with sheath fluid (#171-000055; Bio-Rad) to prepare for bead analyses before processing experimental plates.
2.6. Cytokine data analysis and presentation
Cytokine data were analyzed using Bio-Plex Manager™ 3.0 software (Bio-Rad). Several models were available to create standard curves (e.g. point-to-point (semi-log), linear (semi-log), point-to-point, cubic spline, linear, logistic 4PL, and 5PL). A five-parameter logistic regression model (5PL) with weighting was used (when six or more standards were found to be accurate (i.e., within 70–130% of expected value)) to create standard curves because it gives the greatest dynamic range for each standard curve. On rare occasions when less than six standards were accurate, a 4PL model was used. The 5PL model follows a five-parameter logistic regression as illustrated by Eq. (1) (Baud, 1993)
| (1) |
where a is the response at zero concentration, b the slope, c the midpoint or concentration at the midrange, d the response at maximal or infinite concentration, and m the asymmetry factor. Automatic weighting of points on the standard curve were performed for the 5PL regression. Eight weighting models (linear variance or coefficient of variance, logarithmic variance or coefficient of variance, exponential variance or coefficient of variance, and power law variance or coefficient of variance) are available to the user. These models utilize two coefficients (A and B) that are defined by the software or user. Coefficient A was automatically calculated using the manufacturer’s settings. Coefficient B was equal to 1.8, another recommendation of the manufacturer. The power law variance model of weighting with these two coefficients was used for all serum and tissue assays since it was best matched to the standard curves by the software.
This regression model allows for better handling of asymmetry (a common cause of poor fit for four-parameter logistic regression (Baud, 1993)), poor replicates, and outliers (e.g., by use of weighting) in the standard curves (Nix and Wild, 2001). “Goodness of fit” for all standard curves was assessed by two methods (Davies, 2001; Nix and Wild, 2001). The first consisted of standard value back-calculation (defined as (observed/expected) × 100). Calculated values within 70–130% of expected levels were accepted. The second method of curve evaluation consisted of determining the recovery level of positive controls. Positive control recovery values (also defined as (observed/expected)×100) assessed the assay’s overall calibration (Davies, 2001). If they fell between 80 and 120% of expected values, they were accepted as appropriate by the manufacturer as suggested by Davies (2001). The functional sensitivity for assays was considered as the lowest concentration achieved in the assay whose coefficient of variation (CV) was ≤20% (Davies, 2001).
Standards, positive controls and serum samples were expressed as pg/ml. However, tissue sample values in pg/ml values were multiplied by 2 for expression as pg/mg of total tissue protein since tissue samples originally consisted of total protein aliquots adjusted to 500 μg of total protein per ml. To emphasize this latter point and ease comparison to serum or culture media data we express tissue values as both pg/ml and pg/mg total protein throughout the paper.
2.7. Kainic acid-induced seizures
Seizure activity raises brain tissue levels of various cytokines (Jankowsky and Patterson, 1999; Vezzani et al., 2002). Accordingly, we used a well-established animal model of temporal lobe epilepsy (TLE) (Avanzini et al., 1998) to illustrate the feasibility and utility of multiplexed immunoassays to detect cytokine changes from seizures or related injury in hippocampal tissue. TLE was induced by an intraperitoneal injection of kainic acid (10 mg/kg (Sigma) in PBS at 5 mg/ml). Sham controls received similar volume intraperitoneal injections of PBS. For intraperitoneal injections, all animals were briefly anesthetized with inhalational halothane (5% mixed with air) in a closed vessel to minimize potential restraint stress.
Seizure severity was quantitated for the next 5 h according to a scale proposed by Racine (1972) and modified here to include death: 0, normal behavior; 1, immobility; 2, forelimb and/or tail extension, rigid posture; 3, repetitive movements, head bobbing; 4, rearing and falling; 5, continuous rearing and falling; 6, tonic–clonic seizures; 7, death. Animals were monitored continuously for 5 h and given a score every 20 min that represented the highest seizure severity for that period. The maximal score each hour was used to indicate seizure severity. In the event that animals experienced continuous seizures (i.e., levels 5 or 6) for 20 min, a rescue dose of intraperitoneal sodium pentobarbital (30 mg/kg) was available for injection. If the latter treatment was not effective in stopping seizures after another 20 min, animals would be removed from the study and euthanized via inhalational halothane overdose (Araki et al., 2002; Zhang et al., 2002). No animals required these latter interventions.
2.8. Statistics
SigmaStat 2.0 (SPSS, Chicago, IL) was used for descriptive statistics, t-test and analysis of variance (ANOVA) with post-hoc testing. All data was plotted using SigmaPlot 8.0 (SPSS, Chicago, IL). Data are listed as mean±standard error of the mean and rounded to whole digits. Thus, standard error of means less than 0.5 are listed as “0”.
3. Results
3.1. Serum corticosterone
Corticosterone can influence expression of cytokines with brain (Dinkel et al., 2003; Goujon et al., 1997; John and Buckingham, 2003; Nadeau and Rivest, 2003) and so can be expected to be a sensitive and general reflection of animal status when serum and tissues are harvested for cytokine analyses. We initially used a standard strategy for processing brain tissue for immunohistochemical analyses of inflammatory markers (Caggiano and Kraig, 1996, 1998; Caggiano et al., 1996). However in these preliminary studies, animals from a modestly enriched environment had corticosterone levels of 150 ± 25 ng/ml (n = 9), values that might be more consistent with stress (Diamond et al., 1992) than with environmental enrichment (Kempermann et al., 2002; Kraig et al., 2003) (Fig. 1). As a result, we changed our harvest procedures to a group-anesthesia format that would be conducive for use with animals housed as a group in typical environmental enrichment paradigms (Kempermann et al., 2002; Kraig et al., 2003). Animals harvested using this protocol had serum corticosterone levels of 62 ± 4 ng/ml (n = 9), values that while elevated from normal are consistent with environmental enrichment (Kempermann et al., 2002; Kraig et al., 2003). This is more than a two-fold and significant (P < 0.007, Mann–Whitney rank sum) decrease in mean corticosterone level and nearly a six-fold drop in variance. Thus, the group anesthesia strategy used here for measurement of serum corticosterone (and thus most likely brain cytokines) would be derived from unstressed animals.
Fig. 1.
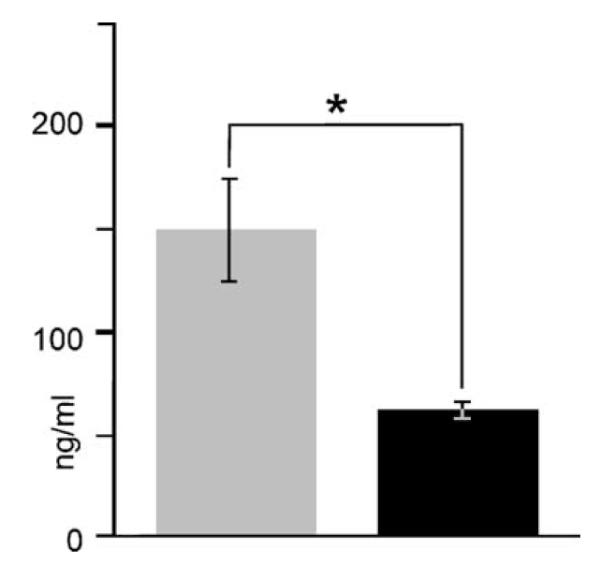
Reduction in serum corticosterone to normal range by use of a group-anesthesia paradigm. Initially, animals were harvested individually as might be done for standard processing of brain for immunohistochemical studies of brain inflammation. However, this procedure triggered a markedly elevated serum corticosterone level (gray bar) of 150±25 ng/ml with a wide variance (n = 9). Since serum corticosterone can influence expression of cytokines, we established a method for harvesting serum and brain tissue that included a serum corticosterone level (black bar) that was within a normal range (62 ± 4 ng/ml; n = 9) for enriched animals. This is more than a two-fold and significant (P < 0.007, Mann–Whitney rank sum) drop in mean corticosterone level and nearly a six-fold drop in variance. It consisted of anesthetizing all animals within a cage as a group and then holding them under anesthesia as each animal was sequentially processed for collection of truncal blood and brain tissue.
3.2. Immunoassays
3.2.1. Single analyte measurements
Our next goal was to define an optimal measurement strategy for serum and tissue cytokines. Here, we found that serum and tissue processing could influence not only the levels of cytokine measured but also the total protein recovered for tissues.
For example, the effect of filtering plus diluting and only diluting serum on the magnitude of IL-1β detection is shown in the left hand and the right hand histograms in Fig. 2A, respectively. Diluting serum (i.e., histograms to left in (A)) at 1:1 (black), 1:4 (gray), 1:8 (white), 1:16 (hatched lines) (n= 3 per dilution) increased the levels of IL-1β measured by bead-based immunoassay at all dilutions. Furthermore, filtering (n = 4) serum increased the amount of IL-1β detected at all dilutions compared to non-filtered counterparts (right hand histograms in A), significantly (*P < 0.001; Student’s t test). Finally, the impact of dilution was also most significant at 1:8 (*P < 0.004–0.001; ANOVA with post-hoc Tukey). Finally, the mean intra assay CV involving the above measurements was 8% for standards and 2% for filtered and diluted samples. The CV for non-filtered and diluted samples was 6%. The mean inter-assay CV (n = 2 plates) for standards and samples was 5 and 13%, respectively.
Fig. 2.
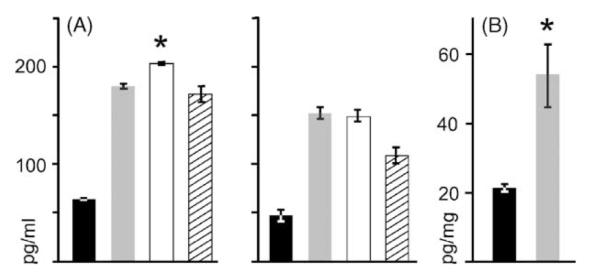
Optimization of serum and tissue sample preparation for bead-based immunoassays. The effect of filtering and diluting serum on magnitude of IL-1β detection is shown in left hand and right hand histograms in (A). Affect of tissue preparation on IL-1β detected is shown in histogram in (B). Diluting serum (i.e., histograms to left in (A)) at 1:1 (black), 1:4 (gray), 1:8 (white), 1:16 (hatched lines) (n = 3 per dilution) increased the levels of IL-1β measured by bead-based immunoassay at all dilutions. Furthermore, filtering (n = 4) serum increased amount of IL-1β detected at all dilutions compared to non-filtered counterparts (right hand histograms in A), significantly (*P < 0.001). Finally, the impact of dilution was also most significant at 1:8 (*P < 0.004–0.001). Brain tissue (B) was either homogenized manually (black) or with an ultrasonic disruptor (gray). Use of the latter prompted a significantly greater (P < 0.003) recovery of IL-1β from matched tissue homogenate samples.
Preparation of tissue for IL-1β analyses influenced both the amount of total protein recovered and the level of IL-1β measured from normalized tissue lysate samples. Sonication of brain samples resulted on average in a two-fold greater recovery of total protein compared to that seen with manual disruption of tissues (both sample varieties at analogous initial wet weights). Furthermore, disruption of tissue samples by sonication was associated with a significantly (P < 0.003; Student’s t-test) greater mean level of measured IL-1β compared to manually disrupted brain tissue (Fig. 2B). Finally, the mean intra-assay CV involving the above measurements was 8% for standards 12% for experimental samples. The mean inter-assay CV (n = 2 plates) for standards and experimental samples was 4 and 7%, respectively.
Optimized methods derived from results obtained for single cytokine measurements were applied to multiplexed nine-cytokine studies. These included strategies for animal care, serum and tissue processing, and device start-up routines. For example with the latter, we obtained more reproducible results with the Bio-Plex™ system if we completed three wash cycles (using deionized water to clean tubing and one priming cycle with sheath fluid) prior to processing experimental plates. Although not systematically studied, we found the above start-up procedure produced (1) less bead aggregation; (2) standards and positive controls that were commonly within expected ranges; and (3) experimental samples with less variance. Taken together these results suggest optimal assay sensitivity, reproducibility and accuracy may be achieved by this increased washing and priming procedure coupled to the manufacturer’s recommended practices.
3.2.2. Multiplexed, nine-cytokine measurements
Several indices showed that accurate and sensitive assays with a wide dynamic range could be performed reproducibly with multiplexed, nine-cytokine analyses of serum and brain tissue using Bio-Rad bead-based assays (Table 2). First, “goodness of fit” measures showed that simultaneous measurements of nine-cytokines could be made reproducibly and accurately. For example, “goodness of fit” (i.e., a measure of accuracy defined as calculations of ((observed/expected) concentrations × 100) was within the 70–130% range of expected levels for 7.1 ± 0.7 (n = 72) of eight values of each standard curve involving serum and 6.3 ± 0.7 (n = 72) of eight values of standard curves for tissue. Second (Table 2), serum assays were wide-ranged and extended 3–4 orders of magnitude from 2–4 to 16 000–32 000 pg/ml with only IFN-γ (2–4000 pg/ml) and GM-CSF (2–8000 pg/ml) showing a more narrowed range (Fig. 3). Tissue assays were somewhat more limited (Fig. 4) but here too assays extended over 3–4 orders of magnitude. Third, positive control values were similarly reproducible (Table 2). For example only 7% of the samples run (e.g., 4 of 54 serum and tissue assays at 100, 800 and 4000 pg/ml) did not fall within the accepted recovery of 80–120% of expected values. Those outside this range were; IL-2 tissue at 800 pg/ml; IL-4 tissue and serum at 4000 pg/ml; and IFN-γ, serum at 4000 pg/ml. The arbitrary appearance of these values suggests that these outliers were random and not a systematic indication of poor multiplexed assay performance. Thus, they are probably best regarded as a general index to help suggest nine-cytokine multiplexed assays can be 93% accurate. Finally, the mean intra assay CV (for serum and tissue standards and experimental samples) was 8% and the mean inter-assay CV (n = 4 plates) for standards and experimental samples was 4 and 7%, respectively.
Table 2.
Bio-plex™ nine-cytokine measurement characteristics
| Analyte | Sensitivity and range (pg/ml) |
Positive controls (pg/ml) (% recovery) |
||||||
|---|---|---|---|---|---|---|---|---|
| Serum | Tissue | 100 |
800 |
4000 |
||||
| Serum | Tissue | Serum | Tissue | Serum | Tissue | |||
| IL-1α | 2–16 000 | 4–16 000 | 91 | 96 | 102 | 104 | 96 | 108 |
| IL-1β | 2–32 000 | 2–4000 | 96 | 97 | 100 | 110 | 98 | 99 |
| IL-2 | 2–16 000 | 2–16 000 | 99 | 85 | 103 | 170 | 97 | 85 |
| IL-4 | 2–32 000 | 2–4000 | 101 | 90 | 96 | 110 | 142 | 134 |
| IL-6 | 2–32 000 | 8–32 000 | 100 | 109 | 101 | 111 | 105 | 96 |
| IL-10 | 2–32 000 | 50–32 000 | 98 | 90 | 116 | 111 | 102 | 104 |
| TNF-α | 4–32 000 | 2–32 000 | 81 | 98 | 93 | 111 | 91 | 108 |
| IFN-γ | 2–4000 | 4–32 000 | 87 | 107 | 117 | 106 | 57 | 106 |
| GM-CSF | 2–8000 | 4–4000 | 101 | 91 | 100 | 111 | 95 | 91 |
Fig. 3.

Standard curves for nine targeted serum cytokines measured via bead-based immunoassay. Data were generated from standards containing nine cytokines at concentrations of 2, 4, 8, 50, 500, 4000, 16 000 and 32 000 pg/ml using replicates of two for each concentration. Standard curves were calculated with Bio-Plex Manager™ software using a five-parameter logistic regression formula with weighting (power law variance). Curves demonstrate the sensitivity, sensitivity over the range, range (i.e., linear over 3–5 orders of magnitude) and ability for simultaneous measurement of at least nine cytokines from 50 μl of serum (Table 2). Eight of nine cytokines had a low-end sensitivity to 2 pg/ml and TNF-α at 4 pg/ml. High-end limit was 32 000 pg/ml for five cytokines (IL-1β, IL-4, IL-6, IL-10, and TNF-α), 16 000 pg/ml for two cytokines (IL-1α and IL-2), and 4000–8000 pg/ml for IFN-γ and GM-CSF.
Fig. 4.

Standard curves for nine targeted brain tissue cytokines measured via bead-based immunoassay. Data were generated from standards containing nine cytokines at concentrations of 2, 4, 8, 50, 500, 4000, 16 000 and 32 000 pg/ml using replicates of two for each concentration. Standard curves were calculated with Bio-Plex Manager™ software using a five-parameter logistic regression with weighting regression formula (power law variance). Curves demonstrate the sensitivity, sensitivity over the range, range and ability for simultaneous to measure at least nine cytokines from tissue samples as small as 50 μl in volume. Low-end sensitivity for tissue samples was 2 pg/ml for IL-1β, IL-2, IL-4, and TNF-α; 4 pg/ml for IFN-γ and GM-CSF; and 8 and 50 pg/ml for IL-6 and IL-10, respectively. Measurements were linear over 3–4 orders of magnitude. The high-end limit was 32 000 pg/ml for four cytokines (IL-6, IL-10, TNF-α and IFN-γ), 16 000 pg/ml for two cytokines (IL-1α, and IL-2) and 4000 pg/ml for three cytokines (IL-1β, IL-4 and GM-CSF).
3.2.3. Seizure-induced cytokine changes
Animals injected with KA to induce TLE showed typical clinical changes for the 5 h observation period immediately after injections (Fig. 5). Animals were initially only immobile and then showed levels of seizure severity that ranged from 3 to 4 (on the Racine scale). Two animals died abruptly in association with seizures, a mortality rate consistent with KA-induced seizures in rats (Avanzini et al., 1998). Nonetheless, three animals were sufficient to illustrate serum and hippocampal tissue cytokine changes from TLE (Fig. 6). Sham control animals had serum levels of IL-1α, IL-1β, IL-2, IL-6, IL-10, IFN-γ, and TNF-α of 39±10, 136±25, 76±11, 249±66, 1346±127, 31±4, and 613±72 pg/ml, respectively. Seizures for 5 h were associated with a drop in all seven of these serum cytokines to 21±3, 72±4, 35±2, 141±22, 712±80, 29±0, and 470±6 pg/ml, respectively. The reduction of IL-2 and IL-10 was significant (P < 0.02 and 0.01, respectively; Student’s t-test). Serum levels of GM-CSF and IL-4 were too low to measure. On the other hand, all nine cytokines were measured in hippocampal brain tissue with sham control levels for IL-1α, IL-1β, IL-2, IL-4, IL-6, IL-10, GM-CSF, IFN-γ and TNF-α, respectively, at 34±4, 12±1, 11±0, 6±0, 113±3, 405±13, 13 ± 0, 22 ± 1, and 5 ± 5 pg/ml (and, e.g., 2× these values when expressed as pg/mg total protein). These levels changed 5 h after the initiation of KA-induced seizures to 28 ± 0, 17 ± 1, 10 ± 0, 5 ± 0, 143 ± 12, 306 ± 19, 11 ± 0 17±0 and 8±0 pg/ml, respectively (2× these values when expressed as pg/mg total protein). IL-1β, IL-6 and TNF-α were greater than sham control levels following seizures. IL-1β was significantly elevated (P < 0.01; Student’s t-test). The remaining (i.e., six of nine) analytes (IL-1α, IL-2, IL-4, IL-10, GM-CSF and IFN-γ) were reduced from sham control levels with IL-4, IL-10 and GM-CSF significantly less (P < 0.002, 0.01, 0.04, respectively; Student’s t-test).
Fig. 5.
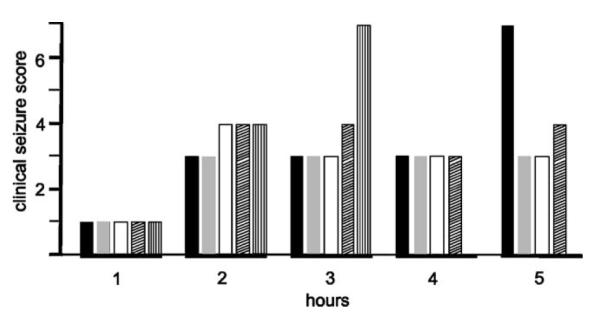
Seizure severity after kainic acid injections. Histograms show hourly seizure severity for 5 h after a single intraperitoneal injection of kainic acid in five rats (indicated by black, gray, white, diagonally hatched and vertically hatched lines, respectively). Clinical seizure severity scoring ranging from “0” for normal animals and “7” for death are detailed in the text. Note, animal death from seizures occurred at 4 and 5 h.
Fig. 6.
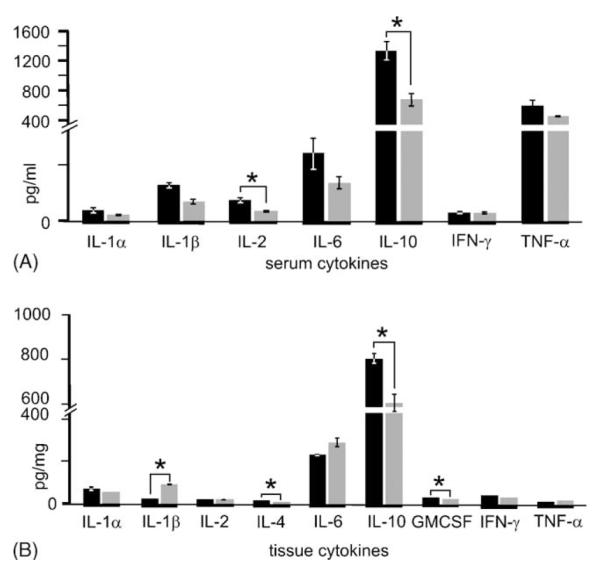
Serum and hippocampal tissue cytokine changes from kainic acid-induced seizures. Serum (A) and hippocampal tissue (B) cytokine measurements were made from three sham control animals (black) and three experimental animals (gray) to illustrate the ability of the Bio-Plex™ system to make sensitive and reproducible experimental measurements. Serum cytokine levels were greater than 2 pg/ml for seven of nine cytokines and thus measurable changes from seizures could be determined for these analytes. Only IL-4 and GM-CSF were too low for measurement. In all measured instances, kainic acid triggered a reduction in serum cytokines 5 h after injections that reached a significant difference (*) compared to sham controls for IL-2 and IL-10 (P < 0.02 and 0.01, respectively; Student’s t-test). Hippocampal brain tissue cytokine levels were greater than the 2–50 pg/ml low-end sensitivity of the assays (see Table 2). In addition 5 h after kainic acid injections, three of nine analytes were greater than sham controls (IL-1β, IL-6 and TNF-α) with IL-1β being significantly greater (P < 0.01; Student’s t-test). Also, six analytes (IL-1α, IL-2, IL-4, IL-10, GM-CSF and IFN-γ) were reduced from sham control levels with IL-4 and IL-10 significantly (P< 0.002 and 0.04, respectively) less.
4. Discussion
We demonstrate methods for simultaneous sensitive, accurate and reliable means for measurement of at least nine exemplary (e.g., cytokine) proteins from rat serum and brain tissue using bead-based immunoassay technology. Our results for serum indicate that seven of nine analytes (i.e., IL-1α, IL-1β, IL-2, IL-4, IL-6, IL-10 and TNF-α) showed a sensitivity and range of 2–4 to 16 000–32 000 pg/ml while IFN-γ and GM-CSF, respectively, showed a sensitivity and range of 2 to 4000–8000 pg/ml. Tissue measurements were nearly as good. For example, four of nine analytes (i.e., IL-1α, IL-2, TNF-α, and IFN-γ) showed a sensitivity and range of 2–4 pg/ml (4–8 pg/mg) to 16 000–32 000 pg/ml (32 000–64 000 pg/mg). Two of nine analytes (e.g., IL-6 and IL-10) were nearly the same showing sensitivity and range of 8 and 50 pg/ml, respectively, to 32 000 pg/ml (64 000 pg/mg). Only IL-4 and GM-CSF showed a comparatively reduced range of 2–4 to 4000 pg/ml (8000 pg/mg). Furthermore, accurate (e.g., intra-assay covariance of 8%) and reliable (e.g., inter-assay covariance 4–7%) measurements were demonstrated for both rat serum and tissue. Finally, other preliminary work indicates that similarly sensitive, accurate and reproducible measurements can be obtained using other brain samples including those from rat hippocampal organotypic cultures (Kunkler et al., 2003), specific-cell enriched samples (e.g., ~100 cells) obtained via laser microscope dissection (Kraig et al., 2002) as well as mouse hippocampus from whole animals (Kraig et al., 2003).
The above results were achieved by following directions provided by the manufacturer with several important additions. First, as noted below, animal care and harvesting of tissues were expressly designed to minimize stress. Second, serum samples were filtered and diluted because this universally increased target protein recovery, an “unmasking effect” that may relate to removal of proteins that could trigger non-specific binding to capture and detection antibodies (Earley et al., 2002). Third, tissue samples were homogenized with sonication. Fourth, in addition to the normal start-up procedures (i.e., a series of fluidic functions) suggested by the manufacturer, we completed three wash cycles (using deionized water to clean tubing) and one priming cycle. Performing these steps reduced the incidence of bead aggregation—and resultant poor assay performance.
4.1. Advantages of multiplexed bead-based immunoassays for protein quantification
Measurements of targeted proteins via bead-based immunoassays can be made from total protein samples of similar magnitude to that used for traditional Western blot and ELISA procedures. While for convenience here we used total protein adjusted to 500 μg/ml (or 25 μg protein for 50 μl samples), we have preliminary experience that indicates this can be reduced to at least 15 ng per sample or well (Kraig et al., 2002), a level considerably less than that used per lane for Western blots. For example using techniques as detailed here, we have successfully made multiplexed measurements for eight cytokines from as few as 100 CA3 pyramidal neurons obtained via laser microscope dissection (Kraig et al., 2002). Brain gray matter contains 83% water, 7.5% protein, 1% RNA, 0.25% DNA and 6% lipids (McIlwain and Bachelard, 1971). Since, a single cell contains about 20 pg of RNA (Schober et al., 2001), 100 CA3 pyramidal cells contain about 15 ng of total protein. Thus, up to 100 targeted proteins can be quantified from bead-based immunoassays with protein sample sizes 100–1000× less than that typically used per lane (or sample) for Western blots or ELISA’s. Use of bead-based immunoassays for proteins reduces by several-fold the time, extent of experiments as well as related costs needed to dissect complex interactions among related signaling molecules because of its ability to multiplex targeted measurements. Perhaps the most serious drawback to application of multiplexed bead-based immunoassays centers on the financial cost per plate potentially lost if samples are run incorrectly. Accordingly, meticulous attention to protocol detail is essential.
4.2. Exemplary cytokine and changes
The cytokines (IL-1α, IL-1β, IL-2, IL-4, IL-6, IL-10, GM-CSF, IFN-γ, and TNF-α) in our measurement panel are often categorized as principally either pro-inflammatory (i.e., IL-1α, IL-1β, IL-2, IL-6, GM-CSF, IFN-γ, and TNF-α) or anti-inflammatory (i.e., IL-4 and IL-10) mediating signaling molecules (Oppenheim and Feldmann, 2001). Our intent here is not to detail mechanisms for their actions in serum or brain tissue. Instead, as with other cytokines, these 9 cytokines are individually pleiotropic and variably pleiotropic in combination with other cytokines (e.g., see Oppenheim and Feldmann, 2001). Thus, their simultaneous quantification from individual samples was used as an example of the potential utility of multiplexed proteomic measurements for deciphering brain function in health and disease.
Excessive amounts of corticosterone can be released from rodent adrenal glands in response to stress (for review seeSapolsky, 2003), which can influence cytokine expression in both the blood (Oppenheim and Feldmann, 2001) and brain (Dinkel et al., 2003). Furthermore, rodent corticosterone levels show a diurnal variation (D’Agostino et al., 1982) as do cytokines in humans (Petrovsky and Harrison, 1998) and so, presumably rats. Thus, it is imperative that the potential for unintentional corticosterone variations be minimized through appropriate experimental design when studying cytokines. Such a paradigm of animal handling, anesthesia and tissue harvesting was used here and confirmed to result in plasma corticosterone levels normal for enriched animals with small inter animal variations. This result has been shown by others with use of similar experimental paradigms. For example, circadian variation in plasma corticosterone can range between 10 ng/ml (a.m.) and 230 ng/ml (p.m.) in male rats with a level of 40 ng/ml at 10 a.m. (i.e., the harvest time for all measurements in this study) (D’Agostino et al., 1982). Environmental enrichment (i.e., enhanced opportunities for social, intellectual and volitional physical activity (van Praag et al., 2000)) typically triggers a rise in plasma corticosterone from that seen in non-enriched animals (e.g., ~20 ng/ml) to about 60 ng/ml (Kempermann et al., 2002), a level consistent with that seen in our studies (i.e., 62 ng/ml). Elevated plasma corticosterone from enrichment is thought by some to reflect only increased aggression of group-housed animals (Haemisch et al., 1994). However, it seems more likely the elevation is physiological since the elevation of corticosterone is diurnal—with an even greater elevation compared to non-enriched animals in the p.m. (de Jong et al., 2000; Kraig et al., 2003). Such phasically elevated corticosterone is known to improve learning and memory (De Kloet et al., 1999; Diamond et al., 1992), a principal neurological consequence of environmental enrichment (Duffy et al., 2001).
Cytokine levels for multiplexed bead-based immunoassays reported here at baseline (i.e., sham conditions) for serum and brain are generally consistent with those reported in the literature using single ELISA’s. Specific variations may be due to factors that include differences in animal handling; differences in sample preparation; differences in choice of antibody pairs; and differences in laboratory techniques. For example, control animals here were exposed to a minimally enriched social and intellectual environment. In addition, they were given an intraperitoneal (sham) NaCl injection 5 h before sampling serum and brain. These maneuvers conceivably could alter cytokine expression. Nonetheless, published cytokine values from ELISA procedures on control or normal animals support measurements made here and thus further confirm, not only the methodology employed but also the results obtained by the multiplexed bead-based immunoassays as used here. Serum levels for IL-1α, IL-1β, IL-6 and TNF-α were found to be 39, 136, 249 and 31 pg/ml, respectively. ELISA-based values for these cytokines in the literature are all somewhat lower with IL-1α, IL-1β, IL-6 and TNF-α at <10 pg/ml (Dinarello, 2001), 14, 50 pg/ml (Borrell et al., 2002), and 2 pg/ml (Shandra et al., 2002), respectively. On the other hand, published ELISA-based values for cytokines in our nine-member panel for brain tissue are even closer to measurements reported here. Dinkel et al. (2003) report values for IL-1β, and TNF-α of 25 and 6, respectively, which agree well with our values of 24 and 10 pg/mg protein. Similarly,Allan et al. (2001) report IL-1α at 35 pg/mg and IL-6 at 150 pg/mg, while we report these cytokine levels in brain at 64 and 226 pg/mg, respectively.
Few measurements of cytokine protein changes in serum or brain are available after seizures. More frequently mRNA changes are reported (e.g., see Jankowsky and Patterson, 1999; Vezzani et al., 2002), which mirror our protein changes here. For example, mRNA’s for IL-1β, IL-6 and TNF-α rise promptly after the induction of KA-induced seizures (Vezzani et al., 2002) though sham injected animals were not used for comparison. We show that 5 h after the induction of KA-induced seizures IL-1β is significantly elevated above sham controls, while IL-4, IL-10 and GM-CSF are significantly less. Comparisons for the latter in the literature are unavailable. However, our intent here by measuring changes from seizures is to begin to demonstrate the type of differential data that might be expected from simultaneous measurement of multiple related protein species. Finally, all serum cytokines measured declined compared to sham controls after KA-induced seizures, some significantly. This most likely reflects a generalized suppressive effect of seizure-induced elevated corticosterone on cytokine expression (Oppenheim and Feldmann, 2001).
5. Conclusions
Bead-based assays can be highly versatile to specific experimental design. For example, a nine-cytokine, commercially available kit was chosen here to be exemplary of the capacity for multiplexed quantification proteins. However, evaluation of user-determined protein targets can be achieved by creating appropriate specific probes sets (de Jager et al., 2003) using commercially available bead coupling kits (e.g., Amine Coupling Kit (#171-406001) from Bio-Rad). Furthermore, bead-based assays can be designed, not only using immunoassays, but also around enzyme, receptor ligand and DNA hybridization capture molecules, further extending their utility (Dunbar et al., 2003; Earley et al., 2002).
Gallagher and Appenzeller (1999) suggest that the predominant approach to scientific inquiry has been reductionist. They go on to suggest that shortcomings of this approach are increasingly apparent but might be mitigated by an “integrative agenda.” This notion is echoed by del Zoppo et al. (2000) who note that simultaneous measurement of multiple proteins may provide improved insight to brain function in health and disease. The availability of sensitive, accurate and reliable bead-based assay technology should help to bring these suggestions to experimental reality.
Acknowledgements
This work was supported by grants from the National Institute of Neurological Disorders and Stroke (NS-19108 and NS-045923) as well grants from the American Heart Association (Bugher award to RPK and SDG to PEK). The authors would like to extend their gratitude to Jim Stejskal for his assistance and technical expertise with serum collection and corticosterone assays and Marcia Kraig for her continued assistance with this work.
References
- Allan SM, Harrsion DC, Read S, Collins B, Parsons AA, Philpott K, Rothwell NJ. Selective increases in cytokine expression in the rat brain in response to striatal injection of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate and interleukin-1. Mol Brain Res. 2001;93:180–9. doi: 10.1016/s0169-328x(01)00211-x. [DOI] [PubMed] [Google Scholar]
- Araki T, Simon RP, Taki W, Lan JQ, Henshall DC. Characterization of neuronal death induced by focally evoked limbic seizures in the C57BL/6 mouse. J Neurosci Res. 2002;69:614–21. doi: 10.1002/jnr.10356. [DOI] [PubMed] [Google Scholar]
- Avanzini G, Moshé SL, Schwartzkroin PA, Engel J., Jr . Animal models of localization-related epilepsy. In: Engel J Jr, Pedley TA, editors. Epilepsy—a comprehensive textbook. Lippincott-Raven; New York: 1998. pp. 427–42. [Google Scholar]
- Ban E, Milon G, Prudhomme N, Fillion G, Haour F. Receptors for interleukin-1 (α and β) in mouse brain: mapping and neuronal localization in hippocampus. Neuroscience. 1991;43:21–30. doi: 10.1016/0306-4522(91)90412-h. [DOI] [PubMed] [Google Scholar]
- Ban EM, Sarlieve LL, Haour FG. Interleukin-1 binding sites on astrocytes. Neuroscience. 1993;52:725–33. doi: 10.1016/0306-4522(93)90421-b. [DOI] [PubMed] [Google Scholar]
- Baud M. Data analysis, mathematical modeling. In: Masseyeff RF, editor. Methods of immunological analysis: fundamentals. vol. 1. VCH; New York: 1993. pp. 656–71. [Google Scholar]
- Borrell J, Vela JM, Arevalo-Martin A, Molina-Holgado E, Guaza C. Prenatal immune challenge disrupts sensorimotor gating in adult rats: implications for the etiopathogenesis of schizophrenia. Neuropsychopharmacology. 2002;26:204–15. doi: 10.1016/S0893-133X(01)00360-8. [DOI] [PubMed] [Google Scholar]
- Breder CD, Dinarello CA, Saper CB. Interleukin-1 immunoreactive innervation of the human hypothalamus. Science. 1988;240:321–4. doi: 10.1126/science.3258444. [DOI] [PubMed] [Google Scholar]
- Caggiano AO, Kraig RP. Eicosanoids and nitric oxide influence reactive gliosis from spreading depression in microglia but not astrocytes. J Comp Neurol. 1996;369:93–108. doi: 10.1002/(SICI)1096-9861(19960520)369:1<93::AID-CNE7>3.0.CO;2-F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caggiano AO, Kraig RP. Neuronal nitric oxide synthase expression is induced in neocortical astrocytes following spreading depression. J Cereb Blood Flow Met. 1998;18:75–87. doi: 10.1097/00004647-199801000-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caggiano AO, Breder CD, Kraig RP. Long-term elevation of cyclooxygenase-2 but not lipoxygenase, in regions synaptically distant from spreading depression. J Comp Neurol. 1996;376:447–62. doi: 10.1002/(SICI)1096-9861(19961216)376:3<447::AID-CNE7>3.0.CO;2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Agostino J, Vaeth GF, Henning SJ. Diurnal rhythm of total and free concentrations of serum corticosterone in the rat. Acta Endocrinol (Copenhagen) 1982;100:85–90. doi: 10.1530/acta.0.1000085. [DOI] [PubMed] [Google Scholar]
- Dasso J, Lee J, Bach H, Mage RG. A comparison of ELISA and flow microsphere-based assays for quantification of immunoglobins. J Immunol Meth. 2002;263:22–33. doi: 10.1016/s0022-1759(02)00028-5. [DOI] [PubMed] [Google Scholar]
- Davies C. Concepts. In: Wild D, editor. The immunoassay handbook. 2nd ed Nature Publishing Group; New York: 2001. pp. 78–110. [Google Scholar]
- de Jager W, Velthius H, Prakken BJ, Kuis W, Rijkers GT. Simultaneous detection of 15 human cytokines in a single sample of stimulated peripheral blood mononuclear cells. Clin Diag Lab Immunol. 2003;10:133–9. doi: 10.1128/CDLI.10.1.133-139.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jong I, Prelle IT, van de Burgwal JA, Lambooij E, Korte SM, Blokhuis HJ, et al. Effects of environmental enrichment on behavioral responses to novelty, learning and memory, and the circadian rhythm in cortisol in growing pigs. Physiol Behav. 2000;68:571–8. doi: 10.1016/s0031-9384(99)00212-7. [DOI] [PubMed] [Google Scholar]
- De Kloet ER, Oitzel MS, Joëls M. Stress and cognition: are corticosteroids good or bad guys? TINS. 1999;22:422–6. doi: 10.1016/s0166-2236(99)01438-1. [DOI] [PubMed] [Google Scholar]
- del Zoppo G, Ginis I, Hallenbeck JM, Iadecola C, Wang X, Feuerstein GZ. Inflammation and stroke: putative role for cytokines, adhesion molecules and iNOS in brain response to ischemia. Brain Pathol. 2000;10:95–112. doi: 10.1111/j.1750-3639.2000.tb00247.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond DM, Bennett MC, Fleshner M, Rose GM. Inverted-U relationship between the level of peripheral corticosterone and the magnitude of hippocampal primed burst potentiation. Hippocampus. 1992;2:421–30. doi: 10.1002/hipo.450020409. [DOI] [PubMed] [Google Scholar]
- Dinarello CA. Biologic basis for interleukin-1 in disease. Blood. 1996;87:2095–147. [PubMed] [Google Scholar]
- Dinarello CA. IL-1α. In: Oppenheim JJ, Feldmann M, editors. Cytokine reference. 3rd ed Academic Press; San Diego: 2001. pp. 308–18. [Google Scholar]
- Dinkel K, MacPherson A, Sapolsky RM. Novel glucocorticoid effects on acute inflammation in the CNS. J Neurochem. 2003;84:705–16. doi: 10.1046/j.1471-4159.2003.01604.x. [DOI] [PubMed] [Google Scholar]
- Dombrowski SM, Hilgetag CC, Barbas H. Quantitative architecture distinguishes prefrontal cortical systems in the rhesus monkey. Cereb Cortex. 2001;11:975–88. doi: 10.1093/cercor/11.10.975. [DOI] [PubMed] [Google Scholar]
- Duffy SN, Craddock KJ, Abel T, Nguyen PV. Environmental enrichment modifies the PKA-dependence of hippocampal LTP and improves hippocampus-dependent memory. Learn Mem. 2001;8:26–34. doi: 10.1101/lm.36301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunbar SA, Vander Zee CA, Oliver KG, Karem KL, Jacobson JW. Quantitative multiplexed detection of bacterial pathogens: DNA and protein applications of the Luminex LabMAP™ System. J Microbiol Meth. 2003;53:245–52. doi: 10.1016/s0167-7012(03)00028-9. [DOI] [PubMed] [Google Scholar]
- Earley MC, Vogt RF, Jr, Shapiro HM, Mandy FF, Kellar KL, Bellisario R, et al. Report from a workshop on multianalyte microsphere assays. Cytometry. 2002;50:239–42. doi: 10.1002/cyto.10140. [DOI] [PubMed] [Google Scholar]
- Fontana A, Kristensen F, Dubs R, Gemsa D, Weber E. Production of prostaglandin E and interleukin-1 like factor by cultured astrocytes and C6 glioma cells. J Immunol. 1982;129:2413–9. [PubMed] [Google Scholar]
- Gallagher R, Appenzeller T. Beyond reductionism. Science. 1999;284:79. [Google Scholar]
- Giulian D, Baker TJ, Shih N, Lachman LB. Interleukin-1 of the central nervous system is produced by amoeboid microglia. J Exp Med. 1986;164:594–604. doi: 10.1084/jem.164.2.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goujon E, Laye S, Parnet P, Dantzer R. Regulation of cytokine gene expression in the central nervous system by glucocorticoids: mechanisms and functional consequences. Psychoneuroendocrionol. 1997;22(Suppl 1):S75–80. doi: 10.1016/s0306-4530(97)00009-7. [DOI] [PubMed] [Google Scholar]
- Haemisch A, Voss T, Gärtner K. Effects of environmental enrichment on aggressive behavior, dominance hierarchies, and endocrine states in male DBA/2J mice. Physiol Behav. 1994;56:1041–8. doi: 10.1016/0031-9384(94)90341-7. [DOI] [PubMed] [Google Scholar]
- Hori T, Oka T, Hosoi M, Abe M, Oka K. Hypothalamic mechanisms of pain modulatory actions of cytokines and prostaglandin E2. Ann NY Acad Sci. 2000;917:106–20. doi: 10.1111/j.1749-6632.2000.tb05375.x. [DOI] [PubMed] [Google Scholar]
- Jankowsky JL, Patterson PH. Differential regulation of cytokine expression following pilocarpine-induced seizure. Exp Neurol. 1999;159:333–46. doi: 10.1006/exnr.1999.7137. [DOI] [PubMed] [Google Scholar]
- Harry G Jean, Bruccoleri A, d’Hellencourt C Lefebvre. Differential modulation of hippocampal chemical-induced injury response by ebselen, pentoxifylline, and TNFalpha-, IL-1alpha-, and IL-6-neutralizing antibodies. J Neurosci Res. 2003;73:526–36. doi: 10.1002/jnr.10653. [DOI] [PubMed] [Google Scholar]
- John CD, Buckingham JC. Cytokines: regulation of the hypothalamopituitary-adrenocortical axis. Curr Opin Pharmacol. 2003;3:78–84. doi: 10.1016/s1471-4892(02)00009-7. [DOI] [PubMed] [Google Scholar]
- Kellar KL, Iannone MA. Multiplexed microsphere-based flow cytometric assays. Exp Hematol. 2002;30:1227–37. doi: 10.1016/s0301-472x(02)00922-0. [DOI] [PubMed] [Google Scholar]
- Kellar KL, Kalwar RR, Dubois KA, Crouse D, Chafin WD, Kane BE. Multiplexed fluorescent bead-based immunoassays for quantitation of human cytokines in serum and culture supernatants. Cytometry. 2001;45:27–36. doi: 10.1002/1097-0320(20010901)45:1<27::aid-cyto1141>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- Kempermann G, Gast D, Gage FH. Neuroplasticity in old age: sustained fivefold induction of hippocampal neurogenesis by long-term environmental enrichment. Ann Neurol. 2002;52:135–43. doi: 10.1002/ana.10262. [DOI] [PubMed] [Google Scholar]
- Kraig RP, Kunkler PE, Fedynyshyn J. Multiple and simultaneous proteomic measures of inflammatory changes from excitotoxic injury in hippocampal organ cultures. Soc Neurosci Abst. 2002 [Session #95.6] [Google Scholar]
- Kraig RP, Hulse RE, Kunkler PE, Langan G. Environmental enrichment (EE) may be neuroprotective by modulating synaptic activity (SA) via pro- & anti-inflammatory mediators (IMs) Soc Neurosci Abst. 2003 [Session #737.15] [Google Scholar]
- Kunkler PE, Hulse RE, Kraig RP. Spreading depression (SD) may worsen & lessen stroke injury by intrinsic pro- & anti-inflammatory mediator profile changes. Soc Neurosci Abst. 2003 [Session #951.4] [Google Scholar]
- Lechan RM, Toni R, Clark BD, Cannon JG, Shaw AR, Dinarello CA, et al. Immunoreactive interleukin-1 beta localization in rat forebrain. Brain Res. 1990;514:135–40. doi: 10.1016/0006-8993(90)90445-h. [DOI] [PubMed] [Google Scholar]
- Linde HW. The physics and chemistry of general anesthetics. In: Soma LR, editor. The textbook of veterinary anesthesia. Williams and Wilkins; Baltimore: 1971. pp. 30–49. [Google Scholar]
- Lynch MA. Interleukin-1 beta exerts a myriad of effects in the brain and in particular in the hippocampus: analysis of some of these actions. Vitam Horm. 2002;64:185–219. doi: 10.1016/s0083-6729(02)64006-3. [DOI] [PubMed] [Google Scholar]
- McIlwain H, Bachelard HS. Biochemistry and the central nervous system. Churchill Livningstone; Edinburgh: 1971. [Google Scholar]
- Nadeau S, Rivest S. Glucocorticoids play a fundamental role in protecting brain during innate immune response. J Neurosci. 2003;23:5536–44. doi: 10.1523/JNEUROSCI.23-13-05536.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nix B, Wild D. Calibration curve-fitting. In: Wild D, editor. The immunoassay handbook. 2nd ed Nature Publishing Group; New York: 2001. pp. 198–210. [Google Scholar]
- Oppenheim JJ, Feldmann M. Introduction to the role of cytokines in innate host defense and adaptive immunity. In: Opennheim JJ, Feldmann M, editors. Cytokine reference. Academic Press; New York: 2001. pp. 3–20. [Google Scholar]
- Patel HC, Boutin H, Allan SM. Interleukin-1 in the brain—mechanisms of action in acute neurodegeneration. Ann NY Acad Sci. 2003;992:39–47. doi: 10.1111/j.1749-6632.2003.tb03136.x. [DOI] [PubMed] [Google Scholar]
- Petrovsky N, Harrison LC. The chronobiology of human cytokine production. Int Rev Immunol. 1998;16:635–49. doi: 10.3109/08830189809043012. [DOI] [PubMed] [Google Scholar]
- Prabhakar U, Eirikis E, Davis HM. Simultaneous quantification of proinflammatory cytokines in human plasma using LabMAP™ assay. J Immunol Meth. 2002;260:207–18. doi: 10.1016/s0022-1759(01)00543-9. [DOI] [PubMed] [Google Scholar]
- Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroenceph Clin Neurophysiol. 1972;32:281–94. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- Rothwell NJ, Luheshi GN. Interleukin-1 in the brain: biology, pathology and therapeutic target. TINS. 2000;23:618–25. doi: 10.1016/s0166-2236(00)01661-1. [DOI] [PubMed] [Google Scholar]
- Sapolsky R. Taming stress. Sci Am. 2003;289:86–95. doi: 10.1038/scientificamerican0903-86. [DOI] [PubMed] [Google Scholar]
- Schneider H, Pitossi F, Balschun D, Wagner A, del Rey A, Besedovsky HO. A neuromodulatory role of interleukin-1beta in the hippocampus. Proc Natl Acad Sci. 1998;23:7778–83. doi: 10.1073/pnas.95.13.7778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schober MS, Min Y-N, Chen YQ. Serial analysis of gene expression in a single cell. BioTechniques. 2001;31:1240–2. [PubMed] [Google Scholar]
- Shandra AA, Godlevsky LS, Vastyanov RS, Oleinik AA, Konovalenko VL, Rapoport EN, et al. The role of TNF-alpha in amygdala kindled rats. Neurosci Res. 2002;42:147–53. doi: 10.1016/s0168-0102(01)00309-1. [DOI] [PubMed] [Google Scholar]
- Song Y, Simonyi K, Thomas R, Zhang A, Huang I, Aguilera A, et al. Simultaneous quantitation of 5 phosphorylated proteins using the Bio-Plex Protein Array System. Bio-Rad Bull. 2000:2632. [Google Scholar]
- van Praag H, Kempermann G, Gage FH. Neural consequences of environmental enrichment. Nat Rev Neurosci. 2000;1:191–8. doi: 10.1038/35044558. [DOI] [PubMed] [Google Scholar]
- Vane JR, Bakhle YS, Botting RM. Cyclooxygenase 1 and 2. Annu Rev Pharmacol Toxicol. 1998;38:97–120. doi: 10.1146/annurev.pharmtox.38.1.97. [DOI] [PubMed] [Google Scholar]
- Vezzani A, Moneta D, Richichi C, Aliprandi M, Borrows SJ, Ravizza T, et al. Functional role of inflammatory cytokines and antiinflammatory molecules in seizures and epileptogenesis. Epilepsia. 2002;43(Suppl 5):30–5. doi: 10.1046/j.1528-1157.43.s.5.14.x. [DOI] [PubMed] [Google Scholar]
- Vignali DAA. Multiplexed particle-based flow cytometric assays. J Immunol Meth. 2000;243:243–55. doi: 10.1016/s0022-1759(00)00238-6. [DOI] [PubMed] [Google Scholar]
- Zhang X, Cui SS, Wallace AE, Hannesson DK, Schmued LC, Saucier DM, et al. Relations between brain pathology and temporal lobe epilepsy. J Neurosci. 2002;22:6052–61. doi: 10.1523/JNEUROSCI.22-14-06052.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]