

Abstract
Background and purpose:
Although the amino acid sequences of rat and human 5-hydroxytryptamine (5-HT) and noradrenaline (NA) transporters (i.e. SERT and NET) are highly homologous, species differences exist in the inhibitory effects of drugs acting at these transporters. Therefore, comparison of the potencies of drugs acting at SERT and NET in native human and rat neocortex may serve to more accurately predict their clinical profile.
Experimental approach:
Synaptosomes prepared from fresh human and rat neocortical tissues were used for [3H]-5-HT and [3H]-NA saturation and competition uptake experiments. The drugs tested included NA reuptake inhibitors (desipramine, atomoxetine and (S,S)-reboxetine), 5-HT reuptake blockers (citalopram, fluoxetine and fluvoxamine) and dual 5-HT/NA reuptake inhibitors (duloxetine and milnacipran).
Key results:
In saturation experiments on synaptosomal [3H]-5-HT and [3H]-NA uptake, the dissociation constants did not indicate species differences although a smaller density of both SERT and NET was observed in human tissues. In competition experiments with the various drugs, marked species differences in their potencies were observed, especially at SERT. The rank order of selectivity ratios (SERT/NET) in human neocortex was as follows: citalopram ≥ duloxetine = fluvoxamine ≥ fluoxetine > milnacipran > desipramine = atomoxetine > (S,S)-reboxetine. Significant species differences in these ratios were observed for duloxetine, atomoxetine and desipramine.
Conclusions and implications:
This study provides the first compilation of drug potency at native human neocortical SERT and NET. The significant species differences (viz., human vs. rat) in drug potency suggest that the general use of rodent data should be limited to predict clinical efficacy or profile.
Keywords: atomoxetine; desipramine; (S,S)-reboxetine; citalopram; fluoxetine; fluvoxamine; duloxetine; milnacipran; noradrenaline uptake; 5-hydroxytryptamine uptake; synaptosomes; neocortex
Introduction
The transporters for noradrenaline (NA) and 5-hydroxytryptamine (5-HT), NET and SERT, respectively, represent established targets for many of the clinically relevant antidepressant drugs, commonly referred to as NA or 5-HT reuptake inhibitors (viz., NRI and SRI). The resulting increase and prolonged action of NA and 5-HT in the synaptic cleft appear to represent the basic mechanism for the clinical efficacy of these drugs (Frazer, 2001; Berton and Nestler, 2006), especially as major depression is classically linked to deficiencies in the monoamine neurotransmitters 5-HT, NA and dopamine (DA). The importance of these monoaminergic systems for the vulnerability of the central nervous system to become ‘depressed’ has recently been confirmed (Ruhéet al., 2007). A discrepancy exists, however, between the well-known delay (i.e. 2–3 weeks) in the clinical onset of antidepressant drug effect and the inhibition of NET and/or SERT. This discrepancy is most likely explained by adaptive changes of pre- and post-synaptic receptors, including the transition from supersensitive to normosensitive presynaptic autoreceptors, which account for the ultimate therapeutic effects of antidepressant drugs (see Göthert and Schlicker, 1997).
The human NET (hNET) was the first monoamine neurotransmitter transporter for which the amino acid sequence was identified by expression cloning (Pacholczyk et al., 1991). Subsequently, the rat NET (rNET), hSERT and rat SERT (rSERT) were identified by two research groups (Blakely et al., 1991; Ramamoorthy et al., 1993; Bruss et al., 1997). Although species variations of NET and SERT tend to be minimal [e.g. the amino acid sequence of rNET is 93% similar to that of hNET (Bruss et al., 1997)], this high homology does not necessarily exclude the existence of pharmacological differences between rat and hNET and SERT respectively. For instance, in transfected cells, a higher affinity of tricyclic antidepressants has been reported for the hSERT than for the rSERT (Barker et al., 1994). The significance or predictive value of results from in vitro studies, including those carried out in isolated cell lines, appears generally to be limited as regards the clinical situation. Despite these limitations, however, data obtained from both cell culture and animal experiments are typically extrapolated to human pharmacology. Species differences in drug potency, as an intervening or complicating variable, are often not considered in this extrapolation of results to the human patient. Therefore, the use of human native and fresh brain tissues to study the effects of NRI and SRI is important for accurate characterization of their pharmacological action in man.
In the present study, we determined and compared the potencies of various NRI, SRI and dual 5-HT and NA reuptake inhibitors (SNRI) using native NET and SERT of human and rat neocortex. The native dopamine transporter (DAT) was not studied, as preliminary experiments indicated low and variable uptake rates of DA being consistent with low levels of DAT detected in the neocortex of mammals (e.g. Wheeler et al., 1993; Chalon et al., 2006). To our knowledge, this is the first time the potencies of these drugs have been obtained in neocortical synaptosomes prepared from fresh specimens of human neocortex. These drugs included both classical compounds (i.e. the tricyclic antidepressant desipramine, the SRI citalopram, fluoxetine and fluvoxamine) and more recently developed agents (i.e. the SNRI duloxetine and milnacipran, the NRI atomoxetine and (S,S)-reboxetine). Note that some of these drugs, depending on their transporter selectivity, are used to treat fibromyalgia and other pain states, attention-deficit/hyperactivity disorder and not primarily depression.
Methods
Tissue sources
Human neocortical tissue specimens were obtained during surgical treatment of drug-resistant epilepsy or of non-epileptogenic brain tumours. Every patient signed a declaration of consent before the operation as requested by the local Ethics Committee. After pre-medication with midazolam, anaesthesia was induced with propofol and fentanyl. Cisatracurium was used for muscle relaxation. Patients received cefuroxim as an intraoperative one-time prophylactic antibiotic. The tissue was carefully removed in a gentle and nearly atraumatic manner and immediately placed in ice-cold saline to ensure viability. The neocortical samples were obtained from a total of 30 patients (14 male, 16 female; mean age: 36.8 years; youngest: 12 years; oldest: 69 years) and included frontal, temporal and parietal areas. The white matter, or macroscopically identified adherent tumorous or otherwise lesioned tissue parts, was separated (and discarded) from the grey matter that contained all six neocortical layers after preparation. The neocortical tissue was then rinsed with ice-cold physiological buffer [composition (mM): NaCl (121), KCl (1.8), CaCl2 (1.3), KH2PO4 (1.2), MgSO4 (1.2), NaHCO3 (25), glucose (10), ascorbic acid (0.06), saturated with 95% O2/5% CO2; pH 7.4] and processed immediately.
Wistar rats (200–300 g, n= 28; University of Freiburg, Freiburg, Germany) were maintained according to institutional policies and guidelines. All efforts were made to reduce the number of animals used and to minimize animal suffering. Animals were killed by decapitation under CO2 anaesthesia. Each brain was quickly removed and rinsed with ice-cold buffer (composition same as above). Neocortical grey matter was isolated and synaptosomes were prepared as described below.
Preparation of synaptosomes
Freshly prepared human (0.8–1.0 g) and rat (0.5–0.8 g) neocortical tissue samples were homogenized in 10 volumes (w/v) of ice-cold sucrose (0.32 M)/HEPES (2.5 mM) buffer (pH 7.4). The following centrifugation steps were carried out: The initial homogenate was centrifuged at 1000 gfor 10 min at 4°C (Heraeus Biofuge 28RS; Osterode, Germany). The resultant supernatant was separated and centrifuged again at 10 000 g for 10 min at 4°C. The supernatant from this centrifugation was discarded, and the synaptosomal pellet was resuspended in ice-cold buffer to obtain a final protein concentration of ∼180 µg (human synaptosomes) or ∼120 µg (rat synaptosomes) per assay tube. Protein content was determined by the method of Lowry et al. (1951).
[3H]-noradrenaline and [3H]-5-hydroxytryptamine uptake assays
Assays were carried out in physiological buffer (composition same as above) containing pargyline (5 µM) to inhibit monoamine oxidase activity and metabolism of the 3H-neurotransmitter. Specific uptake was defined as total uptake minus uptake in the presence of reboxetine or (+)-oxaprotiline (10 µM) (NA uptake), and fluoxetine (10 µM) (5-HT uptake).
For a possible false labelling by [3H]-5-HT or [3H]-NA of dopaminergic terminals (Feuerstein et al., 1986; Lupp et al., 1992) to be avoided, respectively, particular attention was paid to choose inhibitors of NA uptake or of 5-HT uptake, which do not influence the DAT (e.g. Löffler et al., 2006). According to this study, (+)-oxaprotiline and fluvoxamine decreased the accumulation of [3H]-dopamine in the neocortex of rats and humans. This is also true when fluoxetine instead of fluvoxamine or when reboxetine instead of (+)-oxaprotiline is used respectively (own unpublished results).
Saturation uptake experiments
Saturation characteristics of NA and 5-HT uptake were determined by diluting specific concentrations of [3H]-NA and [3H]-5-HT with varying amounts of the corresponding unlabelled compound to obtain final concentrations ranging from 1 nM to 10 µM ([3H]-NA/NA) and from 1 nM to 3.2 µM ([3H]-5-HT/5-HT) respectively. The assay was started by adding 100 µL synaptosomal suspension to 880 µL buffer followed by an 18 min incubation step at ambient temperature. Uptake was then initiated by adding 20 µL of the mixture of radiolabelled and unlabelled ligand. This second incubation period was for 20 min at 37°C to attain equilibrium. Non-specific uptake was determined for each ligand mixture concentration by using the appropriate uptake inhibitor (10 µM, see above).
Reactions were terminated by rapid filtration through glass fibre filters (GF/B, Whatman GmbH, Dassel, Germany) previously soaked in buffer containing 0.1% polyethylenimine, by using a 96-well cell harvester (Brandel M96, Gaithersburg, MD, USA). The filters were then rapidly washed with 3 mL of ice-cold buffer and transferred into scintillation vials. The washing buffer for the NA uptake experiments was pre-adjusted to pH 6.0 (with HCl), effectively reducing non-specific binding of NA to the filter. After addition of a liquid scintillation cocktail (3 mL, Ultima Gold; Packard Bioscience, Groningen, Netherlands), the filters were shaken thoroughly for 1 h. Radioactivity of the filters was determined by using a liquid scintillation analyser (Tri-Carb 2100TR; Packard Instruments, Meriden, CT, USA).
Competition uptake experiments
The assay was started by adding 100 µL synaptosomal suspension to 750 µL assay buffer and 100 µL of competing drug (concentration range: from 1 pM up to 1 mM) or assay buffer (for control). After an 18 min incubation at ambient temperature, [3H]-NA or [3H]-5-HT (50 µL) was added to the assay for a final concentration of 10 nM and 5 nM respectively. The second incubation period and additional details of the assay were as described above. Non-specific uptake was determined for each experiment.
Calculations and statistics
Results are given as parameter estimates and 95% confidence intervals (CI95). Significant differences in the parameter estimates between humans and rats were assumed when the corresponding CI95 did not overlap (Gardner and Altman, 1986). Significance differences between two mean values were assessed with the Student's t-test. The minimal level of significance was P≤ 0.05 (two-tail criterion).
Note, however, that statistically significant differences may not always be pharmacologically relevant. Therefore, differences of less than 0.5 log units were not assumed to have functional implications.
Saturation and inhibition curves were generated by non-linear regression analysis (JMP 8.0, SAS Institute, Heidelberg, Germany; for applied functions, see Steffens and Feuerstein, 2004). The estimated parameters were (i) Umax, the asymptotic maximum of uptake [i.e. the specific uptake as pmol·mg−1 protein into synaptosomes that equals the number of uptake sites·mg−1 protein for a pure bimolecular reaction between transporter and its substrate (5-HT or NA)]; (ii) pEC50, that is, the negative log10 of the concentration of substrate required to reach 50% of Umax; (iii) pIC50, the negative log10 of the concentration of the inhibitor required to inhibit the uptake of neurotransmitter by 50%; (iv) pKd, the negative log10 of the dissociation constant (Kd) between substrate and the corresponding transporter; (v) pKi, the negative log10 of the inhibition constant (Ki) between the competing drug and the transporter; (vi) Imax, the asymptotic maximum of relative uptake inhibition (range of 0 to 1); and (vii) the slope factor c that estimates the existence of a bimolecular reaction between the NA or 5-HT transporter and their ligands (i.e. substrate, inhibitor) (Feuerstein and Limberger, 1999). An estimate of c near unity allows us to assume that the inhibitor binds either at the same uptake site as the substrate (viz., acting as a competitive antagonist), or at a distinct site that allosterically modifies the affinity of the respective substrates for the NA and 5-HT transporters. Note that increasing the number of parameters to be estimated by non-linear regression analysis of the same number of data points may increase the variations of these estimates. Thus, the variances of three parameters, for example, pIC50, Imax and c, are often larger than those of only two parameters estimated simultaneously, for example, Ki and Imax.
As previously described (Steffens and Feuerstein, 2004), the estimate of the dissociation constant between the transporter and inhibitor was based on the difference between the descriptive IC50 value and Ki, mechanistically analogous to the dissociation constant. The Cheng–Prusoff equation (Cheng and Prusoff, 1973) was used to convert the IC50 to Ki, and this was introduced into the equation (2) of Steffens and Feuerstein to yield the following function:
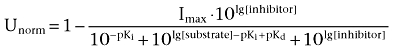 |
(1) |
Materials
The radiolabelled substances were l-[ring-2,5,6-3H]-noradrenaline (1.92 TBq·mmol−1; DuPont, Dreieich, Germany) and [1,2-3H(N)]-5-hydroxytryptamine (1.11 TBq·mmol−1; Perkin-Elmer, Wiesbaden, Germany). Other substances included citalopram hydrobromide, desipramine hydrochloride, fluoxetine hydrochloride, fluvoxamine maleate, 5-hydroxytryptamine hydrochloride, milnacipran hydrochloride, l-noradrenaline hydrochloride, and pargyline hydrochloride (Sigma-Aldrich, Taufkirchen, Germany); (S,S)-reboxetine methanesulphonate (Pfizer, Ann Arbor, MI, USA); (+)-oxaprotiline hydrochloride (Novartis, Basel, Switzerland); and reboxetine mesylate (Tocris, Bristol, UK). Atomoxetine hydrochloride and duloxetine hydrochloride were isolated from capsules/tablets obtained commercially. The substances were initially dissolved in water (10 mM stock) and then further diluted with assay buffer.
Results
Saturation uptake experiments
The saturation uptake experiments determined the kinetic parameters of SERT and NET in human and rat neocortical synaptosomes respectively. The pEC50, Umax and slope factor c values for 5-HT and NA uptake into synaptosomes from both species are given in Table 1. Specific uptake of [3H]-5-HT and [3H]-NA for either human or rat synaptosomes yielded pEC50 values in the high nanomolar range. Because these pEC50 estimates are assumed to represent pKd values due to c near 1 (see ‘Discussion’), species differences were not evident in the affinity of [3H]-5-HT and [3H]-NA to their respective transporters.
Table 1.
Parameters of saturation experiments on [3H]-5-hydroxytryptamine and [3H]-noradrenaline uptake into human and rat neocortical synaptosomes
| pEC50 | Umax (pmol·mg−1protein) | c | |
|---|---|---|---|
| [3H]-5-HT | |||
| Human | 7.32 [6.72, 7.74] | 16.27 [10.27, 29.60]*** | 0.99 [0.80, 1.23] |
| Rat§ | 7.56 [7.45, 7.66] | 39.98 [36.66, 43.72] | 0.91 [0.85, 0.97] |
| [3H]-NA | |||
| Human | 6.46 [5.67, 6.85] | 4.68 [2.73, 12.52]*** | 1.17 [0.88, 1.56]** |
| Rat | 6.64 [6.35, 6.87] | 35.85 [29.72, 44.20] | 0.72 [0.66, 0.79] |
Values given are estimates [CI95] (n≥ 3 independent experiments, at least six concentrations of drug/experiment, each concentration in six replicates).
A significant difference from the corresponding value for rat neocortex is indicated by asterisks
P≤ 0.01,
P≤ 0.001).
Data are from Steffens and Feuerstein (2004).
In contrast, the number of uptake sites (Umax) for [3H]-5-HT and [3H]-NA was significantly lower in human (59 and 87% respectively) than in rat neocortical synaptosomes. Because three of the slope factors c (i.e. Hill coefficients) were near unity, single NA- and 5-HT-uptake sites were assumed for the corresponding bimolecular substrate–transporter interactions. The slope factor c for specific NA uptake in the rat was, however, below unity.
Effect of various reuptake inhibitor drugs on [3H]-5-hydroxytryptamine uptake
The inhibitory effects of the tested drugs on [3H]-5-HT uptake into human and rat neocortical synaptosomes, respectively, are presented in Table 2A.
Table 2.
(A) Parameters of inhibition experiments with various reuptake inhibitor drugs on [3H]-5-hydroxytryptamine uptake into human and rat neocortical synaptosomes. (B) Parameters of inhibition experiments with various reuptake inhibitor drugs on [3H]-noradrenaline uptake into human and rat neocortical synaptosomes
| pIC50 | Imax | c | |
|---|---|---|---|
| A | |||
| Citalopram | |||
| Human | 8.44 (8.32, 8.56)*** | 0.88 (0.85, 0.92) | 1.04 (0.83, 1.35) |
| Rat | 8.78 (8.68, 8.88) | 0.97 (0.93, 1.01) | 1.21 (0.96, 1.57) |
| Fluvoxamine | |||
| Human§ | 7.96 (7.74, 8.15)** | 0.95 (0.89, 1.03) | 0.84 (0.60, 1.24) |
| Rat§ | 8.32 (8.21, 8.43) | 1.00 (0.97, 1.04) | 0.96 (0.798, 1.18) |
| Duloxetine | |||
| Human | 9.26 (9.09, 9.41)** | 0.95 (0.91, 1.00) | 0.43 (0.38, 0.50) |
| Rat | 8.81 (8.52, 9.04) | 1.05 (0.97, 1.15) | 0.44 (0.36, 0.54) |
| Fluoxetine | |||
| Human | 7.85 (6.57, 8.30) | 1.09 (0.91, 1.70) | 0.95 (0.28, −) |
| Rat | 7.48 (7.38, 7.58) | 1.00 (0.95, 1.08) | 1.01 (0.95, 1.08) |
| Milnacipran | |||
| Human | 7.80 (7.66, 7.93)*** | 0.93 (0.88, 0.99) | 0.95 (0.77, 1.20) |
| Rat | 7.39 (7.25, 7.53) | 1.01 (0.95, 1.07) | 0.79 (0.64, 1.00) |
| Atomoxetine | |||
| Human | 7.67 (7.51, 7.81)*** | 0.93 (0.88, 1.00) | 1.00 (0.74, 1.49) |
| Rat | 6.77 (6.65, 6.89) | 1.00 (0.94, 1.04) | 1.13 (0.88, 1.49) |
| Desipramine | |||
| Human | 7.16 (6.93, 7.34)*** | 0.96 (0.88, 1.06) | 0.80 (0.58, 1.12) |
| Rat | 5.97 (5.67, 6.11) | 1.02 (0.91, 1.27) | 1.19 (−, −) |
| (S,S)-Reboxetine | |||
| Human | 4.55 (4.18, 4.79)** | 1.11 (0.97, 1.38) | 0.88 (0.66, 1.24) |
| Rat | 5.08 (4.85, 5.21) | 0.89 (0.77, 1.12) | 1.35 (1.01, 1.77) |
| B | |||
| Citalopram | |||
| Human | 5.03 (2.57, 5.73) | 0.98 (0.68, 3.28) | 0.66 (0.34, 1.35) |
| Rat | 5.35 (5.21, 5.46) | 1.01 (0.94, 1.10) | 0.92 (0.75, 1.14) |
| Fluvoxamine | |||
| Human | 5.62 (5.29, 5.88)*** | 1.06 (0.94, 1.21) | 1.24 (0.75, 2.48) |
| Rat | 6.17 (6.07, 6.29) | 0.80 (0.73, 0.87) | 1.97 (1.42, 2.79) |
| Duloxetine | |||
| Human | 6.89 (5.44, 7.43) | 1.04 (0.80, 1.84) | 0.71 (0.34, 1.55) |
| Rat | 7.40 (7.26, 7.53) | 0.91 (0.86, 0.98) | 0.88 (0.70, 1.11) |
| Fluoxetine | |||
| Human | 5.94 (4.45, 7.14) | 0.90 (0.43, 3.09) | 0.66 (−, −) |
| Rat | 6.45 (6.07, 6.65) | 0.95 (0.83, 1.18) | 0.86 (0.57, 1.34) |
| Milnacipram | |||
| Human | 7.81 (7.31, 8.25) | 0.90 (0.71, 1.12) | 1.07 (0.58, 3.21) |
| Rat | 7.44 (7.31, 7.55) | 0.82 (0.78, 0.88) | 0.95 (0.77, 1.19) |
| Atomoxetine | |||
| Human | 9.51 (9.17, 9.83) | 0.92 (0.82, 1.03) | 0.83 (0.54, 1.39) |
| Rat | 9.58 (9.41, 9.74) | 0.90 (0.87, 0.94) | 0.55 (0.44, 0.71) |
| Desipramine | |||
| Human | 9.13 (8.83, 9.40) | 0.88 (0.77, 1.01) | 0.85 (0.58, 1.40) |
| Rat | 9.33 (9.11, 9.56) | 0.95 (0.89, 1.02) | 1.04 (0.65, 1.77) |
| (S,S)-Reboxetine | |||
| Human | 8.37 (7.89, 8.76)** | 0.87 (0.73, 1.05) | 0.96 (0.54, 2.04) |
| Rat | 8.98 (8.93, 9.04) | 0.75 (0.73, 0.77) | 1.83 (1.53, 2.27) |
Values given are estimates [CI95] (n≥ 2 independent experiments, at least six concentrations of drug/experiment, each concentration in six replicates).
A significant difference from the corresponding value for rat neocortex is indicated by asterisks
P≤ 0.01,
P≤ 0.001).
Data from Lieb et al. (2005).
The inhibition by each compound was concentration dependent and is exemplified by the concentration–inhibition curve for atomoxetine in both species (Figure 1). The pIC50, Imax and c values are listed in Table 2A. All pIC50 values, with the exception of fluoxetine, were significantly different between human and rat neocortical synaptosomes (see also Figure 1). The pIC50-values for desipramine and atomoxetine were relevantly higher (i.e. by more than 0.5 log units) in human than in rat neocortex, contrasting with the pIC50 value for (S,S)-reboxetine that was relevantly higher in rat. All Imax values approximated to unity with no evidence of species differences. The values of the slope factor c differed from unity for (S,S)-reboxetine in rat and for duloxetine in both species. The slope factors c varied markedly around unity for citalopram, fluoxetine and fluvoxamine in rat and for atomoxetine in both rat and human.
Figure 1.
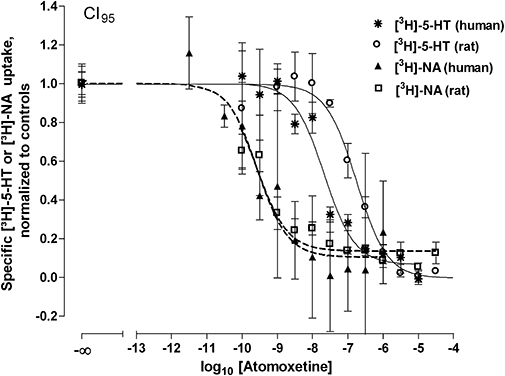
Inhibition of [3H]-5-hydroxytryptamine ([3H]-5-HT) and [3H]-noradrenaline ([3H]-NA) uptake into human and rat neocortical synaptosomes by atomoxetine. Synaptosomes were incubated (20 min, 37°C) with 5 nM [3H]-5-HT or 10 nM [3H]-NA in the presence of various concentrations of atomoxetine. Nonspecific uptake was determined by using 10 µM fluoxetine (5-HT) or 10 µM reboxetine (NA). Results are given as means with CI95 (n≥ 6 determinations) with fitted curves according to function (2) of Steffens and Feuerstein (2004).
On the basis of an assumed c= 1, a pKd for [3H]-5-HT of 7.32 (human) and 7.56 (rat, see Table 1) and a concentration of [3H]-5-HT of 5 nM in the uptake inhibition experiments, the pKi values for the different reuptake inhibitors were calculated (see ‘Methods’) and presented in Table 3.
Table 3.
Estimates of pKi for various drugs inhibiting [3H]-5-hydroxytryptamine and [3H]-noradrenaline uptake into human and rat neocortical synaptosomes
| pKi (5-HT) | pKi (NE) | |
|---|---|---|
| Citalopram | ||
| Human | 8.48 (8.37, 8.60)*** | 5.35 (4.88, 5.85) |
| Rat | 8.83 (8.73, 8.93) | 5.39 (5.30, 5.49) |
| Fluvoxamine | ||
| Human | 8.04 (7.87, 8.21)*** | 5.56 (5.30, 5.82)*** |
| Rat | 8.39 (8.29, 8.50) | 6.07 (5.91, 6.23) |
| Duloxetine | ||
| Human | 9.55 (9.41, 9.70)*** | 7.02 (6.55, 7.47) |
| Rat | 9.14 (8.97, 9.32) | 7.45 (7.34, 7.56) |
| Fluoxetine | ||
| Human | 7.90 (7.50, 8.33) | 6.23 (5.46, 7.15) |
| Rat | 7.54 (7.45, 7.64) | 6.52 (6.36, 6.68) |
| Milnacipran | ||
| Human | 7.85 (7.73, 7.97)*** | 7.80 (7.38, 8.23) |
| Rat | 7.49 (7.37, 7.62) | 7.47 (7.36, 7.58) |
| Atomoxetine | ||
| Human | 7.71 (7.58, 7.85)*** | 9.56 (9.26, 9.86) |
| Rat | 6.83 (6.71, 6.96) | 9.62 (9.49, 9.75) |
| Desipramine | ||
| Human | 7.24 (7.10, 7.40)*** | 9.19 (8.96, 9.43) |
| Rat | 6.00 (5.70, 6.14) | 9.35 (9.13, 9.58) |
| (S,S)-Reboxetine | ||
| Human | 4.68 (4.54, 4.81)** | 8.40 (8.06, 8.75)*** |
| Rat | 4.94 (4.79, 5.08) | 8.99 (8.92, 9.08) |
Values given are estimates [CI95] (n≥ 2 independent experiments, at least 6 concentrations of drug/experiment, each concentration in six replicates).
A significant difference from the corresponding value for rat neocortex is indicated by asterisks
P≤ 0.01,
P≤ 0.001).
All drugs, with the exception of fluoxetine, gave significantly different pKi values for [3H]-5-HT uptake between the human and rat. Relevant potency differences, however, were only seen for desipramine and atomoxetine. The SNRI duloxetine was the most potent compound, having Ki values less than 1 nM in both human and rat neocortical synaptosomes (Table 3).
The rank order of inhibitory potency of the drugs for hSERT was as follows: duloxetine > citalopram > fluvoxamine ≥ fluoxetine ≥ milnacipran ≥ atomoxetine > desipramine > (S,S)-reboxetine. The rank potency order for rSERT was similar: duloxetine > citalopram > fluvoxamine > fluoxetine ≥ milnacipran > atomoxetine > desipramine > (S,S)-reboxetine.
Effect of various reuptake inhibitors on [3H]-noradrenaline uptake
The inhibitory effects of the tested drugs on [3H]-NA uptake into human and rat neocortical synaptosomes, respectively, are listed in Table 2B. All drugs produced concentration-dependent inhibitory effects on [3H]-NA uptake, again similar to atomoxetine (Figure 1).
The pIC50, Imax and c values are given in Table 2B. Fluvoxamine and (S,S)-reboxetine were the only drugs with pIC50 estimates differing by more than 0.5 log units between species. Some of the maxima of relative inhibition (Imax) of [3H]-NA uptake into human synaptosomes, as indicated for citalopram, fluoxetine and duloxetine, varied markedly around unity. Similar high variations around unity were also noted for the slope factor c, including desipramine and fluoxetine (both human and rat), and citalopram, fluvoxamine, atomoxetine, (S,S)-reboxetine, duloxetine and milnacipran (human). Despite the high variability of c for fluvoxamine, atomoxetine and (S,S)-reboxetine in the rat, these slope factors were different from unity.
The apparent pKi values (Table 3) for all reuptake inhibitor drugs were estimated, assuming c= 1, pKd for [3H]-NA/NA of 6.46 (human neocortex) or 6.64 (rat neocortex, Table 1) and the concentration of [3H]-NA (10 nM) in the uptake inhibition experiments (Table 1). Significant and relevant differences in pKi values for [3H]-NA uptake inhibition between human and rat were only observed for fluvoxamine and (S,S)-reboxetine. The NRI atomoxetine was the most potent compound (subnanomolar range) in inhibiting [3H]-NA uptake in both species, whereas citalopram was the least potent drug (Table 3).
The rank order of inhibitory potency of the drugs for hNET was as follows: atomoxetine ≥ desipramine > (S,S)-reboxetine ≥ milnacipran ≥ duloxetine ≥ fluoxetine ≥ fluvoxamine ≥ citalopram. The rank potency order for rNET was similar: atomoxetine ≥ desipramine > (S,S)-reboxetine > milnacipran ≥ duloxetine > fluoxetine > fluvoxamine > citalopram.
Selectivity profile of reuptake inhibitor drugs
Selectivity ratios [pKi(5-HT) minus pKi(NA)] of the various reuptake inhibitor drugs, greater than or less than unity (i.e. significant differences in pKi estimates; compare Harms, 1983) across transporter and species, are depicted in Figure 2.
Figure 2.
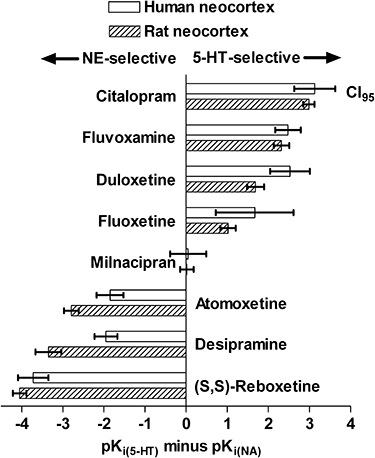
Selectivity profile of several reuptake inhibitor drugs for [3H]-5-hydroxytryptamine ([3H]-5-HT) and [3H]-noradrenaline ([3H]-NA) uptake into human and rat synaptosomes. The selectivity index was defined as pKi (5-HT) minus pKi (NA), corresponding to Ki(5-HT)/Ki(NA).
The rank order of selectivity varied primarily as a function of the 3H-monoamine substrate rather than species. For hSERT the order was citalopram ≥ duloxetine = fluvoxamine ≥ fluoxetine, being very similar to that for rSERT: citalopram > fluvoxamine > duloxetine > fluoxetine. For hNET, the order was (S,S)-reboxetine > desipramine = atomoxetine, again very similar to that for rNET: (S,S)-reboxetine > desipramine > atomoxetine. In both species, desipramine, (S,S)-reboxetine and atomoxetine exhibited markedly higher potency as inhibitors of [3H]-NA than of [3H]-5-HT uptake, whereas all the other compounds (except milnacipran) were selective for [3H]-5-HT uptake. (S,S)-Reboxetine was clearly the most selective NRI in both species, and citalopram was the most selective SRI. Milnacipran was non-selective on SERT and NET and can be classified as a dual-action inhibitor. Significant species differences for the reuptake inhibitor drugs were most evident for desipramine, atomoxetine and duloxetine (Figure 2).
Discussion
In the present study, possible pharmacological differences between human and rat neocortical SERT and NET were assessed by measuring the selectivity and potency of several reuptake inhibitor drugs. This is the first study, to our knowledge, that directly compares the effects of various NRI, SRI and SNRI on [3H]-5-HT and [3H]-NA uptake in synaptosomes prepared from freshly obtained human and rat neocortical tissues.
Because differences in assay conditions between laboratories (e.g. radiolabelled substrate) should only minimally affect the comparison of parameter estimates in different studies (Cheng and Prusoff, 1973) and because mechanistically elucidated parameters are to be preferred over descriptive parameters (Feuerstein and Limberger, 1999; Feuerstein and Sauermann, 2005), Ki and Kd values were calculated rather than IC50 and EC50 values. The calculation of these dissociation constants requires the assumption of bimolecular reactions between the transporter and its ligand or substrate (Feuerstein and Limberger, 1999). A slope factor c around unity suggests that such a bimolecular reaction occurs, which allows the assumption that EC50= pKd. As shown in Table 1, however, the c value for specific [3H]-NA uptake into rat synaptosomes was below unity and, thus, precludes assuming bimolecularity. This means that the pEC50 estimate may not exactly reflect a pKd in the case of rat neocortex and that the interpretation of Umax as number of uptake sites·mg−1 protein must be considered with caution. The difficulty of comparing, for instance, IC50 estimates between laboratories can be illustrated for fluoxetine and desipramine at SERT and NET. Langer et al. (1980) published rather different pIC50s from our data: 6.25 (SERT, rat hypothalamic slices) versus 7.48 (SERT, rat neocortical synaptosomes); 5.32 (SERT, rat hypothalamic slices) versus 5.97 (SERT, rat neocortical synaptosomes); 5.45 (NET, rat hypothalamic slices) versus 6.45 (NET, rat neocortical synaptosomes); 7.70 (NET, rat hypothalamic slices) versus of 9.33 (NET, rat neocortical synaptosomes). These values differ significantly; the exact reasons for such discrepancies are unknown although assay conditions, source of transporter (i.e. slice vs. synaptosomes) and shapes of concentration–inhibition curves can all be considered essential factors.
The pKd values, for the substrates 5-HT and NA and their respective transporters SERT and NET, did not significantly differ between human and rat neocortical synaptosomes. As Kd reflects the degree of spatial fit between a drug and the three-dimensional receptor surface, the similarity of the pKd values for 5-HT and NA transport into neocortical synaptosomes suggests a highly homologous substrate binding site in neocortical hSERT and rSERT, and hNET and rNET respectively. Indeed, the amino acid sequence of rNET is 93% similar to that of hNET (Bruss et al., 1997), and rSERT resembles hSERT by 92% (Ramamoorthy et al., 1993).
In comparison with the present data for the SERT Kd, [i.e. 48 nM (human) and 28 nM (rat)], Mann and Hrdina (1992) reported a higher Kd (210 nM; incubation time of 2 min) for [3H]-5-HT uptake into rat hypothalamic synaptosomes. Wood (1987), however, reported a Kd of 72 nM with rat neocortical synaptosomes (incubation time of 4 or 6 min), approximating the current results (incubation time of 20 min). Because the affinity of the accumulation of 5-HT increases with the incubation time over the first few minutes (see Wood, 1987), the short incubation period of Mann and Hrdina may explain their higher 1/affinity estimate. A recent estimate of the pKm on [3H]-NA uptake into rat neocortical synaptosomes was 6.66 (Jeannotte and Sidhu, 2008), nearly identical to our rat pKd value of 6.64.
In contrast to the interspecies similarity of our pKd values, the number of uptake sites of·mg−1 protein (Umax) for both SERT and NET was significantly lower in human than in rat neocortical synaptosomes, suggesting that the density of both transporters in human tissue is considerably less than in rat tissue. The above-mentioned papers (Wood, 1987; Mann and Hrdina, 1992; Jeannotte and Sidhu, 2008) do not provide Vmax or Umax estimates obtained at equilibrium conditions for comparison with our Umax values. This is because the Vmax values of these studies reflect incubation times of ≤6 min with the respective 3H-monoamine (as compared with 20 min in the present investigation).
As regards the inhibitory effects of the reuptake inhibitor drugs on hSERT, rSERT, hNET and rNET, all substances reduced the uptake of both [3H]-5-HT and [3H]-NA into human and rat neocortical synaptosomes in a concentration-dependent manner (refer to Figure 1). The IC50 values, or the corresponding Ki values, of these drugs using rat neocortex were generally consistent with literature values (Hyttel, 1994; Sanchez and Hyttel, 1999; Hajos et al., 2004; Zhou, 2004; Stahl et al., 2005). The present study, in contrast to some of these cited, gave attention to the shape of the concentration–response curves and the estimates of the slope factor c. If the slope factor resembles unity, then there is a high probability of a bimolecular reaction between the SERT or NET and the inhibitor of these transporters (Feuerstein and Limberger, 1999; Feuerstein and Sauermann, 2005). The probability of a pure bimolecular reaction is also dependent on the range of the CI95 encompassing unity (see Table 2). Only a narrow CI95 of c that includes unity is compatible with the law of mass action describing a bimolecular reaction (see Feuerstein and Limberger, 1999).
Among the SRI tested in the present study, citalopram was the most potent, and fluoxetine was the least potent inhibitor of [3H]-5-HT uptake. Our data for SRIs in rat neocortical synaptosomes are consistent with literature values for IC50 (Frazer, 2001) and affinity (Richelson and Pfenning, 1984; Sanchez and Hyttel, 1999). The slightly higher affinity of citalopram at rSERT, compared with hSERT, agrees with previous results from binding assays (Plenge and Mellerup, 1991). Fluvoxamine also had a significantly lower affinity to hNET, an observation further supported by binding studies that compared affinities of rat and human neocortical NET with NET-transfected cells (Owens et al., 1997). All tested SRIs were selective to varying degrees at inhibiting [3H]-5-HT uptake. The rank order of this SERT selectivity did not differ between human and rat neocortex. Citalopram was the most selective SRI, and fluoxetine was the least, entirely consistent with results from animal studies (Richelson and Pfenning, 1984; Hyttel, 1994; Frazer, 2001).
Duloxetine and milnacipran block the reuptake of both [3H]-5-HT and 3H]-NA with differing selectivity (Stahl et al., 2005). In the present study, milnacipran blocked both [3H]-5-HT and [3H]-NA uptake with similar potency and, therefore, without selectivity. Duloxetine, in contrast, was both more potent and SERT selective. Our data on the potency of these drugs in both species are in agreement with binding and uptake studies (Wong et al., 1993; Beique et al., 1998; Bymaster et al., 2001; Stahl et al., 2005). The equivalent potency of milnacipran at the hSERT and hNET is consistent with previous results from binding and uptake studies on cells transfected with these transporters (Vaishnavi et al., 2004). A preferential action of duloxetine at SERT has also been observed functionally in vivo by using an electrophysiological paradigm and in ex vivo uptake studies (Kasamo et al., 1996).
As expected, desipramine was more potent at inhibiting [3H]-NA than [3H]-5-HT uptake, corroborating results of other researchers (Richelson and Pfenning, 1984; Hyttel, 1994; Sanchez and Hyttel, 1999; Frazer, 2001). Although desipramine was more potent on [3H]-NA uptake, its pKi for [3H]-5-HT uptake differed substantially between human and rat with notably higher affinity for hSERT. The higher potency of desipramine at the hSERT (Ki= 58 nM) is consistent with previous findings using hSERT-transfected cells (Barker et al., 1994; Barker and Blakely, 1996; Owens et al., 1997). This observation suggests that only a minor portion of desipramine's clinical efficacy reflects 5-HT uptake inhibition. Surprisingly, however, although desipramine was NET-selective in both species, the 90-fold higher selectivity for hNET over hSERT contrasted sharply to the >2200-fold higher NET selectivity in rats (Figure 2). This higher selectivity of desipramine for the rNET also agrees with results from binding studies comparing neocortical rNET and cells transfected with hNET (Owens et al., 1997). In contrast, Harms (1983) did not find a significant difference in selectivity for desipramine between human and rat brain slices. The discrepancy between the present results and those of Harms probably relates to the use of slices in the latter study rather than synaptosomes, and the condition of the human tissue (post-mortem vs. fresh). Thus, our results emphasize an important species difference in the selectivity of desipramine.
Some NRI drugs (e.g. reboxetine, atomoxetine) have been developed to treat a variety of psychiatric disorders including depression and attention-deficit/hyperactivity disorder (Zhou, 2004). In the present study, the NRI atomoxetine displayed the highest NET pKi (Table 3), whereas the selectivity to inhibit [3H]-NA uptake was considerably higher in rat neocortical synaptosomes (Figure 2). In uptake studies in cells transfected with cloned hNET and hSERT, atomoxetine exhibited a higher pKi at the NET (Zhou, 2004). Note also that atomoxetine, although binding with high affinity to NET, also inhibits [3H]-5-HT uptake with nanomolar potency and there are significant, relevant differences between human and rat neocortex (19 nM vs. 148 nM, respectively; Table 3). The selectivity of atomoxetine for NET and its slightly lower affinity for the 5-HT uptake site was also confirmed in binding studies (Gehlert et al., 1995).
The NRI reboxetine has also been shown to exhibit a high affinity for the hNET and rNET (Wong et al., 2000; Hajos et al., 2004). The (S,S)-enantiomer is even more potent and selective at the NET than the racemate (i.e. (R,R)- and (S,S)-enantiomers of reboxetine; Zhou (2004). In the present study, the pKi value of (S,S)-reboxetine (8.99) for the rat rNET (Table 3) was nearly identical to that observed in binding studies (9.0; Hajos et al., 2004) and higher than that reported for the racemate tested in rat hypothalamic synaptosomes (8.1; Hajos et al., 2004). The pKi of (S,S)-reboxetine (8.40) observed in human neocortex in the present investigation (Table 3) is, however, higher than that reported for reboxetine in Madin-Darby canine kidney cells expressing hNET (7.96; Wong et al., 2000; Hajos et al., 2004). In contrast, (S,S)-reboxetine was found to be an extremely weak inhibitor of [3H]-5-HT uptake in both species (Table 3). The selectivity of (S,S)-reboxetine for hNET and rNET was 5248-fold and 11 220-fold, being very similar to previous findings for rNET (Hajos et al., 2004). All of these results support the contention (Hajos et al., 2004; Zhou, 2004) that (S,S)-reboxetine is one of the most potent and selective NRI known or available. Finally, (S,S)-reboxetine also exhibits a relevant species difference as indicated by its higher pKi value for [3H]-NA uptake in the rat neocortex (Table 3).
In conclusion, the effects of various reuptake inhibitor drugs on SERT and NET function have been characterized for the first time by using native, fresh human neocortical tissue. The targets of these drugs are considered to be SERT and NET localized to neocortical 5-hydroxytrytaminergic and noradrenergic axon terminals. As shown here, however, relevant species differences can exist for these drugs even though the endogenous substrates, 5-HT and NA, are bound with the same affinity by SERT and NET of both species. Therefore, pharmacological inhibition of [3H]-NA and [3H]-5-HT uptake in rat neocortical synaptosomes cannot always be extrapolated to similar properties in human brain. Moreover, the present findings may help to predict more precisely the profile of antidepressants in their clinical applications.
However, the present paper does not deal with DA transport blockers, and we must not forget that a decrease in dopaminergic transmission may also be one of the neurochemical alterations in depression (for a further discussion of dopamine in the context of antidepressant therapy, see Feuerstein, 2007).
Glossary
Abbreviations:
- 5-HT
5-hydroxytryptamine
- DAT
dopamine transporter
- NA
noradrenaline
- NET
noradrenaline transporter
- NRI
noradrenaline reutake inhibitor
- SERT
5-hydroxytryptamine transporter
- SNRI
(dual) 5-hydroxytryptamine/noradrenaline reuptake inhibitor
- SRI
5-hydroxytryptamine reuptake inhibitor
References
- Barker EL, Blakely RD. Identification of a single amino acid, phenylalanine 586, that is responsible for high affinity interactions of tricyclic antidepressants with the human serotonin transporter. Mol Pharmacol. 1996;50:957–965. [PubMed] [Google Scholar]
- Barker EL, Kimmel HL, Blakely RD. Chimeric human and rat serotonin transporters reveal domains involved in recognition of transporter ligands. Mol Pharmacol. 1994;46:799–807. [PubMed] [Google Scholar]
- Beique JC, Lavoie N, de Montigny C, Debonnel G. Affinities of venlafaxine and various reuptake inhibitors for the serotonin and norepinephrine transporters. Eur J Pharmacol. 1998;349:129–132. doi: 10.1016/s0014-2999(98)00241-6. [DOI] [PubMed] [Google Scholar]
- Berton O, Nestler EJ. New approaches to antidepressant drug discovery: beyond monoamines. Nat Rev Neurosci. 2006;7:137–151. doi: 10.1038/nrn1846. [DOI] [PubMed] [Google Scholar]
- Blakely RD, Berson HE, Fremeau RT, Jr, Caron MG, Peek MM, Prince HK, et al. Cloning and expression of a functional serotonin transporter from rat brain. Nature. 1991;354:66–70. doi: 10.1038/354066a0. [DOI] [PubMed] [Google Scholar]
- Bruss M, Porzgen P, Bryan-Lluka LJ, Bönisch H. The rat norepinephrine transporter: molecular cloning from PC12 cells and functional expression. Brain Res Mol Brain Res. 1997;52:257–262. doi: 10.1016/s0169-328x(97)00267-2. [DOI] [PubMed] [Google Scholar]
- Bymaster FP, Dreshfield-Ahmad LJ, Threlkeld PG, Shaw JL, Thompson L, Nelson DL, et al. Comparative affinity of duloxetine and venlafaxine for serotonin and norepinephrine transporters in vitro and in vivo, human serotonin receptor subtypes, and other neuronal receptors. Neuropsychopharmacology. 2001;25:871–880. doi: 10.1016/S0893-133X(01)00298-6. [DOI] [PubMed] [Google Scholar]
- Chalon S, Hall H, Saba W, Garreau L, Dollé F, Halldin C, et al. Pharmacological characterization of (E)-N-(4-fluorobut-2-enyl)-2beta-carbomethoxy-3beta-(4′-tolyl)nortropane (LBT-999) as a highly promising fluorinated ligand for the dopamine transporter. J Pharmacol Exp Ther. 2006;317:147–152. doi: 10.1124/jpet.105.096792. [DOI] [PubMed] [Google Scholar]
- Cheng Y-C, Prusoff WH. Relationship between the inhibition constant (KI) and the concentation of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- Feuerstein TJ. Presynaptic receptors for dopamine, histamine and serotonin. Handb Exp Pharmacol. 2007;2008:289–338. doi: 10.1007/978-3-540-74805-2_10. [DOI] [PubMed] [Google Scholar]
- Feuerstein TJ, Limberger N. Mathematical analysis of the control of neurotransmitter release by presynaptic receptors as a supplement to experimental data. Naunyn Schmiedebergs Arch Pharmacol. 1999;359:345–359. doi: 10.1007/pl00005361. [DOI] [PubMed] [Google Scholar]
- Feuerstein TJ, Sauermann W. What is the opposite of a receptor reserve? Int J Pharmacol. 2005;1:195–202. [Google Scholar]
- Feuerstein TJ, Hertting G, Lupp A, Neufang B. False labelling of dopaminergic terminals in the rabbit caudate nucleus: uptake and release of [3H]-5-hydroxytryptamine. Br J Pharmacol. 1986;88:677–684. doi: 10.1111/j.1476-5381.1986.tb10250.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazer A. Serotonergic and noradrenergic reuptake inhibitors: prediction of clinical effects from in vitro potencies. J Clin Psychiatry. 2001;62(Suppl. 12):16–23. [PubMed] [Google Scholar]
- Gardner MJ, Altman DG. Confidence intervals rather than P values: estimation rather than hypothesis testing. Br Med J (Clin Res Ed) 1986;292:746–750. doi: 10.1136/bmj.292.6522.746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehlert DR, Schober DA, Gackenheimer SL. Comparison of (R)-[3H]tomoxetine and (R/S)-[3H]nisoxetine binding in rat brain. J Neurochem. 1995;64:2792–2800. doi: 10.1046/j.1471-4159.1995.64062792.x. [DOI] [PubMed] [Google Scholar]
- Göthert M, Schlicker E. Regulation of 5-HT release in the CNS by presynaptic 5-HT autoreceptors and by 5-HT heteroreceptors. In: Baumgarten HG, Göthert M, editors. Serotonergic Neurons and 5-HT Receptors in the CNS. Berlin, Heidelberg and New York: Springer; 1997. pp. 307–350. [Google Scholar]
- Hajos M, Fleishaker JC, Filipiak-Reisner JK, Brown MT, Wong EH. The selective norepinephrine reuptake inhibitor antidepressant reboxetine: pharmacological and clinical profile. CNS Drug Rev. 2004;10:23–44. doi: 10.1111/j.1527-3458.2004.tb00002.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms HH. The antidepressant agents desipramine, fluoxetine, fluvoxamine and norzimelidine inhibit uptake of [3H]noradrenaline and [3H]5-hydroxytryptamine in slices of human and rat cortical brain tissue. Brain Res. 1983;275:99–104. doi: 10.1016/0006-8993(83)90421-3. [DOI] [PubMed] [Google Scholar]
- Hyttel J. Pharmacological characterization of selective serotonin reuptake inhibitors (SSRIs) Int Clin Psychopharmacol. 1994;9(Suppl. 1):19–26. doi: 10.1097/00004850-199403001-00004. [DOI] [PubMed] [Google Scholar]
- Jeannotte AM, Sidhu A. Regulated interactions of the norepineprhine transporter by the actin and microtubule cytoskeletons. J Neurochem. 2008;105:1668–1682. doi: 10.1111/j.1471-4159.2008.05258.x. [DOI] [PubMed] [Google Scholar]
- Kasamo K, Blier P, de Montigny C. Blockade of the serotonin and norepinephrine uptake processes by duloxetine: in vitro and in vivo studies in the rat brain. J Pharmacol Exp Ther. 1996;277:278–286. [PubMed] [Google Scholar]
- Langer SZ, Moret C, Raisman R, Dubocovich ML, Briley M. High-affinity [3H]imipramine binding in rat hypothalamus: association with uptake of serotonin but not of norepinephrine. Science. 1980;210:1133–1135. doi: 10.1126/science.7444441. [DOI] [PubMed] [Google Scholar]
- Lieb K, Fiebich BL, Herpfer I, Mantovani M, Löffler M, Feuerstein TJ. No modulatory effect of neurokinin-1 receptor antagonists on serotonin uptake in human and rat brain synaptosomes. Eur Neuropsychopharmacol. 2005;15:641–646. doi: 10.1016/j.euroneuro.2005.04.013. [DOI] [PubMed] [Google Scholar]
- Löffler M, Bubl B, Huethe F, Hubbe U, McIntosh JM, Jackisch R, et al. Dopamine release in human neocortical slices: characterization of inhibitory autoreceptors and of nicotinic acetylcholine receptor-evoked release. Brain Res Bull. 2006;68:361–373. doi: 10.1016/j.brainresbull.2005.09.018. [DOI] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- Lupp A, Bär KI, Lücking CH, Feuerstein TJ. Different effects of serotonin (5-HT) uptake blockers in caudate nucleus and hippocampus of the rabbit: role of monoamine oxidase in dopaminergic terminals. Psychopharmacology. 1992;106:118–126. doi: 10.1007/BF02253598. [DOI] [PubMed] [Google Scholar]
- Mann CD, Hrdina PD. Sodium dependence of [3H]paroxetine binding and 5-[3H]hydroxytryptamine uptake in rat diencephalon. J Neurochem. 1992;59:1856–1861. doi: 10.1111/j.1471-4159.1992.tb11020.x. [DOI] [PubMed] [Google Scholar]
- Owens MJ, Morgan WN, Plott SJ, Nemeroff CB. Neurotransmitter receptor and transporter binding profile of antidepressants and their metabolites. J Pharmacol Exp Ther. 1997;283:1305–1322. [PubMed] [Google Scholar]
- Pacholczyk T, Blakely RD, Amara SG. Expression cloning of a cocaine- and antidepressant-sensitive human noradrenaline transporter. Nature. 1991;350:350–354. doi: 10.1038/350350a0. [DOI] [PubMed] [Google Scholar]
- Plenge P, Mellerup ET. [3H]citalopram binding to brain and platelet membranes of human and rat. J Neurochem. 1991;56:248–252. doi: 10.1111/j.1471-4159.1991.tb02588.x. [DOI] [PubMed] [Google Scholar]
- Ramamoorthy S, Bauman AL, Moore KR, Han H, Yang-Feng T, Chang AS, et al. Antidepressant- and cocaine-sensitive human serotonin transporter: molecular cloning, expression, and chromosomal localization. Proc Natl Acad Sci USA. 1993;90:2542–2546. doi: 10.1073/pnas.90.6.2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richelson E, Pfenning M. Blockade by antidepressants and related compounds of biogenic amine uptake into rat brain synaptosomes: most antidepressants selectively block norepinephrine uptake. Eur J Pharmacol. 1984;104:277–286. doi: 10.1016/0014-2999(84)90403-5. [DOI] [PubMed] [Google Scholar]
- Ruhé HG, Mason NS, Schene AH. Mood is indirectly related to serotonin, norepinephrine and dopamine levels in humans: a meta-analysis of monoamine depletion studies. Mol Psychiatry. 2007;12:331–359. doi: 10.1038/sj.mp.4001949. [DOI] [PubMed] [Google Scholar]
- Sanchez C, Hyttel J. Comparison of the effects of antidepressants and their metabolites on reuptake of biogenic amines and on receptor binding. Cell Mol Neurobiol. 1999;19:467–489. doi: 10.1023/a:1006986824213. [DOI] [PubMed] [Google Scholar]
- Stahl SM, Grady MM, Moret C, Briley M. SNRIs: their pharmacology, clinical efficacy, and tolerability in comparison with other classes of antidepressants. CNS Spectr. 2005;10:732–747. doi: 10.1017/s1092852900019726. [DOI] [PubMed] [Google Scholar]
- Steffens M, Feuerstein TJ. Receptor-independent depression of DA and 5-HT uptake by cannabinoids in rat neocortex – involvement of Na(+)/K(+)-ATPase. Neurochem Int. 2004;44:529–538. doi: 10.1016/j.neuint.2003.08.009. [DOI] [PubMed] [Google Scholar]
- Vaishnavi SN, Nemeroff CB, Plott SJ, Rao SG, Kranzler J, Owens MJ. Milnacipran: a comparative analysis of human monoamine uptake and transporter binding affinity. Biol Psychiatry. 2004;55:320–322. doi: 10.1016/j.biopsych.2003.07.006. [DOI] [PubMed] [Google Scholar]
- Wheeler DD, Edwards AM, Ondo JG. Dopamine uptake in five structures of the brain: comparison of rate, sodium dependence and sensitivity to cocaine. Neuropharmacol. 1993;32:501–508. doi: 10.1016/0028-3908(93)90176-4. [DOI] [PubMed] [Google Scholar]
- Wong DT, Bymaster FP, Mayle DA, Reid LR, Krushinski JH, Robertson DW. LY248686, a new inhibitor of serotonin and norepinephrine uptake. Neuropsychopharmacology. 1993;8:23–33. doi: 10.1038/npp.1993.4. [DOI] [PubMed] [Google Scholar]
- Wong EH, Sonders MS, Amara SG, Tinholt PM, Piercey MF, Hoffmann WP, et al. Reboxetine: a pharmacologically potent, selective, and specific norepinephrine reuptake inhibitor. Biol Psychiatry. 2000;47:818–829. doi: 10.1016/s0006-3223(99)00291-7. [DOI] [PubMed] [Google Scholar]
- Wood MD. Examination of the relationship between the uptake site for 5-hydroxytryptamine and the high affinity binding site for [3H]imipramine. II. The role of sodium ions. Neuropharmacology. 1987;26:1081–1085. doi: 10.1016/0028-3908(87)90251-6. [DOI] [PubMed] [Google Scholar]
- Zhou J. Norepinephrine transporter inhibitors and their therapeutic potential. Drugs Future. 2004;29:1235–1244. doi: 10.1358/dof.2004.029.12.855246. [DOI] [PMC free article] [PubMed] [Google Scholar]