

Abstract
Background and purpose:
The association between torcetrapib and its off-target effects on blood pressure suggested a possible class-specific effect. The effects of dalcetrapib (RO4607381/JTT-705) and torcetrapib on haemodynamics and the renin-angiotensin-aldosterone system (RAAS) were therefore assessed in a rat model.
Experimental approach:
Arterial pressure (AP) and heart rate were measured by telemetry in normotensive and spontaneously hypertensive rats (SHR) receiving torcetrapib 10, 40 or 80 mg·kg−1·day−1; dalcetrapib 100, 300 or 500 mg−1·kg·day−1; or vehicle (placebo) for 5 days. Expression of RAAS genes in adrenal gland, kidney, aorta and lung from normotensive rats following 5 days' treatment with torcetrapib 40 mg·kg−1·day−1, dalcetrapib 500 mg·kg−1·day−1 or vehicle was measured by quantitative polymerase chain reaction.
Key results:
Torcetrapib transiently increased mean AP in normotensive rats (+3.7 ± 0.1 mmHg), whereas treatment in SHR resulted in a dose-dependent and sustained increase [+6.5 ± 0.6 mmHg with 40 mg·kg−1·day−1 at day 1 (P < 0.05 versus placebo)], which lasted over the treatment period. No changes in AP or heart rate were observed with dalcetrapib. Torcetrapib, but not dalcetrapib, increased RAAS-related mRNAs in adrenal glands and aortas.
Conclusions and implications:
In contrast to torcetrapib, dalcetrapib did not increase blood pressure or RAAS-related gene expression in rats, suggesting that the off-target effects of torcetrapib are not a common feature of all compounds acting on cholesteryl ester transfer protein.
Keywords: CETP inhibitor, torcetrapib, dalcetrapib, blood pressure, SHR
Introduction
Cholesteryl ester transfer protein (CETP) mediates the transfer of cholesteryl ester (CE) in exchange for triglyceride (TG) between lipoproteins, thereby playing a key role in the physiological process of reverse cholesterol transfer and removal of excess cholesterol (Tall, 1993; Barter et al., 2003). The net impact of CETP activity on cholesterol metabolism leads to decreased high-density lipoprotein cholesterol (HDL-C) and increased low-density lipoprotein cholesterol (LDL-C) levels, with a concomitant reduction in both HDL and LDL particle size (Barter and Kastelein, 2006). Agents that act upon CETP, preventing the transfer of CE and TG, have the potential to increase HDL-C levels and decrease LDL-C levels, thus promoting an anti-atherogenic lipoprotein phenotype (Barter and Kastelein, 2006).
The clinical development of the CETP inhibitor torcetrapib (Pfizer, New York, USA) was suspended due to increased mortality and cardiovascular events in the Investigation of Lipid Level Management to Understand its Impact in Atherosclerotic Events (ILLUMINATE) study (Barter et al., 2007). Off-target effects on blood pressure and plasma aldosterone, indicating activation of the renin-angiotensin-aldosterone system (RAAS), may have contributed to the toxicity of torcetrapib (Barter et al., 2007). These effects have raised concerns over the whole class of drugs that act by inhibiting the activity of CETP, and questions were raised as to whether the off-target effects seen with torcetrapib are compound specific or common to all drugs that act on CETP.
Dalcetrapib (formerly known as RO4607381/JTT-705) differs from torcetrapib in its physicochemical properties (molecular weights: 389.6 and 600.4 respectively; computed log octanol/water partition coefficients: –7 and –9 respectively) and structure (Figure 1). Torcetrapib and dalcetrapib decrease CETP activity by different mechanisms. Dalcetrapib forms a bond with cysteine 13 of CETP (Okamoto et al., 2000), most likely located in the lipophilic tunnel of the CETP molecule (Qiu et al., 2007), without inducing the formation of a stable CETP-HDL complex at therapeutic plasma concentrations (Niesor et al., 2008). In contrast, torcetrapib increases the binding affinity of CETP to HDL and the potency for inhibition of CE transfer is correlated with the ability to form a non-productive complex between CETP and HDL (Clark et al., 2006). In phase II clinical trials, dalcetrapib has been shown to increase HDL-C levels without affecting blood pressure (de Grooth et al., 2002; Kuivenhoven et al., 2005; Stein et al., 2009).
Figure 1.
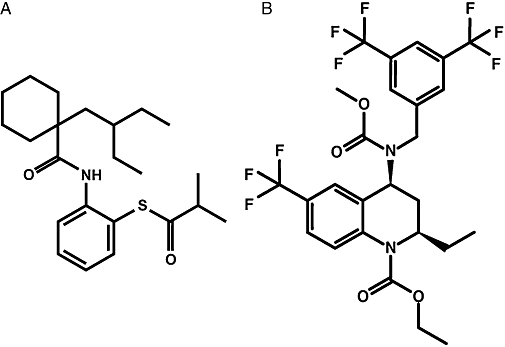
Structure of (A) dalcetrapib and (B) torcetrapib.
The haemodynamic effects of torcetrapib and dalcetrapib were evaluated in normotensive and spontaneously hypertensive rats (SHR). As rats are naturally deficient in CETP, this enables direct comparison of off-target effects by agents that target CETP. The SHR with dysregulation of blood pressure is an accepted model of human essential hypertension and is commonly used for assessing anti-hypertensive compounds (Lerman et al., 2005; Potenza et al., 2005; Yang et al., 2005); the normotensive rat is able to compensate or adapt to haemodynamic changes and is a less sensitive model for blood pressure increases but can be used to study the direct changes and compensatory mechanisms triggered by tested compounds. The potential mechanism of any blood pressure increase was investigated by analysing the expression of RAAS-related genes in various tissues.
Methods
Animals
All animal care and experimental procedures were in accordance with Roche guidelines and national/international guidelines for animal care. Studies were conducted in male normotensive Wistar rats [HanBrl: WIST(SPF), RCC Ltd., Füllingsdorf, Switzerland] and SHR (Charles River Laboratories, Sulzfeld, Germany) weight 240–260 g.
Haemodynamic effects
In one study, normotensive rats and SHR received torcetrapib (10, 40 or 80 mg·kg−1·day−1) or vehicle (placebo) by oral gavage (n= 6–8 per group). In another study, SHR received dalcetrapib (100, 300 or 500 mg·kg−1·day−1) or vehicle by oral gavage (n= 6–8 per group). Treatment was given for five consecutive days followed by a 10 day recovery period.
Arterial pressure (AP), systolic AP, diastolic AP and heart rate were measured continuously via radio-telemetry before, during and after the treatment. Telemetric blood pressure transmitters [TA11PA-C40, Data Sciences International (DSI), St. Paul, MN, USA] were implanted under aseptic conditions in rats anaesthetized with a mixture of 6 mg·kg−1 xylazine (Rompun, Bayer, Provet, Lyssach, Switzerland) and 40 mg·kg−1 ketamine (Ketasol-100, Gräub, Bern, Switzerland) given intraperitoneally. Briefly, a midline abdominal incision was made and a fluid-filled sensor catheter was inserted into the descending aorta above the iliac bifurcation. The tip of the telemetric catheter was positioned in the abdominal aorta caudal to the renal arteries. Rats were allowed to recover for at least 7 days. A receiver board (RPC-1 or RLA 1020, DSI) placed under the animal cages monitored the transmitter signals. The telemetry signal was processed using a multiplexer (Data Exchange Matrix, DSI) and analysed using Dataquest ART software (DSI), which calculated and stored mean values over a 10 s interval every 10 min, with a sampling rate of 1000 Hz. Means of AP values recorded over a 1 h period before, during and after treatment were calculated.
Pharmacokinetic analysis
A group of four rats per dose of torcetrapib and dalcetrapib was treated in parallel with telemetered animals for pharmacokinetic analysis. Torcetrapib plasma concentrations were determined using liquid chromatography with tandem mass spectrometry (LC-MS/MS), which had a quantification limit of 10.0 ng·mL−1. For dalcetrapib, plasma concentrations were determined using LC-MS/MS with a quantification limit of 5.0 ng·mL−1.
RAAS-related gene expression
Quantitative real-time polymerase chain reaction (qPCR) was carried out to determine changes in expression of several RAAS-related genes: angiotensinogen (AOGEN), angiotensin-converting enzyme (ACE), angiotensin receptor 1 (AT1), preproendothelin (preproET1), endothelin-converting enzyme (ECE), endothelin receptor A (ETA), endothelin receptor B (ETB), serum/glucocorticoid regulated kinase 1 (SGK1), steroidogenic acute regulatory protein (StAR), scavenger receptor class B type 1 (SR-B1) and endothelial nitric oxide synthase (eNOS; NOS3). Male normotensive Wistar rats (240–260 g) received torcetrapib 40 mg·kg−1, dalcetrapib 500 mg·kg−1 or vehicle daily for 5 days by oral gavage (n= 6). Similarly, male SHR (240–260 g) received torcetrapib 40 mg·kg−1 or vehicle daily for 5 days by oral gavage (n= 6). Two hours after the last gavage on day 5, rats were killed by decapitation under light isoflurane anaesthesia. Adrenal glands, kidneys, aortas and lungs were removed and placed either in liquid nitrogen and kept frozen at –70°C or in RNAlater (Ambion, Austin, TX, USA) and incubated at 4°C for 24 h. Total RNA extraction was then performed using the FastRNA Pro Green Kit (Qbiogene Ltd., Beckenham, UK) according to the manufacturer's instructions. Further mRNA purification was then performed using TRIzol Plus (Invitrogen, Basel, Switzerland) including an RNAse free DNAse I treatment. cDNA was synthesized using cDNA Synthesis System (Roche Applied Science) and purified using QIAquick PCR Purification Kit (QIAGEN, Hombrechtikon, Switzerland). Primer Express software (PE Applied Biosystems, Foster City, CA, USA). PrimerExpress software (PE Applied Biosystems, Foster City, CA, USA) was used to design qPCR primer pairs based on mRNA sequences from PubMed (Table 1). Real time (RT)-PCR was performed using QuantiTect SYBR Green RT-PCR Kit (QIAGEN) according to the manufacturer's instructions. Gene expression analysis was performed using a Rotor-Gene 6000 RT analyser and software (Corbettt Life Science, Sydney, Australia). Analysis was conducted using the Δ-Δcycle threshold method, which determined fold changes in gene expression relative to a vehicle-treated sample. Each analysis reaction was performed in duplicate, with two samples per condition. Gene expression was normalized to glyceraldehyde-3-phosphate dehydrogenase, which was used as an internal reference gene.
Table 1.
Forward and reverse primers
| Gene | Oligo name | Forward primer | Oligo name | Reverse primer |
|---|---|---|---|---|
| GAPDH | rGAPDH_1032F | GCAACAGCGTGGTGGACCT | rGAPDH_1096R | TGGGTGGTCCAGGGTTTCTTA |
| AOGEN | rANG_1547F | GCAAAAATCAGTGCCTTCACCC | rANG_1674R | AAACAAACCCTCACCCCAGGAG |
| ACE | rACE_F | TTGACGTGAGCAACTTCCAG | rACE_R | CAGATCAGGCTCCAGTGACA |
| AT1 | rAT1A_F | CTCAAGCCTGTCTACGAAAATGAG | rAT1_R | TAGATCCTGAGGCAGGGTGAAT |
| preproET1 | rET1-F | TCTGGGTCAACACTCCCG | rET1_R | AGGATCGCTTAGACCTAGAAGGG |
| ECE | rECE_4351F | AAAGAGCCCTCCTCTCAAGCC | rECE_4413R | CAGTTCCAAACCCCATGTCCT |
| ETA | rET1Ar_960F | AACTCAAGCAGCGTCGAGAG | rET1Ar_1020R | AGGGCGAAGATGACAACCAA |
| ETB | rET1Br_F | TTTAAGTCGTGTTTGTGCTGC | rET1Br-R | AAGCAGGATTTCTTCTTCTC |
| SGK1 | rSGK1_F | TAGCAATCCTCATCGCTTTC | rSGK1_R | GAGTTGTTGGCAAGCTTCTG |
| StAR | rStar_444F | AGGAAAGCCAGCAGGAGAATG | rStar_499R | CACACCTGGCACCACCTTACT |
| SR-B1 | rSRB1_768F | ATGGGACTTCCGGGCAGAT | rSRB1_802R | CAGCGAGGATTCGGGTGTC |
| eNOS | reNOS_F | AAGTGGGCAGCATCACCTAC | reNOS_R | CTGGGAACCACTCCTTTTGA |
AOGEN, angiotensinogen; ACE, angiotensin-converting enzyme; AT1, angiotensin receptor type 1; preproET1, preproendothelin; ECE, endothelin-converting enzyme; ETA, endothelin receptor A; ETB, endothelin receptor B; eNOS, endothelial nitric oxide synthase; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; SGK1, serum/glucocorticoid regulated kinase 1; SR-B1, scavenger receptor class B type 1; StAR, steroidogenic acute regulatory protein.
Data analysis
Statistical analysis of haemodynamic results was conducted by analysis of variance for repetitive measurements followed by Dunnett's test. Blood pressure changes vs. placebo were analysed using Student's t-test. Any difference vs. placebo of P < 0.05 was considered statistically significant. Data are expressed as mean ± s.e. of the mean. Statistical analyses of gene expression data were performed using Student's unpaired t-test. The criterion for significant gene expression changes between the study groups was P < 0.05. Data are expressed as mean (± s.e. mean) fold change compared with vehicle-treated animals.
Materials
Dalcetrapib and torcetrapib were synthesized by Roche. Dalcetrapib was formulated as a micro-emulsion in 0.5% methylcellulose (Metolose SM-1500, Shin-Etsu Chemical Co. Ltd., Tokyo, Japan). Due to its non-homogeneity in methylcellulose, torcetrapib was formulated as a micro-emulsion in polyethylene glycol-15-hydroxystearate (Solutol HS 15, BASF, Ludwigshafen, Germany), medium chain triglyceride (MCT, Roche Galenics, Belvedere, NJ, USA) and water (GSP). Placebo was Solutol HS 15/MCT (BASF) vehicle for torcetrapib and 0.5% methylcellulose for dalcetrapib. Drug/molecular target nomenclature conforms to guidelines in Alexander et al. (2008).
Results
Haemodynamic effects in SHR
In SHR, baseline values for mean AP were similar for all treatment groups (placebo: 145 ± 2 mmHg; torcetrapib 10 mg·kg−1·day−1: 142 ± 2 mmHg; torcetrapib 40 mg·kg−1·day−1: 147 ± 5 mmHg; torcetrapib 80 mg·kg−1·day−1: 142 ± 3 mmHg) and consistent with hypertension. An increase in mean AP was observed in SHR treated with torcetrapib vs. placebo on day 1 ( Figure 2); a significant increase was observed in the 40 mg·kg−1·day−1 group (P < 0.05) (Table 2). Significant increases were also observed for systolic AP and diastolic AP following treatment with torcetrapib 40 mg·kg−1·day−1 (P < 0.05) (Table 2). Once torcetrapib treatment was stopped, mean AP rapidly returned to baseline levels (Figure 3). The increase in mean AP induced by torcetrapib treatment was not associated with significant changes in heart rate (Table 2).
Figure 2.
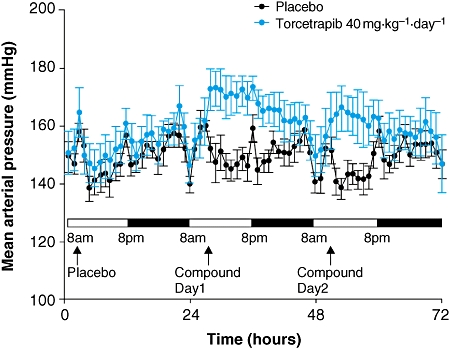
Mean (±s.e. mean) arterial pressure in spontaneously hypertensive rats from the torcetrapib group and placebo group administered placebo at day 0, then torcetrapib 40 mg·kg−1·day−1 or placebo at days 1 and 2.
Table 2.
Changes (mean ± s.e. mean) in AP and heart rate in SHR and normotensive rats treated with torcetrapib on day 1 (data expressed as difference from placebo group)
| Torcetrapib dose | Mean AP (mmHg) | Systolic AP (mmHg) | Diastolic AP (mmHg) | Heart rate (bpm) |
|---|---|---|---|---|
| SHR | ||||
| 10 mg·kg−1·day−1 | 2.7 ± 0.8 | 2.7 ± 0.8 | 2.5 ± 0.8 | 7.1 ± 1.9 |
| 40 mg·kg−1·day−1 | 6.5 ± 0.6* | 7.3 ± 0.9* | 5.3 ± 0.6* | 7.2 ± 2.2 |
| 80 mg·kg−1·day−1 | 7.8 ± 3.9 | 7.8 ± 3.5 | 7.9 ± 4.5 | 2.8 ± 3.4 |
| Normotensive rats | ||||
| 10 mg·kg−1·day−1 | 1.4 ± 0.9 | 1.9 ± 1.0 | 1.0 ± 0.8 | 4.0 ± 3.7 |
| 40 mg·kg−1·day−1 | 3.7 ± 1.0 | 4.9 ± 1.0* | 2.3 ± 1.0 | −0.3 ± 4.7 |
| 80 mg·kg−1·day−1 | 2.6 ± 1.4 | 3.3 ± 1.5 | 1.7 ± 1.1 | −10.7 ± 6.6 |
P < 0.05 vs. placebo.
AP, arterial pressure; bpm, beats per minute; SHR, spontaneously hypertensive rat.
Figure 3.
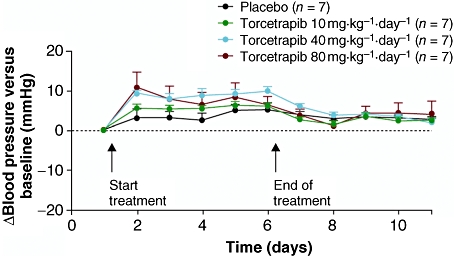
Time course of change in mean (± s.e. mean) arterial pressure from baseline of torcetrapib compared with placebo in spontaneously hypertensive rats.
In the dalcetrapib study, baseline values for mean AP in SHR were similar for all treatment groups (placebo: 152 ± 13 mmHg; dalcetrapib 100 mg·kg−1·day−1: 151 ± 7 mmHg; dalcetrapib 300 mg·kg−1·day−1: 152 ± 6 mmHg; dalcetrapib 500 mg·kg−1·day−1: 148 ± 10 mmHg). There were no significant changes in mean AP vs. placebo with the three dalcetrapib doses tested (Figure 4). After 5 days of treatment with dalcetrapib 500 mg·kg−1·day−1, there was no significant alteration of mean AP (0.8 ± 2.2 mmHg). A trend towards a minor decrease in heart rate was observed with increasing dalcetrapib dose at day 5, which was significant with dalcetrapib 500 mg·kg−1·day−1 (P < 0.05) [dalcetrapib 100 mg·kg−1·day−1: +2 ± 2 beats per minute (bpm); dalcetrapib 300 mg·kg−1·day−1: –7 ± 2 bpm; dalcetrapib 500 mg·kg−1·day−1: –11 ± 2 bpm]. These changes were considered minor based on the rapid heart rate of SHR (approximately 350 bpm).
Figure 4.
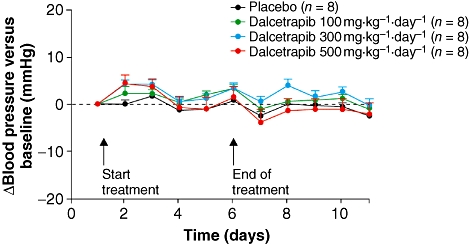
Time course of change in mean (± s.e. mean) arterial pressure from baseline of dalcetrapib compared with placebo in spontaneously hypertensive rats.
Plasma drug concentrations were found to be in a similar range or higher to those observed in clinical studies for torcetrapib and dalcetrapib respectively (Clark et al., 2004; Roche, data on file). In SHR, torcetrapib 40 mg·kg-1·day-1 led to plasma exposure of ∼1800 ng·mL-1, measured 5 h post dosing, and AUC ∼100 ng·mL−1·h−1. Dalcetrapib plasma exposures were measured during the 24 h period at cessation of 5 days of treatment. For dalcetrapib 500 mg·kg−1·day−1, AUC values were 37 300 ng·mL−1·h−1 and Cmax was 3590 ng·mL−1.
Haemodynamic effects in normotensive rats
In normotensive rats, baseline values for mean AP were similar for all treatment groups (placebo: 98 ± 0 mmHg; torcetrapib 10 mg·kg−1·day−1: 101 ± 1 mmHg; torcetrapib 40 mg·kg−1·day−1: 111 ± 1 mmHg; torcetrapib 80 mg·kg−1·day−1: 103 ± 2 mmHg). Following torcetrapib administration, mean AP increased relative to placebo, with simultaneous increases in diastolic and systolic AP. After 1 day of treatment there was a significant increase in systolic AP with torcetrapib 40 mg·kg−1·day−1 (P < 0.05) (Table 2; Figure 5). Torcetrapib 40 mg·kg−1·day−1 and 80 mg·kg−1·day−1 increased mean AP to a similar extent, with the slightly higher effect on mean AP at a dose of 40 mg·kg−1·day−1 compared with 80 mg·kg−1·day−1 within the range of in vivo variability, suggesting that the elevation of mean AP reaches a plateau above 40 mg·kg−1·day−1. Increases in mean, diastolic, and systolic AP were more pronounced and lasted for a longer period on the first day of treatment, following which the hypertensive effects of torcetrapib were smaller and lasted for a shorter time period with each additional day of treatment, over the 5 days of investigation (data not shown). There were no significant changes in heart rate vs. placebo (Table 2).
Figure 5.
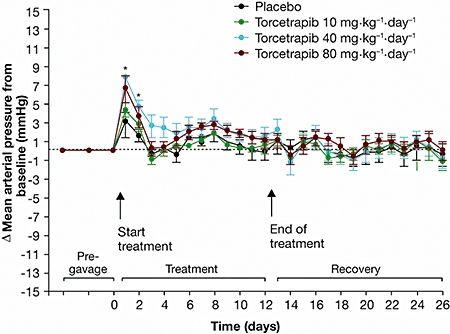
Time course of change in mean (± s.e. mean) arterial pressure from baseline of torcetrapib compared with placebo in normotensive rats.
RAAS gene expression
Expression of RAAS-related genes was investigated in several tissues. Increases in such expression were observed in the adrenal glands of normotensive rats following torcetrapib administration for 5 days. In particular, large fold increases were observed in AT1 receptors, AOGEN, ACE, ECE, SGK1 and endothelin-1, measured as preproET1, compared with placebo (all P < 0.05; Figure 6A). SR-B1, responsible for selective cellular uptake of cholesterol from HDL-C, was also markedly upregulated. Torcetrapib also increased expression of RAAS-related genes in aortic samples, particularly endothelin 1 and ACE. No significant changes in RAAS-related gene expression were observed in kidney or lung samples from normotensive rats treated with torcetrapib.
Figure 6.
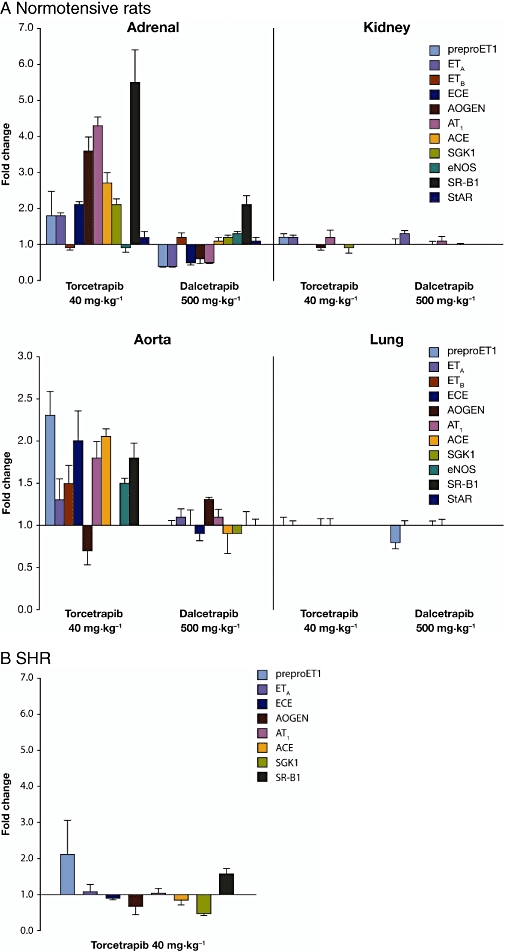
Mean (± s.e. mean) changes in renin-angiotensin-aldosterone system-related gene expression in (A) tissue samples from normotensive rats after 5 days of treatment with torcetrapib or dalcetrapib and (B) samples of adrenal gland from spontaneously hypertensive rats after 5 days of treatment with torcetrapib (all fold changes greater than 1.5-fold are significant at P < 0.05). AOGEN, angiotensinogen; ACE, angiotensin converting enzyme; AT1, angiotensin receptor type 1; preproET1, preproendothelin; ECE, endothelin-converting enzyme; ETA, endothelin receptor A; ETB, endothelin receptor B; eNOS, endothelial nitric oxide synthase; SGK1, serum/glucocorticoid regulated kinase 1; SR-B1, scavenger receptor class B type 1; StAR, steroidogenic acute regulatory protein.
The effect of torcetrapib on RAAS-related gene expression in adrenal tissue from SHR was minimal compared with the response in normotensive rats. In SHR, the maximum mean fold increase was observed for preproET1 (Figure 6B). Additionally, torcetrapib was associated with a slight but non-significant increase in plasma aldosterone levels in both normotensive and SHR models (Figure 7). No significant changes in RAAS-related gene expression were observed in any of the tissue samples taken from normotensive rats following administration of dalcetrapib 500 mg·kg−1·day−1 for 5 days (Figure 6A). The largest change in mRNA levels following dalcetrapib treatment was in SR-B1 expression in adrenal tissue, where a twofold increase was observed.
Figure 7.
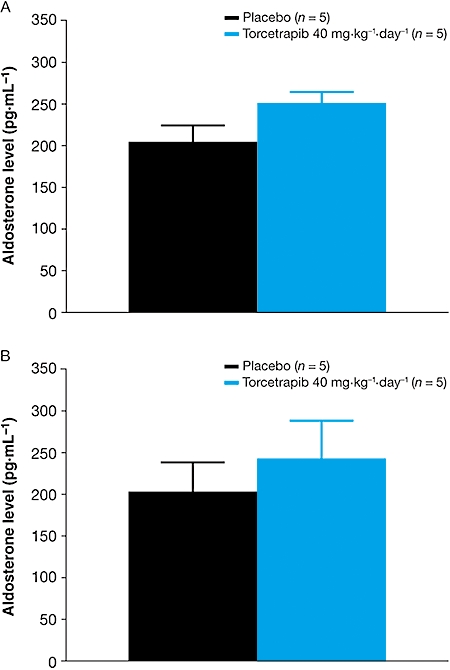
Mean (± s.e. mean) plasma aldosterone levels in (A) normotensive and (B) spontaneously hypertensive rats following 5 days' treatment with torcetrapib 40 mg·kg−1·day−1 or placebo.
Discussion
The failure of torcetrapib, possibly due to its effects on blood pressure (Barter et al., 2007; Bots et al., 2007; Kastelein et al., 2007; Nissen et al., 2007) and plasma aldosterone levels (Barter et al., 2007), raised questions as to whether these safety concerns were compound specific or common to all agents that act upon CETP. We therefore compared torcetrapib and dalcetrapib in a series of preclinical studies to evaluate the acute haemodynamic effects of both agents in rats, a species that does not express CETP, the doses selected reflecting the observation that rats are less sensitive to the blood pressure-raising effects of torcetrapib than primates (DePasquale et al., 2007).
The SHR is an accepted model for human essential hypertension and is standard for assessing anti-hypertensive compounds (Lerman et al., 2005; Potenza et al., 2005; Yang et al., 2005). In the SHR, torcetrapib-induced increase in blood pressure was marked and sustained for as long as the compound was administered. In contrast, dalcetrapib had no effect on blood pressure in this model, implying that the torcetrapib-induced hypertensive effects are not common to all agents that target CETP. It is relevant to note that as rats do not express CETP, the blood pressure effect of torcetrapib cannot be related to inhibition of this therapeutic target. In fact, a selective effect of torcetrapib on blood pressure unrelated to CETP inhibition is further supported by a limited structure-activity study where analogues of torcetrapib devoid of CETP inhibitory activity increased blood pressure to the same or even greater extent than those more potent in inhibiting CETP (DePasquale et al., 2007).
While the SHR is a clinically predictive model for assessing the potential for changes in AP, independent of perturbation of the activities of CETP, it was also of interest to examine whether agents that increase blood pressure in this model have similar effects in the normotensive rat where blood pressure regulation is intact. Treatment of normotensive Wistar rats with torcetrapib caused a dose-dependent and immediate increase in blood pressure (mean, systolic and diastolic AP). This effect was, however, transient, disappearing over a 5 day treatment period, demonstrating tachyphylaxis and involvement of compensatory mechanisms with repeated doses (in agreement with a poster presentation at the American Heart Association Scientific Sessions, 2007; DePasquale et al., 2007). There was no significant change in heart rate, suggesting the absence of direct baroreflex adaptation. Indeed, because of the high sensitivity of the SHR this model is currently used to screen CETP inhibitors which lack the acute hypertensive effect of torcetrapib.
To further investigate the potential hypertensive and compensatory mechanisms for the effect of torcetrapib but not dalcetrapib, we analysed the expression of genes associated with RAAS, as this would provide key evidence to support the effects on blood pressure (Duriez, 2007). Torcetrapib increased the expression of specific RAAS-related genes in adrenal gland and aortic tissue. The preferential upregulation of RAAS genes in the adrenal glands suggests that the off-targets of torcetrapib occur primarily via effects on this tissue. Critical changes in expression of AOGEN, ACE and angiotensin AT1 receptor may be associated with an increase in angiotensin II signalling in adrenal glands, and this is likely to be linked to an increase in aldosterone synthesis. It is also of interest to note the significant effects of torcetrapib on the expression of endothelin 1, measured as preproET1, in adrenal tissue (1.8-fold increase) and more importantly in aortic samples (2.3-fold increase), suggesting a role in vascular tissue as well as the adrenal glands. Similar effects were not seen with dalcetrapib. In addition to the effects on RAAS gene upregulation discussed here, Capponi et al. (2008) and Stein et al. (2009) have reported that torcetrapib but not dalcetrapib increases aldosterone secretion and expression of the aldosterone synthase gene CYP11B2 in the well-characterized human adrenocarcinoma cell line H295R, providing further evidence that torcetrapib is a potent steroidogenic agent on adrenal glomerulosa cells.
Results of the present study corroborate findings from preclinical studies where torcetrapib but not anacetrapib was associated with an acute increase in blood pressure in a number of animal species, indicating that the hypertensive effects noted with torcetrapib are not common to all agents that act on CETP (Forrest et al., 2008). The hypertensive effect of torcetrapib was associated with an acute increase in plasma levels of aldosterone and was not observed in adrenalectomized rats; in addition, torcetrapib but not anacetrapib was shown to cause the release of aldosterone from rat primary adrenocortical cells in vitro (Forrest et al., 2008). In this study, torcetrapib did not have a direct action on vascular tone, as it did not contract isolated arterial vascular segments in vitro and did not enhance responses to other vasoactive agents. Further investigations revealed that the acute increase in blood pressure associated with torcetrapib treatment is not likely to be mediated via aldosterone; indeed the mechanism of this effect is yet unknown (Forrest et al., 2008). The increase in aldosterone and RAAS activation associated with torcetrapib could be a contributory factor to the adverse cardiovascular events reported in clinical trials of torcetrapib (Forrest et al., 2008; Vergeer et al., 2008). Indeed, post hoc analyses of the ILLUMINATE and Investigation of Lipid Level Management Using Coronary Ultrasound to Assess Reduction of Atherosclerosis by CETP Inhibition and HDL Elevation studies reported that an increase in aldosterone levels was observed in the torcetrapib group compared with the placebo group (Barter et al., 2007; Nicholls et al., 2008). Furthermore, findings from the Rating Atherosclerotic Disease Change by Imaging with a New CETP Inhibitor trial demonstrated that the use of RAAS inhibitors aggravated the blood pressure increase observed (Vergeer et al., 2008). Indeed, initiation of RAAS inhibitors for blood pressure increase after the start of torcetrapib did not contribute to lower end-of-study blood pressure readings. In contrast, the initiation of diuretics resulted in reduced blood pressure levels. The authors suggest that this is consistent with a renin- and angiotensin-independent, direct mineralocorticoid effect by torcetrapib contributing to a volume overload.
With regard to RAAS-related gene expression, the present study found, interestingly, that gene expression was also increased in aortic tissue, albeit to a lesser extent than in adrenal glands. Previous studies have suggested that atherosclerotic vessels in particular may be highly sensitive to locally produced angiotensin II (Nickenig and Böhm, 1997; Brasier, et al., 2002). Hence, upregulation of RAAS components within the vessel wall may contribute to plaque destabilization, partly or wholly independent of circulating RAAS activity (Brasier, et al., 2002).
The exact mechanism by which torcetrapib increases RAAS expression remains to be established. The concomitant upregulation of SR-B1 may in fact be secondary to the increased demand for cholesterol needed for steroid hormone synthesis, since adrenal SR-B1 is the major acceptor for HDL-delivered cholesterol to adrenal cells (Williams et al., 2002).
In the present study, dalcetrapib had no effect on blood pressure in SHR and on RAAS-related gene expression in either adrenal glands or aortic tissue in normotensive rats. Similarly, dalcetrapib has been shown to have no acute effect on blood pressure in normotensive animal models (Roche data on file). As gene expression changes are strongest in the normotensive model, this precluded the need to examine the effect of dalcetrapib on gene expression in SHR.
In line with these observations, no clinically relevant effects on blood pressure have been observed in phase II trials of up to 12 weeks duration conducted in patients with mild hyperlipidaemia (de Grooth et al., 2002), patients with type II dyslipidaemia treated with pravastatin (Kuivenhoven et al., 2005) or in a pooled analysis of five trials in patients at high risk of coronary heart disease (Stein et al., 2009).
In conclusion, the present findings indicate that cardiovascular off-target effects, manifested as acute or sustained increased blood pressure and activation of the RAAS, reported with torcetrapib but not dalcetrapib, is most likely to be specific to torcetrapib and not common to other agents that decrease CETP activity.
Acknowledgments
Support for this study was provided by F. Hoffmann-La Roche Ltd. The authors acknowledge assistance from Jacques Mizrahi, Robert Wolfgang for telemetry work, and the Department of Vascular and Metabolic Disease at F. Hoffmann-La Roche Ltd, Basel.
Editorial assistance was provided by Teresa Haigh for Prime Healthcare during the preparation of this report and funded by F. Hoffmann-La Roche Ltd.
Glossary
Abbreviations:
- AP
arterial pressure
- bpm
beats per minute
- CE
cholesteryl ester
- CETP
cholesteryl ester transfer protein
- HDL-C
high-density lipoprotein cholesterol
- LC-MS/MS
liquid chromatography with tandem mass spectrometry
- LDL-C
low-density lipoprotein cholesterol
- qPCR
quantitative real-time polymerase chain reaction
- RAAS
renin-angiotensin-aldosterone system
- SHR
spontaneously hypertensive rats
- SR-B1
scavenger receptor class B type 1
- TG
triglyceride
Conflict of interest
ESG Stroes has no relationships to disclose. JJP Kastelein receives research grants from F. Hoffmann-La Roche Ltd, Novartis, Pfizer, AstraZeneca, ISIS, MSD and Schering-Plough. A Bénardeau, O Kuhlmann, D Blum, LA Campos, RG Clerc and EJ Niesor are employees of F. Hoffmann-La Roche Ltd.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC) Br J Pharmacol. (3rd edn.) 2008;153(Suppl. 2):S1–S209. doi: 10.1038/sj.bjp.0707746. (2008 revision. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barter PJ, Brewer HB, Jr, Chapman MJ, Hennekens CH, Rader DJ, Tall AR. Cholesteryl ester transfer protein: a novel target for raising HDL and inhibiting atherosclerosis. Arterioscler Thromb Vasc Biol. 2003;23:160–167. doi: 10.1161/01.atv.0000054658.91146.64. [DOI] [PubMed] [Google Scholar]
- Barter PJ, Caulfield M, Eriksson M, Grundy SM, Kastelein JJ, Komajda M, et al. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med. 2007;357:2109–2122. doi: 10.1056/NEJMoa0706628. [DOI] [PubMed] [Google Scholar]
- Barter PJ, Kastelein JJ. Targeting cholesteryl ester transfer protein for the prevention and management of cardiovascular disease. J Am Coll Cardiol. 2006;47:492–499. doi: 10.1016/j.jacc.2005.09.042. [DOI] [PubMed] [Google Scholar]
- Bots ML, Visseren FL, Evans GW, Riley WA, Revkin JH, Tegeler CH, et al. Torcetrapib and carotid intima-media thickness in mixed dyslipidaemia (RADIANCE 2 study): a randomised, double-blind trial. Lancet. 2007;370:153–160. doi: 10.1016/S0140-6736(07)61088-5. [DOI] [PubMed] [Google Scholar]
- Brasier AR, Recinos A, III, Eledrisi MS. Vascular inflammation and the renin-angiotensin system. Arterioscler Thromb Vasc Biol. 2002;22:1257–1266. doi: 10.1161/01.atv.0000021412.56621.a2. [DOI] [PubMed] [Google Scholar]
- Capponi AM, Clerc RG, Campos L, Perez A, Rincon Garriz JM, Maugeais C, et al. No increase in the in vitro production of aldosterone or the expression of CYP11B2 with the CETP modulator dalcetrapib (RO4607381/JTT-705), in contrast with torcetrapib [abstract] Circulation. 2008;118:S452. [Google Scholar]
- Clark RW, Sutfin TA, Ruggeri RB, Willauer AT, Sugarman ED, Magnus-Aryitey G, et al. Raising high-density lipoprotein in humans through inhibition of cholesteryl ester transfer protein: an initial multidose study of torcetrapib. Arterioscler Thromb Vasc Biol. 2004;24:490–497. doi: 10.1161/01.ATV.0000118278.21719.17. [DOI] [PubMed] [Google Scholar]
- Clark RW, Ruggeri RB, Cunningham D, Bamberger MJ. Description of the torcetrapib series of cholesteryl ester transfer protein inhibitors, including mechanism of action. J Lipid Res. 2006;47:537–552. doi: 10.1194/jlr.M500349-JLR200. [DOI] [PubMed] [Google Scholar]
- DePasquale M, Knight D, Loging W, Morehouse L, Winter S, Blasi E, et al. Mechanistic studies of hemodynamics with a series of cholesterol ester transfer protein inhibitors. Circ Res. 2007;101:1209–1210. [Google Scholar]
- Duriez P. CETP inhibition. Lancet. 2007;370:1882–1883. doi: 10.1016/S0140-6736(07)61788-7. [DOI] [PubMed] [Google Scholar]
- Forrest MJ, Bloomfield D, Briscoe RJ, Brown PN, Cumiskey A-M, Ehrhart J, et al. Torcetrapib-induced blood pressure elevation is independent of CETP inhibition and is accompanied by increased circulating levels of aldosterone. Br J Pharmacol. 2008;154:1465–1473. doi: 10.1038/bjp.2008.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Grooth GJ, Kuivenhoven JA, Stalenhoef AF, de Graaf J, Zwinderman AH, Posma JL, et al. Efficacy and safety of a novel cholesteryl ester transfer protein inhibitor, JTT-705, in humans: a randomized Phase II dose response study. Circulation. 2002;105:2159–2165. doi: 10.1161/01.cir.0000015857.31889.7b. [DOI] [PubMed] [Google Scholar]
- Kastelein JJ, van Leuven SI, Burgess L, Evans GW, Kuivenhoven JA, Barter PJ, et al. Effect of torcetrapib on carotid atherosclerosis in familial hypercholesterolemia. N Engl J Med. 2007;356:1620–1630. doi: 10.1056/NEJMoa071359. [DOI] [PubMed] [Google Scholar]
- Kuivenhoven JA, de Grooth GJ, Kawamura H, Klerkx AH, Wilhelm F, Trip MD, et al. Effectiveness of inhibition of cholesteryl ester transfer protein by JTT-705 in combination with pravastatin in type II dyslipidemia. Am J Cardiol. 2005;95:1085–1088. doi: 10.1016/j.amjcard.2004.12.064. [DOI] [PubMed] [Google Scholar]
- Lerman LO, Chade AR, Sica V, Napoli C. Animal models of hypertension: an overview. J Lab Clin Med. 2005;146:160–173. doi: 10.1016/j.lab.2005.05.005. [DOI] [PubMed] [Google Scholar]
- Nicholls SJ, Tuzcu EM, Brennan DM, Tardif JC, Nissen SE. Cholesteryl ester transfer protein inhibition, high-density lipoprotein raising, and progression of coronary atherosclerosis: insights from ILLUSTRATE (Investigation of Lipid Level Management Using Coronary Ultrasound to Assess Reduction of Atherosclerosis by CETP Inhibition and HDL Elevation) Circulation. 2008;118:2506–2514. doi: 10.1161/CIRCULATIONAHA.108.790733. [DOI] [PubMed] [Google Scholar]
- Nickenig G, Böhm M. Regulation of the angiotensin AT1 receptor expression by hypercholesterolemia. Eur J Med Res. 1997;2:285–289. [PubMed] [Google Scholar]
- Niesor EJ, von der Marck E, Brousse M, Maugeais C. Inhibition of cholesteryl ester transfer protein: different in vitro characteristics of RO4607381/JTT-705 and torcetrapib [abstract. Atherosclerosis. 2008;199:231. [Google Scholar]
- Nissen SE, Tardif JC, Nicholls SJ, Revkin JH, Shear CL, Duggan WT, et al. Effect of torcetrapib on the progression of coronary atherosclerosis. N Engl J Med. 2007;356:1304–1316. doi: 10.1056/NEJMoa070635. [DOI] [PubMed] [Google Scholar]
- Okamoto H, Yonemori F, Wakitani K, Minowa T, Maeda K, Shinkai H. A cholesteryl ester transfer protein inhibitor attenuates atherosclerosis in rabbits. Nature. 2000;406:203–207. doi: 10.1038/35018119. [DOI] [PubMed] [Google Scholar]
- Potenza MA, Marasciulo FL, Chieppa DM, Brigiani GS, Formoso G, Quon MJ, et al. Insulin resistance in spontaneously hypertensive rats is associated with endothelial dysfunction characterized by imbalance between NO and ET-1 production. Am J Physiol Heart Circ Physiol. 2005;289:H813–H822. doi: 10.1152/ajpheart.00092.2005. [DOI] [PubMed] [Google Scholar]
- Qiu X, Mistry A, Ammirati MJ, Chrunyk BA, Clark RW, Cong Y, et al. Crystal structure of cholesteryl ester transfer protein reveals a long tunnel and four bound lipid molecules. Nat Struct Mol Biol. 2007;14:106–113. doi: 10.1038/nsmb1197. [DOI] [PubMed] [Google Scholar]
- Stein EA, Stroes ESG, Steiner G, Buckley BM, Capponi AM, Burgess T, et al. Safety and tolerability of dalcetrapib. Am J Cardiol. 2009;104:82–91. doi: 10.1016/j.amjcard.2009.02.061. [DOI] [PubMed] [Google Scholar]
- Tall AR. Plasma cholesteryl ester transfer protein. J Lipid Res. 1993;34:1255–1274. [PubMed] [Google Scholar]
- Vergeer M, Bots ML, van Leuven SI, Basart DC, Sijbrands EJ, Evans GW, et al. Cholesteryl ester transfer protein inhibitor torcetrapib and off-target toxicity: a pooled analysis of the rating atherosclerotic disease change by imaging with a new CETP inhibitor (RADIANCE) trials. Circulation. 2008;118:2515–2522. doi: 10.1161/CIRCULATIONAHA.108.772665. [DOI] [PubMed] [Google Scholar]
- Williams DL, Wong JS, Hamilton RL. SR-BI is required for microvillar channel formation and the localization of HDL particles to the surface of adrenocortical cells in vivo. J Lipid Res. 2002;43:544–549. [PubMed] [Google Scholar]
- Yang L, Gao YJ, Lee RM. The effects of quinapril and atorvastatin on artery structure and function in adult spontaneously hypertensive rats. Eur J Pharmacol. 2005;518:145–151. doi: 10.1016/j.ejphar.2005.05.009. [DOI] [PubMed] [Google Scholar]