

Abstract
Peripheral tissue injury is associated with changes in protein expression in sensory neurons that may contribute to abnormal nociceptive processing. We used cultured dorsal root ganglion (DRG) neurons as a model of axotomized neurons to investigate early changes in protein expression after nerve injury. Comparing protein levels immediately after DRG dissociation and 24 h later by proteomic differential expression analysis, we found a substantial increase in the levels of the neurotrophin-inducible protein VGF (nonacronymic), a putative neuropeptide precursor. In a rodent model of nerve injury, VGF levels were increased within 24 h in both injured and uninjured DRG neurons, and the increase persisted for at least 7 d. VGF was also upregulated 24 h after hindpaw inflammation. To determine whether peptides derived from proteolytic processing of VGF participate in nociceptive signaling, we examined the spinal effects of AQEE-30 and LQEQ-19, potential proteolytic products shown previously to be bioactive. Each peptide evoked dose-dependent thermal hyperalgesia that required activation of the mitogen-activated protein kinase p38. In addition, LQEQ-19 induced p38 phosphorylation in spinal microglia when injected intrathecally and in the BV-2 microglial cell line when applied in vitro. In summary, our results demonstrate rapid upregulation of VGF in sensory neurons after nerve injury and inflammation and activation of microglial p38 by VGF peptides. Therefore, VGF peptides released from sensory neurons may participate in activation of spinal microglia after peripheral tissue injury.
Introduction
Sensory neurons respond to peripheral tissue damage, such as nerve injury or inflammation, with changes in protein expression and functional properties that contribute to altered nociceptive processing in the spinal cord (Woolf and Costigan, 1999; Campbell and Meyer, 2006; White et al., 2007). Spinal microglia participate in the development of inflammatory and nerve injury-induced hypersensitivity through mechanisms mediated in part by the mitogen-activated protein kinase (MAPK) p38 (Jin et al., 2003; Svensson et al., 2003; Raghavendra et al., 2004; Tsuda et al., 2004, 2005; Boyle et al., 2006). Within sensory neurons, changes in expression levels have been reported for a number of signaling mediators, including neuropeptides, neurotrophins, and chemokines (Fukuoka et al., 1998; Woolf and Costigan, 1999; Karchewski et al., 2002; Zhang and De Koninck, 2006). The upregulation of signaling mediators released from sensory neurons in spinal cord may contribute to microglial activation within the dorsal horn after peripheral tissue damage.
In this study, we used cultured adult dorsal root ganglia (DRG) as a model system of axotomized neurons to investigate early changes in protein expression after nerve injury. Relative quantitation of protein levels was done using iTRAQ (for isobaric tag for relative and absolute quantitation), a mass spectrometry-based method for proteomic differential expression analysis (Li et al., 2007; Lund et al., 2007). Among the proteins identified as upregulated in this analysis, we selected for additional study the neurosecretory protein VGF (nonacronymic) based on evidence that its expression is induced by neuronal lesions and neurotrophins (Levi et al., 2004). VGF, a 617 aa protein related to the chromogranins, has the characteristics of a neuropeptide precursor, but the precise identities of bioactive VGF-derived peptides and the putative receptors that mediate their action are unknown. VGF has been implicated in neuroplasticity associated with depression as well as learning and memory, and, in cultured hippocampal neurons, VGF-derived peptides potentiated synaptic activity (Alder et al., 2003; Hunsberger et al., 2007; Thakker-Varia et al., 2007; Bozdagi et al., 2008). In this study, we addressed the hypotheses that VGF is upregulated in sensory neurons after peripheral tissue injury and that VGF-derived peptides have pronociceptive spinal effects. Our results demonstrate rapid VGF upregulation after nerve injury and inflammation and show that VGF-derived peptides evoke p38-dependent thermal hyperalgesia and p38 activation in microglial cells.
Materials and Methods
All procedures conform to guidelines of the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of the University of Minnesota.
DRG cultures
Primary cultures of rat DRG neurons were prepared using methods similar to those described previously (Khasabova et al., 2004). Briefly, DRG were isolated from all spinal levels of four adult male rats (125–200 g, Sprague Dawley; Harlan), enzymatically dissociated in collagenase-D (1.5 mg/ml, two 45 min incubations; Roche Diagnostics) and triturated through fire-constricted Pasteur pipettes. For proteomic and Western blot analysis, all cells from one rat were divided in half and plated in two 10 cm glass Petri dishes coated with poly-l-lysine (10 μg/ml; Sigma-Aldrich) and laminin (10 μg/ml; Sigma-Aldrich) in F-12/Ham's medium containing 10% fetal bovine serum (plating density, 50,000–80,000 cells per dish). Cells from one of the dishes were harvested ∼1.5 h after plating (t0), and cells from the other dish were harvested 24 h later (t24). For immunohistochemistry, glass coverslips were placed into 10 cm glass Petri dishes before coating with poly-l-lysine and laminin and removed either 1.5 h (t0) or 24 h (t24) after plating, rinsed twice with PBS, incubated for 20 min in fixative (4% paraformaldehyde and 0.2% picric acid in 0.1 m phosphate buffer, pH 6.9), and rinsed in three changes of PBS before proceeding with immunohistochemistry.
Proteomic analysis of DRG cultures
Cells were washed with PBS and detached by gentle pipetting of the buffer and agitation at 4°C. Recovery of neurons was verified visually. The cells were collected by centrifugation at 3000 × g for 15 min, frozen in liquid nitrogen, and stored until sample preparation. The cells were separated into subcellular fractions using ProteoExtract Subcellular Proteome Extraction kit (Calbiochem), and the proteins in the cytoplasmic and nuclear fractions were prepared for mass spectrometric analysis by precipitation using cold acetone. Proteins from the cytoplasmic and nuclear fractions of both time points were denatured, reduced and cysteine alkylated before digestion with trypsin, and labeled with the iTRAQ reagents (Applied Biosystems) following the directions of the manufacturer. The labeled tryptic peptides in the four samples (114, t0 cytoplasmic fraction; 115, t24 cytoplasmic fraction; 116, t0 nuclear fraction; and 117, t24 nuclear fraction) were combined and separated by two-dimensional liquid chromatography (2D LC) using strong cation exchange on a Magic 2002 HPLC system (Michrom BioResourses), followed by C18 reverse-phase LC (Dionex/LC Packings capillary LC system) online with a QSTAR Pulsar i Mass Spectrometer (Applied Biosystems) as described by Lund et al. (2007). The tandem mass spectrometry (MS/MS) data were analyzed using ProteinPilot Software 2.0.1 (Applied Biosystems/MDS Sciex) with the Paragon Algorithm (Shilov et al., 2007) and searched against a nonredundant database of human, mouse, and rat proteins compiled from National Center for Biotechnology Information on March 9, 2007 that included common contaminants (59,817 total proteins) for the identification and quantification of proteins. ProteinPilot assigns confidence of protein identification based on the highest confidence peptide used to identify the protein, and peptide confidence is a measure of sequence accuracy determined by MS/MS sequence coverage and the number of other potential sequence matches. The resulting set of identified proteins, which included corresponding gene identifiers and expression values, was uploaded into Ingenuity Pathways Analysis (IPA) (Ingenuity Systems) and annotated. Each gene identifier was mapped to its corresponding gene object in the Ingenuity Pathways Knowledge Base. Additional analysis was focused on proteins with iTRAQ ratios >2. For proteins with ratios that showed more than a twofold increase but were identified with <95% confidence, the identification was confirmed by manual inspection of the peptide spectra. In addition, ratios with p > 0.05 (assigned by ProteinPilot) were verified by manual inspection of the iTRAQ reporter regions fragmented from the peptides.
Western blot analysis of DRG cultures
Cells were homogenized for 5 s on ice in extraction buffer (TBS, pH 7.4, 1% Triton X-100, 10 mm EDTA, and 10 mm EGTA) in the presence of protease inhibitors (Complete Mini, EDTA-free; Roche) using a glass homogenizer. The supernatant was collected and mixed with 4× loading buffer (3:1 sample/loading buffer; NuPage; Invitrogen), heated for 10 min at 70°C, and loaded into wells of 4–12% Bis-Tris gels (NuPage; Invitrogen). The gels underwent electrophoresis at constant voltage (160 V) for 1 h, and proteins were transferred from the gel onto polyvinylidene fluoride membranes for 1 h using constant amperage (250 mA). The membranes were incubated as follows: (1) in blocking buffer [TBS, pH 7.4, containing 0.2% I-Block reagent (Applied Biosystems) and 0.1% Tween 20] overnight at 4°C; (2) in anti-VGF primary antisera (1:10,000; generated in house as described below) overnight at 4°C; (3) washed four times for 30 min in blocking buffer; (4) in horseradish peroxidase-conjugated secondary antibodies (1:10,000; Jackson ImmunoResearch) at room temperature for 2 h; (5) washed four times for 30 min in TBS containing 0.1% Tween 20 and two times for 30 min in TBS; and (6) in chemiluminescent substrate (Pierce Supersignal) for 5 min. The membranes were then exposed to Kodak X-O-Mat Blue film (Eastman Kodak).
Models of persistent pain
Spinal nerve ligation.
Spinal nerve ligation (SNL) was performed on adult male Sprague Dawley rats (150–200 g; Harlan) similar to previously described procedures (Kim and Chung, 1992). Under isoflurane anesthesia, the left L5 transverse process was exposed and removed, and the L5 spinal nerve was tightly ligated with 6-0 silk suture. Sham surgery consisted of removal of the transverse process and visualization of the L5 spinal nerve without touching it. Induction of hypersensitivity was measured using von Frey filaments and the up–down method as described previously (Chaplan et al., 1994). Animals were killed by perfusion fixation 1, 3, or 7 d after surgery.
Inflammation.
Complete Freund's adjuvant (CFA) inflammation was induced as described previously (Wenk et al., 2006). Under isoflurane anesthesia, 100 μl of CFA (1:1 emulsion in saline; Sigma) was injected subcutaneously into the plantar surface of the right hindpaw of adult male Sprague Dawley rats (150–200 g; Harlan). The animals were killed by perfusion fixation 24 h after CFA injection.
VGF peptides
The following VGF-derived peptides were used in this study: AQEE-30, corresponding to the last 30 aa (588-AQEEADAEER RLQEQEELENYIEHVLLHRP-617) of the VGF protein; AQEE-11, corresponding to the first 11 aa of AQEE-30; and LQEQ-19, corresponding to the last 19 aa of AQEE-30. The peptides were individually synthesized and purified by HPLC (Biomedical Genomics Center, University of Minnesota, Minneapolis, MN).
Behavioral testing of the effects of VGF peptides
VGF peptides were administered intrathecally in conscious adult male mice (20–25 g) as described (Hylden and Wilcox, 1981). A 30-gauge, 0.5-inch needle connected to a 50 μl Luer-hub Hamilton syringe was used to deliver 5 μl of saline (vehicle), AQEE-30, LQEQ-19, or AQEE-11. Baseline tail-flick (TF) latency data in response to water immersion (49°C) was collected on all mice before the injection and at selected time points after injection. In some cases, LQEQ-19- and saline-injected mice were killed by perfusion fixation immediately after behavioral testing. TF latency data was analyzed by first calculating delta TF values (Experimental TF latency − Baseline TF latency) and using those values in the formula for percentage inhibition for the mean of each dose group [(Control delta TF latency − Experimental delta TF latency)/(Control delta TF latency) × 100]. In some experiments, the following inhibitors were administered as a 5 min pretreatment: 7-NI (7-nitroindazole), 10 nmol (Tocris Biosciences); l-NAME (N-ω-nitro-l-arginine methyl ester), 100 nmol (Sigma-Aldrich); MK801 [(+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine maleate], 10 nmol (Sigma-Aldrich); GF109203X (2-[1-(3-dimethylaminopropyl)-1H-indol-3-yl]-3-(1H-indol-3-yl)maleimide), 1 and 10 nmol (Tocris Biosciences); KT5720 [(9S,10R,12R)-2,3,9,10,11,12-hexahydro-10-hydroxy-9-methyl-1-oxo- 9,12-epoxy-1H-diindolo[1,2,3-fg:3′,2′,1′-kl]pyrrolo[3,4-i][1,6]benzodiazocine-10-carboxylicacid hexyl ester], 1.7 nmol (Sigma-Aldrich); U0126 [1,4-diamino-2,3-dicyano-1,4-bis(o-aminophenylmercapto)butadiene], 2.5 nmol (Tocris Biosciences); SB202190 [4-(4-fluorophenyl)-2-(4-hydroxyphenyl)-5-(4-pyridyl)1H-imidazole], 0.1, 1, and 2.5 nmol (Tocris Biosciences); and SB600125, 2.5 nmol (Tocris Biosciences). The selected doses were similar to doses used previously in similar experimental paradigms (Inoue and Ueda, 2000; Sakurada et al., 2002; Wu et al., 2006; Gabra et al., 2007; Komatsu et al., 2007). 7-NI, l-NAME, MK801, GF109203X, and KT5720 were dissolved with 0.9% normal saline, pH 7.2. U0126, SB202190, and SB600125 were dissolved in DMSO and diluted to a final concentration of 5% DMSO with 0.9% normal saline, pH 7.2.
Immunohistochemistry
For preparation of DRG or spinal cord tissue sections, rats or mice were deeply anesthetized with isoflurane and perfused via the heart with calcium-free Tyrode's solution (in mm: 116 NaCl, 5.4 KCl, 1.6 MgCl2.6H20, 0.4 MgSO4.7H2O, 1.4 NaH2PO4, 5.6 glucose, and 26 Na2HCO3), followed by fixative (described above), and finally with 10% sucrose in PBS. DRG and spinal cords were removed and incubated in 10% sucrose overnight at 4°C. Tissues were cryostat sectioned (14 μm) and thaw mounted onto gelatin-coated slides. Coverslips with cultured cells or tissue sections were preabsorbed in blocking buffer (PBS containing 0.3% Triton X-100, 1% BSA, and 1% normal donkey serum) for 30 min, incubated in primary antisera listed below (coverslips for 4 h at room temperature; tissue sections overnight at 4°C), rinsed with PBS three times for 10 min, incubated in cyanine 3-conjugated or cyanine 2-conjugated secondary antisera (1:300 and 1:100, respectively; Jackson ImmunoResearch) for 1 h at room temperature, washed with PBS, and mounted onto slides or coverslipped using PBS/glycerol containing 0.1% p-phenylenediamine (Sigma). In some cases, DRG sections were counterstained with YOYO-1 (Invitrogen) before coverslipping.
Primary antisera for immunohistochemistry
VGF antisera.
At the onset of the immunohistochemical experiments, several commercial VGF antisera (Santa Cruz Biotechnology) were evaluated for their ability to label cultured DRG neurons. Adequate labeling in the neuronal cell bodies was obtained with the N-terminal antiserum anti-VGF (D-20) diluted at 1:300. This antiserum was used for quantitative image analysis in DRG cultures and DRG tissue sections (see below). Subsequently, it was noted that this antiserum did not label the processes of cultured neurons and that the labeling in superficial dorsal horn of spinal cord sections was very weak. Therefore, we generated antisera to the VGF C terminus using previously described methods (Vulchanova et al., 1997). Briefly, the peptide AQEE-30 was conjugated to bovine thyroglobulin (Sigma) using glutaraldehyde. The peptide conjugate (1 mg/ml) was emulsified with an equal volume of Freund's adjuvant (Difco) and injected into female guinea pigs (n = 4; Harlan) at 2 week intervals (0.5 mg of peptide for initial and 0.25 mg of peptide for subsequent immunizations). Staining with anti-VGF antisera was blocked by preabsorption of the antisera with the cognate peptide (10 μg/ml). In addition, the specificity of the antisera was ascertained by Western blot analysis, which showed bands consistent with previous reports (Fig. 1), as well as by double labeling with the N-terminal antisera in DRG, which showed an overlapping pattern of staining (data not shown). The C-terminal VGF antisera (1:1000) were used for quantitative image analysis in spinal cord. Additionally, the quantification in DRG cultures was repeated with the C-terminal antisera, yielding the same results as the N-terminal antisera.
Figure 1.

Upregulation of VGF immunoreactivity in cultured DRG neurons. A, In Western blot analysis, samples from t24 DRG cultures show a band at ∼90 kDa, which corresponds to the unprocessed form of VGF. The lower-molecular-weight bands most likely correspond to proteolytic fragments of VGF. Consistent with VGF increase at t24 compared with t0, VGF-immunoreactive bands at t0 are nearly below the detection limit. B, C, Cultured DRG neurons fixed at t0 and double labeled with anti-VGF (B) and anti-PGP (C), which shows all neurons in the field of view. D, E, Cultured DRG neurons fixed at t24 and double labeled with anti-VGF (D) and anti-PGP (E). The number of VGF-positive neurons increased at t24 (D).
Other primary antisera.
The following other primary antisera were used for immunohistochemistry: mouse anti-protein gene product (PGP) (1:1000; Biogenesis); rabbit anti-TrkA (1:1000; Advanced Targeting Systems); guinea pig anti-P2X3 (1:500) (Vulchanova et al., 1997); goat anti-ATF3 (1:300; Santa Cruz Biotechnology); rabbit anti-calcitonin gene-related peptide (CGRP) (1:1000; ImmunoStar); rabbit anti-phosphorylated (phospho)-p38 (1:100; Cell Signaling Technology); mouse anti-neuronal-specific nuclear protein (NeuN) (1:1000; Millipore Bioscience Research Reagents); mouse anti-GFAP (1:1000; Cell Signaling Technology); rat anti-CD68 (1:100; AbD Serotec).
Quantitative image analysis
During image analysis, the observer was blinded to the experimental treatments.
Cultured DRG neurons.
Nine coverslips from three different DRG cultures (three coverslips per culture) were analyzed for each time point. Stacks of PGP9.5/VGF double-labeled confocal images (three per coverslip) collected with a Bio-Rad MRC-1024 microscope were analyzed using NIH ImageJ. Neurons, identified based on their PGP9.5 labeling, were outlined, and the average intensity of VGF immunoreactivity (IR) of each neuron was measured. For each of the three cultures, 18 VGF-negative cells (two per image) were identified from the t0 images (negative cells were often hard to identify in t24 images). The threshold intensity for identifying VGF-positive neurons for t0 and t24 of each culture was defined as the average intensity of these negative cells + 2 SDs.
DRG neurons in tissue sections.
For each DRG, five sections equally spaced throughout its length were triple labeled with anti-VGF (N-terminal), YOYO-1, and either anti-TrkA or anti-P2X3. Stacks of triple-labeled confocal images (nine per DRG) were analyzed using NIH ImageJ. Neurons were identified and outlined based on their Nissl-like YOYO-1 labeling, and cell body area and the average intensity of VGF staining of each neuron was measured. Three VGF-negative neurons were identified in each image, and the threshold intensity for identifying VGF-positive neurons was defined as the average of the three unlabeled neurons + 2 SDs. Similarly, the average intensity of TrkA and P2X3 staining was measured, and the threshold intensity for identifying positive neurons was determined based on the average intensity of three unlabeled neurons.
VGF-IR and CGRP-IR in rat spinal cord.
Quantitative analysis was performed at two levels of the L5 spinal segment that were separated by ∼1 mm. The results from these two levels were similar. In sections double labeled for VGF and CGRP, confocal images of dorsal horn were analyzed using NIH ImageJ. For each spinal cord, a threshold intensity, corresponding to the average intensity of unlabeled regions within the dorsal horn, was selected to separate VGF labeling from background. The number of pixels with intensity above threshold (thresholded area) was measured in the superficial laminae of the right and left dorsal horn. To eliminate variability between animals attributable to tissue processing, labeling on the left side (operated in SNL and sham animals) was normalized to the right side. For each spinal cord, data were collected from three sections, averaged, and expressed as percentage change [i.e., (ipsilateral − contralateral)/contralateral × 100]. CGRP-IR in dorsal horn was analyzed as described for VGF-IR.
phospho-p38-IR in mouse spinal cord.
Dorsal horn images were collected with a confocal microscope (Olympus FluoView 1000 IX2). For each image, a threshold intensity, corresponding to the maximum intensity of an unlabeled region, was selected to separate phospho-p38 labeling from background. The pixel area of phospho-p38-IR above background was normalized to the spinal cord area included in the image and expressed as percentage thresholded area. For each animal, percentage thresholded area was determined by adding percentage thresholded area of both left and right dorsal horns from three spinal cord sections.
Microglial cultures
The BV-2 microglial cell line (a kind gift from Dr. Jonathan Godbout, The Ohio State University, Columbus, OH) was maintained in DMEM supplemented with 10% FBS. Cells were treated with LQEQ-19 or vehicle (serum-free DMEM) for 2 min. For Western blot, treated cells were lysed [20 mm Tris, 150 mm NaCl, pH 8, and 1% (v/v) Triton X-100] in the presence of protease inhibitors (Complete Mini, EDTA-free; Roche,) and phosphatase inhibitors (Halt Phosphatase Inhibitor Cocktail; Thermo Fisher Scientific) on ice for 1 h. The samples were centrifuged (40,000 × g, 4°C, 30 min) and loaded (60 μg/lane) onto 4–12% Bis-Tris gels (Invitrogen) and run at 130 V for 90 min. Next proteins were transferred to nitrocellulose membranes and treated as follows: (1) incubated in blocking buffer [Tris-buffered saline, 0.05% (v/v) Tween 20, 5% (w/v) nonfat milk] for 120 min at room temperature; (2) washed with TBS containing Tween 20 (four times for 10 min); (3) incubated in anti-phospho-p38 (1:700; Cell Signaling Technology) in TBS containing 0.5% Tween 20 and 5% BSA at 4°C for 12 h; (4) washed in TBS containing 0.5% Tween 20 (four times for 10 min); (5) incubated in goat anti-rabbit-IRDye800CW antibody (Li-Cor Biotechnology) for 90 min at room temperature; (6) washed in TBS containing 0.5% Tween 20 (three times for 10 min) and TBS (two times for 10 min). The membrane was then scanned with the Li-Cor Biotechnology Odyssey imager. The fluorescence intensities of the phosphorylated p38 signals were measured, and the membrane was stripped by NewBlot Nitro Stripping Buffer (Li-Cor Biotechnology) for 4 min. Removal of antibodies was verified by rescanning of the membrane. The membrane was then reprobed as above except with rabbit anti-p38 as the primary antibody (1:1000, catalog #2831; Cell Signaling Technology). The phospho-p38 intensities were normalized to their corresponding total p38 intensities and then normalized to the control treatment (0 nm LQEQ-19) values. For immunohistochemistry, cells were grown to confluence on coverslips and, after treatment, were processed as described above for DRG cultures.
Results
Proteomic analysis of cultured sensory neurons
We postulated that, over the first 24 h after dissociation and plating, cultured adult sensory neurons undergo changes in protein expression that are similar to those occurring in axotomized neurons. Cytoplasmic and nuclear fractions obtained from DRG cultures at t0 and t24 were compared using iTRAQ-based quantification of relative abundance combined with 2D LC-MS/MS. A database search using spectra detected by 2D LC-MS/MS identified 3569 distinct peptides. These peptides were mapped to 537 proteins (≥66% confidence), of which 481 were identified with >95% confidence. Quantification of protein ratios in the four samples was based on the ratios of the iTRAQ reporter tags for each identified peptide (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). The data were further examined using IPA. Of the 537 identified proteins, 436 were annotated in the IPA Knowledge Base. The majority of these proteins (∼80%) were annotated as cytoplasmic, and only 16% were annotated as nuclear. A large number of cytoplasmic proteins was detected in the nuclear fractions, preventing adequate evaluation of changes in nuclear proteins. Therefore, subsequent analysis was focused on changes in protein levels in the cytoplasmic fractions.
The ratio cytoplasmic t24/cytoplasmic t0 indicated that the majority of quantified proteins (∼60%) were changed <1.5-fold between t0 and t24, indicating consistency of sample preparation at the two time points. The proteins that were increased more than twofold after 24 h in culture are shown in Table 1. The potential functional relevance of these proteins was inferred by an IPA network analysis-guided literature review. However, confirmation of upregulation and analysis of actual functional relevance requires validation by molecular and cellular biology methods. We focused on elucidating the role of VGF, which demonstrated the largest change in expression. The neurosecretory protein VGF was identified by a single peptide, and iTRAQ relative quantification showed a 45-fold increase in the levels of this peptide at t24, suggesting upregulation of VGF in the DRG cultures (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
Table 1.
Proteins showing more than twofold increase at t24 compared with t0
| Protein name | Fold increase | Potential functional relevance |
|---|---|---|
| Oxidative stress | ||
| Ferritin heavy polypeptide 1 | 6.68* | Protective during oxidative stress (Chiba et al., 2007). |
| Thioredoxin reductase 1 | 2.64# | Protective during oxidative stress (Xia et al., 2003); upregulated in DRG at 14 d in a model of HIV neuropathy (Maratou et al., 2009). |
| Prothymosin α | 2.56* | Regulates apoptosis and oxidative stress (Karapetian et al., 2005; Fujita and Ueda, 2007); upregulated in DRG 14 d after SNTa(Maratou et al., 2009). |
| Membrane–cytoskeletal signaling | ||
| Myristoylated alanine-rich C-kinase substrate-like 1, MARCKSL1 | 2.12# | Protein kinase C (PKC) substrate, which binds actin, calmodulin, and acidic phospholipids. |
| Brain abundant membrane attached signal protein 1 (BASP1/CAP-23) | 2.28## | PKC substrate, which binds actin, calmodulin, and acidic phospholipids; upregulated in DRG 3, 6, and 14 d after sciatic nerve crush (Mason et al., 2002). |
| Gravin/A kinase anchor protein (AKAP12) | 2.68# | Scaffold protein that interacts with PKA, PKC, and actin; expressed in unmyelinated sensory neurons (Irmen et al., 2008). |
| Tropomyosin 4 | 2.17### | Neuronal tropomyosin found in growth cones and presynaptic terminals (Had et al., 1994); upregulated 7 d after SNL (Komori et al., 2007). |
| Thymosin β4 | 2.03### | Actin-sequestering protein; promotes wound healing and repair after hypoxia (Goldstein et al., 2005); upregulated in DRG 14 d after SNT (Maratou et al., 2009). |
| Transcription regulation | ||
| APEX nuclease | 3.95# | DNA repair and regulation of transcription factors (Vasko et al., 2005; Qu et al., 2007). |
| Y box binding protein 1 (YBX1) | 3.27# | Transcription factor and translation regulator (Kohno et al., 2003); upregulated in DRG 14 d after SNT (Maratou et al., 2009). |
| Neuropeptide signaling | ||
| CGRP precursor | 5.19* | CGRP is expressed in nociceptive sensory neurons. Complex regulation in nerve injury, see Discussion. |
| Neurosecretory protein VGF | 45.35# | Putative precursor of signaling peptides; upregulated 3 d after sciatic nerve axotomy and 14 d after SNL (Costigan et al., 2002;Wang et al., 2002; Valder et al., 2003). |
| Other | ||
| Diazepam binding inhibitor (DBI) | 2.33* | Stimulation of steroid synthesis. DBI expressed in satellite cells, expression not altered 3 d after sciatic nerve transection (Karchewski et al., 2004); upregulated in DRG 14 d after SNT (Maratou et al., 2009). |
| Proteasome P20 subunit (PSMB7) | 2.13# | Protein degradation. Proteasome activity affects nerve injury induced hypersensitivity (Moss et al., 2003; Ossipov et al., 2007). |
| α-Galactosidase | 2.21## | Glycolipid and glycoprotein degradation. Mutations associated with Fabry's disease. Downregulated in DRG 14 d after SNT (Maratou et al., 2009). |
| Palmytoyl-protein thioesterase 1 | 2.38# | Lysosomal enzyme involved in degradation of lipoproteins. Mutations associated with neurodegeneration (Gardiner, 2000). |
| Acetyl-coenzyme A acetyltransferase 2 | 2.68## | Peroxysomal enzyme involved in fatty acid metabolism. |
| Cell cycle exit and neuronaldifferentiation 1 (CEND1) | 2.87## | Function described by Politis et al. (2007). |
| LSM3 homolog, U6 small nuclear RNA associated | 2.12# | Upregulated in DRG 14 d after SNT (Maratou et al., 2009). Function described by Beggs (2005). |
Proteins showing more than twofold increase at t24 compared with t0. Relative protein abundance was determined using iTRAQ combined with 2D LC-MS/MS. Fold increase was determined from ratios of individual peptides assigned to each protein by ProteinPilot. Ratios calculated from three or more peptides and with p < 0.05 are indicated by asterisk (*).
For ratios with p > 0.05, the data was verified by manual inspection as described in Materials and Methods. The table includes ratios obtained from a single peptide (indicated by #) or
the average of two (##) or
more (###) peptides.
aSNT, L5 spinal nerve transection.
VGF is upregulated in cultured sensory neurons
The increase in VGF levels in cultured DRG neurons at t24 was confirmed by Western blot analysis and immunocytochemistry (Fig. 1). Two protein bands, one of ∼90 kDa and one of ∼70 kDa were seen in homogenates from cells maintained in culture for 24 h but not in homogenates from cells that were harvested shortly after plating. Immunocytochemical analysis of the cultures indicated that the number of VGF-positive neurons, expressed as the percentage of neurons identified by the pan-neuronal marker PGP 9.5, was significantly increased at t24 compared with t0 (20 ± 4% at t0 compared with 82 ± 7% at t24; p < 0.05, t test). The quantification of the percentage of VGF-positive neurons was not confounded by a change in the total number of neurons over the 24 h period because the total number of PGP-positive cells at t0 and t24 (91 ± 7 and 86 ± 15, respectively) was not significantly different. Similar immunocytochemical results were obtained with the C-terminal VGF antiserum and the commercially available N-terminal antiserum.
VGF is upregulated in DRG after nerve injury
We predicted that changes in protein expression in cultured neurons would parallel nerve injury-induced changes in the L5 SNL model. Therefore, the expression of VGF was examined immunohistochemically in L5 DRG 24 h after SNL. As shown in Figure 2, the number of VGF-positive neurons in L5 DRG of SNL animals was significantly increased compared with sham-operated and naive animals. One day after SNL, 80% of L5 DRG neurons were VGF positive compared with 37% in sham-operated animals and 20% in naive animals. Analysis of colocalization showed overlap of VGF-IR with both TrkA and P2X3 labeling (Fig. 3). Of the VGF-positive neurons, 51.9 ± 1.4% were TrkA positive and 52.3 ± 3.6% were P2X3 positive; conversely, 90.8 ± 2.3% of the P2X3-positive and 70.7 ± 3.5% of the TrkA-positive neurons in the injured L5 DRG were also VGF positive. Size distribution analysis indicated that the subset of VGF-positive cells in injured L5 DRG included small/medium-sized and large (diameter >25 μm) neurons (Table 2). The significant increase in VGF-IR was maintained 7 d after SNL. Notably, whereas the number of small/medium-sized VGF-positive neurons at that time was decreased relative to day 1, the number of large VGF-positive neurons was slightly increased (Table 2).
Figure 2.
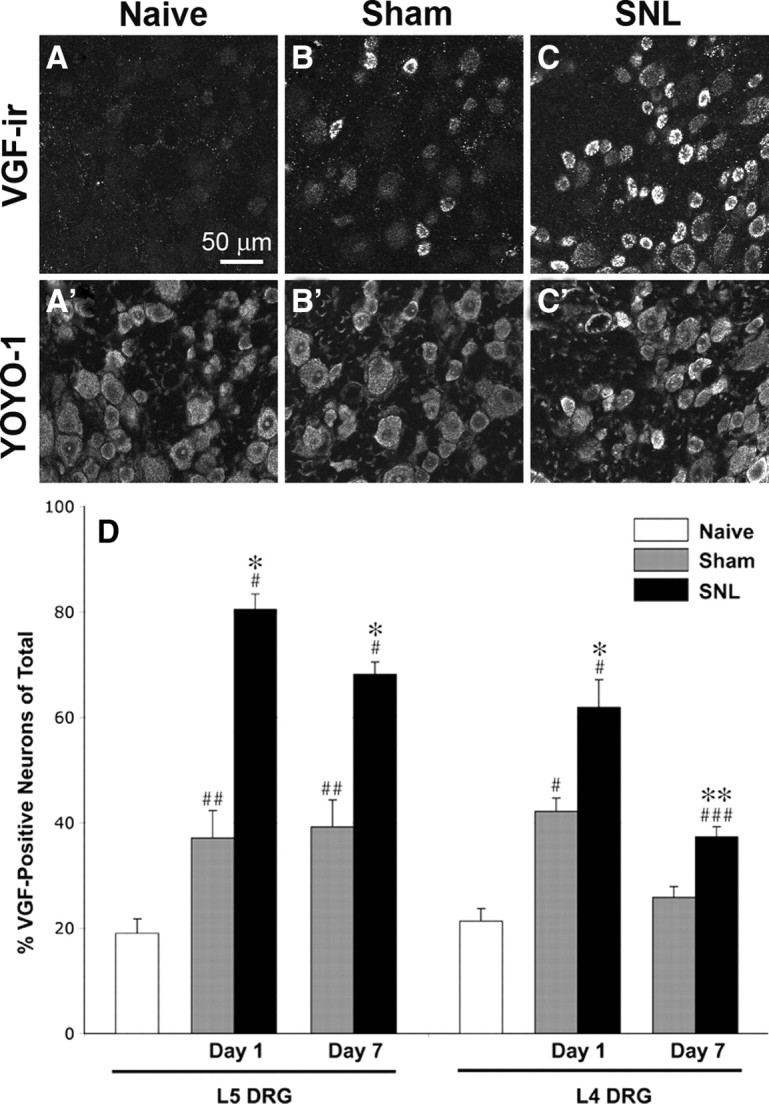
Nerve injury-induced upregulation of VGF in DRG neurons. A–C, VGF-IR increased dramatically 1 d after SNL (C) and to a lesser extent after sham surgery (B). Corresponding images of YOYO-1 labeling (A′–C′) show all cells in the fields of view shown in A–C. D, Quantitative analysis showed a significant increase in the number of VGF-positive neurons 1 and 7 d after SNL in both injured L5 and uninjured L4 DRG. Sham surgery resulted in a more moderate but significant increase in the number of VGF-positive neurons in L5 DRG at day 1 and day 7 and in L4 DRG at day 1. Significant changes are indicated by * when compared with sham and # when compared with naive (*p < 0.001; **p < 0.05; #p < 0.001; ##p < 0.005; ###p < 0.01; one-way ANOVA and Student–Newman–Keuls test for multiple comparisons; n = 4 in each group).
Figure 3.

Colocalization of VGF-IR with TrkA-IR and P2X3-IR in L5 DRG 1 d after SNL. The pie chart insets show the proportions of single-labeled VGF-positive cells (green), single-labeled TrkA-positive (A, red) or P2X3-positive (B, red) cells, and double-labeled cells (yellow). A, Colocalization of VGF-IR and TrkA-IR. The proportion of double-labeled cells (yellow) represents 51.9 ± 1.4% of the VGF-positive neurons (green + yellow) and 70.7 ± 3.5% of the TrkA-positive neurons (red + yellow). B, Colocalization of VGF-IR and P2X3-IR. The proportion of double-labeled cells (yellow) represents 52.3 ± 3.6% of the VGF-positive neurons (green + yellow) and 90.8 ± 2.3% of the TrkA-positive neurons (red + yellow).
Table 2.
Size distribution of VGF-positive neurons after SNL
| Small/medium (diameter <25 μm), of total | VGF+ neurons,% of small/medium | Large (diameter >25 μm), % of total | VGF+ % of large | |
|---|---|---|---|---|
| L5 | ||||
| Day 1 | 78.2 ± 2.1 | 86 ± 1.6* | 22.4 ± 2.3 | 59.8 ± 5.8*** |
| Day 7 | 76.9 ± 3.5 | 64.6 ± 3.3* | 20.8 ± 3.9 | 75.8 ± 3.1*** |
| L4 | ||||
| Day 1 | 65.9 ± 1.8 | 65.1 ± 5.9** | 34.7 ± 1.5 | 40.8 ± 3.3 |
| Day 7 | 65.8 ± 3.7 | 40.3 ± 3.9** | 36.5 ± 4.6 | 30.2 ± 7 |
The percentage of small/medium and large VGF-positive neurons in L5 and L4 DRG was compared between day 1 and day 7. At day 7, there was a significant decrease in the number of small/medium VGF-positive neurons in both L5 (*p < 0.005, t test) and
L4 (**p < 0.05, t test) DRG.
The number of large VGF-positive neurons at day 7 was significantly increased (***p < 0.05, one-tailed t test) in L5 DRG and decreased in L4 DRG.
In addition to the injured L5 DRG neurons, VGF was upregulated in uninjured L4 DRG neurons after SNL (Fig. 2D). The number of VGF-positive cells was significantly increased in L4 DRG both 1 and 7 d after SNL compared with sham controls and naive animals. Similar to L5 DRG, at day 1, approximately half (53.3 ± 4.4%) of the VGF-positive neurons were also TrkA positive (supplemental Table 1, available at www.jneurosci.org as supplemental material).
It was possible that, in L4 DRG of SNL animals and DRG of sham-operated animals, the VGF upregulation was restricted to neurons inadvertently injured during the surgery. To address this possibility, we examined immunohistochemically the colocalization of VGF and the nerve injury-inducible transcription factor ATF3 (Fig. 4). As described previously (Shortland et al., 2006), we found that ATF3-IR was present in virtually all L5 neurons of SNL animals as well as in a limited number of neurons within L4 and sham DRG. In the injured L5 neurons, we observed a nearly complete overlap of VGF-IR and ATF3-IR. However, in L4 DRG of SNL animals, there were many VGF-positive cells that were ATF3 negative. Similarly, in sham DRG, VGF-IR was not restricted to ATF3-IR neurons. These results show that VGF was upregulated in injured as well as uninjured neurons.
Figure 4.
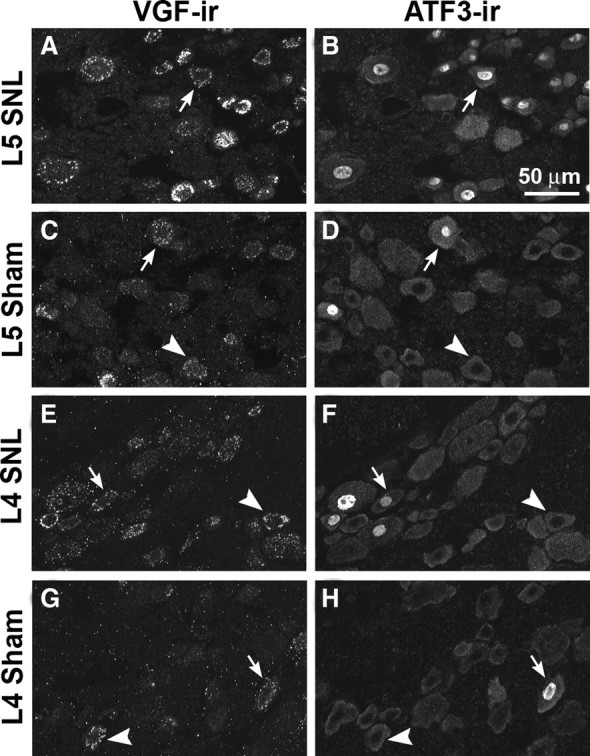
Relationship of VGF- and ATF3-IR in L5 and L4 DRG 1 d after SNL or sham surgery. In double-labeled sections, VGF- and ATF3-IR overlapped substantially in injured L5 DRG (A, B). In sham L5 DRG (C, D), in L4 DRG from SNL animals (E, F), and in sham L4 DRG (G, H), VGF-IR was present in ATF3-positive and ATF3-negative neurons. Arrows show examples of colocalization of VGF-IR and ATF3-IR. Arrowheads show examples of VGF-positive neurons without ATF3-IR in their nuclei.
VGF is upregulated in spinal cord after nerve injury
VGF-IR also increased in the superficial dorsal horn of the spinal cord after SNL. VGF immunofluorescence was measured in the superficial dorsal horn of three sections of L5 spinal cord from four different animals 3 d after SNL surgery. VGF-IR was significantly increased within the dorsal horn ipsilateral to SNL compared with sham surgery and naive animals (Fig. 5). In the same sections, there was no difference in CGRP-IR in the three experimental groups (data not shown). To determine whether VGF-IR increased in central terminals of primary afferent neurons or in intrinsic spinal cord neurons, we examined the relationship of VGF-IR to CGRP-IR, which in dorsal horn is localized exclusively in primary afferent terminals. The colocalization of VGF-IR and CGRP-IR in the superficial dorsal horn ipsilateral to SNL was increased compared with spinal cord from naive animals, consistent with elevation in VGF levels in central terminals of primary afferent neurons after SNL (Fig. 5E–J). It should be noted that the abundance of VGF-IR in spinal cord as revealed in these experiments by our C-terminal antiserum may be underestimated if differential proteolytic processing of VGF (Brancia et al., 2005) results in fragments that are not recognized by this antiserum.
Figure 5.

Changes in VGF-IR in L5 segment of spinal cord after SNL. A–C, Three days after SNL, VGF-IR appeared increased within the dorsal horn ipsilateral to SNL (A) compared with sham surgery (B) and naive animals (C). D, Quantitative analysis indicated a significant increase in VGF-IR ipsilateral to SNL compared with sham surgery and naive spinal cord (one-way ANOVA and Student–Newman–Keuls multiple comparisons test, p < 0.05; n = 4 for each group). E–J, Compared with naive spinal cord (F, H, J), VGF-IR (red) was increased after SNL (E, G, I), whereas CGRP-IR (green) appeared unchanged at this time point. The colocalization of VGF- and CGRP-IR in superficial dorsal horn was also increased after SNL as indicated by the presence of more yellow and orange puncta ipsilateral to SNL (I) compared with naive spinal cord (J).
VGF is upregulated after CFA-induced inflammation
The increased number of VGF-IR neurons in DRG from sham animals suggested that VGF might also be induced in inflammatory conditions because the sham surgery is accompanied by substantial tissue damage and inflammation. Therefore, we examined changes in VGF-IR 24 h after CFA-induced hindpaw inflammation. CFA inflammation resulted in a more modest but still significant increase in the number of VGF-expressing neurons in L5 DRG (28.2 ± 0.3% in CFA L5 DRG compared with 19.0 ± 2.8% in naive L5 DRG; p < 0.05, t test). In L5 DRG from CFA-treated animals, 65.5 ± 5.2% of the VGF-positive neurons were also TrkA positive (supplemental Table 1, available at www.jneurosci.org as supplemental material). Thus, VGF expression in sensory neurons appears to be induced by a spectrum of manipulations that includes varying degrees of inflammation and nerve injury.
VGF-derived peptides evoke thermal hyperalgesia
Previous studies have reported biological activity of the VGF C-terminal peptides TLQP-62, AQEE-30, and LQEQ-19 (Alder et al., 2003; Succu et al., 2005). To determine whether they have a role in nociceptive processing, AQEE-30 and LQEQ-19 were injected intrathecally into naive adult mice (Fig. 6). In the warm-water (49°C) tail-immersion assay, mice show baseline tail-flick latencies in the range of 6–9 s (x = 7.77 ± 0.06 s, mean ± SEM, n = 156, for data shown in Figs. 6, 7). After intrathecal delivery, AQEE-30 evoked a partial thermal hyperalgesia by 10 min, which was fully expressed by 30 min and persisted for at least 60 min and up to 90 min at the highest dose (Fig. 6, Table 3). The induction of thermal hyperalgesia was dose dependent, as evidenced by a significant difference in the magnitude of the hyperalgesia evoked by the different doses at the 30 min time point (one-way ANOVA, F(3,20) = 3.48; p < 0.05, Tukey's post hoc test). However, there was no statistical difference between the magnitudes of the thermal hyperalgesia evoked by the two highest doses (3 nmol, −1.3 ± 0.19 s; 0.3 nmol, −1.7 ± 0.34 s; p > 0.05; n = 6).
Figure 6.

VGF-derived peptides evoke thermal hyperalgesia. A, Intrathecal injection of AQEE-30 dose dependently evoked thermal hyperalgesia in mice as detected by the warm-water (49°C) tail-immersion assay. The hyperalgesia persisted for up to 60 min (0.03, 0.3, and 3 nmol) and 90 min at the highest dose (3 nmol). Data are expressed as the reduction in withdrawal latency compared with baseline for each individual mouse (delta). B, Thermal hyperalgesia was evoked by intrathecal injection of LQEQ-19, which corresponds to the C-terminal 19 aa of AQEE-30, but not by AQEE-11, which corresponds to the leading 11 aa of AQEE-30. AQEE-11 and LQEQ-19 were tested at a dose (3 nmol) equimolar to the highest dose of AQEE-30.
Figure 7.

The p38 MAPK inhibitor SB202190 dose dependently reversed AQEE-30 and LQEQ-19 evoked thermal hyperalgesia. The inhibitor was injected intrathecally as a 5 min pretreatment before injection of equieffective doses of AQEE-30 (0.3 nmol) or LQEQ-19 (1 nmol). Thermal hyperalgesia was measured 30 and 10 min after intrathecal injection of AQEE-30 and LQEQ-19, respectively. Data are expressed as percentage inhibition of the maximum thermal hyperalgesia (AQEE-30, −2.29 s; LQEQ-19, −2.43 s) detected in a separate control group pretreated with saline.
Table 3.
Impact of high intrathecal dose of peptide on tail-flick latencies (TFL)
| Peptide | n | Dose (i.t.) | Baseline TFL (s) | 10 min | 30 min | 90 min |
|---|---|---|---|---|---|---|
| AQEE-30 | 6 | 3 nmol | 7.5 s ± 0.27 | 7.2 s ± 0.15* | 6.2 s ± 0.23** | 6.5 s ± 0.48** |
| LQEQ-19 | 6 | 3 nmol | 7.4 s ± 0.47 | 5.7 s ± 0.13** | 6.0 s ± 0.67** | 7.5 s ± 0.39 |
| AQEE-11 | 6 | 3 nmol | 7.7 s ± 0.33 | 7.5 s ± 0.31 | 7.7 s ± 0.35 | 7.6 s ± 0.36 |
*p < 0.05, significant difference from baseline.
**p < 0.01, significant difference from baseline.
LQEQ-19, which corresponds to the last 19 aa of AQEE-30, evoked hyperalgesia of similar magnitude (−1.4 ± 0.43; n = 6) but with a more rapid onset to full expression and a shorter duration. In contrast, AQEE-11, which corresponds to the leading 11 aa of AQEE-30, had no effect on tail-withdrawal latency (Fig. 6, Table 3).
VGF-derived peptides activate p38 MAPK in microglia
Several pharmacological agents known to interfere with pathways involved in pain processing were evaluated for inhibition of the thermal hyperalgesia evoked by AQEE-30 and LQEQ-19. These included inhibitors of nitric oxide synthase (7-NI, 10 nmol; l-NAME, 100 nmol), NMDA receptor (MK801, 10 nmol), protein kinase C (GF109203X, 1 and 10 nmol), protein kinase A (KT5720, 1.7 nmol), and MAPK (U0126, 2.5 nmol; SB202190, 0.1, 1, and 2.5 nmol; SB600125, 2.5 nmol). Inhibition of the spinal effects of the VGF peptides was achieved only by the p38 inhibitor SB202190, which dose dependently reversed AQEE-30 (0.3 nmol) and LQEQ-19 (1 nmol) evoked thermal hyperalgesia (Fig. 7). The thermal hyperalgesia evoked by these doses was equivalent between the two peptides (AQEE-30, −2.4 ± 0.23 s; LQEQ-19, −2.3 ± 0.37 s; p > 0.05, Student's t test; n = 6 per group) and was not statistically different from the thermal hyperalgesia evoked in the previous experiment (Fig. 7) with the same doses and time points (Student's t test, p > 0.05). Administration of SB202190 alone had no effect on thermal withdrawal latencies.
Activation of p38 MAPK was examined immunohistochemically in spinal cords obtained at the time of peak hyperalgesia (10 min) from mice injected intrathecally with LQEQ-19 or saline (Fig. 8). In spinal cords from LQEQ-19-injected mice, we observed a nearly threefold increase in immunofluorescence for phosphorylated p38, measured as pixel area with staining intensity above background (percentage thresholded area, 0.36 ± 0.08 in LQEQ-19-treated compared with 0.13 ± 0.04 in saline-treated animals; p < 0.05, one-tailed t test; n = 3 for each group) (Fig. 8A–C). Immunoreactivity for phosphorylated p38 did not overlap with labeling for the neuronal marker NeuN and the astrocyte marker GFAP. However, it colocalized extensively with labeling for CD68, a marker for reactive microglia, suggesting that phosphorylated p38 was localized in spinal cord microglia (Fig. 8D–G). To determine whether LQEQ-19 has direct effects on microglia, we exposed the BV-2 microglial cell line to the peptide. This treatment resulted in the concentration-dependent increase of p38 phosphorylation in the cells (Fig. 9).
Figure 8.
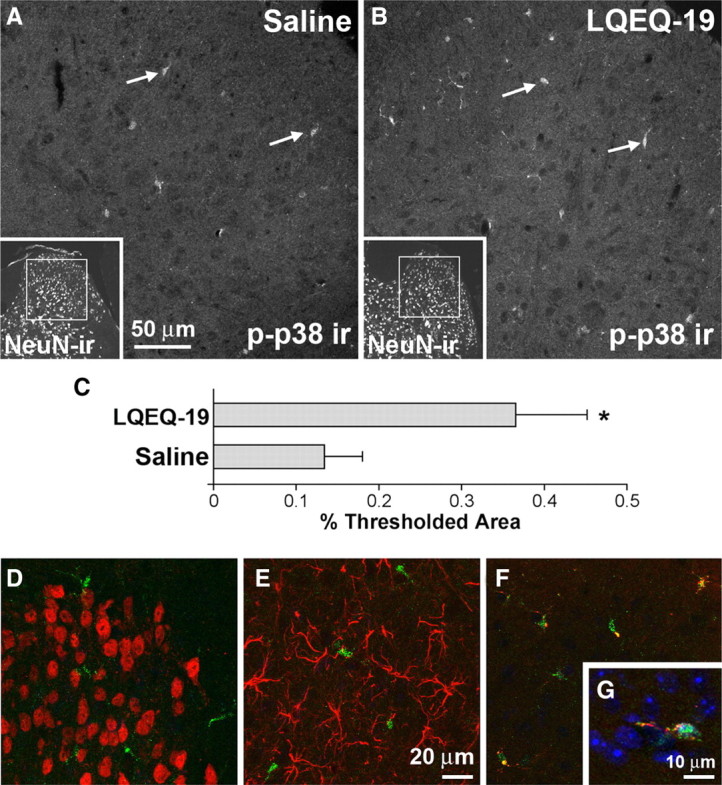
Increased phosphorylation of p38 MAPK in spinal microglia after intrathecal injection of LQEQ-19. A, B, Phospho-p38 (p-p38) immunofluorescence was increased 10 min after LQEQ-19 injection (3 nmol) compared with saline injection. Arrows show examples of phospho-p38-positive spinal microglia. Insets show NeuN staining of the same sections at lower magnification and indicate the approximate location of the high-magnification images within the dorsal horn. C, Quantitative image analysis of phospho-p38 immunofluorescence (percentage thresholded area, 0.36 ± 0.08 in LQEQ-19-treated compared with 0.13 ± 0.04 in saline-treated animals; p < 0.05, one-tailed t test; n = 3 for each group). D–F, Double labeling of spinal cord sections from LQEQ-19 injected mice for phospho-p38 (C–E, green) and NeuN (C, red), GFAP (D, red), and CD68 (E, red). Colocalization, appearing as yellow puncta in E, was seen only with anti-phospho-p38 and anti-CD68. G, A cell labeled for phospho-p38 (green), CD68 (red), and the nuclear stain DAPI (4′,6′-diamidino-2-phenylindole) (blue). Labeling for phospho-p38 is seen in the nucleus of the cell as well as in the cytoplasm, in which it overlaps with anti-CD68 staining.
Figure 9.
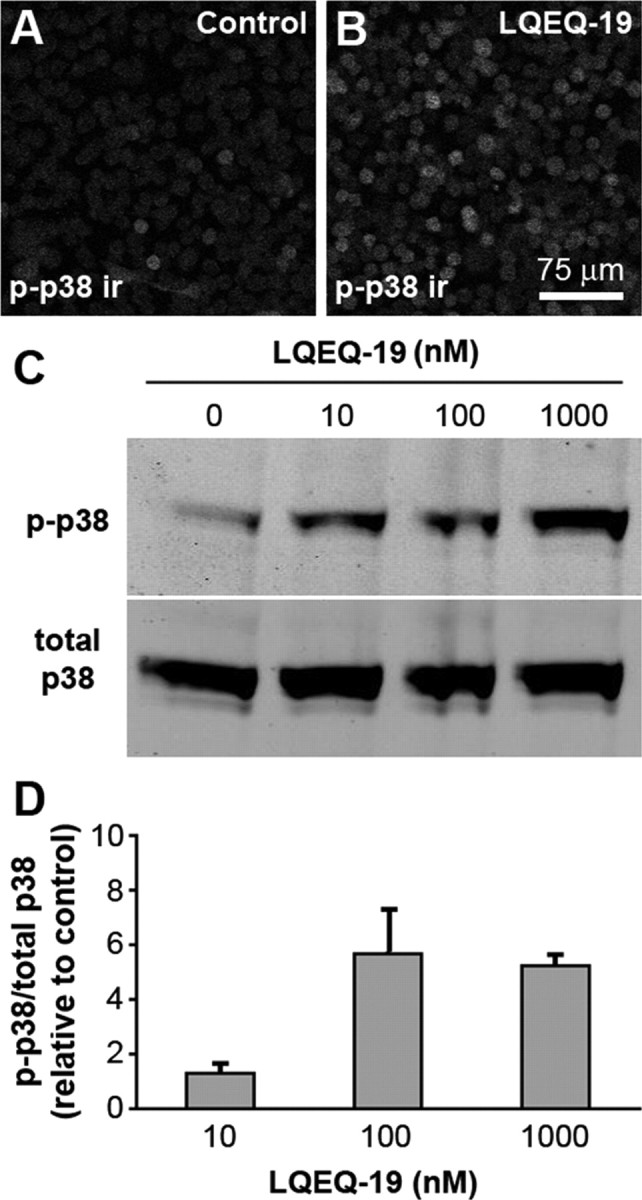
Treatment of BV-2 microglial cells with LQEQ-19 resulted in increased p38 phosphorylation. A, B, The number of phospho-p38-positive (p-p38) cells is increased after incubation of the cells with LQEQ-19 (1 μm) for 2 min. C, D, Dose-dependent increase in the relative levels of phospho-p38 after LQEQ-19 treatment. In D, relative phospho-p38 was expressed as phospho-p38/total p38 and normalized relative to control values. The data represent averages of three experiments.
Discussion
The present study contributes to the characterization of a novel signaling system within spinal cord pain pathways. The potential relevance of VGF was first inferred from proteomic differential expression analysis of an in vitro model of injured sensory neurons. Subsequent experiments provided evidence for upregulation of VGF after nerve injury and inflammation and for the functional relevance of VGF peptides to spinal nociceptive processing.
Proteomic analysis
Large-scale expression analysis has been used to study global changes under conditions of chronic pain, leading to identification of genes with uncharacterized functions in pain signaling (Costigan et al., 2002; Wang et al., 2002; Valder et al., 2003; Komori et al., 2007). We used a proteomic approach to analyze early changes in protein levels within cultured DRG neurons, which were used as a model of axotomized neurons. Several of the proteins that were increased more than twofold are associated with oxidative stress, consistent with the hypoxia accompanying preparation of primary cultures. The transcription regulators APEX1 and YBX1 have also been linked to protection from oxidative stress (Kohno et al., 2003; Vasko et al., 2005; Qu et al., 2007). Moreover, IPA network analysis showed that APEX1, YBX1, and prothymosin α are regulated by casein kinase II, suggesting that the upregulation of these proteins may reflect activation of a common network that involves this enzyme. Another group of upregulated proteins is associated with membrane– cytoskeletal signaling at growth cones and presynaptic terminals and possibly with regulation of second-messenger availability (Laux et al., 2000; Diviani and Scott, 2001; Sundaram et al., 2004). For example, MARCKSL1 and BASP1/CAP-23 may control the phosphoinositol-(4,5)biphosphate-dependent regulation of channels contributing to sensory neuron sensitization, including TRPV1 and TRPA1 (Akopian et al., 2007; Lishko et al., 2007; Lukacs et al., 2007). Our proteomic analysis also indicated upregulation of two neuropeptide precursors. The upregulation of CGRP precursor in the first 24 h of DRG culture is consistent with increases in CGRP in cultured trigeminal neurons and in some models of nerve injury (Kuris et al., 2007; Zheng et al., 2008). Previous studies have shown that, in the SNL model, the number of CGRP-positive neurons in injured ganglia was unchanged 24 h after injury (Lee et al., 2001) and substantially decreased at 7 d (Fukuoka et al., 1998; Lee et al., 2001; Hammond et al., 2004). VGF upregulation in DRG after nerve injury has been detected previously by microarray analysis (Costigan et al., 2002; Wang et al., 2002; Valder et al., 2003) and was described recently in the spared nerve injury model (Moss et al., 2008).
Upregulation of VGF after nerve injury and inflammation
The increase in VGF levels in cultured DRG neurons was confirmed by Western blot and immunocytochemical analysis. This increase was paralleled by VGF upregulation in the SNL model of nerve injury. Within 24 h of SNL, VGF-IR was present in nearly all injured L5 and over half of uninjured L4 DRG neurons. The time course and level of upregulation clearly distinguish VGF from other neuropeptides known to be increased after nerve injury (Zhang et al., 1995; Fukuoka et al., 1998; Shortland et al., 2006). For example, 1 d after SNL, galanin was present in ∼40% of injured L5 DRG neurons and NPY was nearly absent, whereas the levels of both peptides were unchanged in uninjured L4 neurons (Shortland et al., 2006). VGF labeling colocalized with both TrkA and P2X3 labeling, suggesting that the factors governing VGF induction are not restricted to the NGF- or GDNF-dependent subsets of sensory neurons. The rapid increase in VGF levels after SNL in both injured and uninjured sensory neurons and its persistence 7 d after the nerve injury are consistent with involvement of VGF in both the development and maintenance of nerve-injury-induced hypersensitivity. Within 24 h of SNL, injured myelinated and uninjured unmyelinated fibers develop spontaneous activity (for review, see Ringkamp and Meyer, 2005). The increased VGF expression in injured large neurons and uninjured small neurons suggests that increased release of VGF peptides from spontaneously active fibers may contribute to abnormal nociceptive processing in dorsal horn during the development of hypersensitivity. Moreover, VGF upregulation in large neurons persists 7 d after injury, consistent with continued contribution of large afferents to the maintenance of hypersensitivity (Ossipov et al., 2000; Sun et al., 2001).
Because VGF is a putative neuropeptide precursor (Levi et al., 2004), VGF-derived peptides are likely transported to the central terminals of DRG neurons. Therefore, VGF upregulation in DRG after SNL was expected to result in increased levels of VGF-IR within the superficial dorsal horn of spinal cord. Quantitative analysis in spinal cord demonstrated a significant increase in VGF-IR ipsilateral to SNL, and the increased colocalization of VGF- and CGRP-IR in superficial dorsal horn is consistent with elevated VGF levels within central terminals of sensory neurons. Therefore, increased spinal release of VGF peptides from sensory neurons may provide an early signal for peripheral nerve injury. Increased VGF expression after nerve injury has also been shown in spinal neurons (Moss et al., 2008), suggesting additional functions of VGF peptides in spinal plasticity.
The number of VGF-IR DRG neurons also increased after sham surgery but to a level significantly lower than SNL. This increase is likely attributable to tissue damage at the surgical site. The sham surgery includes removal of the vertebral transverse process and overlaying muscles, causing inflammation and injury to fibers innervating these tissues. Consistent with tissue damage after sham surgery, ATF3 expression was reported in 40% of sham L5 DRG 1 and 14 d after surgery (Shortland et al., 2006). We also observed an inflammation-induced increase in the number of VGF-positive neurons after hindpaw injection of CFA. Together, these results suggest that VGF expression in sensory neurons may be regulated by a spectrum of manipulations, which include varying degrees of inflammation and nerve injury.
VGF peptides evoke thermal hyperalgesia through activation of microglial p38 MAPK
The 617 aa sequence of VGF contains nearly a dozen potential cleavage sites. Functional effects have been reported for several potential proteolytic products contained within the C-terminal 62 aa portion of VGF (Alder et al., 2003; Succu et al., 2005). Recently, TLPQ-21, a VGF-derived peptide located immediately upstream from AQEE-30 within the VGF sequence, was shown to modulate inflammatory pain (Rizzi et al., 2008), and TLPQ-62, a peptide that includes both TLPQ-21 and AQEE-30, produced mechanical allodynia when administered intrathecally in rat (Moss et al., 2008). Our observations of dose-dependent thermal hyperalgesia evoked by intrathecal injection of AQEE-30 and LQEQ-19 provide additional evidence for a role of VGF peptides in nociceptive processing. Notably, the hyperalgesia evoked by VGF peptides exceeded in magnitude and duration the effect of NMDA in the same experimental paradigm (Kitto et al., 1992; Roberts et al., 2005). Although the tail-withdrawal paradigm used in our studies is a measure of acute hyperalgesia rather than hypersensitivity associated with persistent pain, Moss et al. (2008) demonstrated TLPQ-62-induced mechanical and cold allodynia, consistent with a role of VGF peptides in nerve injury-induced hypersensitivity.
The signaling mechanisms of VGF-derived peptides are completely uncharacterized. Therefore, we examined several signaling pathways for their involvement in the spinal effects of AQEE-30 and LQEQ-19. The thermal hyperalgesia was inhibited dose dependently by the potent and selective p38 MAPK inhibitor SB202190. The activation of p38 by intrathecal injection of LQEQ-19 was confirmed by immunohistochemistry. LQEQ-19-dependent p38 phosphorylation was seen predominantly in spinal microglia, and LQEQ-19 induced p38 phosphorylation in the microglial cell line BV-2. These findings suggest that AQEE-30 and LQEQ-19 may act directly on spinal microglia and contribute to microglial activation. It remains to be determined whether the longer C-terminal peptide TLPQ-62 also mediates p38 activation in spinal microglia.
Spinal microglia participate in the development of inflammatory and nerve injury-induced hypersensitivity in part through the activation of p38. Several mediators have been implicated in the mechanisms of p38 activation, including TNFα, MCP-1, and fractalkine (Svensson et al., 2005; Zhang and De Koninck, 2006; Clark et al., 2007; Zhang et al., 2007; Zhuang et al., 2007; Milligan et al., 2008). Our results suggest that VGF peptides released from sensory neurons may also participate in activation of spinal microglia. Under normal conditions, VGF levels in sensory neurons are low, suggesting limited contribution to acute pain signaling. After nerve injury or inflammation, the combination of rapid VGF upregulation and increased excitability of sensory neurons is likely to potentiate the release of VGF peptides within the dorsal horn, in which they would be able to activate microglial p38.
It is presently unclear which functionally relevant VGF peptides are produced in vivo in sensory neurons and spinal cord. Candidates include TLPQ-62 and shorter fragments derived from it (i.e., TLPQ-21, AQEE-30, and LQEQ-19). The excitatory effects of TLPQ-62 on dorsal horn neurons may be underlined by microglia-dependent sensitization after action of the C-terminal portion of TLPQ-62 (i.e., AQEE-30 and LQEQ-19) as well as by activity of its N terminus (i.e., TLPQ-21), analogous to the effects in hippocampal slices, which were not mimicked by the C-terminal peptides (Bozdagi et al., 2008; Moss et al., 2008). The pattern of VGF processing can vary among different cell types and sometimes even within the same cell type (Brancia et al., 2005). Therefore, differential proteolytic processing of VGF within sensory neurons after different types of tissue damage may generate signaling peptides with distinct contributions to spinal pain mechanisms.
In conclusion, this study contributes to the increasing body of evidence for a role of VGF in neuroplasticity and provides the first description of a signaling pathway activated by VGF peptides. Our results suggest a novel action of VGF peptides as potential neuro-glial messengers after nerve injury and inflammation, a discovery driven by proteomic differential expression analysis and validated through anatomical and functional experiments.
Footnotes
This work was supported by National Institute on Drug Abuse/National Institutes of Health Grants K01 DA017236 (L.V.) and T32 DA07234 (P.D.B.). We thank Galina Kalyuzhnaya and Kristin Krebs for technical assistance, Dr. Jonathan Godbout for his kind gift from BV-2 cells, and Drs. Robert Elde, Alice Larson, Virginia Seybold, and Laura Stone for valuable insights. We are also grateful to Dr. LeeAnn Higgins and the Center for Mass Spectrometry and Proteomics for assistance with proteomic data collection.
References
- Akopian AN, Ruparel NB, Jeske NA, Hargreaves KM. Transient receptor potential TRPA1 channel desensitization in sensory neurons is agonist dependent and regulated by TRPV1-directed internalization. J Physiol. 2007;583:175–193. doi: 10.1113/jphysiol.2007.133231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alder J, Thakker-Varia S, Bangasser DA, Kuroiwa M, Plummer MR, Shors TJ, Black IB. Brain-derived neurotrophic factor-induced gene expression reveals novel actions of VGF in hippocampal synaptic plasticity. J Neurosci. 2003;23:10800–10808. doi: 10.1523/JNEUROSCI.23-34-10800.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle DL, Jones TL, Hammaker D, Svensson CI, Rosengren S, Albani S, Sorkin L, Firestein GS. Regulation of peripheral inflammation by spinal p38 MAP kinase in rats. PLoS Med. 2006;3 doi: 10.1371/journal.pmed.0030338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozdagi O, Rich E, Tronel S, Sadahiro M, Patterson K, Shapiro ML, Alberini CM, Huntley GW, Salton SR. The neurotrophin-inducible gene Vgf regulates hippocampal function and behavior through a brain-derived neurotrophic factor-dependent mechanism. J Neurosci. 2008;28:9857–9869. doi: 10.1523/JNEUROSCI.3145-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brancia C, Nicolussi P, Cappai P, La Corte G, Possenti R, Ferri GL. Differential expression and seasonal modulation of VGF peptides in sheep pituitary. J Endocrinol. 2005;186:97–107. doi: 10.1677/joe.1.05992. [DOI] [PubMed] [Google Scholar]
- Campbell JN, Meyer RA. Mechanisms of neuropathic pain. Neuron. 2006;52:77–92. doi: 10.1016/j.neuron.2006.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Clark AK, Yip PK, Grist J, Gentry C, Staniland AA, Marchand F, Dehvari M, Wotherspoon G, Winter J, Ullah J, Bevan S, Malcangio M. Inhibition of spinal microglial cathepsin S for the reversal of neuropathic pain. Proc Natl Acad Sci U S A. 2007;104:10655–10660. doi: 10.1073/pnas.0610811104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costigan M, Befort K, Karchewski L, Griffin RS, D'Urso D, Allchorne A, Sitarski J, Mannion JW, Pratt RE, Woolf CJ. Replicate high-density rat genome oligonucleotide microarrays reveal hundreds of regulated genes in the dorsal root ganglion after peripheral nerve injury. BMC Neurosci. 2002;3:16. doi: 10.1186/1471-2202-3-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diviani D, Scott JD. AKAP signaling complexes at the cytoskeleton. J Cell Sci. 2001;114:1431–1437. doi: 10.1242/jcs.114.8.1431. [DOI] [PubMed] [Google Scholar]
- Fukuoka T, Tokunaga A, Kondo E, Miki K, Tachibana T, Noguchi K. Change in mRNAs for neuropeptides and the GABA(A) receptor in dorsal root ganglion neurons in a rat experimental neuropathic pain model. Pain. 1998;78:13–26. doi: 10.1016/S0304-3959(98)00111-0. [DOI] [PubMed] [Google Scholar]
- Gabra BH, Kessler FK, Ritter JK, Dewey WL, Smith FL. Decrease in N-methyl-d-aspartic acid receptor-NR2B subunit levels by intrathecal short-hairpin RNA blocks group I metabotropic glutamate receptor-mediated hyperalgesia. J Pharmacol Exp Ther. 2007;322:186–194. doi: 10.1124/jpet.107.120071. [DOI] [PubMed] [Google Scholar]
- Goldstein AL, Hannappel E, Kleinman HK. Thymosin beta4: actin-sequestering protein moonlights to repair injured tissues. Trends Mol Med. 2005;11:421–429. doi: 10.1016/j.molmed.2005.07.004. [DOI] [PubMed] [Google Scholar]
- Hammond DL, Ackerman L, Holdsworth R, Elzey B. Effects of spinal nerve ligation on immunohistochemically identified neurons in the L4 and L5 dorsal root ganglia of the rat. J Comp Neurol. 2004;475:575–589. doi: 10.1002/cne.20209. [DOI] [PubMed] [Google Scholar]
- Hunsberger JG, Newton SS, Bennett AH, Duman CH, Russell DS, Salton SR, Duman RS. Antidepressant actions of the exercise-regulated gene VGF. Nat Med. 2007;13:1476–1482. doi: 10.1038/nm1669. [DOI] [PubMed] [Google Scholar]
- Hylden JL, Wilcox GL. Intrathecal substance P elicits a caudally-directed biting and scratching behavior in mice. Brain Res. 1981;217:212–215. doi: 10.1016/0006-8993(81)90203-1. [DOI] [PubMed] [Google Scholar]
- Inoue M, Ueda H. Protein kinase C-mediated acute tolerance to peripheral mu-opioid analgesia in the bradykinin-nociception test in mice. J Pharmacol Exp Ther. 2000;293:662–669. [PubMed] [Google Scholar]
- Irmen CP, Siegel SM, Carr PA. Localization of SSeCKS in unmyelinated primary sensory neurons. J Brachial Plex Peripher Nerve Inj. 2008;3:8. doi: 10.1186/1749-7221-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin SX, Zhuang ZY, Woolf CJ, Ji RR. p38 mitogen-activated protein kinase is activated after a spinal nerve ligation in spinal cord microglia and dorsal root ganglion neurons and contributes to the generation of neuropathic pain. J Neurosci. 2003;23:4017–4022. doi: 10.1523/JNEUROSCI.23-10-04017.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karchewski LA, Gratto KA, Wetmore C, Verge VM. Dynamic patterns of BDNF expression in injured sensory neurons: differential modulation by NGF and NT-3. Eur J Neurosci. 2002;16:1449–1462. doi: 10.1046/j.1460-9568.2002.02205.x. [DOI] [PubMed] [Google Scholar]
- Khasabova IA, Harding-Rose C, Simone DA, Seybold VS. Differential effects of CB1 and opioid agonists on two populations of adult rat dorsal root ganglion neurons. J Neurosci. 2004;24:1744–1753. doi: 10.1523/JNEUROSCI.4298-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Chung JM. An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain. 1992;50:355–363. doi: 10.1016/0304-3959(92)90041-9. [DOI] [PubMed] [Google Scholar]
- Kitto KF, Haley JE, Wilcox GL. Involvement of nitric oxide in spinally mediated hyperalgesia in the mouse. Neurosci Lett. 1992;148:1–5. doi: 10.1016/0304-3940(92)90790-e. [DOI] [PubMed] [Google Scholar]
- Kohno K, Izumi H, Uchiumi T, Ashizuka M, Kuwano M. The pleiotropic functions of the Y-box-binding protein, YB-1. Bioessays. 2003;25:691–698. doi: 10.1002/bies.10300. [DOI] [PubMed] [Google Scholar]
- Komatsu T, Sakurada C, Sasaki M, Sanai K, Tsuzuki M, Bagetta G, Sakurada S, Sakurada T. Extracellular signal-regulated kinase (ERK) and nitric oxide synthase mediate intrathecal morphine-induced nociceptive behavior. Neuropharmacology. 2007;52:1237–1243. doi: 10.1016/j.neuropharm.2007.01.003. [DOI] [PubMed] [Google Scholar]
- Komori N, Takemori N, Kim HK, Singh A, Hwang SH, Foreman RD, Chung K, Chung JM, Matsumoto H. Proteomics study of neuropathic and nonneuropathic dorsal root ganglia: altered protein regulation following segmental spinal nerve ligation injury. Physiol Genomics. 2007;29:215–230. doi: 10.1152/physiolgenomics.00255.2006. [DOI] [PubMed] [Google Scholar]
- Kuris A, Xu CB, Zhou MF, Tajti J, Uddman R, Edvinsson L. Enhanced expression of CGRP in rat trigeminal ganglion neurons during cell and organ culture. Brain Res. 2007;1173:6–13. doi: 10.1016/j.brainres.2007.07.073. [DOI] [PubMed] [Google Scholar]
- Laux T, Fukami K, Thelen M, Golub T, Frey D, Caroni P. GAP43, MARCKS, and CAP23 modulate PI(4,5)P(2) at plasmalemmal rafts, and regulate cell cortex actin dynamics through a common mechanism. J Cell Biol. 2000;149:1455–1472. doi: 10.1083/jcb.149.7.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WT, Sohn MK, Park SH, Ahn SK, Lee JE, Park KA. Studies on the changes of c-fos protein in spinal cord and neurotransmitter in dorsal root ganglion of the rat with an experimental peripheral neuropathy. Yonsei Med J. 2001;42:30–40. doi: 10.3349/ymj.2001.42.1.30. [DOI] [PubMed] [Google Scholar]
- Levi A, Ferri GL, Watson E, Possenti R, Salton SR. Processing, distribution, and function of VGF, a neuronal and endocrine peptide precursor. Cell Mol Neurobiol. 2004;24:517–533. doi: 10.1023/b:cemn.0000023627.79947.22. [DOI] [PubMed] [Google Scholar]
- Li KW, Miller S, Klychnikov O, Loos M, Stahl-Zeng J, Spijker S, Mayford M, Smit AB. Quantitative proteomics and protein network analysis of hippocampal synapses of CaMKIIalpha mutant mice. J Proteome Res. 2007;6:3127–3133. doi: 10.1021/pr070086w. [DOI] [PubMed] [Google Scholar]
- Lishko PV, Procko E, Jin X, Phelps CB, Gaudet R. The ankyrin repeats of TRPV1 bind multiple ligands and modulate channel sensitivity. Neuron. 2007;54:905–918. doi: 10.1016/j.neuron.2007.05.027. [DOI] [PubMed] [Google Scholar]
- Lukacs V, Thyagarajan B, Varnai P, Balla A, Balla T, Rohacs T. Dual regulation of TRPV1 by phosphoinositides. J Neurosci. 2007;27:7070–7080. doi: 10.1523/JNEUROSCI.1866-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund TC, Anderson LB, McCullar V, Higgins L, Yun GH, Grzywacz B, Verneris MR, Miller JS. iTRAQ is a useful method to screen for membrane-bound proteins differentially expressed in human natural killer cell types. J Proteome Res. 2007;6:644–653. doi: 10.1021/pr0603912. [DOI] [PubMed] [Google Scholar]
- Maratou K, Wallace VC, Hasnie FS, Okuse K, Hosseini R, Jina N, Blackbeard J, Pheby T, Orengo C, Dickenson AH, McMahon SB, Rice AS. Comparison of dorsal root ganglion gene expression in rat models of traumatic and HIV-associated neuropathic pain. Eur J Pain. 2009;13:387–398. doi: 10.1016/j.ejpain.2008.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason MR, Lieberman AR, Grenningloh G, Anderson PN. Transcriptional upregulation of SCG10 and CAP-23 is correlated with regeneration of the axons of peripheral and central neurons in vivo. Mol Cell Neurosci. 2002;20:595–615. doi: 10.1006/mcne.2002.1140. [DOI] [PubMed] [Google Scholar]
- Milligan ED, Sloane EM, Watkins LR. Glia in pathological pain: a role for fractalkine. J Neuroimmunol. 2008;198:113–120. doi: 10.1016/j.jneuroim.2008.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss A, Ingram R, Koch S, Theodorou A, Low L, Baccei M, Hathway GJ, Costigan M, Salton SR, Fitzgerald M. Origins, actions and dynamic expression patterns of the neuropeptide VGF in rat peripheral and central sensory neurones following peripheral nerve injury. Mol Pain. 2008;4:62. doi: 10.1186/1744-8069-4-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossipov MH, Lai J, Malan TP, Jr, Porreca F. Spinal and supraspinal mechanisms of neuropathic pain. Ann N Y Acad Sci. 2000;909:12–24. doi: 10.1111/j.1749-6632.2000.tb06673.x. [DOI] [PubMed] [Google Scholar]
- Qu J, Liu GH, Huang B, Chen C. Nitric oxide controls nuclear export of APE1/Ref-1 through S-nitrosation of cysteines 93 and 310. Nucleic Acids Res. 2007;35:2522–2532. doi: 10.1093/nar/gkl1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghavendra V, Tanga FY, DeLeo JA. Complete Freunds adjuvant-induced peripheral inflammation evokes glial activation and proinflammatory cytokine expression in the CNS. Eur J Neurosci. 2004;20:467–473. doi: 10.1111/j.1460-9568.2004.03514.x. [DOI] [PubMed] [Google Scholar]
- Ringkamp M, Meyer RA. Injured versus uninjured afferents: Who is to blame for neuropathic pain? Anesthesiology. 2005;103:221–223. doi: 10.1097/00000542-200508000-00002. [DOI] [PubMed] [Google Scholar]
- Rizzi R, Bartolomucci A, Moles A, D'Amato F, Sacerdote P, Levi A, La Corte G, Ciotti MT, Possenti R, Pavone F. The VGF-derived peptide TLQP-21: a new modulatory peptide for inflammatory pain. Neurosci Lett. 2008;441:129–133. doi: 10.1016/j.neulet.2008.06.018. [DOI] [PubMed] [Google Scholar]
- Roberts JC, Grocholski BM, Kitto KF, Fairbanks CA. Pharmacodynamic and pharmacokinetic studies of agmatine after spinal administration in the mouse. J Pharmacol Exp Ther. 2005;314:1226–1233. doi: 10.1124/jpet.105.086173. [DOI] [PubMed] [Google Scholar]
- Sakurada S, Orito T, Sakurada C, Sato T, Hayashi T, Mobarakeh JI, Yanai K, Onodera K, Watanabe T, Sakurada T. Possible involvement of tachykinin NK(1) and NMDA receptors in histamine-induced hyperalgesia in mice. Eur J Pharmacol. 2002;434:29–34. doi: 10.1016/s0014-2999(01)01523-0. [DOI] [PubMed] [Google Scholar]
- Shilov IV, Seymour SL, Patel AA, Loboda A, Tang WH, Keating SP, Hunter CL, Nuwaysir LM, Schaeffer DA. The Paragon Algorithm, a next generation search engine that uses sequence temperature values and feature probabilities to identify peptides from tandem mass spectra. Mol Cell Proteomics. 2007;6:1638–1655. doi: 10.1074/mcp.T600050-MCP200. [DOI] [PubMed] [Google Scholar]
- Shortland PJ, Baytug B, Krzyzanowska A, McMahon SB, Priestley JV, Averill S. ATF3 expression in L4 dorsal root ganglion neurons after L5 spinal nerve transection. Eur J Neurosci. 2006;23:365–373. doi: 10.1111/j.1460-9568.2005.04568.x. [DOI] [PubMed] [Google Scholar]
- Succu S, Mascia MS, Melis T, Sanna F, Melis MR, Possenti R, Argiolas A. Pro-VGF-derived peptides induce penile erection in male rats: Involvement of paraventricular nitric oxide. Neuropharmacology. 2005;49:1017–1025. doi: 10.1016/j.neuropharm.2005.05.015. [DOI] [PubMed] [Google Scholar]
- Sun H, Ren K, Zhong CM, Ossipov MH, Malan TP, Lai J, Porreca F. Nerve injury-induced tactile allodynia is mediated via ascending spinal dorsal column projections. Pain. 2001;90:105–111. doi: 10.1016/s0304-3959(00)00392-4. [DOI] [PubMed] [Google Scholar]
- Sundaram M, Cook HW, Byers DM. The MARCKS family of phospholipid binding proteins: regulation of phospholipase D and other cellular components. Biochem Cell Biol. 2004;82:191–200. doi: 10.1139/o03-087. [DOI] [PubMed] [Google Scholar]
- Svensson CI, Marsala M, Westerlund A, Calcutt NA, Campana WM, Freshwater JD, Catalano R, Feng Y, Protter AA, Scott B, Yaksh TL. Activation of p38 mitogen-activated protein kinase in spinal microglia is a critical link in inflammation-induced spinal pain processing. J Neurochem. 2003;86:1534–1544. doi: 10.1046/j.1471-4159.2003.01969.x. [DOI] [PubMed] [Google Scholar]
- Svensson CI, Schäfers M, Jones TL, Powell H, Sorkin LS. Spinal blockade of TNF blocks spinal nerve ligation-induced increases in spinal P-p38. Neurosci Lett. 2005;379:209–213. doi: 10.1016/j.neulet.2004.12.064. [DOI] [PubMed] [Google Scholar]
- Thakker-Varia S, Krol JJ, Nettleton J, Bilimoria PM, Bangasser DA, Shors TJ, Black IB, Alder J. The neuropeptide VGF produces antidepressant-like behavioral effects and enhances proliferation in the hippocampus. J Neurosci. 2007;27:12156–12167. doi: 10.1523/JNEUROSCI.1898-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuda M, Mizokoshi A, Shigemoto-Mogami Y, Koizumi S, Inoue K. Activation of p38 mitogen-activated protein kinase in spinal hyperactive microglia contributes to pain hypersensitivity following peripheral nerve injury. Glia. 2004;45:89–95. doi: 10.1002/glia.10308. [DOI] [PubMed] [Google Scholar]
- Tsuda M, Inoue K, Salter MW. Neuropathic pain and spinal microglia: a big problem from molecules in “small” glia. Trends Neurosci. 2005;28:101–107. doi: 10.1016/j.tins.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Valder CR, Liu JJ, Song YH, Luo ZD. Coupling gene chip analyses and rat genetic variances in identifying potential target genes that may contribute to neuropathic allodynia development. J Neurochem. 2003;87:560–573. doi: 10.1046/j.1471-4159.2003.02016.x. [DOI] [PubMed] [Google Scholar]
- Vasko MR, Guo C, Kelley MR. The multifunctional DNA repair/redox enzyme Ape1/Ref-1 promotes survival of neurons after oxidative stress. DNA Repair (Amst) 2005;4:367–379. doi: 10.1016/j.dnarep.2004.11.006. [DOI] [PubMed] [Google Scholar]
- Vulchanova L, Riedl MS, Shuster SJ, Buell G, Surprenant A, North RA, Elde R. Immunohistochemical study of the P2X2 and P2X3 receptor subunits in rat and monkey sensory neurons and their central terminals. Neuropharmacology. 1997;36:1229–1242. doi: 10.1016/s0028-3908(97)00126-3. [DOI] [PubMed] [Google Scholar]
- Wang H, Sun H, Della Penna K, Benz RJ, Xu J, Gerhold DL, Holder DJ, Koblan KS. Chronic neuropathic pain is accompanied by global changes in gene expression and shares pathobiology with neurodegenerative diseases. Neuroscience. 2002;114:529–546. doi: 10.1016/s0306-4522(02)00341-x. [DOI] [PubMed] [Google Scholar]
- Wenk HN, Brederson JD, Honda CN. Morphine directly inhibits nociceptors in inflamed skin. J Neurophysiol. 2006;95:2083–2097. doi: 10.1152/jn.00394.2005. [DOI] [PubMed] [Google Scholar]
- White FA, Jung H, Miller RJ. Chemokines and the pathophysiology of neuropathic pain. Proc Natl Acad Sci U S A. 2007;104:20151–20158. doi: 10.1073/pnas.0709250104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf CJ, Costigan M. Transcriptional and posttranslational plasticity and the generation of inflammatory pain. Proc Natl Acad Sci U S A. 1999;96:7723–7730. doi: 10.1073/pnas.96.14.7723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu HE, Sun HS, Cheng CW, Tseng LF. p38 mitogen-activated protein kinase inhibitor SB203580 reverses the antianalgesia induced by dextro-morphine or morphine in the mouse spinal cord. Eur J Pharmacol. 2006;550:91–94. doi: 10.1016/j.ejphar.2006.08.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, De Koninck Y. Spatial and temporal relationship between monocyte chemoattractant protein-1 expression and spinal glial activation following peripheral nerve injury. J Neurochem. 2006;97:772–783. doi: 10.1111/j.1471-4159.2006.03746.x. [DOI] [PubMed] [Google Scholar]
- Zhang J, Shi XQ, Echeverry S, Mogil JS, De Koninck Y, Rivest S. Expression of CCR2 in both resident and bone marrow-derived microglia plays a critical role in neuropathic pain. J Neurosci. 2007;27:12396–12406. doi: 10.1523/JNEUROSCI.3016-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Shi TJ, Ji RR, Zhang YZ, Sundler F, Hannibal J, Fahrenkrug J, Hökfelt T, Zhang Y. Expression of pituitary adenylate cyclase-activating polypeptide in dorsal root ganglia following axotomy: time course and coexistence. Brain Res. 1995;705:149–158. doi: 10.1016/0006-8993(95)01150-1. [DOI] [PubMed] [Google Scholar]
- Zheng LF, Wang R, Xu YZ, Yi XN, Zhang JW, Zeng ZC. Calcitonin gene-related peptide dynamics in rat dorsal root ganglia and spinal cord following different sciatic nerve injuries. Brain Res. 2008;1187:20–32. doi: 10.1016/j.brainres.2007.10.044. [DOI] [PubMed] [Google Scholar]
- Zhuang ZY, Kawasaki Y, Tan PH, Wen YR, Huang J, Ji RR. Role of the CX3CR1/p38 MAPK pathway in spinal microglia for the development of neuropathic pain following nerve injury-induced cleavage of fractalkine. Brain Behav Immun. 2007;21:642–651. doi: 10.1016/j.bbi.2006.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]