

Abstract
Background and purpose:
Theoretically, three α1-adrenoceptor subtypes can interact at the signalling level to alter vascular contraction or at the molecular level to alter each other's cellular location. The α1A/B-adrenoceptor knockout mouse (α1A/B-KO) was used to study the isolated α1D-adrenoceptor to consider these potential interactions in native tissue.
Experimental approach:
Pharmacological analysis of carotid and mesenteric arteries employed wire myography and fluorescent ligand binding (α1-adrenoceptor ligand BODIPY FL-prazosin, QAPB).
Key results:
α1A/B-KO carotid had clear α1D-adrenoceptor-induced contractions. In WT carotid α1D-adrenoceptor dominated but all three α1-subtypes participated. α1A/B-KO mesenteric had α1D-adrenoceptor responses with high sensitivity and small maximum, explaining how α1D-adrenoceptor could determine agonist sensitivity in WT. In both arteries α1A/B-KO fluorescence levels were reduced but pharmacologically more consistent with ‘pure’α1D-adrenoceptors. α1D-Adrenoceptor binding in α1A/B-KO was observed on the cell surface and intracellularly and was present in a high proportion of smooth-muscle cells in both strains, regardless of artery type.
Conclusions and implications:
‘Pure’α1D-adrenoceptor pharmacology in α1A/B-KO provides a quantitative standard. Functionally, the α1D- and α1A-adrenoceptors produce additive responses and do not significantly compensate for each other. α1D-Adrenoceptor contributes to sensitivity even in resistance arteries. In α1A/B-KO, the loss of α1A- and α1B-adrenoceptors is reflected by a general decrease in fluorescence, but similar binding distribution to WT indicates that the α1D-adrenoceptor location in native smooth-muscle cells is not influenced by other α1-adrenoceptors. Equivalent levels of receptors did not correspond to equivalent responses. In conclusion, α1-subtypes do not interact but provide independent alternative signals for vascular regulation.
Keywords: vascular smooth muscle, adrenoceptor, α1A/B-adrenoceptor knockout, wire myography, fluorescent ligand binding, confocal microscopy
Introduction
Three α1-adrenoceptor subtypes (α1A, α1B and α1D) exist, as defined by pharmacological analysis and molecular cloning (Alexander et al., 2008). With more than one receptor present in each tissue (Hrometz et al., 1999; Daly et al., 2002; Deighan et al., 2005) and the limited number of highly selective compounds available, the analysis of the α1-adrenoceptors in native tissue, and ultimately their role in adrenergic regulation of blood pressure, is complex.
α1D-Adrenoceptor-mediated contraction has been examined previously in arteries in which it predominates, such as the mouse carotid (Daly et al., 2002; Deighan et al., 2005) and aorta (Yamamoto and Koike, 2001; Daly et al., 2002). However, the presence of the other α1-subtypes may influence potency estimates of agonists and antagonists in these preparations. In arteries in which another α1-subtype predominates, the contribution of the α1D-adrenoceptor can be obscured. By isolating the α1D-adrenoceptor in the double α1A-/α1B-adrenoceptor knockout mouse (α1A/B-KO), the present study will examine its role as a ‘minor’ subtype in the mesenteric artery, in addition to obtaining potency values for agonists and antagonists at the dominant α1D-adrenoceptor in the carotid.
Some studies of receptor localization in isolated cells have found that, while receptors are found both on the external plasma membrane and in the endoplasmic reticulum, the main cellular location of the α1A- and α1D-adrenoceptors is intracellular (Mackenzie et al., 2000; Chalothorn et al. 2002; Hague et al. 2004; Wright et al. 2008) and the α1B-adrenoceptor is predominantly found on the cell surface (McCune et al., 2000; Hague et al., 2004). However, other studies in isolated cells have shown that all three α1-subtypes are located at both sites with no apparent differences (Daly et al., 1998; Hrometz et al., 1999; Pediani et al., 2000; Morris et al., 2004). Thus, there is no obvious guide as to what to expect in native tissues. We recently showed that receptor localization can be studied in native arterial tissue by using fluorescent ligand binding and that, in live, unfixed arteries from the α1B/D-KO, the α1A-adrenoceptor was present on the cell surface and intracellularly (Methven et al., 2009). Of the three α1-adrenoceptors, our understanding of the cellular location of the α1D-adrenoceptor has been most limited. The present study provides the opportunity to clarify this and relate receptor localization to function. This is particularly important because immunocytochemistry has not been proved of great utility for α1-adrenoceptor localization, and two recent papers have challenged their specificity (Jensen et al., 2009; Pradidarcheep et al., 2009).
Thus, the aim of the present study was to employ the α1A/B-KO to establish the pharmacological properties of the α1D-adrenoceptor in native tissue and, by referral to the wild-type mouse (WT), whether the other α1-subtypes interact with the α1D-adrenoceptor to alter vascular contraction or cellular location.
Methods
Animals used and vessel preparation
All animal care and this investigation conformed to the provisions of the UK Animals (Scientific procedures) Act 1986. Experimental protocols and rationale have been described in detail previously (Methven et al., 2009).
α1A/B-KO mice were generated by cross-breeding single knockout mice of the α1A-adrenoceptor (α1A-KO) and α1B-adrenoceptor (α1B-KO), as described previously (O'Connell et al., 2003). WT littermates were used as control mice, and all mice were of C57Bl6/J background and were bred and genotyped at the University of Glasgow.
It should be noted that pregnancy rate in knockouts of the α1A-adrenoceptor was markedly reduced. This is in agreement with Sanbe et al. (2007), who reported that the reduction in pregnancies was a result of male infertility, by impaired contraction of the vas deferens, and ruled out sperm abnormalities or differences in sexual behaviour as potential causes.
Male mice, 4–5 months old, weighing between 27 and 46 g (WT: 30–38 g; α1A-KO: 29–41 g; α1B-KO: 31–46 g; α1AB-KO: 27–45 g), were used. Mice were maintained on a 12:12-h light/dark schedule at 22–25°C with 45–65% humidity and fed ad libitum on a standard rodent diet and provided drinking water. The mice were killed by carbon dioxide asphyxiation before first-order mesenteric, and carotid arteries were isolated by using a dissection microscope.
Experimental protocols for functional pharmacology
Two-millimetre sections of artery were mounted in physiological salt solution at 37°C on a four-chamber wire myograph (Danish Myotechnology, Aarhus, Denmark). Following equilibration periods and application of 250-mg passive tension, a start-up procedure using 10 µM of one of the three agonists [the ‘subtype non-selective’α1-adrenoceptor agonist, phenylephrine; an agonist ‘selective’ for the α1A-adrenoceptor, A-61603 (N-(5-[4,5-dihydro-1H-imidazol-2-y]-2-hydroxy-5,6,7,8-tetrahydronaphthalen-1-yl)methanesulphonamide) (Knepper et al., 1995); or 5-hydroxytryptamine] was performed. The viability of the endothelium was tested by using 3 µM acetylcholine. Data were recorded by using Powerlab and Chart (version 5.0) (ADInstruments, Chalgrove, UK).
Cumulative concentration–response curves were produced for each agonist (half-log increments 1 nM to 30 µM or 100 µM). Subtype selective antagonists were incubated for 30 min: prazosin 1nM, 10nM and 100nM (non-selective α1-adrenoceptor antagonist); BMY7378 (8-(2-[4-(2-methoxyphenyl)-1-piperazinyl]ethyl)-8-azaspiro[4.5]decane-7,9-dione dihydrochloride) 10 nM, 30 nM and 100 nM [α1D-adrenoceptor-selective antagonist (Goetz et al., 1995; Saussy et al. 1996)]; and RS100-329 (5-methyl-3-[3-[4-[2-(2,2,2,-trifluroethoxy)phenyl]-1- piperazinyl]propyl]-2,4-(1H,3H)-pyrimidinedione hydrochloride) 10 nM and 100 nM (α1A-adrenoceptor-selective antagonist (Williams et al., 1999).
Experimental protocols for visualization of smooth muscle cell α1-adrenoceptors
BODIPY FL-prazosin (QAPB) incubations
In the WT and α1A/B-KO, 3- to 5-mm segments of live unfixed arteries were incubated with QAPB (100 nM for 120 min) at room temperature (21°C).
Non-fluorescent antagonists were used to compete for QAPB binding sites in segments from the same animals as QAPB controls. Once QAPB equilibrium was established (60 min Pediani et al., 2005), segments were coincubated with an antagonist for 60 min: prazosin (100 nM); rauwolscine (100 nM); RS100-329 (100 nM) or BMY7378 (100 nM). The concentration of each antagonist was consistent with the concentration of those selected previously (Methven et al., 2009).
Arteries were mounted in the incubation solutions with a coverslip (No. 1.5) on top. The mesenteric artery was mounted intact, while the carotid artery was sliced open with a single-edged razor blade and laid flat (Miquel et al., 2005).
Optical sections were collected on a Bio-Rad Radiance 2100 Confocal Laser Scanning System (Bio-Rad, Hemel Hempstead, Hetfordshire, UK) at an excitation/emission of 488/515 nm for QAPB. Arteries were visualized by using ×40 oil immersion objective (numerical aperture 1.0). Optimal laser intensity, gain (contrast) and offset (brightness) were determined and then kept constant for each artery. Photomultiplier sensitivities were set such that all pixels fell within the range 0 (black) to 255 (white). Carotid arteries were imaged at high power (zoom 8) to include only smooth-muscle cells and avoid areas containing elastic lamina in the media. Mesenteric artery images were collected at low power (zoom 3), which allows coverage of a wider area of exclusively smooth-muscle cells. Arteries were scanned by Z-series (0.5 µm slices) from the internal elastic lamina through the media, producing a stack of approximately 20–30 µm in depth. Each procedure was carried out in triplicate on four different mice per strain.
Data analysis and statistical procedures
All graphical and statistical analysis was performed by using Graph Pad Prism (version 5.0, Graphpad Software, San Diego, CA, USA). Mean data were compared across mouse strains and in the presence and absence of the selected antagonists by using either one-way anova and Bonferroni's post-test or Student's t-test.
Functional pharmacology
Data are expressed as a percentage of the control curve maximum and represent the mean value ± SEM. When a maximum response was achieved, pEC50 values were calculated for control curves and agonist curves in the presence of antagonists.
Quantitative confocal microscopy
Image analysis was performed as described in Methven et al. (2009) using Metamorph software (version 6.1, Molecular Devices, Downingtown, PA, USA). A single plane at a depth of 4–6 µm from the internal elastic lamina of each z-series was selected for analysis to capture only smooth-muscle cells. Average pixel intensity was measured for the entire plane (96 × 96 µm for mesenteric arteries; 36 × 36 µm for carotid arteries) and individual smooth-muscle cells (smooth-muscle cells were outlined and measurements made as a Region of Interest). The average pixel intensity correlated to a greyscale range of 0 (black) to 255 (white), with intermediate intensities being assigned an appropriate grey level.
Drugs and chemicals
Physiological salt solution was of the following composition (in mM): 119 NaCl, 4.7 KCl, 1.2 MgSO4.H2O, 1.2 KH2PO4, 24.9 NaHCO3, 2.5 CaCl2 and 11.1 glucose.
All analytical grade drugs were dissolved and diluted to the required concentration in distilled water (unless otherwise stated). Drugs supplied by Sigma Aldrich (Poole, UK) were phenylephrine, acetylcholine, prazosin, 5-hydroxytryptamine, BMY 7378 (8-(2-[4-(2-methoxyphenyl)-1-piperazinyl]ethyl)-8-azaspiro[4.5]decane-7,9-dione dihydrochloride), by Tocris (Bristol, UK) A-61603 (N-(5-[4,5-dihydro-1H-imidazol-2-y]-2-hydroxy-5,6,7,8-tetrahydronaphthalen-1-yl)methanesulphonamide), RS100-329 (5-methyl-3-[3-[14-[2-(2,2,2,-trifluroethoxy)phenyl]-1- piperazinyl]propyl]-2,4-(1H,3H)-pyrimidinedione hydrochloride), rauwolscine, and by Invitrogen (Paisley, UK) QAPB; this was dissolved in dimethyl sulfoxide and diluted in freshly gassed physiological salt solution at room temperature of 21°C.
All drug and molecular target nomenclature conforms to The British Journal of Pharmacology's ‘Guide to Receptors and Channels’ (Alexander et al., 2008).
Results
Functional pharmacology
Carotid arteries
In the WT and all three knockout strains, the maximum response and sensitivity to 5-hydroxytryptamine were similar (Table 1). This eliminates generic structural or smooth muscle sensitivity changes as a basis for differences subsequently found using drugs acting via α1-adrenoceptors.
Table 1.
Agonist responses in mouse carotid arteries
| Mouse |
Phenylephrine |
A-61603 |
5-HT |
|||
|---|---|---|---|---|---|---|
| Maximum response (g) | pEC50 | Maximum response (g) | pEC50 | Maximum response (g) | pEC50 | |
| WT | 0.32 ± 0.03 | 6.9 ± 0.13 | 0.23 ± 0.02 | 6.1 ± 0.13 | 0.28 ± 0.04 | 6.7 ± 0.29 |
| α1A-KO | 0.29 ± 0.02 | 7.0 ± 0.06 | 0.15 ± 0.01* | 5.7 ± 0.09* | 0.26 ± 0.04 | 7.0 ± 0.14 |
| α1B-KO | 0.27 ± 0.03 | 7.0 ± 0.17 | 0.23 ± 0.03 | 6.4 ± 0.14 | 0.28 ± 0.05 | 6.9 ± 0.16 |
| α1A/B-KO | 0.21 ± 0.01* | 7.1 ± 0.08 | 0.12 ± 0.02** | 5.6 ± 0.11* | 0.27 ± 0.04 | 7.2 ± 0.12 |
Data are expressed as mean ± SEM.
P < 0.05;
P < 0.01 compared to WT (one-way anova, Bonferroni's post test).
α1A-KO, α1A-adrenoceptor knockout; α1A/B-KO, α1A/B-adrenoceptor knockout; α1B-KO, α1B-adrenoceptor knockout; WT, wild type mice.
Phenylephrine (non-selective α1-adrenoceptor agonist) produced concentration-dependent contractions in all mouse strains (Figure 1). In the α1A-KO and α1B-KO, the maximum response was not significantly different compared with the WT but was significantly reduced in the α1A/B-KO (Table 1). Sensitivity to phenylephrine was not significantly different between mouse strains. These data not only confirm that the α1D-adrenoceptor is the main contractile α1-adrenoceptor in the WT but also suggest that the α1A- and/or the α1B-adrenoceptor modulates the phenylephrine-induced response at high concentrations, although this effect was small.
Figure 1.
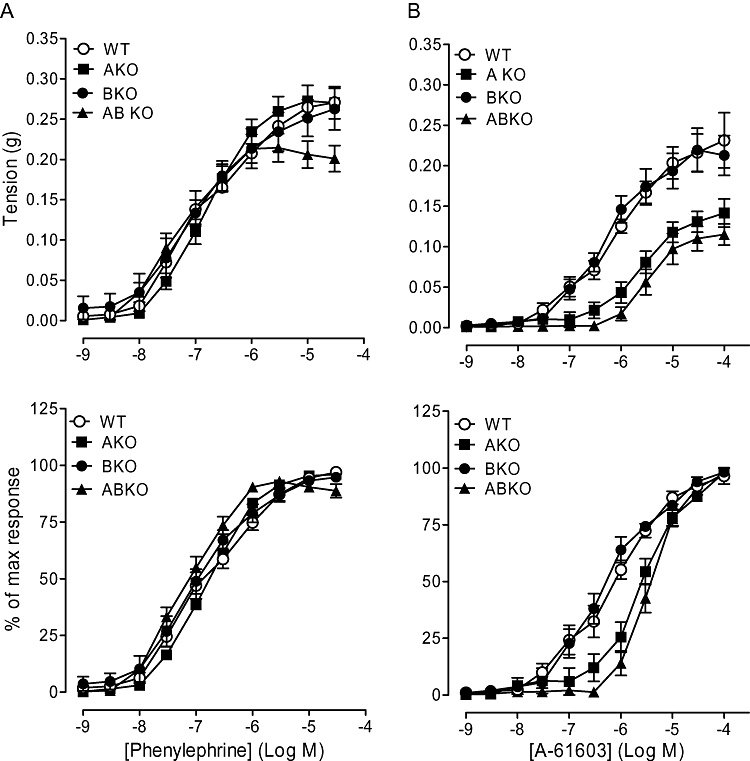
Contractile responses to (A) phenylephrine (n= 6) and (B) A-61603 (n= 6) in carotid arteries of wild type (WT), α1A-adrenoceptor knockout (AKO), α1B-adrenoceptor knockout (BKO) and α1A/B-adrenoceptor knockout (ABKO) mice. Data points are expressed in g tension or percentage of control curve maximum as mean ± SEM.
A-61603-induced, concentration-related contractile response curves in all mouse strains (Figure 1). Both the maximum response and sensitivity to A-61603 were significantly reduced in the α1A- and α1A/B-KO compared with those in the WT and α1B-KO (Table 1). These data suggest that the α1A-adrenoceptor-selective agonist response in the WT consists of α1A- and α1D-adrenoceptor components.
Prazosin competitively antagonized the phenylephrine-induced response in the α1A/B-KO, producing a pA2 consistent with an α1-adrenoceptor-mediated response (Figure 2, Table 2). In the α1A-KO, the phenylephrine response was inhibited by prazosin (Figure 2); 10 nM acted competitively (Table 2) and 100 nM significantly reduced the maximum response achieved within the agonist concentration range used.
Figure 2.
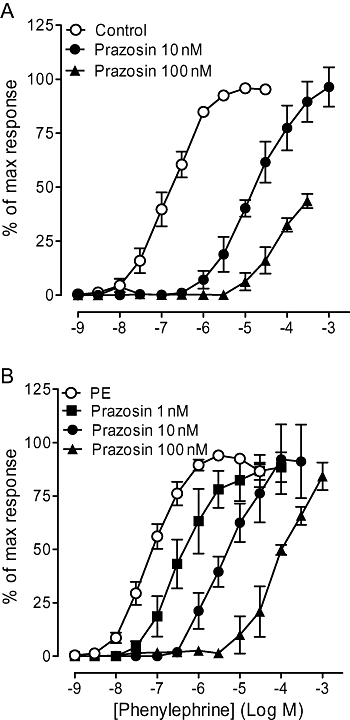
Effects of prazosin on contractile response to phenylephrine in carotid arteries of (A) α1A-adrenoceptor knockout (α1A-KO) (n= 6) and (B) α1A/B-adrenoceptor knockout (α1A/B-KO) (n= 7) mice. Data points are expressed as percentage of control curve maximum as mean ± SEM.
Table 2.
Potency values of antagonists against phenylephrine and A-61603 in mouse carotid arteries
|
Phenylephrine |
A-61603 |
|||||
|---|---|---|---|---|---|---|
| α1A-KO | α1A/B-KO | α1A/B-KO | ||||
| Antagonist | pA2/pKB | Slope | pA2/pKB | Slope | pA2/pKB | Slope |
| Prazosin | 9.3 at 10 nM | – | 9.4 | 1.3 (0.9–1.6) | – | – |
| BMY 7378 | 9.0 at 100 nM | – | 9.4 | 0.8 (0.3–1.8) | 8.5 at 10 nM | – |
| RS100-329 | – | – | 7.8 at 100 nM | – | No shift at 100 nM | |
α1A-KO, α1A-adrenoceptor knockout; α1A/B-KO, α1A/B-adrenoceptor knockout; α1B-KO, α1B-adrenoceptor knockout; WT, wild type mice.
BMY 7378 competitively antagonized the phenylephrine response in the α1A-KO and α1A/B-KO (Figure 3; Table 2). The high pKB (9.0) and pA2 (9.4) produced in the α1A-KO and α1A/B-KO, respectively, showed that the response was mediated by α1D-adrenoceptors. BMY 7378 (10 nM) produced a parallel rightward shift of the A-61603 curve in the α1A/B-KO (Table 2), suggesting that A-61603 produces at least part of its action via α1D-adrenoceptors.
Figure 3.
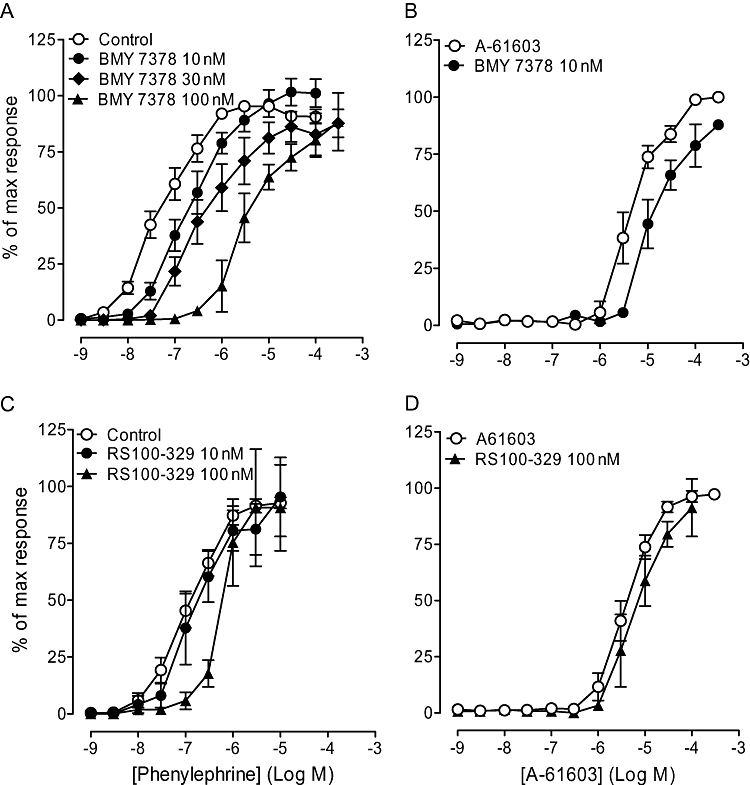
Effects of subtype selective antagonists in mouse carotid arteries of α1A/B-KO. Antagonizing effect of BMY 7378 on contractile response to (A) phenylephrine (n= 7) and (B) A-61603 (n= 6). Antagonizing effect of RS100-329 on contractile response to (C) phenylephrine (n= 6) and (D) A-61603 (n= 6). Data points are expressed as percentage of control curve maximum as mean ± SEM.
In the α1A/B-KO, the phenylephrine-induced response was not significantly affected by RS100-329 (10 nM), but 100 nM shifted the curve rightwards (Figure 3). The consequent low pKB of 7.8 (Table 2) shows the potency of RS100-329 at the α1D-adrenoceptor and is consistent with ligand binding estimates. RS100-329 100 nM was tested further against the A-61603 curve in the α1A/B-KO but had no significant effect (Figure 3). This provides further evidence to show that the action of A-61603 in the α1A/B-KO is via the α1D-adrenoceptor.
Mesenteric arteries
5-Hydroxytryptamine produced concentration-related contractile responses in all three knockout strains, showing no significant differences to the WT (Table 1). This suggests that there are no generic structural or smooth-muscle sensitivity changes in the knockout mice.
In the WT and α1B-KO, a concentration-related contractile response to phenylephrine was observed, with a significantly higher maximum response than in both the α1A-KO and α1A/B-KO (Figure 4; Table 3). Sensitivity to phenylephrine was significantly higher in both the α1A-KO and α1A/B-KO than in the WT and α1B-KO. This not only suggests that, in the mesenteric artery, the α1A-adrenoceptor is the main contractile α1-adrenoceptor but also indicates that the α1D-adrenoceptor but not the α1B-adrenoceptor has a significant contractile role at low concentrations.
Figure 4.
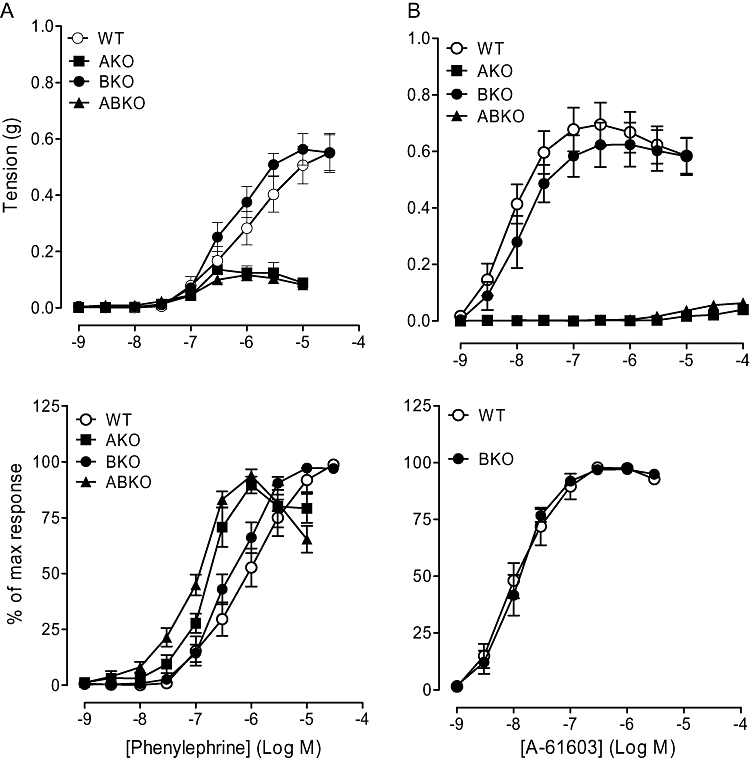
Contractile responses to (A) phenylephrine (n= 7) and (B) A-61603 (n= 6) in mesenteric arteries of wild type (WT), α1A-adrenoceptor knockout (AKO), α1B-adrenoceptor knockout (BKO) and α1A/B-adrenoceptor knockout (ABKO) mice. Data points are expressed in g tension or percentage of control curve maximum as mean ± SEM. (A-61603 data for AKO and ABKO mice have not been expressed as % of own maximum as a result of the small magnitude of the response).
Table 3.
Agonist responses in mouse mesenteric arteries
| Mouse |
Phenylephrine |
A-61603 |
5-HT |
|||
|---|---|---|---|---|---|---|
| Maximum response (g) | pEC50 | Maximum response (g) | pEC50 | Maximum response (g) | pEC50 | |
| WT | 0.56 ± 0.07 | 6.1 ± 0.19 | 0.64 ± 0.10 | 7.9 ± 0.14 | 0.29 ± 0.03 | 6.7 ± 0.06 |
| α1A-KO | 0.19 ± 0.04*** | 6.7 ± 0.07* | 0.04 ± 0.01*** | 5.0 ± 0.12*** | 0.23 ± 0.03 | 6.5 ± 0.03 |
| α1B-KO | 0.58 ± 0.06 | 6.4 ± 0.10 | 0.64 ± 0.08 | 7.9 ± 0.15 | 0.27 ± 0.02 | 6.8 ± 0.17 |
| α1A/B-KO | 0.13 ± 0.02*** | 7.0 ± 0.24*** | 0.07 ± 0.01*** | 5.2 ± 0.17*** | 0.20 ± 0.03 | 6.9 ± 0.06 |
Data are expressed as mean ± SEM.
P < 0.05;
P < 0.001 compared with WT (one-way ANOVA, Bonferroni's post test).
α1A-KO, α1A-adrenoceptor knockout; α1A/B-KO, α1A/B-adrenoceptor knockout; α1B-KO, α1B-adrenoceptor knockout; WT, wild type mice.
Concentration-dependent contractions were obtained to A-61603 in mesenteric arteries of the WT and α1B-KO, but contractions to A-61603 were produced only at high concentrations in the α1A/B-KO and α1A-KO (Figure 4; Table 3). Both maximum response and sensitivity to A-61603 were significantly reduced in the α1A-KO and α1A/B-KO compared with both the WT and α1B-KO. This elimination of the response confirms the dominant α1A-adrenoceptor-mediated contraction in this artery in the WT.
Prazosin (1 nM) caused a rightward displacement of the phenylephrine concentration–response curve without reducing the maximum response in the α1A/B-KO (Figure 5), producing a pKB of 10.3 indicative of an α1-adrenoceptor-mediated response. However, 10 nM and 100 nM produced a non-competitive blockade. Similarly, prazosin (100 nM) reduced the maximum response in the α1A-KO. Because of the low potency of the phenylephrine-induced response, no further antagonist study was performed in the mesenteric artery of either the α1A-KO or the α1A/B-KO.
Figure 5.
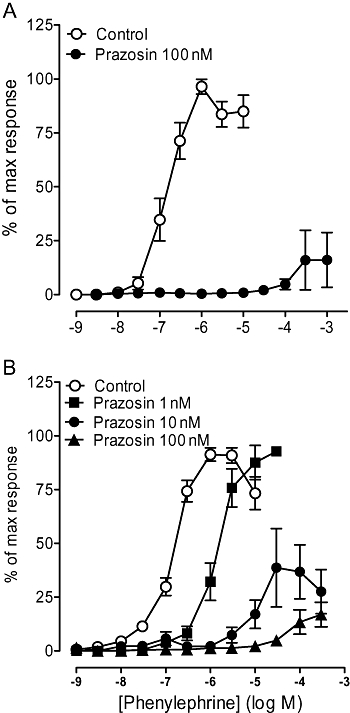
Effects of prazosin on contractile response to phenylephrine in mesenteric arteries of (A) α1A-adrenoceptor knockout (α1A-KO) (n= 6) and (B) α1A/B-adrenoceptor knockout (α1A/B-KO) (n= 7). Data points are expressed as percentage of control curve maximum as mean ± SEM.
Visualization of α1-adrenoceptors using the fluorescent ligand QAPB
Carotid artery
Visually, fluorescent binding to smooth-muscle cells was comparable in the WT and α1A/B-KO, showing a wide distribution throughout the media (Figure 6). However, the intensity of fluorescence of the 36 × 36 µm plane was significantly reduced in the α1A/B-KO compared with the WT (Table 4). Furthermore, the intensity of fluorescence of smooth-muscle cells, when measured separately, was significantly lower in the α1A/B-KO than in the WT (Table 4), indicative of a general decrease in fluorescence in the knockout strains. In both mouse strains, QAPB was observed on the cell surface and perinuclear sites, with evidence of both diffuse and punctate binding. In the α1A/B-KO, this demonstrates that the α1D-adrenoceptor is present in both locations.
Figure 6.
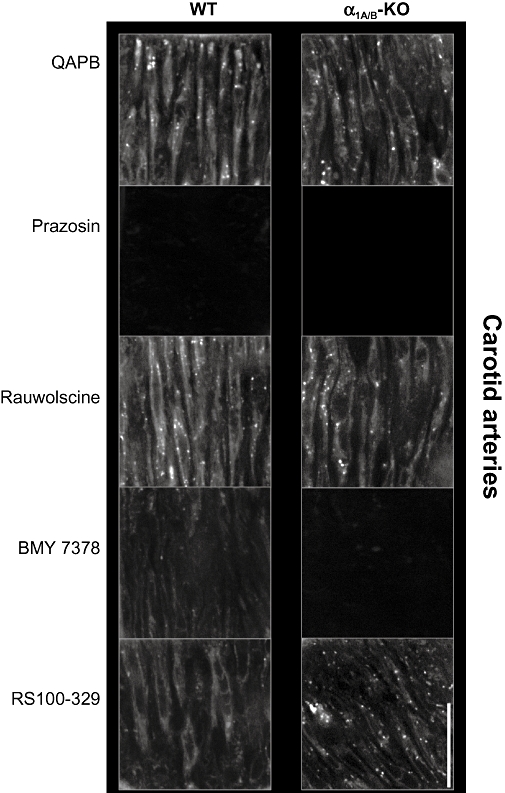
BODIPY FL-prazosin (QAPB) (100 nM) fluorescent ligand binding in mouse carotid arteries. Comparison of wild type (WT) and α1A/B-adrenoceptor knockout (α1A/B-KO) mice and effect of selective antagonists on QAPB fluorescent binding. Non-fluorescent antagonists (100 nM) were used to compete with QAPB after QAPB equilibrium was established. All images were collected under identical conditions of laser intensity, photomultiplier gain (contrast) and offset (brightness). Imaging was performed at high power (zoom 8) to include only smooth-muscle cells and avoid areas containing elastic lamina in the media. An image size of 512 × 512 pixels produced a field size of 36 µm × 36 µm. Calibration bar indicates 10 µm. Each image is representative of those generated from four mice.
Table 4.
Comparison of average pixel intensity of entire image and smooth-muscle cells (SMC) in wild-type (WT) and α1A/B-adrenoceptor knockout (α1A/B-KO) mice
| Mouse strain |
Carotid arteries (n =4) |
Mesenteric arteries (n =4) |
||
|---|---|---|---|---|
| Average pixel intensity of 36×36 µm plane | Average pixel intensity of SMC | Average pixel intensity of 96×96 µm plane | Average pixel intensity of SMC | |
| WT | 50.7 ± 2.0 | 65.5 ± 2.4 | 73.6 ± 3.7 | 93.4 ± 3.5 |
| α1A/B-KO | 30.7 ± 1.2*** | 39.1 ± 1.7** | 28.7 ± 2.2*** | 63.1 ± 3.1** |
Data are expressed as mean ± SEM.
Mesenteric arteries were imaged at low power (zoom 3), which allows coverage of a wider area of exclusively smooth-muscle cells; carotid arteries were imaged at high power (zoom 8) to include only smooth-muscle cells and avoid areas containing elastic lamina in the media.
Individual smooth-muscle cells from a plane from each z-stack were outlined as a Region of interest to measure average pixel intensity.
P < 0.01;
P < 0.001 compared with WT (Student's t-test).
The non-selective α1-adrenoceptor antagonist, prazosin (100 nM), competed with QAPB to significantly reduce fluorescent binding to a background level in the WT and α1A/B-KO (Figure 6; Table 5). No significant difference in fluorescent ligand binding was detected in the presence of the non-selective α2-adrenoceptor antagonist rauwolscine (100 nM) (Figure 6; Table 5). These findings confirm that, at 100nM, QAPB is a selective ligand for α1-adrenoceptors.
Table 5.
Effect of subtype-selective antagonists on fluorescent ligand binding in carotid and mesenteric arteries
| Antagonist |
Average pixel intensity |
|||
|---|---|---|---|---|
|
WT |
α1A/B-KO |
|||
| Carotid arteries (n =4) | Mesenteric arteries (n =4) | Carotid arteries (n =4) | Mesenteric arteries (n =4) | |
| QAPB control | 50.7 ± 2.0 | 73.6 ± 3.7 | 30.7 ± 1.2 | 30.7 ± 1.2 |
| Prazosin | 4.4 ± 0.8*** | 6.8 ± 1.2*** | 7.2 ± 1.3*** | 7.2 ± 1.3*** |
| Rauwolscine | 54.4 ± 2.4 | 67.8 ± 4.5 | 27.7 ± 4.1 | 27.7 ± 4.1 |
| BMY 7378 | 16.2 ± 4.3*** | 27.8 ± 3.8*** | 7.9 ± 0.8*** | 7.9 ± 0.8*** |
| RS100-329 | 24.0 ± 2.5*** | 21.9 ± 1.9*** | 27.2 ± 3.7 | 27.2 ± 3.7 |
Data are expressed as mean ± SEM.
Mesenteric arteries were imaged at low power (zoom 3), which allows coverage of a wider area of exclusively smooth-muscle cells; carotid arteries were imaged at high power (zoom 8) to include only smooth-muscle cells and avoid areas containing elastic lamina in the media.
P < 0.001 compared to QAPB control (one-way ANOVA, Bonferroni's post test).
α1A/B-KO, α1A/B-adrenoceptor knockout; WT, wild type mice.
At 100 nM, BMY 7378 should selectively compete for α1D-adrenoceptors but not the α1A- or α1B-adrenoceptor. QAPB binding was significantly reduced by BMY 7378 in the WT and reduced to a background level in the α1A/B-KO (Figure 6; Table 5), confirming that the α1D-adrenoceptor is present in smooth-muscle cells of this artery in both strains and that only the α1D-adrenoceptor is present in the α1A/B-KO.
In the presence of RS100-329 (100 nM), fluorescent ligand binding was significantly reduced in the WT but not in the α1A/B-KO (Figure 6; Table 5). This is in agreement with the α1A-adrenoceptor being present in the WT and RS100-329 100 nM not having an effect on the α1D-adrenoceptor.
Mesenteric artery
In the WT and α1A/B-KO, the QAPB binding to smooth-muscle cells was similar and was observed throughout the media (Figure 7). In both strains, smooth-muscle cells showing QAPB binding fluoresced brightly, but fluorescence varied between individual cells. Quantitative analysis showed that the intensity of fluorescence of the 96 × 96 µm plane was significantly reduced in the α1A/B-KO compared with the WT (Table 4). The fluorescence of individual smooth-muscle cells was significantly lower in the α1A/B-KO than in the WT (Table 4), consistent with a general decrease of fluorescence in the knockout mice. Evidence of fluorescent ligand binding was observed on the cell membrane and intracellular compartments of smooth-muscle cells, with both diffuse and punctate binding observed intracellularly.
Figure 7.

BODIPY FL-prazosin (QAPB) (100 nM) fluorescent ligand binding in mouse mesenteric arteries. Comparison of wild type (WT) and α1A/B-adrenoceptor knockout (α1A/B-KO) mice and effect of selective antagonists on QAPB fluorescent binding. Non-fluorescent antagonists (100 nM) were used to compete with the fluorescent ligand QAPB after QAPB equilibrium was established. All images were collected under identical conditions of laser intensity, photomultiplier gain (contrast) and offset (brightness). Imaging was performed at low power (zoom 3), which allows coverage of a wide area of exclusively smooth-muscle cells. An image size of 512 × 512 pixels produced a field size of 96 µm × 96 µm. Calibration bar indicates 10 µm. Each image is representative of those generated from four mice.
In both mouse strains, the non-selective α1-adrenoceptor antagonist, prazosin (100 nM), abolished visible QAPB binding and significantly reduced measured fluorescence (Figure 7, Table 5). This, together with the finding that the non-selective α2-adrenoceptor antagonist, rauwolscine (100 nM), had no significant effect (Figure 7, Table 5), showed that QAPB is binding to α1-adrenoceptor sites on smooth-muscle cells.
Following competition with the selective α1D-adrenoceptor antagonist, BMY 7378 (100 nM), QAPB binding was significantly reduced in the WT but was reduced to background levels in the α1A/B-KO (Figure 7; Table 5), showing that the α1D-adrenoceptor is present in the media of both strains of mice.
The selective α1A-adrenoceptor antagonist, RS100-329 (100 nM), significantly reduced fluorescent ligand binding in the WT (Figure 7; Table 5). RS100-329 (100 nM) appeared to be selective for α1A-adrenoceptor sites because the same concentration had no significant effect on QAPB binding in the absence of the α1A-adrenoceptor in the α1A/B-KO.
Discussion and conclusions
The arteries from α1A/B-KO displayed a ‘pure’α1D-adrenoceptor pharmacology that provides a quantitative standard for interpreting studies in other strains of mouse or other species, which are commonly compromised by the presence of other α1-subtypes. The α1D-adrenoceptor-selective antagonist, BMY 7378, became highly potent, and the α1A-adrenoceptor-selective antagonist RS100-329 showed a low potency, in both cases consistent with radioligand binding studies of recombinant receptors.
‘Pure’α1D-adrenoceptor pharmacology in the α1A/B-KO
α1D-Adrenoceptor pharmacology was very clear in the carotid, where BMY 7378 had a pA2 of 9.4 against phenylephrine, in line with pKi values at recombinant α1d-adrenoceptor s (pKi 8.1–9.3) (Goetz et al., 1995; Hieble et al., 1995; Kenny et al., 1995; Saussy et al., 1996; Piascik et al., 1997; Testa et al., 1997; Mackenzie et al., 2000; Yoshio et al., 2001; Deighan et al., 2004) and shows that the lower pKB of 8.3 previously published for the WT (Deighan et al., 2005) was a result of phenylephrine activating multiple α1-subtypes. Similarly, the potent and selective α1A-adrenoceptor antagonist, RS100-329, had the relatively low pKB of 7.8. This is an accurate reflection of this antagonist's potency at the α1D-adrenoceptor and is comparable to its pKi of 7.9 at the cloned α1D-adrenoceptor (Williams et al., 1999; Deighan et al. 2004) and its pKB of 7.9 obtained against phenylephrine in the WT (Methven et al., 2009). Thus, the α1-adrenoceptor-mediated response in the α1A/B-KO carotid artery appears to be representative of a ‘pure’α1D-adrenoceptor population.
Complex α1-adrenoceptor pharmacology in the WT is explained by multiple subtypes
In the carotid artery, the most straightforward way to interpret the pharmacological data in the WT is to work back from the effects of the non-selective α1-adrenoceptor agonist, phenylephrine, in the α1A/B-KO. Sensitivity to phenylephrine was highest in the α1A/B-KO, providing evidence that this agonist has higher potency at the α1D-adrenoceptor than at the α1A-adrenoceptor so that measured sensitivity is reduced when both are present.
Although knocking out the α1A-adrenoceptor alone had no significant effect on the phenylephrine-induced response, knocking out both the α1A- and α1B-adrenoceptors reduced the maximum and the responses to supramaximal concentrations. This was the only evidence from the present study, albeit indirect and a small effect, for any influence of the α1B-adrenoceptor in either artery studied.
The mesenteric arteries of the α1A/B-KO also showed clear characteristics of an α1D-adrenoceptor population, but, in this case, the responses were greatly reduced compared with WT, reflecting the greater contribution of the α1A-adrenoceptor to the maximum contraction. However, responses to phenylephrine in the α1A-KO and α1AB-KO, although reduced in size, showed higher sensitivity than in the WT. This shows that an additional component for phenylephrine in the WT arises from the α1D-adrenoceptor, and explains the complex pharmacology of antagonists versus phenylephrine in the WT.
Thus, although the dominant α1-subtype differed between the conductance and resistance arteries, the response in both vessels contained a significant α1D-adrenoceptor component.
Relevance to the α1L-/α1H-adrenoceptor hypothesis
The tissues studied did not demonstrate low affinity to prazosin. In another study of α1-KO mice, low affinity to prazosin was found in the vas deferens and prostate of the WT, α1B-KO and α1D-KO, and these showed an increased affinity in the α1A-KO (Muramatsu et al., 2008), suggesting that the presence of the α1A-adrenoceptor leads to low affinity for prazosin. However, in our study of the α1A/B-KO, prazosin was highly potent at α1D-adrenoceptors in both mesenteric and carotid arteries, and this was also found at α1A-adrenoceptors in the α1B/D-KO (Methven et al., 2009). This does not support the idea that either of these two receptor subtypes represents a low-affinity ‘α1L-adrenoceptor’.
Interestingly, in the α1A/B-KO and α1A-KO mesenteric arteries, prazosin produced a non-competitive block against the residual phenylephrine-induced response at concentrations of 10 nM and higher. A similar phenomenon was observed in the α1B/D-KO and α1D-KO carotid artery (Methven et al., 2009) and in the α1A-KO prostate, with prazosin (1 nM) against noradrenaline (Muramatsu et al., 2008). In all of these tissues, prazosin shows insurmountable antagonism when the main contractile α1-adrenoceptor is absent. A possible mechanism for such an effect was indicated in a study of recombinant α1A-adrenoceptors, where antagonism exhibited by prazosin (and WB4101) was compatible with a model in which the dissociation of antagonists from the receptor was very slow and was a major factor in their inhibition of the transient phase (intracellular release of Ca2+) of the agonist-mediated response, leading to the appearance of insurmountable antagonism (Pediani et al., 2000). This is characteristic of a system with a low receptor reserve. Because the existence of such a phasic response in smooth muscle was one of the original pointers to α1-subtypes and ligand-directed signalling (McGrath, 1982, 1983, 1985), this may explain part of the mechanism involved.
The resulting exaggerated blockade of the minor contributor could be another explanation for the very varied potency of prazosin in a wide variety of tissues that has been ascribed to the putative presence of receptors with varying affinity for prazosin termed α1L- and α1H-adrenoceptors (Flavahan and Vanhoutte, 1986; Muramatsu et al. 1990).
The difficulty in ascribing any of the cloned α1-adrenoceptors to either of these classes may be explained by the variation in the identity of the more susceptible receptor according to the tissue.
Comparison of the isolated α1A- and α1D-adrenoceptors using double knockouts
The present study with the α1A/B-KO provides a profile of the effects of agonists at α1D-adrenoceptors. This, together with our previous findings in the α1B/D-KO (Methven et al., 2009), illustrates that the carotid artery has a dominant α1D-adrenoceptor response combined with a small but significant α1A-adrenoceptor response at high agonist concentrations. The α1D- and α1A-adrenoceptors summate to produce the overall response, with no evidence of compensation when one or the other is lost. Because the α1A-adrenoceptor has a relatively lower potency than the α1D-adrenoceptor, the latter dominates all responses except at very high concentrations.
In the mesenteric artery, there is an almost converse depiction of a dominant α1A-adrenoceptor response combined with a small but significant α1D-adrenoceptor response, which summate to produce the overall response. However, because the α1D-adrenoceptor-mediated response occurs at low concentrations, it may actually be the more important receptor for physiological concentrations of agonists. This would explain the unexpectedly large effect of α1D-adrenoceptor antagonists against responses to perivascular nerve stimulation (Bexis et al., 2008). It also leads to a confounding effect on selective antagonist potency, as mentioned earlier. Overall, our evidence suggests that, in native tissue, the responses to α1-subtypes are additive and can function independently of each other. So there was no evidence of interactions occurring between α1-adrenoceptors at the signalling level that could alter the function of a particular subtype.
Comparison of the double knockouts also provides insight into the differences between agonists at the receptor subtypes. At the α1A-adrenoceptor, the responses to A-61603 were substantially greater than those to phenylephrine, indicating that phenylephrine is a partial agonist at this receptor. The converse occurs at the α1D-adrenoceptor, where phenylephrine had greater efficacy than A-61603, and, thus, A-61603 is a partial agonist. Indeed, phenylephrine is as much a selective α1D-adrenoceptor agonist as A-61603 is a selective α1A-adrenoceptor agonist.
The α1B-adrenoceptor remains enigmatic as far as function in arteries is concerned. In future studies, the α1A/D-KO may illuminate the function, if any, of the α1B-adrenoceptor in the vasculature. For the moment, a definite action of the α1B-adrenoceptor remains unproven.
α1D-Adrenoceptor location and potential interaction with other subtypes
Two recent papers by independent groups of laboratories (Jensen et al., 2009; Pradidarcheep et al., 2009) have concluded that the commercially available antibodies for α1-adrenoceptor subtypes are not specific. If this can be generalized throughout the literature, there is, in effect, no reliable information on the distribution of α1-adrenoceptors in native tissues. This greatly enhances the value of the present study, where the selectivity of the fluorescent ligand is proven by using selective antagonists. Thus, our data, here on α1D-adrenoceptors and in the works of Methven et al. (2009) on α1A-adrenoceptors, are the only reliable guide to α1-adrenoceptor distribution in blood vessels.
By isolating the α1D-adrenoceptor in the α1A/B-KO, its widespread distribution was evident. The smooth-muscle cells of the α1A/B-KO had a reduced level of fluorescence but had a comparable binding pattern to the WT. This suggests that, in the α1A/B-KO, the overall reduction in fluorescence is a result of the loss of the other α1-subtypes from at least some of the same cells. This provides indirect evidence that different subtypes are present in the same cells, a vital piece of information if molecular interactions between α1-subtypes are to occur. Surprisingly, this has not been demonstrated previously. On the other hand, because the cellular distribution of the fluorescence was similar when the α1D-adrenoceptor alone was present, this also suggests that the distribution of the α1D-adrenoceptor is not regulated by either the α1A- or α1B-adrenoceptor. Furthermore, we found no evidence of an exaggerated internalization of the isolated α1D-adrenoceptor as would be expected from the hypothesis that, through heterodimerization, the α1B-adrenoceptor chaperones the α1D-adrenoceptor to the cell surface (Uberti et al., 2003; Hague et al., 2004). Regardless of whether the α1D-adrenoceptor has a major or minor role in a particular vessel, the α1D-adrenoceptor was found at both the cell membrane and at intracellular sites. The detection of α1-subtypes at both cellular locations is consistent with studies in isolated cells (Daly et al., 1998; Hrometz et al., 1999; Pediani et al., 2000; Morris et al., 2004) and our previous study in native tissue from the α1B/D-KO (Methven et al., 2009). Indeed, it is difficult to see how the situation could be otherwise because α1-adrenoceptors are synthesized inside the cell and need to be located at the plasmalemmal membrane to function and constitutively cycle between the two locations (Morris et al., 2004; Pediani et al., 2005).
Collectively, these data show that the expression of the α1D-adrenoceptor was not altered by the removal of the α1A- and α1B-adrenoceptors at either a cellular or subcellular level.
Relationship between receptor subtype location and function
There was no correlation between which subtype was dominant in mediating contraction and the amount of fluorescent binding detected when that subtype was isolated. Carotid and mesenteric arteries showed similar levels of both receptor subtypes but unequal functional responses. This confirms the poor correlation between mRNA or receptor expression and function. Despite the abundance of all three α1-adrenoceptors being similar, the contribution of each subtype to a functional response varies considerably (Piascik et al., 1995; Yang et al., 1997). There must be another explanation for the functional dominance of each receptor rather than its level of mRNA expression or functional protein. This most probably lies with the efficiency of the signalling process rather than the expression level of the receptor subtype.
Although its functional significance varied between artery types, the distribution of the α1D-adrenoceptor was ubiquitous. This contrasts with the more heterogeneous distribution of the α1A-adrenoceptor (Methven et al., 2009). Stassen et al. (1998) demonstrated that the expression of the α1A-adrenoceptor is dependent on the innervation of arteries, while, in poorly innervated arteries, such as the carotid artery, ‘non-α1A-adrenoceptors’ were dominant. Thus, the distribution of the individual α1-adrenoceptor subtypes may be linked to the level of adrenergic innervation that the arteries receive. The α1D-adrenoceptor-dominant carotid artery is poorly innervated and has a thick medium consisting of several layers of elastic lamina separating smooth-muscle cells layers. Thus, a high number of smooth-muscle cells expressing a functional population of α1D-adrenoceptors would facilitate a cell–cell transmission of excitation. In contrast, the highly innervated, α1A-adrenoceptor-dominant mesenteric artery would be less reliant on a high number of smooth-muscle cells expressing the α1A-adrenoceptor, as depolarization can easily spread through the thin media of this artery.
Therapeutic potential
The present study has demonstrated that the α1D-adrenoceptor exists and is functional in examples of a conductance artery and a resistance artery. In particular, the highly sensitive contractile response of the α1D-adrenoceptor in the mesenteric artery demonstrates a role for the α1D-adrenoceptor in the vasculature and is supported by reduced vasoconstriction to noradrenaline and reduced blood pressure in the α1D-KO (Tanoue et al., 2002). The α1D-adrenoceptor also has a role in the regulation of coronary vasoconstriction (Turnbull et al., 2003). These functions, together with the presence of the α1D-adrenoceptor being linked to the onset of hypertension (Villalobos-Molina and Ibarra, 1999), demonstrate the great importance of the development of more selective agonists and antagonists for research and ultimately for therapeutic use. Furthermore, α1-adrenoceptor antagonists remain extensively in use for the treatment of benign prostatic hyperplasia, without relevant regard to their subtype selectivity. This study provides powerful tools for overcoming the uncertainties of studies with immunocytochemistry or selective agonists or antagonists in native tissues. The α1A/B-KO can be utilized to further our understanding of the expression and physiological or pathophysiological roles of α1D-adrenoceptors and to assess the selectivity of existing and new compounds.
Acknowledgments
The authors thank Mrs Joyce Macmillan for her expert advice and for genotyping the mice. L.M. received generous support from the British Heart Foundation (PG/05/140/20094 and FS/04/034).
Glossary
Abbreviations:
- A-61603
N-(5-[4,5-dihydro-1H-imidazol-2-y]-2-hydroxy-5,6,7,8-tetrahydronaphthalen-1-yl)methanesulphonamide
- α1A/B-KO
α1A/B-adrenoceptor knockout
- BMY 7378
8-(2-[4-(2-methoxyphenyl)-1-piperazinyl]ethyl)-8-azaspiro[4.5]decane-7,9-dione dihydrochloride
- KO
knockout
- QAPB
BODIPY FL-prazosin
- RS100-329
5-methyl-3-[3-[4-[2-(2,2,2,-trifluroethoxy)phenyl]-1-piperazinyl]propyl]-2,4-(1H,3H)-pyrimidinedione hydrochloride
- WT
wild type
References
- Alexander S, Mathie A, Peters J. Guide to receptors and channels (GRAC) Br J Pharmacol. 2008;153:S14. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bexis S, Cleary L, McGrath JC, Tanoue A, Tsujimoto G, Docherty JR. Alpha(1D)-adrenoceptors mediate nerve and agonist-evoked contractions in mouse vas deferens: evidence obtained from knockout technology. Auton Autacoid Pharmacol. 2008;28:81–85. doi: 10.1111/j.1474-8673.2008.00420.x. [DOI] [PubMed] [Google Scholar]
- Chalothorn D, McCune DF, Edelmann SE, Garcia-Cazarin ML, Tsujimoto G, Piascik MT. Differences in the cellular localization and agonist-mediated internalization properties of the alpha 1-adrenoceptor subtypes. Mol Pharmacol. 2002;61:1008–1016. doi: 10.1124/mol.61.5.1008. [DOI] [PubMed] [Google Scholar]
- Daly CJ, Deighan C, McGee A, Mennie D, Ali Z, McBride M, et al. A knockout approach indicates a minor vasoconstrictor role for vascular {alpha}1B-adrenoceptors in mouse. Physiol Genomics. 2002;9:85–91. doi: 10.1152/physiolgenomics.00065.2001. [DOI] [PubMed] [Google Scholar]
- Daly CJ, Milligan CM, Milligan G, Mackenzie JF, McGrath JC. Cellular localization and pharmacological characterization of functioning alpha-1 adrenoceptors by fluorescent ligand binding and image analysis reveals identical binding properties of clustered and diffuse populations of receptors. J Pharmacol Exp Ther. 1998;286:984–990. [PubMed] [Google Scholar]
- Deighan C, Methven L, Naghadeh MM, Wokoma A, MacMillan J, Daly CJ, et al. Insights into the functional roles of alpha1-adrenoceoptor subtypes in mouse carotid arteries using knockout mice. Br J Pharmacol. 2005;144:558–565. doi: 10.1038/sj.bjp.0706089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deighan C, Woollhead A, Colston J, McGrath JC. Hepatocytes from {alpha}1B-adrenoceptor knockout mice reveal compensatory adrenoceptor subtype substitution. Br J Pharmacol. 2004;142:1031–1037. doi: 10.1038/sj.bjp.0705872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavahan NA, Vanhoutte PM. [alpha]1-Adrenoceptor subclassification in vascular smooth muscle. Trends Pharmacol Sci. 1986;7:347–349. [Google Scholar]
- Goetz AS, King HK, Ward SDC, True TA, Rimele TJ, Saussy J. BMY 7378 is a selective antagonist of the D subtype of [alpha]1-adrenoceptors. Eur J Pharmacol. 1995;272:R5–R6. doi: 10.1016/0014-2999(94)00751-r. [DOI] [PubMed] [Google Scholar]
- Hague C, Uberti M, Chen Z, Hall R, Minneman KP. Cell surface expression of alpha 1D-adrenergic receptors is controlled by heterodimerization with alpha 1B-adrenergic receptors. J Biol Chem. 2004;279(15):15541–15549. doi: 10.1074/jbc.M314014200. [DOI] [PubMed] [Google Scholar]
- Hieble JP, Bylund DB, Clarke DE, Eikenburg DC, Langer SZ, Lefkowitz RJ, et al. International Union of Pharmacology. X. Recommendation for nomenclature of alpha 1-adrenoceptors: consensus update. Pharmacol Rev. 1995;47:267–270. [PubMed] [Google Scholar]
- Hrometz SL, Edelmann SE, McCune DF, Olges JR, Hadley RW, Perez DM, et al. Expression of multiple alpha1-adrenoceptors on vascular smooth muscle: correlation with the regulation of contraction. J Pharmacol Exp Ther. 1999;290:452–463. [PubMed] [Google Scholar]
- Jensen BC, Swigart PM, Simpson PC. Ten commercial antibodies for alpha-1-adrenergic receptor subtypes are nonspecific. Naunyn Schmiedebergs Arch Pharmacol. 2009;379:409–412. doi: 10.1007/s00210-008-0368-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenny BA, Chalmers DH, Philpott PC, Naylor AM. Characterization of an alpha 1D-adrenoceptor mediating the contractile response of rat aorta to noradrenaline. Br J Pharmacol. 1995;115:981–986. doi: 10.1111/j.1476-5381.1995.tb15907.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knepper SM, Buckner SA, Brune ME, DeBernardis JF, Meyer MD, Hancock AA. A-61603, a potent alpha 1-adrenergic receptor agonist, selective for the alpha 1A receptor subtype. J Pharmacol Exp Ther. 1995;274:97–103. [PubMed] [Google Scholar]
- Mackenzie JF, Daly CJ, Pediani JD, McGrath JC. Quantitative imaging in live human cells reveals intracellular alpha 1-adrenoceptor ligand-binding sites. J Pharmacol Exp Ther. 2000;294:434–443. [PubMed] [Google Scholar]
- McCune DF, Edelmann SE, Olges JR, Post GR, Waldrop BA, Waugh DJJ, et al. Regulation of the cellular localization and signalling properties of the alpha 1B- and alpha 1D-adrenoceptors by agonists and inverse agonists. Mol Pharmacol. 2000;57:659–666. doi: 10.1124/mol.57.4.659. [DOI] [PubMed] [Google Scholar]
- McGrath JC. Evidence for more than one type of post-junctional [alpha]-adrenoceptor. Biochem Pharmacol. 1982;31:467–484. doi: 10.1016/0006-2952(82)90147-2. [DOI] [PubMed] [Google Scholar]
- McGrath JC. The variety of vascular alpha-adrenoceptors. Trends Pharmacol Sci. 1983;4:14–18. [Google Scholar]
- McGrath JC. Alpha-adrenoceptor agonists and the Ca2+-dependence of smooth muscle contraction: evidence for subtypes of receptors or for agonist-dependent differences in the agonist–receptor interaction? Clin Sci (Lond) 1985;68(Suppl. 10):55s–63s. doi: 10.1042/cs068s055. [DOI] [PubMed] [Google Scholar]
- Methven L, McBride M, Wallace GA, McGrath JC. The alpha-1B/D-adrenoceptor knockout mouse permits isolation of the vascular alpha-1A-adrenoceptor and elucidates its relationship to the other subtypes. Br J Pharmacol. 2009;158:209–224. doi: 10.1111/j.1476-5381.2009.00269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miquel RM, Segura V, Ali Z, D'Ocon P, McGrath JC, Daly CJ. 3-D image analysis of fluorescent drug binding. Mol Imaging. 2005;4:1–13. doi: 10.1162/15353500200504172. [DOI] [PubMed] [Google Scholar]
- Morris DP, Price RR, Smith MP, Lei B, Schwinn DA. Cellular trafficking of human {alpha}1a-adrenergic receptors is continuous and primarily agonist-independent. Mol Pharmacol. 2004;66:843–854. doi: 10.1124/mol.104.000430. [DOI] [PubMed] [Google Scholar]
- Muramatsu I, Morishima S, Suzuki F, Yoshiki H, Anisuzzaman ASM, Tanaka T, et al. Identification of alpha-1L-adrenoceptor in mice and its abolition by alpha-1A-adrenoceptor gene knockout. Br J Pharmacol. 2008;155:1224–1234. doi: 10.1038/bjp.2008.360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muramatsu I, Ohmura T, Kigoshi S, Hashimoto S, Oshita M. Pharmacological subclassification of alpha1-adrenoceptors in vascular smooth muscle. Br J Pharmacol. 1990;99:197–201. doi: 10.1111/j.1476-5381.1990.tb14678.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell TD, Ishizaka S, Nakamura A, Swigart PM, Rodrigo MC, Simpson GL, et al. The {alpha}1A/C- and {alpha}1B-adrenergic receptors are required for physiological cardiac hypertrophy in the double-knockout mouse. J Clin Invest. 2003;111:1783–1791. doi: 10.1172/JCI16100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pediani J, Mackenzie JF, Heeley RP, Daly CJ, McGrath JC. Single-cell recombinant pharmacology: bovine alpha-1a adrenoceptors in rat-1 fibroblasts release intracellular calcium, display subtype-characteristic agonism and antagonism and exhibit an antagonist-reversible inverse concentration-response phase. J Pharmacol Exp Ther. 2000;293:895. [PubMed] [Google Scholar]
- Pediani JD, Colston JF, Caldwell D, Milligan G, Daly CJ, McGrath JC. Beta-arrestin-dependent spontaneous alpha-1a-adrenoceptor endocytosis causes intracellular transportation of alpha-blockers via recycling compartments. Mol Pharmacol. 2005;67:1004. doi: 10.1124/mol.104.008417. [DOI] [PubMed] [Google Scholar]
- Piascik MT, Guarino RD, Smith MS, Soltis EE, Saussy DJ, Perez DM. The specific contribution of the novel alpha-1D adrenoceptor to the contraction of vascular smooth muscle. J Pharmacol Exp Ther. 1995;275:1583–1589. [PubMed] [Google Scholar]
- Piascik MT, Hrometz SL, Edelmann SE, Guarino RD, Hadley RW, Brown RD. Immunocytochemical localization of the alpha-1b adrenergic receptor and the contribution of this and the other subtypes to vascular smooth muscle contraction: analysis with selective ligands and antisense oligonucleotides. J Pharmacol Exp Ther. 1997;283:854–868. [PubMed] [Google Scholar]
- Pradidarcheep W, Stallen J, Labruyère WT, Dabhoiwala NF, Lamers WH. Lack of specificity of commercially available antisera against muscarinergic and adrenergic receptors. Naunyn Schmiedebergs Arch Pharmacol. 2009;379:397–402. doi: 10.1007/s00210-009-0393-0. [DOI] [PubMed] [Google Scholar]
- Sanbe A, Tanaka Y, Fujiwara Y, Tsumura H, Yamauchi J, Cotecchia S, et al. Alpha-1-adrenoceptors are required for normal male sexual function. Brain Res. 2007;152:320–343. doi: 10.1038/sj.bjp.0707366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saussy DJ, Goetz AS, Queen KL, King HK, Lutz MW, Rimele TJ. Structure activity relationships of a series of buspirone analogs at alpha-1 adrenoceptors: further evidence that rat aorta alpha-1 adrenoceptors are of the alpha-1D-subtype. J Pharmacol Exp Ther. 1996;278:136–144. [PubMed] [Google Scholar]
- Stassen FR, Maas RG, Schiffers PM, Janssen GM, De Mey JG. A positive and reversible relationship between adrenergic nerves and alpha-1a adrenoceptors in rat arteries. J Pharmacol Exp Ther. 1998;284:399–405. [PubMed] [Google Scholar]
- Tanoue A, Nasa Y, Koshimizu T, Shinoura H, Oshikawa S, Kawai T, et al. The {alpha}1D-adrenergic receptor directly regulates arterial blood pressure via vasoconstriction. J Clin Invest. 2002;109:765–775. doi: 10.1172/JCI14001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Testa R, Guarneri L, Angelico P, Poggesi E, Taddei C, Sironi G, et al. Pharmacological characterization of the uroselective alpha-1 antagonist Rec 15/2739 (SB 216469): role of the alpha-1l adrenoceptor in tissue selectivity, part II. J Pharmacol Exp Ther. 1997;281:1284–1293. [PubMed] [Google Scholar]
- Turnbull L, McCloskey DT, O'Connell TD, Simpson PC, Baker AJ. Alpha 1-adrenergic receptor responses in alpha 1AB-AR knockout mouse hearts suggest the presence of alpha 1D-AR. AJP Heart Circ Physiol. 2003;284:H1104–H1109. doi: 10.1152/ajpheart.00441.2002. [DOI] [PubMed] [Google Scholar]
- Uberti M, Hall R, Minneman KP. Subtype-specific dimerization of alpha1-adrenoceptors: effects on receptor expression and pharmacological properties. Mol Pharmacol. 2003;64:1379–1390. doi: 10.1124/mol.64.6.1379. [DOI] [PubMed] [Google Scholar]
- Villalobos-Molina R, Ibarra M. Vascular 1D-adrenoceptors: are they related to hypertension? Arch of Med Res. 1999;30:347–352. doi: 10.1016/s0188-0128(99)00047-0. [DOI] [PubMed] [Google Scholar]
- Williams TJ, Blue DR, Daniels DV, Davis B, Elworthy T, Gever JR, et al. In vitro {alpha}1-adrenoceptor pharmacology of Ro 70-0004 and RS-100329, novel {alpha}1A-adrenoceptor selective antagonists. Br J Pharmacol. 1999;127:252–258. doi: 10.1038/sj.bjp.0702541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright CD, Chen Q, Baye NL, Huang Y, Healy CL, Kasinathan S, et al. Nuclear alpha-1-adrenergic receptors signal activated ERK localization to caveolae in adult cardiac myocytes. Circ Res. 2008;103:992–1000. doi: 10.1161/CIRCRESAHA.108.176024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto Y, Koike K. Characterization of alpha1-adrenoceptor-mediated contraction in the mouse thoracic aorta. Eur J Pharmacol. 2001;424:131–140. doi: 10.1016/s0014-2999(01)01134-7. [DOI] [PubMed] [Google Scholar]
- Yang M, Verfurth F, Buscher R, Michel MC. Is alpha-1D-adrenoceptor protein detectable in rat tissues? Naunyn Schmiedebergs Arch Pharmacol. 1997;119:269–277. doi: 10.1007/pl00004966. [DOI] [PubMed] [Google Scholar]
- Yoshio R, Taniguchi T, Itoh H, Muramatsu I. Affinity of serotonin receptor antagonists and agonists to recombinant and native alpha1-adrenoceptor subtypes. Jpn J Pharmacol. 2001;86:189–195. doi: 10.1254/jjp.86.189. [DOI] [PubMed] [Google Scholar]