

Abstract
Background and purpose:
Here we have examined the effects of the novel peptide antagonist N-[1-(2,6-dichlorophenyl)methyl]-3-(1-pyrrolidinylmethyl)-1H-indol-5-yl]aminocarbonyl}-glycinyl-L-lysinyl-L-phenylalanyl-N-benzhydrylamide (K-14585) on proteinase-activated receptor (PAR)2-mediated intracellular signalling events.
Experimental approach:
Using NCTC2544 cells expressing PAR2, we assessed the effects of K-14585 on PAR2-mediated [3H] inositol phosphate accumulation, MAP kinase activation, p65 NFκB phosphorylation and DNA binding and IL-8 production.
Key results:
Pretreatment with K-14585 (5 µM) inhibited [3H] inositol phosphate levels stimulated by PAR2-activating peptide Ser-Leu-Ile-Gly-Lys-Val (SLIGKV-OH) in PAR2-expressing NCTC2544 cells. K-14585 pretreatment did not influence PAR2-mediated extracellular regulated kinase activation but inhibited p38 MAP kinase phosphorylation. At a higher concentration (30 µM), K-14585 alone stimulated p38 MAP kinase activation. These effects were replicated in EAhy926 cells, endogenously expressing PAR2, but not in parental or PAR4-expressing NCTC2544 cells, suggesting these effects were PAR2-dependent. SLIGKV-mediated stimulation of p38 MAP kinase phosphorylation was substantially reduced by the Gq/11 inhibitor YM-254890, without affecting K-14585-mediated phosphorylation. Pretreatment with K-14585 inhibited PAR2-mediated p65 NFκB phosphorylation and NFκB-DNA binding. K-14585 (30 µM) alone stimulated comparable NFκB reporter activity to SLIGKV-OH. K-14585 inhibited SLIGKV-stimulated IL-8 production, but given alone increased IL-8. While SLIGKV-induced IL-8 formation was reduced by both SB203580 and YM-254890, the response to K-14585 was sensitive to SB203580 but not YM-254890.
Conclusions and implications:
These data reveal that K-14585 has a duality of action functioning both as an antagonist and agonist due to either partial agonist actions or possible agonist-directed signalling. The data also suggest two modes of p38 MAP kinase activation emanating from PAR2, one Gq/11-dependent and the other Gq/11-independent.
Keywords: PAR2, Gq/11 signalling, p38 MAP kinase, K-14585 peptide antagonist, inositol phosphate
Introduction
Proteinase-activated receptor-2 (PAR2) is the second member of the proteinase-activated receptor subfamily of G-protein-coupled receptors (see Macfarlane et al., 2001; Ossovskaya and Bunnett, 2004; Kanke et al., 2005). To date, four members of the PAR family have been identified, namely PAR1, PAR2, PAR3 and PAR4 (nomenclature follows Alexander et al., 2008). As with other family members, PAR2 is activated through serine proteolytic cleavage of the amino terminal of the receptor, thus unmasking a tethered ligand that then binds to the receptor causing intramolecular activation (Nystedt et al., 1994). Serine proteases such as trypsin and tryptase serve as the predominant endogenous activators for PAR2, while synthetic activating peptides derived from the tethered ligand sequence such as Ser-Leu-Ile-Gly-Lys-Val (SLIGKV-OH) (human sequence), SLIGRL (murine variant) and substituted peptides including 2fl-LIGKV are also able to stimulate receptor activation without prior cleavage of the receptor (Kawabata et al., 2004).
The expression of PAR2 has been detected in a variety of human tissues including blood vessels, skin, airways, brain and gastrointestinal tract (Macfarlane et al., 2001). In fact, PAR2 activation has been shown to mediate diverse biological functions such as blood haemostasis, skin pigmentation, bronchoconstriction/dilation and inflammation (Ossovskaya and Bunnett, 2004; Kanke et al., 2005). A large body of evidence now supports a role for PAR2 in several disease states including neurogenic inflammation (Steinhoff et al., 2000), arthritis (Ferrell et al., 2003; McIntosh et al., 2007), skin disorders (Kawagoe et al., 2002; Seeliger et al., 2003) and colitis (Cenac et al., 2002; 2003;). Therefore, development of a selective and potent antagonist would be a potentially useful therapy in a number of these conditions. However, in contrast to PAR1, for which high-affinity non-peptide antagonists are now available (Chackalamannil, 2006), the development of PAR2 antagonists is limited to a recently described low-affinity PAR2 antagonist, ENMD-1068 (Kelso et al., 2006). However, a series of peptide compounds, including N-[1-(2,6-dichlorophenyl)methyl]-3-(1-pyrrolidinylmethyl)-1H-indol-5-yl]aminocarbonyl}-glycinyl-L-lysinyl-L-phenylalanyl-N-benzhydrylamide (K-14585), have recently been developed and characterized as moderately potent inhibitors of PAR2-mediated vascular and inflammatory responses (Kanke et al., 2009). The mechanism(s) of their inhibitory action was unclear, especially at the level of intracellular signalling events.
Therefore, in this study we sought to characterize the effects of one of these novel peptide-mimetic PAR2 antagonists, K-14585, on the signalling pathways mediated by the PAR2-activating peptide, SLIGKV-OH. We show here that while K-14585 at low-micromolar concentrations inhibits distinct signalling parameters induced by SLIGKV-OH in NCTC2544 cells stably expressing PAR2, at higher concentrations it has agonist actions. These novel data suggest the potential of either partial agonist activity or different agonist/receptor signalling conformations that mediates different signalling cassettes.
Methods
Cell culture
Human skin epithelial cells NCTC2544 were maintained in M199 medium, 10% (v/v) foetal calf serum, 100 units penicillin·mL−1 and 100 µg streptomycin·mL−1 in a humidified atmosphere containing 5% CO2 at 37°C. NCTC2544 cells stably expressing either human PAR2 (NCTC2544-PAR2) or PAR4 (NCTC2544-PAR4) were maintained in complete M199 medium containing additionally 400 µg·mL−1 of Geneticin to maintain selection pressure and passaged using Versene. A clone expressing both PAR2 and an NFκB reporter plasmid (NCTC2544-PAR2-NFκB cells) were grown in M199 supplemented with 400 µg·mL−1 Geneticin and 5 µg·mL−1 Blasticidin S. (Macfarlane et al., 2005). The endothelial cell hybrid line EAhy926 was cultured as outlined previously (Paul et al., 2000).
Inositol phosphate assay
The accumulation of [3H] inositol phosphates (inositol mono- bis- tris- and tetrakis-phosphates; IP1–4) as an indirect index of inositol trisphosphate formation was assayed as described previously (Plevin et al., 1994). NCTC2544-PAR2 cells were grown to confluence in 24-well plates and serum-starved to make them quiescent for 18 h with serum-free M199 medium supplemented with myo-[2-3H-(N)]-inositol (0.5 µCi·mL−1; 1 Ci = 37 GBq). Cells were pre-incubated with LiCl (10 mM) for 15 min prior to adding the appropriate concentrations of K-14585 for a further 30 min. Cells were then stimulated with SLIGKV (30 µM) for 60 min. Following this, cells were washed in ice-cold phosphate-buffered saline (PBS), and inositol phosphates were extracted by partition in 2:1 methanol/chloroform. After further addition of deionized water and chloroform, the inositol phosphate-containing aqueous phase was transferred into vials containing pre-washed Dowex formate anion exchange resin. Total inositol phosphates ([3H]-IP1–4) were eluted using elution buffer (1 M ammonium formate, 0.1 M formic acid in distilled water). One millilitre of the elution solution was mixed with 4 mL of scintillant fluid (Optiphase Hi-safe™) and quantified using liquid β-scintillation counter (PerkinElmer, Waltham, MA, USA).
Western blotting of phosphorylated MAP kinase and p65 NFκB
Proteins (10 µg per lane) were separated by 10% SDS-PAGE and transferred onto nitrocellulose (Costar, Buckinghamshire, UK). The membranes were blocked for non-specific binding for 2 h in 2% (w/v) bovine serum albumin (BSA) diluted in NATT buffer [50 mM Tris-HCl, 150 mM NaCl, 0.2% (v/v) Tween-20]. The blots were then incubated overnight with 5–10,00 dilution (depending upon optimum titre) of primary antibody phospho-ERK (extracellular regulated kinase), p38 or NFκB (serine 536) diluted in 0.2% (w/v) BSA in NATT buffer. The blots were washed with NATT buffer for 90 min and incubated with HRP-conjugated secondary antibody [20 ng·mL−1 in 0.2% (w/v) BSA diluted in NATT buffer] for 90 min. After a further 90 min wash, the blots were subjected to ECL reagent and exposed to Kodak X-ray film. Blots were routinely re-probed for total ERK, p38 and NFκB as appropriate.
Electrophoretic mobility shift assay (EMSA)
NCTC2544 cells were grown to confluence on 6-well plates and serum-starved overnight. Cells were then incubated with the appropriate concentrations of K-14585 for 30 min before being stimulated with SLIGKV (30 µM) for a further 60 min. Reactions were terminated by washing cells twice with ice-cold PBS. Cells were then removed by scraping and transferred to Eppendorf tubes. Nuclear extracts were prepared as previously described (Schreiber et al., 1989) and the protein content of the recovered samples then determined by means of Bradford assay. Nuclear extracts (5 µg) were incubated in binding buffer [10 mM Tris-HCl pH 7.5, 4% (v/v) glycerol, 1 mM MgCl2, 0.5 mM EDTA, 0.5 mM DTT, 50 mM NaCl, 50 µg·mL−1 poly(dI-dC).poly(dI-dC)] for 30 min before the addition of 1 µL (50 000 cpm) of 32P-labelled double-stranded NFκB consensus oligonucleotide [5′-AGT TGA GGG GAC TTT CCC AGG C-3′] (Promega, Southampton, UK) for a further 30 min. Following this, 1 µL of 10× gel loading buffer (250 mM Tris-HCl pH 7.5, 0.2% (w/v) bromophenol blue, 40% (v/v) glycerol) was added to samples, and protein/DNA complexes were resolved by non-denaturing electrophoresis on 5% (w/v) acrylamide slab gels. Gels were initially pre-run in (0.5×) Tris-borate-EDTA buffer (TBE) for 30 min at 100 V and subsequent to loading of samples electrophoresis maintained at 100 V for 45–60 min. Gels were dried and NFκB/probe complexes visualized by autoradiography.
NFκB reporter activity assay
NCTC2544-PAR2-NFκB cells were grown to confluency in 96-well plates (flat, white with clear bottom, Costa, the Netherlands) and serum-starved for 18 h before incubating the cells with appropriate concentrations of K-14585 for 30 min. Subsequently, cells were stimulated with SLIGKV (30 µM) for 4 h. Stimulation was terminated by aspiration of the medium; followed by washing twice with ice-cold PBS. Cells were then assayed for luciferase activity using the Steady Glo® kit (Promega) according to the manufacturer's instructions.
IL-8elisa
NCTC2544-PAR2 cells were grown to confluence in a 12-well plate and incubated at 37°C and humidified with 5% CO2. Cells were then stimulated for 6 h with agents as required. Following stimulation supernatant was collected from the plates and cells were discarded. The 96-well plates were prepared with capture antibody specific for IL-8 (1:2000) and incubated overnight at 4°C. Capture antibody was aspirated and replaced with blocking buffer, which was made according to the manufacturer's instructions. On day of the assay, blocking buffer was aspirated and replaced with IL-8 standards (0–1 ng) and samples that were diluted 1:10 in assay buffer. Once prepared, the plate was incubated at 37°C for 1 h. Following incubation, plate was washed three times in manufacturer's wash buffer and incubated with detection antibody specific to IL-8 (1:2500). The plate was incubated for 90 min at room temperature, after which it was again washed and incubated with Streptavidin HRP conjugate antibody (1:2000) and incubated at room temperature for 1 h. The plate was washed again (×3), and antibody replaced with the recommended substrate (tetramethylbenzidine, 6 mg·mL−1), and incubated at room temperature for 30 min. Following colour change, manufacturers stop solution was added and the plate read at 450 nm.
Data analysis
Where experimental data are shown as gels these represent one of at least three experiments. Densitometry measurement was performed by using Scion Image programme. Luciferase experiments were performed at least three times as quadruplicate. Data were expressed as mean ± SEM. Statistical analysis was performed by one-way anova with Dunnett's post hoc test (P < 0.05, P < 0.01).
Materials
All materials used were of highest grade available and were purchased from Sigma-Aldrich Co. Ltd. (Poole, UK) unless otherwise stated. Foetal calf serum, Medium 199 with Earl's salt supplement (M199), G418 were purchased from Invitrogen (Paisley, UK). Antibodies against phospho-ERK, ERK, p38 MAP kinase and p65 NFκB were obtained from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA); phospho-p65 NFκB from New England Biolab (UK) and phospho-p38 MAP kinase from GE Healthcare UK, Ltd (Little Chalfont, UK). HRP-conjugated antibodies were from Invitrogen. K-14585 was a kind gift from Kowa Ltd., Tokyo, Japan. YM-254890 was a kind gift of Astellas Pharma. Inc, Tokyo, Japan.
Synthesis of PAR2 antagonist
The peptide mimetic PAR2 antagonist, K-14585, was synthesized at Kowa Tokyo New Drug Research Laboratories as outlined previously (Kanke et al., 2009). The chemical structure was confirmed by nuclear magnetic resonance and mass spectrometry. The purity of compound (>95%) was determined by high-performance liquid chromatography. The compound was dissolved in DMSO, and aliquots were kept at −20°C until use. The human PAR2-activating peptide, SLIGKV-OH, were synthesized and purified (>95%) by high-performance liquid chromatography, and the structures were confirmed by mass spectrometry.
Results
We selected K-14585 for the study as it was found to be found to be the most potent test compound (Kanke et al., 2009) and examined a number of PAR2-mediated signalling cascades in NCTC2544-PAR2 cells (Kanke et al., 2001). Figure 1 shows that SLIGKV (30 µM) induced an increase of approximately sixfold of basal control in the accumulation of total [3H] inositol phosphate in NCTC2544-PAR2 cells. This response was significantly inhibited by K-14585 at 5 and 30 µM giving 67 ± 4.6% and 68.4 ± 10.4% (n= 4) inhibition of the peptide response. Notably, K-14585 at all the concentrations tested did not significantly alter the basal level of total [3H] inositol phosphate accumulation. Studies also sought to determine whether trypsin-mediated [3H]-IP accumulation could be similarly effected. Pretreatment of the cells with concentrations of K-14585 up to 30 µM gave a small but significant inhibition of trypsin-stimulated [3H]-IP accumulation; however, this was as little as 20–30% maximum (fold response; trypsin 30 nM = 12.3 ± 2.1 trypsin plus K-14585 (30 µM) = 9.6 ± 1.6 n= 3, P < 0.05). This pattern of inhibition is in keeping with data obtained previously for this compound (Kanke et al., 2009).
Figure 1.

Inhibitory effect of K-14585 on proteinase-activated receptor (PAR)2-mediated [3H] inositol phosphates (IP) accumulation in NCTC2544-PAR2 cells. NCTC2544-PAR2 cells pre-labelled with 0.5 µCi·mL−1 of [3H]-2-myoinositol overnight and pre-incubated with 10 mM LiCl were pretreated with vehicle (0.1% DMSO) (-) or 1–30 µM of N-[1-(2,6-dichlorophenyl)methyl]-3-(1-pyrrolidinylmethyl)-1H-indol-5-yl]aminocarbonyl}-glycinyl-L-lysinyl-L-phenylalanyl-N-benzhydrylamide (K-14585) for 30 min prior to stimulation with 30 µM Ser-Leu-Ile-Gly-Lys-Val (SLIGKV-OH) for an additional 45 min. Samples were assessed for [3H]-IP accumulation as outlined in the Methods. Each value represents the mean ± SEM of at least three experiments. *P < 0.05 compared with SLIGKV-OH stimulation.
We then investigated the effects of K-14585 on PAR2-mediated phosphorylation of ERK and p38 MAP kinase, using Western blotting to determine if there were any differences in sensitivity to inhibition by K-14585. SLIGKV-OH (30 µM) stimulated the phosphorylation of ERK in NCTC2544-PAR2 cells, producing an increase of 8.9 ± 0.4-fold in activity (Figure 2). This response, however, was not significantly affected by pretreatment of the cells with K-14585. Interestingly, K-14585 (30 µM) alone, when added to cells, was able to stimulate a small increase in ERK activation, resulting in a 2.8 ± 1.1-fold increase in phosphorylation.
Figure 2.

The effect of N-[1-(2,6-dichlorophenyl)methyl]-3-(1-pyrrolidinylmethyl)-1H-indol-5-yl]aminocarbonyl}-glycinyl-L-lysinyl-L-phenylalanyl-N-benzhydrylamide (K-14585) on Ser-Leu-Ile-Gly-Lys-Val (SLIGKV-OH)-stimulated phosphorylation of extracellular regulated kinase (ERK) in NCTC2544-PAR2 cells. NCTC2544-PAR2 cells were serum-starved and incubated with 1–30 µM of K-14585 for 30 min prior to stimulation with 30 µM SLIGKV-OH for an additional 5 min. Samples were assessed for ERK phosphorylation or total ERK expression as outlined in the Methods. Blots were quantified by densitometry and each value represents the mean ± SEM of at least 3–4 experiments.
A similar but more striking result was obtained for p38 MAP kinase (Figure 3). The SLIGKV-OH (30 µM) caused a marked increase in the phosphorylation of p38 MAP kinase. This response was observed to be inhibited at 5 µM of K-14585 whereas at 30 µM no inhibition was observed. Surprisingly, at this higher concentration, K-14585 alone was observed to increase p38 MAP kinase phosphorylation, to a small but significant extent (P < 0.05), although not as great as that produced by SLIGKV-OH alone.
Figure 3.
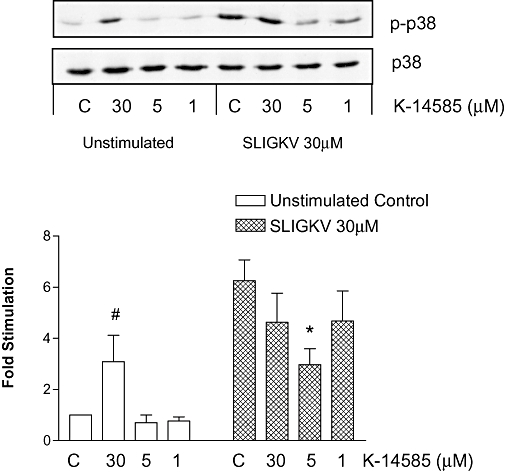
Dual effect of N-[1-(2,6-dichlorophenyl)methyl]-3-(1-pyrrolidinylmethyl)-1H-indol-5-yl]aminocarbonyl}-glycinyl-L-lysinyl-L-phenylalanyl-N-benzhydrylamide (K-14585) on Ser-Leu-Ile-Gly-Lys-Val (SLIGKV-OH)-stimulated p38 MAP kinase phosphorylation in NCTC2544-PAR2 cells. NCTC2544-PAR2 cells were serum-starved and incubated with 1–30 µM of K-14585 for 30 min prior to stimulation with 30 µM SLIGKV-OH for an additional 5 min. Samples were assessed for p38 MAP kinase phosphorylation or total p38 MAP kinase expression as outlined in the Methods. Blots were quantified by densitometry and each value represents the mean ± SEM, n= 4. *P < 0.05 compared with SLIGKV stimulation, #P < 0.05 from control values.
In order to confirm that the effects of K-14585 on SLIGKV-stimulated signalling parameters measured in NCTC2544-PAR2 were solely due to its effect on PAR2, we carried out similar experiments in the parental cell line, NCTC2544 (Figure 4A). Stimulation of the cells with SLIGKV-OH (30 µM) did not induce the phosphorylation of ERK or p38 MAPK or p65 NFκB. Compound K-14585, at all concentrations tested, also did not elicit any effects on the parameters measured, suggesting its actions are indeed PAR2-specific. NCTC2544 express moderate amounts of PAR1 (Kawabata et al., 2004) suggesting no effect of K-14585 upon this subtype. Furthermore, K-14585 had no discernable effect upon basal or thrombin-mediated p38 MAP kinase phosphorylation in cells overexpressing PAR4 (Figure 4B).
Figure 4.

Lack of effect of N-[1-(2,6-dichlorophenyl)methyl]-3-(1-pyrrolidinylmethyl)-1H-indol-5-yl]aminocarbonyl}-glycinyl-L-lysinyl-L-phenylalanyl-N-benzhydrylamide (K-14585) in parental NCTC2544 cells and proteinase-activated receptor (PAR)4 NCTC2544 cells. Parental NCTC2544 cells (A) or PAR4-expressing cells (B) were serum-starved and incubated with 1–30 µM of K-14585 for 30 min prior to stimulation with 30 µM Ser-Leu-Ile-Gly-Lys-Val (SLIGKV-OH) (A) or 3 U·mL−1 thrombin (B) for an additional 5 min. Samples were assessed for extracellular regulated kinase (A), p38 MAP kinase (A and B) or p65 NFκB phosphorylation (A) as indicated and as outlined in the Methods. Each blot is representative of at least two others.
While our studies suggest that the effect of K-14585 is likely to be PAR2-specific we assessed effects in EAhy926 cells that endogenously express PAR2 (Paul et al., 2000). In this cell line we found that SLIGKV-OH stimulated an increase in [3H] inositol phosphates, which was significantly reduced (P < 0.05) following pre-incubation with K-14585 at concentrations up to 30 µM (Figure 5A). However, when assessing p38 phosphorylation we found that, while pre-incubation with a low concentrations of K-14585 (5 µM) was able to inhibit stimulation in response to SLIGKV-OH (P < 0.05, n= 4), at a much higher concentration (30 µM), K-14585 given alone was able to stimulate a significant increase in phosphorylation of p38 (Figure 5B). This again links the effects of K-14585 with the expression of PAR2.
Figure 5.

The effects of N-[1-(2,6-dichlorophenyl)methyl]-3-(1-pyrrolidinylmethyl)-1H-indol-5-yl]aminocarbonyl}-glycinyl-L-lysinyl-L-phenylalanyl-N-benzhydrylamide (K-14585) on [3H] inositol phosphates (IP) accumulation and p38 MAP kinase are replicated in EAhy926 cells. EAhy926 cells were serum-starved and incubated with 1–30 µM of K-14585 for 30 min prior to stimulation with 30 µM Ser-Leu-Ile-Gly-Lys-Val (SLIGKV-OH) for an additional 60 min (A) or 5 min (B). Samples were assessed for [3H]-IP accumulation (A) and p38 MAP kinase phosphorylation or total p38 MAP kinase expression (B) as outlined in the Methods. Blots were quantified by densitometry and each value represents the mean ± SEM from three experiments. *P < 0.05 compared with peptide stimulation.
We sought to investigate the activation of p38 MAP kinase by K-14585 further, by assessing the involvement of upstream intermediates in the activation of p38 MAP kinase (Figure 6). Cells were pre-incubated with the Gq/11 inhibitor YM-254890 (Takasaki et al., 2004). At a concentration of 100 nM, which essentially abolished [3H] inositol phosphates accumulation in PAR2 stimulated cells (Goon Goh et al., 2008), SLIGKV-OH-mediated stimulation of p38 MAP kinase was partially inhibited by incubation with YM-254890. However, phosphorylation of p38 MAP kinase in response to K-14585 was not significantly inhibited. This strongly suggests the presence of two PAR2-dependent pathways activating p38 MAP kinase, one dependent upon Gq/11 and the other Gq/11-independent, which can be activated by K-14585.
Figure 6.

N-[1-(2,6-dichlorophenyl)methyl]-3-(1-pyrrolidinylmethyl)-1H-indol-5-yl]aminocarbonyl}-glycinyl-L-lysinyl-L-phenylalanyl-N-benzhydrylamide (K-14585) activated p38 MAP kinase in a Gq/11-independent manner in NCTC2544-PAR2 cells. NCTC2544-PAR2 cells were serum-starved and incubated with 100 nM of YM-254890 for 30 min prior to stimulation with 30 µM K-14585 or Ser-Leu-Ile-Gly-Lys-Val (SLIGKV-OH) for an additional 5 min. Samples were assessed for phosphor-p38 MAP kinase or total p38 levels as outlined in the Methods. Blots were quantified by densitometry and each value represents the mean ± SEM from three experiments. **P < 0.05 compared with peptide stimulation.
Work from our laboratory has previously shown that PAR2 stimulates NFκB activity at the levels of NFκB-DNA binding and transcriptional activity (Kanke et al., 2001; Macfarlane et al., 2005). We therefore examined the effects of K-14585 on PAR2-mediated NFκB activation (Figure 7). SLIGKV (30 µM) induced the phosphorylation of p65 NFκB (Figure 7A). Pretreatment of the cells with K-14585 (all concentrations tested) significantly attenuated the response produced. Alone, K-14585 did not significantly alter basal p65 NFκB phosphorylation at any concentrations tested. NFκB-DNA binding activity was similarly affected (Figure 7B). Binding in response to SLIGKV-OH was significantly reduced by 30 and 5 µM of K-14585 with 83 ± 6% inhibition and 52 ± 4% inhibition respectively. Again, K-14585 (30 µM) alone did not enhance the basal PAR2-mediated NFκB-DNA binding activity. TNFα (20 ng·mL−1) was employed as a negative control in this study and as expected, NFκB-DNA binding activity stimulated by TNFα remained unaffected in the presence of 30 µM K-14585.
Figure 7.

Inhibition of PAR2-mediated p65 NFκB phosphorylation and DNA binding by N-[1-(2,6-dichlorophenyl)methyl]-3-(1-pyrrolidinylmethyl)-1H-indol-5-yl]aminocarbonyl}-glycinyl-L-lysinyl-L-phenylalanyl-N-benzhydrylamide (K-14585) in NCTC2544-PAR2 cells. NCTC2544-PAR2 cells were serum-starved and incubated with 1–30 µM of K-14585 for 30 min prior to stimulation with 30 µM Ser-Leu-Ile-Gly-Lys-Val (SLIGKV-OH) for an additional 30 (A) or 60 min (B). Samples were assessed for p65 NFκB serine 536 phosphorylation (A) or NFκB-DNA binding (B) as outlined in the Methods. Blots were quantified by densitometry and each value represents the mean ± SEM. *P < 0.05 compared with peptide stimulation; **P < 0.01; n= 4.
As K-14585 appeared to inhibit both PAR2-stimulated NFκB phosphorylation and DNA binding, we then sought to evaluate K-14585 on PAR2-induced NFκB-mediated transcriptional activity. As shown in Figure 8A, SLIGKV-OH (30 µM) caused a substantial increase in NFκB-linked luciferase reporter activity in NCTC2544-PAR2-NFκB cells. This response was significantly reduced by 5 µM K-14585 (70 ± 14% inhibition), with no significant alteration observed following pretreatment of the cells with 30 and 1 µM K-14585. Surprisingly, compound K-14585 (30 µM) alone also induced a marked response, the magnitude of which was comparable with that induced by SLIGKV-OH. Our data suggest that K-14585 was able to stimulate the phosphorylation of p38 MAP kinase and increase NFκB reporter activity; hence it is possible that these pathways are linked. However, pretreatment with the p38 MAP kinase inhibitor SB203580 (10 µM) did not appear to inhibit the NFκB-transcriptional activity stimulated by K-14585 or SLIGKV-OH (Figure 8B).
Figure 8.

N-[1-(2,6-dichlorophenyl)methyl]-3-(1-pyrrolidinylmethyl)-1H-indol-5-yl]aminocarbonyl}-glycinyl-L-lysinyl-L-phenylalanyl-N-benzhydrylamide (K-14585) activated p38 MAP kinase-independent NFκB reporter activity in NCTC2544-PAR2-NFκB cells. NCTC2544-PAR2 cells stably expressing a 3× NFκB reporter construct were pre-incubated with 1–30 µM of K-14585 for 30 min prior to stimulation with 30 µM Ser-Leu-Ile-Gly-Lys-Val (SLIGKV-OH) for an additional 4 h (A). In (B) cells were pre-incubated with 10 µM SB203580 30 min prior to addition of SLIGKV-OH (30 µM) or K-14585 (30 µM) for a further 4 h. Samples were assessed for luciferase activity as outlined in the Methods. Each value represents the mean ± SEM. *P < 0.05 compared with peptide stimulation, n= 4. #P < 0.05 compared with unstimulated control.
We also examined the effects of K-14585 on functional cellular responses in terms of IL-8 production (Figure 9). SLIGKV-OH alone stimulated IL-8 production over 8 h, equivalent to a 7.6 ± 0.9-fold increase of the unstimulated output (Figure 9A). Pre-incubation with K-14585 reduced SLIGKV-OH-mediated IL-8 formation at 5 and 10 µM, however at 30 µM K-14585 significantly enhanced the response. Exposure to K-14585 alone at 30 µM stimulated IL-8 production as well as SLIGKV-OH (8 ± 0.4-fold; n= 6) (Figure 9B,C) consistent with its duality of action. In considering the roles of signalling pathways in the actions of SLIGKV-OH or K-14585, we examined the contribution of p38 MAP kinase and Gq/11 pathways. Pre-incubation with the p38 MAP kinase inhibitor, SB203580, significantly reduced IL-8 production in response to SLIGKV-OH or K-14858 (Figure 9B). In contrast, only IL-8 production in response to SLIGKV-OH was sensitive to inhibition by YM-254890, and the response to K-14585 was not significantly affected (SL + YM = 73 ± 5% inhibition; K-14585 + YM = 7 ± 4% inhibition) (Figure 9C).
Figure 9.

N-[1-(2,6-dichlorophenyl)methyl]-3-(1-pyrrolidinylmethyl)-1H-indol-5-yl]aminocarbonyl}-glycinyl-L-lysinyl-L-phenylalanyl-N-benzhydrylamide (K-14585) stimulated p38 MAP kinase-dependent but Gq/11-independent IL-8 production in NCTC2544-PAR2 cells. NCTC2544-PAR2 cells were serum-starved and incubated with 5–30 µM of K-14585 for 30 min prior to stimulation with 30 µM Ser-Leu-Ile-Gly-Lys-Val (SLIGKV-OH) for an additional 8 h (A). In (B and C) cells were pre-incubated with either SB203580 (10 µM) or YM-254890 (100 nM) prior to stimulation with SLIGKV-OH or K-14585 for an additional 8 h. Samples were assessed for IL-8 production as outlined in the Methods. Each value represents the represents the mean ± SEM, (n= 4). *P < 0.05 compared with peptide stimulation; **P < 0.01.
Discussion
This study has addressed the inhibition of PAR2-mediated signalling using the novel compound K-14585, a putative PAR2 competitive antagonist (Kanke et al., 2009). Despite the fact that PAR2 exhibits pro-inflammatory roles in various pathological conditions, a decade after the receptor's discovery there is still no potent PAR2 antagonist available. Recently we have utilized the compound ENMD-1068 (Kelso et al., 2006) and while it has PAR2-antagonistic activity, its potency is low with effects in the low millimolar range. Compound K-14585 is a novel synthetic peptide derived from the PAR2-tethered ligand sequence and has recently been characterized for its effects upon PAR2-mediated receptor binding and a number of peptide-induced functional responses (Kanke et al., 2009). Therefore, we sought to investigate the inhibitory effects of compound K-14585 on different PAR2-coupled signalling effector systems.
The effects of compound K-14585 on PAR2-mediated inositol trisphosphate (IP3) production (measured indirectly as total [3H] inositol phosphates; IP1–4), MAP kinase activation and NFκB signalling were examined in clone G cells, a keratinocyte cell line that stably expresses PAR2. The data presented here showed that compound K-14585 at concentrations up to 30 µM significantly reduced the total inositol phosphate production stimulated by the PAR2 peptide. As inositol phosphate formation is an early event in the PAR2 signalling cascade, this is likely to reflect receptor inhibition. In contrast, K-14585 was only moderately effective against trypsin-mediated [3H] inositol phosphate accumulation, a result consistent with other responses such as NFκB reporter activity (Kanke et al., 2009). This difference in ability to inhibit protease and activating peptide stimulated responses is a characteristic of this family of receptor and for PAR1 only recently have antagonists been shown to inhibit successfully both PAR1-activating peptide and thrombin responses (Andrade-Gordon et al., 1999; Damiano et al., 2003; Maryanoff et al., 2003). It is likely that differences in the three dimensional structure of the tethered ligand relative to the peptide in aqueous solution makes inhibition of the latter easier to achieve and is a clear limitation in the current development of PAR2 antagonist drugs. Alternatively it may suggest that the peptide SLIGKV-OH binds to a site partially distinct from the tethered ligand binding site and that K-14585 only competes for binding to this site. Nevertheless, K-14585 appears to be more potent than ENMD-1068, which has a potency in the low millimolar range (Kelso et al., 2006).
While pretreatment with lower concentrations of K-14585 was able to inhibit SLIGKV-stimulated intracellular signalling events including [3H] inositol phosphate accumulation (Figure 1) and NFκB activation (Figure 7), we found that the compound at higher concentrations was able to activate p38 MAP kinase and to a lesser extent ERK. These findings were replicated in cells endogenously expressing PAR2 but not in the parental NCTC2544 cells suggesting a degree of selectivity. These results also suggest the potential for K-14585 to have a possible partial agonist activity such that at lower concentrations antagonism is observed but at higher concentrations activation results. Alternatively, some sort of additional interaction with the receptor occurs at higher concentrations. The first scenario, however, is less likely as at a high concentration of K-14585, no activation of either [3H] inositol phosphate accumulation, phosphorylation of p65 NFκB or NFκB-DNA binding was observed. The effect was clearly pathway-specific suggesting two modes of interaction with the receptor, one as an antagonist, the other as an activator. Recent analysis of agonist/receptor interactions supports this second interpretation (Baker and Hill, 2007). Studies show that receptors such as the β1-adrenoceptor may have two ligand binding sites, orthosteric and allosteric, each with differing pharmacological properties (Granneman, 2001; Molenaar, 2003). These different agonist/receptor conformations have the potential to couple to different signalling pathways. For example, propranolol is an inverse agonist at the level of cAMP accumulation, but a partial agonist at the level of ERK activation, an action that is G-protein-independent (Azzi et al., 2003). Similar types of dual G-protein-dependent and independent coupling to ERK and other intermediates have recently been demonstrated for PAR2 (Wang and DeFea, 2006; Kumar et al., 2007) but not as described here, as a consequence of distinct ligand/receptor interactions. It is possible that K-14585 could activate PAR2 to promote epidermal growth factor receptor transactivation (Darmoul et al., 2004); however, our studies show that PAR2-mediated activation of kinase signalling was not affected by epidermal growth factor receptor inhibitors. Thus, further experimentation is required using better pharmacological tools to probe this phenomenon.
Nevertheless, the potential of agonist/receptor-specific signalling pathways was further exemplified by examining more closely Gq/11-dependent p38 MAP kinase activation. While activation by SLIGKV-OH was partly inhibited by the Gq/11 inhibitor YM-254890 (Takasaki et al., 2004) suggesting some role for Gq/11, the response to K-14585 was not, suggesting two different modes for p38 MAP kinase activation emanating from the receptor. We have previously tried to assess the involvement of Gq/11 in PAR2 signalling indirectly by examining the role of Ca2+-dependent protein kinase C in the regulation of ERK and p38 MAP kinase phosphorylation (Macfarlane et al., 2005), and these studies strongly suggest no role for Gq/11 in this regard. However, these studies utilized high concentrations of the agonist trypsin, which might preferentially activate Gq/11-independent signalling. The concentration of SLIGKV-OH used in the current study was low, 30 µM, allowing a Gq/11 dependency to be revealed for p38 MAP kinase. The lack of Gq/11 dependency was also observed using the novel peptide 2f-LIGKV-OH (results not shown), suggesting that certain PAR2 peptide substitutions might generate a specific ligand/receptor receptor conformation and may explain the more prolonged action of 2f-LIGKV-OH in vivo (Ferrell et al., 2003), presumed previously to be due to resistance of the substituted peptide to degradation (Kawabata et al., 2004). Furthermore, the Gq/11-dependent component did not extend to ERK activation, SLIGKV-OH stimulation of ERK activation was not inhibited by YM-254890 (results not shown), and this would be in keeping with the dependency of PAR2 ERK signalling upon β-arrestins (Wang and DeFea, 2006; Kumar et al., 2007), similar to that observed with for other G-protein-coupled receptors such as the vasopressin V2 (Charest et al., 2007) and angiotensin AT1 (Wei et al., 2003) receptors. K-14585 alone caused a small, twofold to threefold increase, in ERK activation and this activation was found to be wholly Gq/11-independent.
When assessing the NFκB pathway, the dual effects of K-14585 was also revealed. K-14585 was able to strongly inhibit both p65 NFκB phosphorylation and NFκB-DNA binding. Both these events are regulated by upstream activation of the inhibitory κB kinases (Kanke et al., 2001), and we have previously demonstrated that inhibitory κB kinase activation is, in turn, likely to be regulated by Ca2+-dependent protein kinase C-mediated signalling (Macfarlane et al., 2005). This again might reflect competition for a specific peptide-mediated activation of this pathway. However, while compound K-14585 was able to inhibit DNA reporter activation in response to activating peptide, a reflection of the effects upstream in the NFκB pathway, at 30 µM alone gave a very strong activation of NFκB reporter activity that was equal in magnitude to that obtained with the peptide, again indicative of an dual function at the signalling level. The most parsimonious interpretation of this data was that K-14585 was able to strongly activate p38 MAP kinase and thus modulate reporter activity. Studies have shown that p38 MAP kinase can either directly activate NFκB-DNA binding by an effect upstream in the pathway (Kim et al., 2002) or can modulate NFκB-dependent transcriptional activity, following DNA binding, by phosphorylation of p65 NFκB (Korus et al., 2002; Darieva et al., 2004). However, the selective p38 MAP kinase inhibitor SB203580 that has been characterized recently in our laboratory (Ritchie et al., 2007) was unable to reverse the effect of K-14585 on reporter activity suggesting another mechanism by which K-14585 can influence NFκB transcriptional activation. Interestingly, we found that SB203580 was also without effect upon peptide-stimulated reporter activity, again strongly suggesting that p38 MAP kinase was not involved in the regulation of NFκB transcription in this cell model.
Finally, we tested the effect of K-14585 at the level of IL-8 production shown previously to be regulated by PAR2 (Ramachandran et al., 2007). Once again, IL-8 production induced by SLIGKV-OH was inhibited by lower concentrations of K-14585 but at a higher concentration an increase was observed, again indicative of a dual effect. As both K-14585 and activating peptide-induced IL-8 responses were significantly reduced by the p38 MAP kinase inhibitor SB203580, this suggests that p38 plays an essential role in the regulation of IL-8 in response to PAR2, a finding consistent with recent studies (Yoshida et al., 2007). Again YM-254890 reduced IL-8 induction by SLIGKV but not that induced by K-14585 supporting the idea that K-14585 utilizes a Gq/11-independent pathway. However, the fact that IL-8 production in response to SLIGKV-OH was also reduced by YM-254890 does show that PAR2 coupling to Gq/11 is functionally relevant for IL-8 induction and mediated via p38 MAP kinase. This also suggests that inhibition of Gq/11 in this context may be able to inhibit some of the inflammatory effects of PAR2in vivo.
In conclusion we have demonstrated that the novel PAR2 antagonist K-14585 shows moderate antagonist actions at a number of signalling pathways. However, we have uncovered the potential for this compound to have dual effects such that certain signalling pathways are activated at higher concentrations, indicative of agonist-directed signalling. Specifically, K-14585 was able to inhibit PAR2-mediated Gq/11-dependent pathways and to stimulate Gq/11-independent events, including activation of p38 MAP kinase.
Acknowledgments
This work was supported by The Wellcome Trust. FGG is a recipient of an ORS and University of Strathclyde studentship.
Glossary
Abbreviations:
- K-14585
N-[1-(2,6-dichlorophenyl)methyl]-3-(1-pyrrolidinylmethyl)-1H-indol-5-yl]aminocarbonyl}-glycinyl-L-lysinyl-L-phenylalanyl-N-benzhydrylamide
- PAR
proteinase-activated receptor
- SLIGKV-OH
Ser-Leu-Ile-Gly-Lys-Val
References
- Alexander SP, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 3rd edition. Br J Pharmacol. 2008;153(Suppl 2):S1, S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade-Gordon P, Maryanoff BE, Derian CK, Zhang HC, Addo MF, Darrow AL, et al. Design, synthesis, and biological characterization of a peptide-mimetic antagonist for a tethered-ligand receptor. Proc Natl Acad Sci USA. 1999;96:12257–12262. doi: 10.1073/pnas.96.22.12257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzi M, Charest PG, Angers S, Rousseau G, Kohout T, Bouvier M, et al. Beta-arrestin-mediated activation of MAPK by inverse agonists reveals distinct active conformations for G protein-coupled receptors. Proc Natl Acad Sci USA. 2003;100:11406–11411. doi: 10.1073/pnas.1936664100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker JG, Hill SJ. Multiple GPCR conformations and signalling pathways: implications for antagonist affinity estimates. Trends Pharmacol Sci. 2007;28:374–381. doi: 10.1016/j.tips.2007.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cenac N, Coelho AM, Nguyen C, Compton S, Andrade-Gordon P, MacNaughton WK, et al. Induction of intestinal inflammation in mouse by activation of proteinase-activated receptor-2. Am J Pathol. 2002;161:1903–1915. doi: 10.1016/S0002-9440(10)64466-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cenac N, Garcia-Villar R, Ferrier L, Larauche M, Vergnolle N, Bunnett NW, et al. Proteinase-activated receptor-2-induced colonic inflammation in mice: possible involvement of afferent neurons, nitric oxide, and paracellular permeability. J Immunol. 2003;170:4296–4300. doi: 10.4049/jimmunol.170.8.4296. [DOI] [PubMed] [Google Scholar]
- Chackalamannil S. Thrombin receptor (protease activated receptor-1) antagonists as potent antithrombotic agents with strong antiplatelet effects. J Med Chem. 2006;49:5389–5403. doi: 10.1021/jm0603670. [DOI] [PubMed] [Google Scholar]
- Charest PG, Oligny-Longpre G, Bonin H, Azzi M, Bouvier M. The V2 vasopressin receptor stimulates ERK1/2 activity independently of heterotrimeric G protein signalling. Cell Signal. 2007;19:32–41. doi: 10.1016/j.cellsig.2006.05.020. [DOI] [PubMed] [Google Scholar]
- Damiano BP, Derian CK, Maryanoff BE, Zhang HC, Gordon PA. RWJ-58259: a selective antagonist of protease activated receptor-1. Cardiovasc Drug Rev. 2003;21:313–326. doi: 10.1111/j.1527-3466.2003.tb00124.x. [DOI] [PubMed] [Google Scholar]
- Darieva Z, Lasunskaia EB, Campos MN, Kipnis TL, Da Silva WD. Activation of phosphatidylinositol 3-kinase and c-Jun-N-terminal kinase cascades enhances NF-kappaB-dependent gene transcription in BCG-stimulated macrophages through promotion of p65/p300 binding. J Leukoc Biol. 2004;75:689–697. doi: 10.1189/jlb.0603280. [DOI] [PubMed] [Google Scholar]
- Darmoul D, Gratio V, Devaud H, Laburthe M. Protease-activated receptor 2 in colon cancer: trypsin-induced MAPK phosphorylation and cell proliferation are mediated by epidermal growth factor receptor transactivation. J Biol Chem. 2004;279:20927–20934. doi: 10.1074/jbc.M401430200. [DOI] [PubMed] [Google Scholar]
- Ferrell WR, Lockhart JC, Kelso EB, Dunning L, Plevin R, Meek SE, et al. Essential role for proteinase-activated receptor-2 in arthritis. J Clin Invest. 2003;111:35–41. doi: 10.1172/JCI16913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goon Goh F, Sloss CM, Cunningham MR, Nilsson M, Cadalbert L, Plevin R. G-protein-dependent and -independent pathways regulate proteinase-activated receptor-2 mediated p65 NFkappaB serine 536 phosphorylation in human keratinocytes. Cell Signal. 2008;20:1267–1274. doi: 10.1016/j.cellsig.2008.02.015. [DOI] [PubMed] [Google Scholar]
- Granneman JG. The putative beta4-adrenergic receptor is a novel state of the beta1-adrenergic receptor. Am J Physiol Endocrinol Metab. 2001;280:E199, E202. doi: 10.1152/ajpendo.2001.280.2.E199. [DOI] [PubMed] [Google Scholar]
- Kanke T, Macfarlane SR, Seatter MJ, Davenport E, Paul A, McKenzie RC, et al. Proteinase-activated receptor-2-mediated activation of stress-activated protein kinases and inhibitory kappa B kinases in NCTC 2544 keratinocytes. J Biol Chem. 2001;276:31657–31666. doi: 10.1074/jbc.M100377200. [DOI] [PubMed] [Google Scholar]
- Kanke T, Takizawa T, Kabeya M, Kawabata A. Physiology and pathophysiology of proteinase-activated receptors (PARs): PAR-2 as a potential therapeutic target. J Pharmacol Sci. 2005;97:38–42. doi: 10.1254/jphs.fmj04005x7. [DOI] [PubMed] [Google Scholar]
- Kanke T, Kabeya M, Kubo S, Kondo S, Yasuoka K, Tagashira J, et al. Novel antagonists for proteinase-activated receptor 2: inhibition of cellular and vascular responses in vitro and in vivo. Br J Pharmacol. 2009;158:361, 371. doi: 10.1111/j.1476-5381.2009.00342.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawabata A, Kanke T, Yonezawa D, Ishiki T, Saka M, Kabeya M, et al. Potent and metabolically stable agonists for protease-activated receptor-2: evaluation of activity in multiple assay systems in vitro and in vivo. J Pharmacol Exp Ther. 2004;309:1098–1107. doi: 10.1124/jpet.103.061010. [DOI] [PubMed] [Google Scholar]
- Kawagoe J, Takizawa T, Matsumoto J, Tamiya M, Meek SE, Smith AJ, et al. Effect of protease-activated receptor-2 deficiency on allergic dermatitis in the mouse ear. Jpn J Pharmacol. 2002;88:77–84. doi: 10.1254/jjp.88.77. [DOI] [PubMed] [Google Scholar]
- Kelso EB, Lockhart JC, Hembrough T, Dunning L, Plevin R, Hollenberg MD, et al. Therapeutic promise of proteinase-activated receptor-2 antagonism in joint inflammation. J Pharmacol Exp Ther. 2006;316:1017–1024. doi: 10.1124/jpet.105.093807. [DOI] [PubMed] [Google Scholar]
- Kim SJ, Hwang SG, Shin DY, Kang SS, Chun JS. p38 kinase regulates nitric oxide-induced apoptosis of articular chondrocytes by accumulating p53 via NFkappa B-dependent transcription and stabilization by serine 15 phosphorylation. J Biol Chem. 2002;277:33501–33508. doi: 10.1074/jbc.M202862200. [DOI] [PubMed] [Google Scholar]
- Korus M, Mahon GM, Cheng L, Whitehead IP. p38 MAPK-mediated activation of NF-kappaB by the RhoGEF domain of Bcr. Oncogene. 2002;21:4601–4612. doi: 10.1038/sj.onc.1205678. [DOI] [PubMed] [Google Scholar]
- Kumar P, Lau CS, Mathur M, Wang P, DeFea KA. Differential effects of beta-arrestins on the internalization, desensitization and ERK1/2 activation downstream of protease activated receptor-2. Am J Physiol Cell Physiol. 2007;293:C346, C357. doi: 10.1152/ajpcell.00010.2007. [DOI] [PubMed] [Google Scholar]
- Macfarlane SR, Seatter MJ, Kanke T, Hunter GD, Plevin R. Proteinase-activated receptors. Pharmacol Rev. 2001;53:245–282. [PubMed] [Google Scholar]
- Macfarlane SR, Sloss CM, Cameron P, Kanke T, McKenzie RC, Plevin R. The role of intracellular Ca2+ in the regulation of proteinase-activated receptor-2 mediated nuclear factor kappa B signalling in keratinocytes. Br J Pharmacol. 2005;145:535–544. doi: 10.1038/sj.bjp.0706204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh KA, Plevin R, Ferrell WR, Lockhart JC. The therapeutic potential of proteinase-activated receptors in arthritis. Curr Opin Pharmacol. 2007;7:334–338. doi: 10.1016/j.coph.2007.01.002. [DOI] [PubMed] [Google Scholar]
- Maryanoff BE, Zhang HC, Andrade-Gordon P, Derian CK. Discovery of potent peptide-mimetic antagonists for the human thrombin receptor, protease-activated receptor-1 (PAR-1) Curr Med Chem Cardiovasc Hematol Agents. 2003;1:13–36. doi: 10.2174/1568016033356724. [DOI] [PubMed] [Google Scholar]
- Molenaar P. The ‘state’ of beta-adrenoceptors. Br J Pharmacol. 2003;140:1–2. doi: 10.1038/sj.bjp.0705420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nystedt S, Emilsson K, Wahlestedt C, Sundelin J. Molecular cloning of a potential proteinase activated receptor. Proc Natl Acad Sci USA. 1994;91:9208–9212. doi: 10.1073/pnas.91.20.9208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossovskaya VS, Bunnett NW. Protease-activated receptors: contribution to physiology and disease. Physiol Rev. 2004;84:579–621. doi: 10.1152/physrev.00028.2003. [DOI] [PubMed] [Google Scholar]
- Paul A, Torrie LJ, McLaren GJ, Kennedy C, Gould GW, Plevin R. P2Y receptor-mediated inhibition of tumor necrosis factor alpha -stimulated stress-activated protein kinase activity in EAhy926 endothelial cells. J Biol Chem. 2000;275:13243–13249. doi: 10.1074/jbc.275.18.13243. [DOI] [PubMed] [Google Scholar]
- Plevin R, Kellock NA, Wakelam MJ, Wadsworth R. Regulation by hypoxia of endothelin-1-stimulated phospholipase D activity in sheep pulmonary artery cultured smooth muscle cells. Br J Pharmacol. 1994;112:311–315. doi: 10.1111/j.1476-5381.1994.tb13070.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran R, Sadofsky LR, Xiao Y, Botham A, Cowen M, Morice AH, et al. Inflammatory mediators modulate thrombin and cathepsin-G signaling in human bronchial fibroblasts by inducing expression of proteinase-activated receptor-4. Am J Physiol Lung Cell Mol Physiol. 2007;292:L788, L798. doi: 10.1152/ajplung.00226.2006. [DOI] [PubMed] [Google Scholar]
- Ritchie E, Saka M, Mackenzie C, Drummond R, Wheeler-Jones C, Kanke T, et al. Cytokine upregulation of proteinase-activated-receptors 2 and 4 expression mediated by p38 MAP kinase and inhibitory kappa B kinase beta in human endothelial cells. Br J Pharmacol. 2007;150:1044–1054. doi: 10.1038/sj.bjp.0707150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber E, Matthias P, Muller MM, Schaffner W. Rapid detection of octamer binding proteins with ‘mini-extracts’ prepared from a small number of cells. Nucleic Acids Res. 1989;17:6419. doi: 10.1093/nar/17.15.6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeliger S, Derian CK, Vergnolle N, Bunnett NW, Nawroth R, Schmelz M, et al. Proinflammatory role of proteinase-activated receptor-2 in humans and mice during cutaneous inflammation in vivo. FASEB J. 2003;17:1871–1885. doi: 10.1096/fj.02-1112com. [DOI] [PubMed] [Google Scholar]
- Steinhoff M, Vergnolle N, Young SH, Tognetto M, Amadesi S, Ennes HS, et al. Agonists of proteinase-activated receptor 2 induce inflammation by a neurogenic mechanism. Nat Med. 2000;6:151–158. doi: 10.1038/72247. [DOI] [PubMed] [Google Scholar]
- Takasaki J, Saito T, Taniguchi M, Kawasaki T, Moritani Y, Hayashi K, et al. A novel Gαq/11-selective inhibitor. J Biol Chem. 2004;279:47438–47445. doi: 10.1074/jbc.M408846200. [DOI] [PubMed] [Google Scholar]
- Wang P, DeFea KA. Protease-activated receptor-2 simultaneously directs beta-arrestin-1-dependent inhibition and Galphaq-dependent activation of phosphatidylinositol 3-kinase. Biochemistry. 2006;45:9374–9385. doi: 10.1021/bi0602617. [DOI] [PubMed] [Google Scholar]
- Wei H, Ahn S, Shenoy SK, Karnik SS, Hunyady L, Luttrell LM, et al. Independent beta-arrestin 2 and G protein-mediated pathways for angiotensin II activation of extracellular signal-regulated kinases 1 and 2. Proc Natl Acad Sci USA. 2003;100:10782–10787. doi: 10.1073/pnas.1834556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida N, Katada K, Handa O, Takagi T, Kokura S, Naito Y, et al. Interleukin-8 production via protease-activated receptor 2 in human esophageal epithelial cells. Int J Mol Med. 2007;19:335–340. doi: 10.3892/ijmm.19.2.335. [DOI] [PubMed] [Google Scholar]