

Abstract
In order for neurons to perform their function, they must establish a highly polarized morphology characterized, in most of the cases, by a single axon and multiple dendrites. Herein we find that the evolutionarily conserved protein Kidins220 (kinase D-interacting substrate of 220-kDa), also known as ARMS (ankyrin repeat-rich membrane spanning), a downstream effector of protein kinase D and neurotrophin and ephrin receptors, regulates the establishment of neuronal polarity and development of dendrites. Kidins220/ARMS gain and loss of function experiments render severe phenotypic changes in the processes extended by hippocampal neurons in culture. Although Kidins220/ARMS early overexpression hinders neuronal development, its down-regulation by RNA interference results in the appearance of multiple longer axon-like extensions as well as aberrant dendritic arbors. We also find that Kidins220/ARMS interacts with tubulin and microtubule-regulating molecules whose role in neuronal morphogenesis is well established (microtubule-associated proteins 1b, 1a, and 2 and two members of the stathmin family). Importantly, neurons where Kidins220/ARMS has been knocked down register changes in the phosphorylation activity of MAP1b and stathmins. Altogether, our results indicate that Kidins220/ARMS is a key modulator of the activity of microtubule-regulating proteins known to actively regulate neuronal morphogenesis and suggest a mechanism by which it contributes to control neuronal development.
Keywords: Cell/Neuron, Cell/Differentiation, Cytoskeleton/Microtubules, Neurobiology/Neuroscience, Phosphorylation/Cytoskeletal Proteins, Protein/Protein-Protein Interactions, Axon/Dendrites, Kinase D-interacting Substrate of 220 kDa (Kidins220)/Ankyrin-Repeat-rich Membrane-spanning (ARMS)
Introduction
Neuronal differentiation comprises several steps, among which the acquirement of a polarized axon-dendrite phenotype, with the corresponding asymmetrical distribution of proteins, is crucial. The morphological changes, followed by a neuron in order to polarize and form a single axon and multiple dendrites, are triggered by signaling cascades evoked by both intracellular and extracellular cues (1–4).
Embryonic hippocampal neurons in culture constitute a model to study the mechanisms governing the establishment of polarity (2, 5, 6). These neurons undergo clear morphological changes during in vitro polarization. First, neurons attach to the plate and form lamellipodia and filopodia (stage 1). After several hours, they extend several minor immature neurites of apparent equivalent nature (stage 2) until one of these minor processes extends rapidly and becomes the axon (stage 3). The remaining neurites develop into dendrites (stage 4), after which neurons become morphologically and functionally mature (stage 5) (5, 6).
During the early events of the establishment of polarity in this model, differences in local actin polymerization among the immature neurites play a crucial role in axonal determination (5, 7). In a similar manner, microtubule dynamics influence neuronal polarization, because local microtubule stabilization in one neurite specifies an axonal fate (8). Other known regulators of neuronal polarity and axon specification include proteins involved in polarized trafficking (4, 9, 10). However, how these different molecules are linked to the extracellular cues that may modulate these processes in vivo, and how these diverse processes are coordinated to generate a polarized and mature neuron is only beginning to be understood.
Kidins220 (kinase D-interacting substrate of 220 kDa), also known as ARMS (ankyrin repeat-rich membrane-spanning), is an integral membrane protein associated with lipid rafts, abundant in the developing nervous system and in highly plastic areas of the adult brain (11–13). This protein was first identified as a substrate for protein kinase D (PKD)4 (12). Members of the PKD family serve as regulators of polarized membrane trafficking among other functions (14–17) and modulate the establishment of neuronal polarity and maturation as it has been recently found (10, 18, 19). Kidins220/ARMS was also identified as an effector of neurotrophin and ephrin receptors, both of which play a prominent role in the development of the vertebrate nervous system (20–25). Downstream of neurotrophin receptors, Kidins220/ARMS is required for signaling pathways involved in neurite outgrowth (26–28). Recent studies have also focused on the mechanisms that control Kidins220/ARMS traffic to sites where it might regulate the cellular response to stimuli, such as neurotrophins and ephrins. In this context, it has been demonstrated that Kidins220/ARMS undergoes a kinesin-1-dependent transport linked to neurotrophin signaling (29), whereas PKD1 and -2 also control Kidins220/ARMS traffic (30). Other studies have shown that Kidins220/ARMS localization at the neuromuscular junction, where it enhances EphA4 signaling, is regulated by α-syntrophin (31).
Given the prominent expression of Kidins220/ARMS in the developing nervous system and its role as an effector of molecules that regulate neuronal development, we decided to analyze its functions at early stages during the establishment of neuronal polarity as well as during axonal and dendritic maturation. Herein we show that Kidins220/ARMS regulates polarity establishment and neuronal development. Importantly, we find that Kidins220/ARMS displays a unique ability to interact and modulate the activity of microtubule-regulating molecules that exert a crucial role in the control of neuronal morphogenesis. Our results suggest that Kidins220/ARMS could be controlling neuronal development by modulating the activity of these microtubule-regulating proteins.
EXPERIMENTAL PROCEDURES
Reagents
Texas Red-phalloidin, TRITC-phalloidin, and fluorescein isothiocyanate-phalloidin were from Molecular Probes. Oligonucleotides were purchased from Invitrogen. The BCA reagent was from Pierce, and ECL was from GE Healthcare. All other reagents were from standard suppliers.
Antibodies
The following antibodies were used. Mouse monoclonal anti-β-actin (AC-15), mouse monoclonal anti-α-tubulin (DM 1A), mouse monoclonal anti-tyrosinated tubulin (clone A1.2), mouse monoclonal anti-acetylated tubulin (clone 6-11B-1), mouse monoclonal anti-MAP1b HC (AA6), and goat anti-mouse IgG (A-6531) were all from Sigma; mouse monoclonal anti-βIII tubulin/Tuj1 (MO15013) was from Neuromics Antibodies (Northfield, MA); mouse monoclonal anti-GM130 was from BD Transduction Laboratories (Palo Alto, CA); goat polyclonal anti-MAP1 LCs, recognizing both LC1 and LC2 of MAP1 (C-20), were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA); mouse monoclonal SMI-31 antibody, rabbit polyclonal anti-Ser(P)-16 stathmin (ab47328), and rabbit polyclonal anti-stathmin (ab20022), which recognizes a conserved region in all stathmin proteins (panstathmin), were all purchased from Abcam (Cambridge, UK); rabbit polyclonal anti-Thr(P)-514-CRMP2, total CRMP2, Ser(P)-916-PKD, and total PKD were from Cell Signaling (Danvers, MA); rabbit polyclonal anti-neuronal specific enolase was from ICN Biomedicals (Costa Mesa, CA); rabbit polyclonal anti-stathmin, SCG10 (superior cervical ganglion 10), Sclip (SCG10-like protein), Ser(P)-16-stathmin, and Ser(P)-38 stathmin were a generous gift from Dr. André Sobel (Institut du Fer a Moulin, Paris, France); and rabbit polyclonal anti-MAP2 was a generous gift from Dr Félix Hernández (Centro de Biologia Molecular Severo Ochoa, Madrid, Spain). Kidins220/ARMS rabbit polyclonal and mouse monoclonal antibodies have been previously described (11, 12); horseradish peroxidase-conjugated secondary antibodies were from GE Healthcare; and Alexa 488-, Alexa 594-, and Alexa 647-conjugated secondary antibodies were from Molecular Probes.
Plasmid Constructs
To generate the shRNA vectors against Kidins220/ARMS, the following oligonucleotides were cloned into the pLentiLox3.7 (pLL3.7) vector at the HpaI and XhoI sites as previously described (32). An additional shRNA vector against Kidins220/ARMS was generated in the same way: sh1, 5′-TGC CGG AAC ATA CGT GAA CAT ATT CAA GAG ATA TGT TCA CGT ATG TTC CGG CTT TTT TC-3′ and 5′-TCG AGA AAA AAG CCG GAA CAT ACG TGA ACA TAT CTC TTG AAT ATG TTC ACG TAT GTT CCG GCA-3′. A control shRNA vector (shC) had already been generated (32). The targeted sequences correspond to Kidins220/ARMS rat mRNA at positions 775–795 and 4890–4912 for sh1 and sh2, respectively. All constructs were sequenced.
Culture and Transfection of Neurons and PC12 Cells
Cultures of primary hippocampal neurons were prepared from embryonic day 18 rat embryos as described (33, 34). Dissociated neurons were seeded on coverslips or dishes coated with poly-l-lysine (1 mg/ml) in plating minimal essential medium (minimal essential medium supplemented with 0.6% glucose and 10% horse serum) for 4–6 h until neurons attached. The medium was then replaced by minimal essential medium containing N2 supplement (Invitrogen). Primary cortical neurons were prepared from embryonic day 18 rat embryos by mechanically dissociating the dissected cortices in culture medium (minimal essential medium supplemented with 28.5 mm NaHCO3, 22.2 mm glucose, 0.1 mm glutamine, 5% fetal bovine serum, and 5% horse serum). Neurons were counted and seeded on poly-l-lysine (1 μg/ml)-coated dishes in the same medium. All cultures were incubated at 37 °C in a humidified atmosphere containing 5% CO2. PC12 rat pheochromocytoma cells were cultured in Dulbecco's modified Eagle's medium (Invitrogen), supplemented with 7.5% fetal calf serum, 7.5% horse serum, and 2 mm glutamine. Transfection of neurons in suspension was done using the Nucleofector device (Amaxa, Gaithersburg, MD) according to the manufacturer's instructions. Transfection of neurons seeded over poly-l-lysine-coated coverslips was done in serum-free medium with Lipofectamine 2000 reagent (Invitrogen). Cells were analyzed after different days in vitro (DIV), as specified throughout. PC12 cells were seeded at 50–60% confluence on collagen-coated dishes and were transfected in serum-free medium with Lipofectamine 2000 reagent (Invitrogen), following the manufacturer's instructions.
Yeast Two-hybrid Assays
Yeast two-hybrid screens were performed using the Matchmaker system 3 (BD Biosciences) according to the manufacturer's instructions. The two baits used consisted of Kidins220/ARMS N-terminal ankyrin repeats (amino acids 1–211) and C-terminal region (amino acids 760–1762), each fused in frame with the Gal4 DNA binding domain of the pGBKT7 vector. Each construct was used to screen a mouse brain cDNA library cloned into pACT2 plasmid. Positive colonies were isolated based on their capacity to express the markers ADE2, HIS3, and LacZ.
Immunofluorescence and Confocal Microscopy
Cells grown on coverslips were fixed for 5 min in 4% paraformaldehyde containing 4% sucrose in phosphate-buffered saline at 37 °C. Cells were then permeabilized with 0.2% Triton X-100 in phosphate-buffered saline during 5 min at room temperature. After blocking (5% bovine serum albumin in phosphate-buffered saline for 1 h), cells were incubated with the corresponding primary antibodies, and immunoreactivity was detected with the suitable fluorophore-conjugated secondary antibody before mounting on slides with Mowiol4-88 (Harland Co.). Confocal images were acquired using an inverted Leica TCS SP5 laser confocal microscope with a ×63 plan-achromatic oil immersion objective and processed with the LAS AF Leica application suite and Adobe Photoshop CS2 (Adobe Systems Inc.). All images correspond to the projection of sections from a ∼50-μm z-stack, except for colocalization analysis, where they correspond to 0.5–0.7-μm single sections.
Preparation of Protein Extracts
Hippocampal and cortical neurons were lysed in radioimmune precipitation buffer (25 mm Tris-HCl, pH 7.6, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 150 mm NaCl) with protease and phosphatase inhibitors for 15 min at 4 °C, and lysates were centrifuged for 15 min at 12,000 rpm at 4 °C. This supernatant was considered the soluble fraction. When required, the insoluble pellet was re-extracted by further incubation in radioimmune precipitation buffer for 6 h. The solution was then passed through 0.8-, 0.6-, and 0.5-mm syringes until a non-viscous solution was obtained. These lysates were recentrifuged for 15 min at 12,000 rpm at 4 °C, and the supernatant obtained was considered the re-extracted fraction. Brain lysates were obtained from Wistar rats at different ages by homogenization in radioimmune precipitation buffer using 0.8- and 0.5-mm needles and centrifugation for 30 min at 12,000 rpm at 4 °C. Protein determination in the extracts was done using the BCA reagent. Animal manipulation was performed in compliance with European Community law 86/609/EEC and approved by the “Consejo Superior de Investigaciones Científicas” committee.
Coimmunoprecipitation and Immunoblot Analysis
Lysates from cortical neurons were subjected to immunoprecipitation with the corresponding antibodies bound to either protein A- or protein G-Sepharose beads for 2 h at 4 °C. Immunoprecipitates were washed extensively with radioimmune precipitation buffer, and bound proteins were eluted with Laemmli sample buffer. For immunoblot analysis, total lysates or immunoprecipitates were resolved by SDS-PAGE and transferred to nitrocellulose. Membranes were blocked in TBST (20 mm Tris-HCl, pH 7.6, 137 mm NaCl, 0.05% Tween 20) plus 5% low fat milk powder for 1 h at room temperature and probed with different primary antibodies in blocking solution. After washing in TBST, membranes were incubated with the appropriate secondary antibodies conjugated to peroxidase. Immunoreactive bands were visualized by ECL.
Morphometric Analysis, Quantification of Immunofluorescence, and Statistics
To analyze the neuronal morphology, shRNA-transfected cells were chosen based on GFP expression. All processes were traced, their numbers were counted, and their lengths were measured using the semiautomatic plug-in NeuronJ from the ImageJ software (National Institutes of Health, Bethesda, MD). Axons were defined as Tau1 or SMI-31-positive neurites with a length at least twice that of the cell body and always greater than 100 μm in length at 3 DIV. Dendrites were defined as MAP2-positive neurites. Dendrites were classified as normal if neurons possessed three or more processes longer than 20 μm in length or aberrant if neurons displayed processes shorter than 20 μm or presented a lamellar structure surrounding the cell body instead of a defined neurite. Immunofluorescence signal intensity was quantified using MetaMorph Imaging System software (Universal Imaging Corp.). For Kidins220/ARMS and F-actin fluorescence ratio analysis, the NIH Image software was used to measure the total fluorescence of the area of the neurite tip (marked based on fluorescence channels and phase-contrast images) in each fluorescence channel from each individual image by considering background and total fluorescence for each channel. All data were expressed as mean ± S.E. Comparison between two groups was made using an unpaired Student's t test. Data significantly different from control were represented as follows: ***, p < 0.001; **, p < 0.01; *, p < 0.05.
RESULTS
Kidins220/ARMS Is Localized at the Tip of All Neurites and at the Golgi Apparatus in Cultured Hippocampal Neurons
To define the function of Kidins220/ARMS in neuronal development, we first determined its levels in cultured hippocampal neurons from embryonic day 18 rat embryos at different times (DIV) during their development in vitro. As the hippocampal neurons matured, there was a significant decrease on Kidins220/ARMS signal (Fig. 1A, left). This decline, which correlated with the pattern of expression in rat brain extracts (Fig. 1A, right), suggested a role for this protein in the early phases of neuronal differentiation. To gain insight into this, we next examined Kidins220/ARMS intracellular distribution in low density cultures of hippocampal neurons at different developmental stages. At stage 1 (0 DIV, 6 h), Kidins220/ARMS was distributed within the cell body, enriched in small vesicles and around the nucleus, and along the plasma membrane (Fig. 1B, a). It was also found at filopodia and lamellipodia (Fig. 1B, a). During stage 2 (1.5 DIV; Fig. 1B, b) and stage 3 (2.5 DIV; Fig. 1B, c), neurons also contained Kidins220/ARMS enriched in a juxtanuclear region and in all neurites, where it was particularly concentrated at the growth cones partially colocalizing with F-actin (Fig. 1B, b and c). This enrichment was more prominent at stage 2, where Kidins220/ARMS staining was less intense in the α-tubulin-enriched neurite shaft (Fig. 1C). Additionally, immunofluorescence with the Golgi apparatus marker GM130 confirmed that the majority of Kidins220/ARMS juxtanuclear staining overlapped with this compartment (Fig. 1D; stage 2 is shown). Consistent with the immunoblot results (Fig. 1A) at later stages of differentiation (stages 4 and 5, 4 and >7 DIV), Kidins220/ARMS immunolabeling decreased (not shown).
FIGURE 1.

Kidins220/ARMS expression and distribution in hippocampal neurons in vitro. A, lysates from different developmental stages (DIV) of rat hippocampal neurons in culture or rat brain from embryonic (E), postnatal (P, days), or adult (M; months) animals were immunoblotted with anti-Kidins220/ARMS antibody. β-Actin was used as a control. B, hippocampal neurons were fixed at stage 1 (a), 2 (b), or 3 (c) and immunostained with phalloidin-TRITC (F-Actin, red). Scale bar, 10 μm. C and D, hippocampal neurons were fixed at stage 2 and immunostained with anti-Kidins220/ARMS (green) and α-tubulin (C; red) or the Golgi marker GM130 (D; red). Note that Kidins220/ARMS co-localizes with the Golgi apparatus in the juxtanuclear region. Nuclei stained with DAPI (blue) staining are shown in the merged image. Scale bar, 10 μm.
Notably, we observed that in neurons at stage 2 or stage 2–3 transition, Kidins220/ARMS was not evenly distributed in the growth cones of all immature neurites, being significantly more concentrated on those that showed a higher F-actin staining. We quantified the fluorescence intensity of Kidins220/ARMS and F-actin for each neurite of a population of stage 2 hippocampal neurons (n = 40 neurons, three experiments) and found a direct and significant correlation (Fig. 2). The single neurite tip with the lowest F-actin content had the lowest Kidins220/ARMS concentration, and those neurites with more F-actin had a 3-fold higher intensity for Kidins220/ARMS signal (Fig. 2C).
FIGURE 2.

Correlation between Kidins220/ARMS and F-actin levels in stage 2 neurons. A, stage 2 hippocampal neurons were fixed and immunostained with anti-Kidins220/ARMS (green) and phalloidin-Texas Red (F-Actin, red). Scale bar, 20 μm. B, quantification of the relative fluorescence intensity of Kidins220/ARMS and F-actin for each numbered neurite of a representative stage 2 hippocampal neuron (shown in A). Note the high degree of correlation between the intensities of both signals. C, in a stage 2 hippocampal neuron population, the single neurite tip with lowest actin filament content had the lowest Kidins220/ARMS concentration, and those neurites with more F-actin had a 3.3-fold higher intensity for Kidins220/ARMS signal (n = 40, three independent experiments). Statistical significance was evaluated by the Student's unpaired t test (*, p < 0.05). a.u., arbitrary units.
Early Stage Overexpression of Kidins220/ARMS Hinders the Development of Hippocampal Neurons
To investigate the Kidins220/ARMS role, we performed gain of function experiments by transfecting hippocampal neurons in suspension at 0 DIV with a vector with Kidins220/ARMS cDNA fused to GFP-C terminus (GFP-Kidins220/ARMS). We looked for phenotypic changes in transfected compared with non-transfected neurons in the same culture or with parallel cultures transfected with a vector expressing GFP alone (Fig. 3). GFP-only neurons displayed either the normal stage 1 phenotype, with broad lamellipodia (not shown), or the characteristic multineurite phenotype (Fig. 3A, a). In contrast, a significant percentage of GFP-Kidins220/ARMS-positive neurons displayed small bodies and a rounded morphology when compared with untransfected or control neurons (Fig. 3A, b and c). Quantification analysis showed that at very early stages (1.5 DIV), 69.5% of neurons ectopically expressing Kidins220/ARMS were rounded versus 0.0% of untransfected sister neurons or the GFP-transfected ones (Fig. 3B, GFP n = 184 neurons and GFP-Kidins220/ARMS n = 170 neurons, three experiments). At the same time points, 70.4% of GFP-alone-positive neurons were at stage 2 (Fig. 3B) The percentage of GFP-Kidins220/ARMS-positive neurons that reached stage 1, stage 2, or stage 3 was only 16.7, 12.2, and 1.7%, respectively (Fig. 3B). An example of these is shown in supplemental Fig. 1A (only stage 1 (panel a), and stage 2 (panel b) are shown). Thus, the early expression of GFP-Kidins220/ARMS in neurons transfected at 0 DIV made the cells smaller and less differentiated. This phenotype indicated that the excess of this protein in the cytoplasm of the cells or its ectopic localization is toxic to these neurons, highlighting the importance of its threshold levels at least at this stage. Consistent with overexpression-mediated toxicity, almost no GFP-Kidins220/ARMS-positive cells were found when cultures were fixed at longer times post-transfection. Furthermore, the dramatic reduction of cell body size was accompanied by a tubulin ring surrounding the cell surface (Fig. 3A, c) and fragmentation of the Golgi apparatus, as assessed by immunodetection of GM130 (supplemental Fig. 1B). Yet it is not possible to conclude if Golgi fragmentation/dysfunction caused growth reversion or is simply a consequence of other cellular defects. On the other hand, GFP-Kidins220/ARMS overexpression at later stages by transfecting neuronal cultures at 1 DIV and fixing at 3 DIV (supplemental Fig. 2) or other cell types, such as HEK293T (not shown), did not result in significant changes in cellular adhesion, neuronal shape and size, or increased toxicity. These neurons also presented extensions, showing again here that ectopic expression of this protein is especially deleterious in early neuronal differentiation steps, in a very narrow window during early hippocampal neuronal development in vitro, before the establishment of neuronal polarity. To assess this possibility, we performed loss of function experiments.
FIGURE 3.

Ectopic expression of GFP-Kidins220/ARMS hampers early neuronal development. A, hippocampal neurons transfected after dissection with GFP as control (a) or GFP-Kidins220/ARMS (b and c) were fixed at 1.5 DIV and immunostained for the neuronal marker βIII-tubulin/Tuj1 (blue) and phalloidin-TRITC (F-Actin, red). Scale bar, 10 μm. B, quantification of the number of rounded, stage 1, 2, or 3 neurons after Kidins220/ARMS overexpression relative to control GFP-transfected cells. The data shown are the means ± S.E. of three independent experiments (GFP n = 184 cells, GFP-Kidins220/ARMS n = 170 cells), and statistical significance was evaluated by Student's unpaired t test (***, p < 0.001).
Knockdown of Kidins220/ARMS Induces the Formation of Multiple Axon-like Processes
Kidins220/ARMS levels were knocked down by RNA interference using an expression vector for shRNAs, which co-expresses GFP (see “Experimental Procedures” and Ref. 32). This construct allowed us to label and monitor the transfected cells where interference was taking place. After cloning two specific shRNAs for rat Kidins220/ARMS (sh1 and sh2), the efficiency at reducing its protein levels was first assessed by immunoblotting total lysates from transfected PC12 cells (Fig. 4A). Transfection of the vector containing a nonspecific shRNA sequence was used as a control (shC). Kidins220/ARMS expression was specifically reduced in cells transfected with sh1 and sh2 constructs. Because sh2 transfection resulted in a higher reduction of Kidins220/ARMS levels, we performed most of our quantitative studies using this vector. Kidin220/ARMS sh2-RNA was also able to suppress specific immunostaining in hippocampal neurons (Fig. 4B; see the knockdown of Kidins220/ARMS signal by sh1-RNA in hippocampal neurons in supplemental Fig. 3A). Importantly, a significant percentage of sh2-transfected neurons formed more than one long neurite (Fig. 4, C and D). Neurons transfected with sh1-RNA vector showed a similar result (supplemental Fig. 3B). The axonal nature of these extensions was confirmed by staining with the axonal markers SMI-31 (Fig. 4C) and Tau1 (not shown). Quantification analysis revealed a 3-fold increase in neurons presenting multi-axon-like processes in cultures where Kidins220/ARMS had been down-regulated (Fig. 4D) (9.6% for shC versus 33.2% for sh2; shC n = 148 neurons and sh2 n = 128 neurons, three experiments). To further validate the above results, we next analyzed the effect of Kidins220/ARMS down-regulation on the more differentiated stage 3 neurons. Kidins220/ARMS knockdown produced neurons with longer axonal-like processes compared with shC-transfected cells (Fig. 4E). When these neurons were classified according to the distinct ranges of their axonal lengths, we observed a clear displacement of the curve representing sh2-transfected neurons toward longer axonal ranges (Fig. 4E; n = 100 neurons for both shC and sh2). Considering that Kidins220/ARMS levels are high at early differentiation stages, these last results are compatible with the view that this protein plays a role in preventing neurons from generating more than one axon.
FIGURE 4.

Down-regulation of Kidins220/ARMS in hippocampal neurons induces the formation of multiple axon-like processes. A, PC12 cells were transfected with control shRNA (GFP-shC) or Kidins220/ARMS-specific shRNA (GFP-sh1 and -sh2) vectors. After 96 h, cells were lysed, and Kidin220/ARMS knockdown was examined by immunoblot analysis. Neuronal specific enolase (NSE) levels were used as control. B, hippocampal neurons were transfected with GFP-shC (a) or GFP-sh2 (b) and immunostained for Kidins220/ARMS (red). GFP-sh2 neurons showed decreased Kidins220/ARMS labeling. Merged images are also shown (a and b). Scale bar, 20 μm. C, hippocampal neurons transfected with GFP-shC (a) or GFP-sh2 (b) and fixed at stage 3 were immunostained with the axonal marker SMI-31 (red). Only merged images are shown. Early down-regulation of Kidins220/ARMS increased the number of SMI-31-positive processes per neuron. Scale bar, 20 μm. D, quantification of the percentage of hippocampal neurons bearing several axon-like processes after knocking down Kidins220/ARMS (sh2) compared with control (shC). The data shown are the means ± S.E. of three independent experiments (shC n = 148 cells, sh2 n = 128 cells), and statistical significance was evaluated by Student's unpaired t test (**, p < 0.01). E, frequency histogram of the percentage of hippocampal neurons with distinct axonal lengths in control or in Kidins220/ARMS knockdown cells (shC n = 100 cells, sh2 n = 100 cells).
Knockdown of Kidins220/ARMS Negatively Regulates Normal Development of Dendrites
We then analyzed the effects of Kidins220/ARMS down-regulation on the development of dendrites. After transfecting hippocampal neurons with the control or Kidins220/ARMS shRNA vectors, we analyzed the dendritic morphology at stage 4 by immunostaining with the dendritic marker MAP2 (microtubule-associated protein 2). Fig. 5A shows that whereas the shC neurons displayed normal dendrites (a), sh2 neurons displayed an aberrant dendritic arbor characterized by fewer and shorter MAP2-positive processes (Fig. 5A, b; see the same effect in sh1-RNA-transfected hippocampal neurons in supplemental Fig. 3C).
FIGURE 5.

Down-regulation of Kidins220/ARMS in hippocampal neurons alters normal dendrite development. A, hippocampal neurons were transfected with GFP-shC (a) as control or GFP-sh2 Kidins220/ARMS-specific shRNA (b). After 5 DIV, neurons were fixed and immunostained for the dendritic marker MAP2 (red). Similar to non-transfected neurons, GFP-shC neurons developed normal dendrites, whereas GFP-sh2 neurons displayed aberrant MAP2-positive processes. Compare the green neuron where Kidins220/ARMS has been knocked down with the neighbor neuron. Scale bar, 20 μm. B, quantification of the percentage of neurons at stage 4 bearing normal or aberrant dendritic processes. The data shown are the means ± S.E. of three independent experiments (shC n = 100 cells, sh2 n = 100 cells), and statistical significance was evaluated by Student's unpaired t test (**, p < 0.01).
In order to quantify the percentage of neurons with altered dendritic arbor, we classified them as normal if they possessed three or more dendrites longer than 20 μm or aberrant if they lacked dendrites, their number was significantly lower, and/or their length was shorter than 20 μm. We selected this method based on the fact that after 1 or 2 days in culture, most hippocampal neurons present a single, long “major” process that averaged 80–100 μm in length and several “minor” processes of much shorter length (10–15 μm). Once minor processes attain this length, they exhibit little net elongation, although they remain motile and extend and retract for short distances and later become dendrites (6). We considered that neurons bearing fewer or shorter dendrites or where these processes were not formed presented an abnormal dendritic development. Neurons transfected with shC displayed normal dendrites (Fig. 5B), whereas after Kidins220/ARMS knockdown, the percentage of neurons with normal MAP2-positive dendritic arbor suffered a 6-fold decrease (79.4% for shC versus 12.8% for sh2; shC n = 100 neurons, sh2 n = 100 neurons, three experiments). Such a severe phenotype indicates that Kidins220/ARMS is an important regulator of hippocampal dendritic development.
Kidins220/ARMS Interacts with Microtubule-regulating Proteins that Actively Regulate Neuronal Morphogenesis
In order to gain insight into the molecular mechanisms through which Kidins220/ARMS regulates neuronal polarity and development, we searched for its interactors. We performed two separate yeast two-hybrid screenings of a mouse brain cDNA library using Kidins220/ARMS N- and C-terminal regions as baits (Fig. 6A). Using the N terminus (Kidins220-Ank), we screened 2.4 × 106 clones and identified three positive clones containing full-length SCG10 (superior cervical ganglion 10) and one with full-length Sclip (SCG10-like protein), both members of the stathmin family of proteins (Fig. 6B). We also screened 3.9 × 107 clones with Kidins220/ARMS C terminus (Kidins220-C-ter) and identified, among others, 37 clones corresponding to the two regulatory light chains of MAP1a LC2 and MAP1b LC1 (microtubule-associated proteins 1a and 1b) (Fig. 6C). The identified proteins belong to the superfamily of microtubule-regulating factors and are particularly relevant because they are highly abundant in the developing nervous system and actively regulate the dynamicity of the microtubule network during the successive steps of neuronal morphogenesis (35–40) (recently reviewed in Ref. 41).
FIGURE 6.

Kidins220/ARMS domains used as baits in yeast two-hybrid assays and the interacting proteins identified. A, the 11 ankyrin repeats present at the Kidins220/ARMS N-terminal region (Kidins220-Ank) as well as its C-terminal region containing a proline-rich region, a SAM domain, and a PDZ binding motif (Kidins220-C-ter) were used as baits to screen a mouse brain cDNA library. B, schematic representation of the domain structure for the stathmin family members identified as Kidins220/ARMS-interacting proteins. All members contain the highly conserved regulatory and tubulin binding domains. SCG10, Sclip, and RB3 also possess an N-terminal membrane-anchoring domain absent in stathmin. Three independent clones of SCG10 and one clone of Sclip were identified. C, schematic representation of the domain structure for MAP1a and MAP1b. These proteins consist of a dimer between a heavy chain and a light chain derived from the same polypeptide. A very high number of clones of MAP1 were identified as Kinds220/ARMS-interacting proteins (37 in total).
To verify these interactions in mammalian cells, we performed co-immunoprecipitation experiments from 7 DIV cortical neurons. Given that microtubule-associated proteins and tubulin reside mainly at an insoluble fraction, we lysed the neurons to obtain a soluble fraction and re-extracted the insoluble pellet. We then immunoprecipitated Kidins220/ARMS from both fractions and performed immunoblot analysis for stathmin, SCG10, Sclip, and MAP1 LCs. Kidins220/ARMS interacted with SCG10, Sclip, and MAP1 LCs mainly at the re-extracted fraction, whereas stathmin did not (Fig. 7A, left). We also performed reverse immunoprecipitation and immunoblot analysis and detected that Kidins220/ARMS was present in MAP1 LCs immunoprecipitates (Fig. 7A, right). However, Sclip and SCG10 immunoprecipitates did not show a band corresponding to the molecular weight of Kidins220/ARMS (not shown), indicating that this association was not detectable by reverse immunoprecipitation. Altogether, these data indicate that Kidins220/ARMS specifically and directly binds to SCG10, Sclip, and MAP1 LCs.
FIGURE 7.

Kidins220/ARMS interacts with SCG10, Sclip, and MAP1 in neurons. Cortical neurons were lysed to obtain a soluble fraction, and the insoluble pellet was re-extracted in order to immunoprecipitate Kidins220/ARMS or MAP1 LCs from both fractions (soluble and re-extracted). A, Kidins220/ARMS immunocomplexes were analyzed for the presence of Kidins220/ARMS (positive control) and stathmin, SCG10, Sclip, and MAP1 LCs by immunoblot. As a negative control, soluble lysates were also immunoprecipitated with an antibody against an IgG (Ip IgG). MAP1 LC immunocomplexes were analyzed for the presence of Kidins220/ARMS and MAP1 LCs (positive control) by immunoblot. As a negative control, soluble lysates were also immunoprecipitated with an antibody against an IgG. B, hippocampal neurons fixed at stage 3 were immunostained for Kidins220/ARMS (green) and MAP1 LCs (red), and their colocalization was analyzed by confocal microscopy. Both proteins colocalize predominantly in the axon and its growth cone (see enlargements). C, stage 3 hippocampal neurons were immunostained with anti-Kidins220/ARMS (green) and anti-stathmin (red) antibodies. The inserts show how stathmin and Kidins220/ARMS colocalize in the axonal and dendritic growth cones. A single 0.7-μm section for each channel and their merged image is shown. Scale bar, 20 μm.
We also examined the subcellular distribution of these interactors in stage 3 hippocampal neurons by immunofluorescence using Kidins220/ARMS-, MAP1 LC-, stathmin-, SCG10-, and Sclip-specific antibodies. Confocal microscopy analysis showed MAP1 LCs and Kidins220/ARMS colocalizing predominantly along the axon and at the axonal growth cones (Fig. 7B). Stathmin and Kidins220/ARMS colocalized mainly at the axonal and dendritic growth cones (Fig. 7C), whereas both SCG10 (Fig. 8A) and Sclip (Fig. 8B) where highly enriched together with Kidins220/ARMS in a juxtanuclear region and at the growth cones. Therefore, the spatial distribution of Kidins220/ARMS and MAP1 LCs, SCG10, or Sclip suggests that their association may occur at different subcellular compartments within the neuron.
FIGURE 8.

Kidins220/ARMS colocalizes with SCG10 and Sclip in hippocampal neurons. A and B, hippocampal neurons fixed at stage 3 were immunostained for Kidins220/ARMS (green) and SCG10 or Sclip (red), and their colocalization was analyzed by confocal microscopy. The enlargements show how these two stathmin family members and Kidins220/ARMS colocalize predominantly in the dendritic growth cones and in a juxtanuclear region. A single 0.7-μm section for each channel and their merged image is shown. Scale bar, 20 μm.
Next, we analyzed Kidins220/ARMS association with other microtubule-regulating factors known to participate in the coordination and dynamic reorganization of the microtubule cytoskeleton during dendritic and axonal development, such as MAP2 and CRMP2 (collapsing response mediator protein-2) (42–47). We immunoprecipitated Kidins220/ARMS from 7 DIV cortical neurons and examined the presence of MAP2 and CRMP2. Kidins220/ARMS coimmunoprecipitated with the high molecular weight isoforms of MAP2 (HMW MAP2), but not with CRMP2 (Fig. 9). Finally, we examined Kidins220/ARMS interaction with tubulin and with any of its post-translationally modified forms, acetylated or tyrosinated, characteristic of a more stable or dynamic microtubule cytoskeleton (48–50). Immunoblot analysis showed that βIII tubulin and acetylated and tyrosinated α-tubulin coimmunoprecipitated with Kidins220/ARMS (Fig. 9).
FIGURE 9.
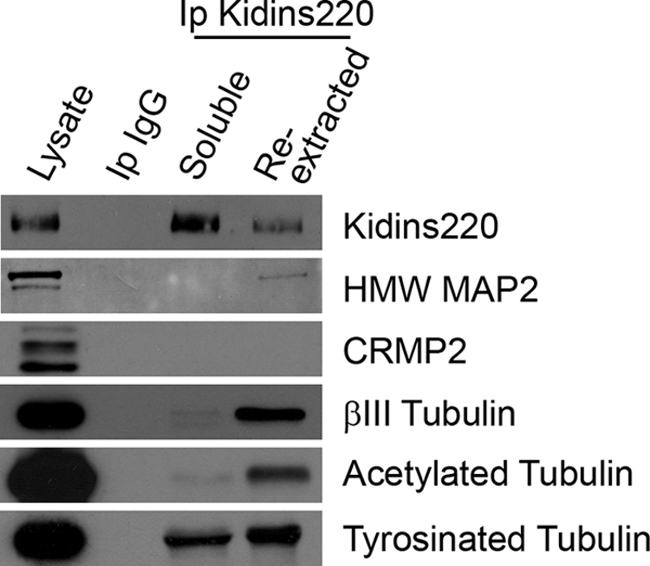
Kidins220/ARMS interacts with MAP2 and tubulin in cortical neurons. Soluble and re-extracted fractions form cortical neurons were used to immunoprecipitate Kidins220/ARMS. As a negative control, similar soluble lysates were incubated with an antibody against a non-relevant IgG chain (Ip IgG). The different immunocomplexes were analyzed for the capacity of Kidins220/ARMS to immunoprecipitate MAP2 and CRMP2 as well as βIII-tubulin or the post-translationally modified (acetylated or tyrosinated) α-tubulin. As a positive control, Kidins220/ARMS is present in its own immunocomplexes.
Kidins220/ARMS Modulates the Activity of MAP1b and Stathmins
The interaction of Kidins220/ARMS with tubulin and members of the stathmin and MAP1 families could indicate that Kidins220/ARMS serves a modulatory function toward the activity/localization of regulators of microtubule dynamics during neuronal morphogenesis. This potential role could contribute to the neuronal phenotype observed after Kidins220/ARMS overexpression and knockdown experiments. To test this hypothesis, we knocked down Kidins220/ARMS in immature hippocampal neurons (using sh1 and sh2 lentiviral vectors and shC as control) and analyzed the levels and activity of some of its associated microtubule-regulatory proteins (Fig. 10). The activity of stathmins and MAP1 over the dynamic reorganization of microtubules is fine tuned by phosphorylation (51–57). MAP1b HC phosphorylation favors more dynamic microtubules, whereas its dephosphorylation promotes microtubule stabilization (53, 57). The stathmin family of phosphoproteins sequester or release tubulin, depending on their phosphorylation state. The activity of stathmins sequestering free tubulin contributes indirectly to microtubule destabilization and is inhibited by phosphorylation at several residues (serines 16, 25, 38, and 63) (51, 52, 55, 56). Therefore, we monitored potential changes in the activity of these regulatory molecules by examining their phosphorylation state after Kidins220/ARMS knockdown. We used two phosphoantibodies, one recognizing phosphorylated Ser-16 in stathmin (Ser(P)-16), a site highly conserved in SCG10 (Ser-50) and Sclip (Ser-50), and the other phosphorylated Ser-38 in stathmin (Ser(P)-38). Additionally, we used the antibody SMI-31, which recognizes the phosphorylated forms of MAP1b HC, Tau, and neurofilament proteins, all of which can be distinguished by immunoblot based on their different molecular weights (58–61). Immunoblot analysis showed that knocking down Kidins220/ARMS resulted in a decrease in Ser(P)-16-stathmin signal but did not alter Ser(P)-38-stathmin or total protein levels of stathmin, SCG10, or Sclip (Fig. 10A). We must point out that the Ser(P)-16-stathmin antibody might also recognize this highly conserved residue also present in SCG10 and Sclip sequence (Ser(P)-50). However, this phosphoantibody only gave a clear signal by Western blot in the band corresponding to stathmin (18 kDa). We could not determine the phosphorylation state of SCG10 or Sclip (20–25 kDa each) in our samples by this method. This result does not exclude the possibility that SCG10 and Sclip are phosphorylated. It is feasible that the protein and/or the phosphorylation levels of these two members are lower than the ones presented by stathmin and escape the threshold of detection by the phosphoantibody in Western blot analysis. Another possibility is that the affinity of this antibody for Ser(P)-50 might differ from the one toward Ser(P)-16. Importantly, lysates from Kidins220/ARMS-knockdown neurons showed a significant decrease in the phosphorylation of the band corresponding to MAP1b HC (320–340 kDa) when using the SMI-31 antibody, accompanied by an increase in total MAP1b HC levels (Fig. 10A, middle panels).
FIGURE 10.

Kidins220/ARMS knockdown modulates the phosphorylation of stathmins and MAP1b-HC. A, hippocampal neurons were transfected with control (shC) or Kidins220/ARMS-specific (sh1 and sh2) shRNA vectors, and Kidins220/ARMS protein levels were determined after 4 DIV by immunoblot analysis. The same lysates were probed for phosphorylated forms of stathmin (Ser(P)-16 (pS16) and Ser(P)-38 (pS38)); total stathmin, SCG10, and Sclip; the phosphorylated form of MAP1b (SMI-31 antibody, 320–340 kDa band); total MAP1b HC and LCs; MAP2; Ser(P)-916 (pS916) PKD; and total PKD as well as for the acetylated and tyrosinated forms of α-tubulin, βIII tubulin, Thr(P)-514 (pT514) CRMP2, and total CRMP2. Neuronal specific enolase (NSE) is shown as a control. The arrowheads indicate immunoblots for which significant signal intensity changes were observed. One representative immunoblot of three independent experiments is shown. B, hippocampal neurons were transfected with GFP-shC (a) as control or GFP-sh2 Kidins220/ARMS-specific shRNA (b) and immunostained with antibodies against Ser(P)-16-stathmin (red). GFP signal served to identify neurons transfected with the different shRNAs. Scale bar, 20 μm. C, quantification of the relative fluorescence intensity for Ser(P)-16-stathmin on GFP-positive (transfected) neurons with respect to GFP-negative (untransfected) cells. Although shC-transfected neurons display a nearly identical intensity of Ser(P)-16-stathmin as untransfected neurons (ratio is almost 1), Ser(P)-16-stathmin decreases significantly in sh2-transfected neurons. The data shown are the means ± S.E. of three independent experiments (shC n = 52 cells, sh2 n = 74 cells), and statistical significance was evaluated by Student's unpaired t test (*, p < 0.05).
We also examined the levels of total PKD as well as its active form (phosphorylated at Ser-916, Ser(P)-916) (62), which is known to regulate neuronal polarity, development, and dendritic arborization (10, 18, 19, 63), but did not find any changes after Kidins220/ARMS knockdown (Fig. 10A, middle panels). Additionally, we analyzed MAP2 and the microtubule-polymerizing factor CRMP2 that also cooperate to control microtubule assembly during neuronal development (42–47). We did not observe significant changes in the protein levels of total MAP2 and total or inactive CRMP (phosphorylated at Thr-514, Thr(P)-514) (47) (Fig. 10A, middle and right panels). The same was true for tubulin and its post-translationally modified forms (acetylated or tyrosinated) (Fig. 10A, right panels).
Immunofluorescence analysis and quantification, using an antibody that recognizes stathmin, SCG10, and Sclip (panstathmin), confirmed that total stathmins signal did not change (supplemental Fig. 4). However, using the Ser(P)-16-stathmin antibody (pStathmin) the result was strikingly different (Fig. 10B, compare a and b). We could quantify that the total fluorescence intensity given by this antibody significantly decreased more than 2-fold (over a 50% reduction) after Kidins220/ARMS knockdown (Fig. 10C; shC n = 52 cells, sh2 n = 74 cells, three experiments). Notably, panstathmin immunostaining showed a distribution enriched at the cell body and at the growth cone (Fig. 10B, a). However, stathmin is mainly cytosolic and presents a low concentration at the neuronal body (Fig. 7B), whereas SCG10 (Fig. 8A) and Sclip (Fig. 8B) highly concentrate precisely at the Golgi apparatus at the cellular soma and at growth cones (64–66). This observation indicates that this antibody is recognizing Ser(P)-16 in stathmin and Ser(P)-50 in SCG10 and Sclip, and its signal is globally reduced after Kidins220/ARMS knockdown. On the other hand, we were not able to detect changes in SMI-31 immunolabeling (not shown), probably due to the ability of this antibody to recognize phosphorylation sites within other proteins that might not be altered by Kidins220/ARMS down-regulation.
These results demonstrate that Kidins220/ARMS knockdown reduces the phosphorylation of both stathmins and MAP1b, thereby modulating the activity of these different microtubule-regulating proteins, whose function in axon formation, elongation, and neuronal morphogenesis is well established (41). Altogether, our results suggest that Kidins220/ARMS could be controlling neuronal development by modulating the activity of these microtubule-regulating proteins.
DISCUSSION
Neurons become highly polarized cells by breaking their initial sphere symmetry and establishing two structurally and functionally distinct compartments, axons and dendrites. Here, we present data strongly indicating that Kidins220/ARMS prevents immature neurites from becoming axons while favoring dendrite development. This effect is concurrent with changes in the activity (determined as phosphorylation state) of microtubule-regulating proteins taking place in neurons where Kidins220/ARMS has been knocked down. The role of these molecules in neuronal development (neuritogenesis, establishment of polarity, elongation, and branching of axons and dendrites) through a complex network that modulates microtubule dynamics is well documented (for a review, see Ref. 41 and references therein). Therefore, our findings constitute the first evidence of Kidins220/ARMS being a unique protein able to interact with and modulate the activity of these two different types of microtubule-regulating proteins (MAP1b and stathmins; see below). In addition, the fact that Kidins220/ARMS participates in different steps taking place during neuronal maturation suggests that it could contribute to (or be a key molecule in) the mechanisms by which microtubule-regulating proteins control neuronal development.
Kidins220/ARMS Exerts an Opposite Effect on Axonal and Dendritic Establishment and Development
Kidins220/ARMS is highly enriched at neuronal growth cones. It is noteworthy that in neurons undergoing stage 2–3 transition, Kidins220/ARMS is significantly less concentrated in growth cones possessing less F-actin. At a certain moment of this developmental stage transition, one of the neurites will contain lower F-actin levels and will become the axon (7). Consequently, this same neurite would also possess lower Kidins220/ARMS levels. This correlation between reduced Kidins220/ARMS levels and axon determination is in accordance with Kidins220/ARMS down-regulation experiments, where we observe the growth of more than one neurite per neuron with axonal characteristics (SMI-31-positive). These results are indicative of a role for Kidins220/ARMS in preventing neurons from generating more than one axon.
Besides its role in polarity establishment and axonal outgrowth, Kidins220/ARMS favors dendrite development because its knockdown produces a severe deleterious effect on dendritic outgrowth. Again, this effect correlates well with the higher levels of Kidins220/ARMS observed in neurites destined to become dendrites. In support of our results, while this manuscript was in preparation, Kidins220/ARMS was found to regulate dendritic branching and spine stability in vivo (67). From these results, we would predict that Kidins220/ARMS gain of function experiments would result in an enhanced dendritic outgrowth and reduced axonal outgrowth. However, Kidins220/ARMS overexpression in young hippocampal neurons results in neurons bearing no neurites, Golgi apparatus fragmentation, and hampered neuronal viability. This phenotype resembles the one of hippocampal neurons overexpressing SCG10, characterized by a round shape (68). At present, we do not know if the cause of this phenotype is due to dramatic changes in microtubule dynamics, which could explain the lack of neuritic extensions, Golgi fragmentation, and toxicity in these young neurons. Forced Kidins220/ARMS expression in our neuronal cultures at later stages or in other cell types, such as HEK293T, did not result in significant changes on cellular shape and size or increased toxicity. Furthermore, other authors have performed Kidins220/ARMS overexpression experiments in PC12 cells (27, 29) or in non-neuronal cells (31) without similar consequences. Thus, the ectopic expression of this protein is especially deleterious during early hippocampal neuronal development in vitro, before the establishment of neuronal polarity. There appears to be a neuron-specific, very early window of time in which Kidins220/ARMS levels and distribution must be tightly regulated for correct neuronal development to occur. In summary, based on the subcellular localization analysis and knockdown phenotype described in this study, we can define Kidins220/ARMS as a regulator of axon specification and dendrite development.
Association of Kidins220/ARMS with PKD and Kinesin-1; Relevance for Neuronal Development
Kidins220/ARMS has been functionally linked to several neuronal polarity and dendritic outgrowth regulators (12, 29), whose association might also be relevant in explaining the phenotype observed after Kidins220/ARMS down-regulation.
We identified Kidins220/ARMS as a PKD1 substrate (12). One of the functions of PKD is to control the fission of trans-Golgi-derived vesicles (17, 69–72). Very recent studies have unveiled a role for PKD in neuronal polarized traffic, its activity being crucial for post-Golgi forward secretory trafficking of dendritic membrane proteins and dendritic outgrowth (18, 19, 63). PKD1 and -2, but not PKD3, have also been described as essential for the establishment and maintenance of hippocampal neuronal polarity (10). Loss of function of these two PKD isoforms leads to the symmetric outgrowth of multiple axon-like processes by a mechanism dependent on its association with the Golgi apparatus and its ability to regulate membrane traffic directionality (10). Because in neurons, Kidins220/ARMS traffic to the cell surface is controlled by PKD1 and -2, but not PKD3 (30), it is noteworthy that loss of function of either Kidins220/ARMS or PKD1/2 gives rise to a similar phenotype, rendering hippocampal neurons bearing multiple axonal processes and devoid of normal dendritic extensions while preserving the integrity of the Golgi apparatus. One can therefore speculate that alterations in Kidin220/ARMS localization and consequently its function at sites where it might be essential for proper neuronal development might be in part responsible for the phenotypes observed after PKD loss of function. Consistent with this idea and with Kidins220/ARMS being downstream of PKD, we observe no changes in total or active PKD after Kidins220/ARMS knockdown. The establishment and maintenance of neuronal polarity involves an intimate orchestration between the traffic of Golgi-derived vesicles and their membranous cargoes along the actin and microtubule cytoskeleton (2, 4, 8, 73–75). We could envisage that PKD-Kidins220/ARMS complexes could be regulating polarized protein traffic, given that Golgi carriers must be transported by molecular motors through the cytoskeletal network. In this sense, it is noteworthy that Kidins220/ARMS traffic has also been linked to molecular motors important for neuron polarity establishment. Bracale et al. (29) identified it as a cargo for kinesin-1 motor complex carriers. Kinesin motors drive the transport of multiple cargoes along microtubule tracks. It is noteworthy that kinesin-1-selective translocation has been shown to mark the initial axonal specification during polarity establishment (9).
Kidins220/ARMS Associates with and Modulates the Activity of Microtubule-regulating Proteins Known to Control Neuronal Morphogenesis
Performing two-hybrid screenings and immunoprecipitation assays, we identified the association of Kidins220/ARMS with microtubule-regulatory proteins. These proteins, MAP1 LCs and two members of the stathmin family of proteins (SCG10 and Sclip) actively control the dynamicity of the microtubule network during different phases of neuronal morphogenesis (recently reviewed in Ref. 41).
Witte et al. (8) have shown that a moderate or severe microtubule destabilization leads to distinct effects on the successive phases of neuronal morphogenesis. For instance, slight microtubule stabilization drastically increases neurite outgrowth without compromising microtubule dynamics and promotes the appearance of two or more axonal processes and at least one or two dendrites, producing a phenotype reminiscent of Kidins220/ARMS knockdown. Conditions of extreme microtubule stabilization completely block microtubule dynamics as well as axonal growth. Therefore, a slight, balanced shift of microtubule dynamics toward more stable microtubules is necessary to induce axon formation. This slight microtubule stabilization can be achieved by coordinating and modulating the activity of microtubule-regulatory proteins. In this sense, several studies have identified that these molecules regulate not only neuronal polarity and axonal and dendritic outgrowth (38, 41, 47, 76, 77) but also other steps of neuronal differentiation (41).
Here we discover the interaction of Kidins220/ARMS with tubulin and microtubule-regulating proteins that modulate microtubule dynamics by different mechanisms. Importantly, one of these regulators we found is MAP1b, a factor that acts directly on microtubules to stabilize them. MAP1b is present in axons, soma, and dendrites, although it is especially prominent in extending axons and their growth cones (39, 78–82). This microtubule-associated protein regulates early axonal outgrowth and establishment of neuronal polarity by modulating microtubule dynamics through its capacity to bind the microtubule lattice and regulate microtubule assembly and stability in in vitro and in vivo models (83–91).
In contrast, the stathmin family of phosphoproteins, the other microtubule regulators we found to associate with Kidins220/ARMS, belong to the tubulin-regulating proteins. Kidins220/ARMS associates with SCG10 and Sclip but not with stathmin, another member of the family. Whereas stathmin is ubiquitous and cytosolic, SCG10 and Sclip are almost exclusive of the nervous system and accumulate at the Golgi apparatus, vesicles along neuritic processes, and growth cones (64–66). This distinct spatial distribution that associates SCG10 and Sclip with membranes is due to the presence of an additional N-terminal region that is absent in stathmin (see Fig. 6). Our colocalization studies show a high enrichment of SCG10, Sclip, and Kidins220/ARMS in the perinuclear region and at growth cones. Therefore, it is conceivable that this membrane-anchoring domain is mediating their specific association with Kidins220/ARMS in these specific subcellular compartments.
Stathmin family factors indirectly modulate the dynamicity and assembly of microtubules by controlling the overall or local free soluble tubulin availability. In this way, they regulate diverse phases of neuronal morphogenesis, depending on the type of neurons studied and environmental cues (68, 92). In cultured hippocampal neurons, Sclip knockdown increases axonal branching, whereas SCG10 down-regulation promotes growth cone expansion (66). In cerebellar Purkinje neurons, stathmin down-regulation or its inactivation by phosphorylation leads to dendritic arborization, whereas the overexpression of stathmin or SCG10 reduces dendritic growth (36).
A recent study analyzed the combined effects of MAP1b and SCG10 on the control of microtubule stability (77). These authors propose that the antagonistic effects of MAP1b and SCG10 and a fine tuning of the balance of these proteins and their activity may be critical for the regulation of neuronal microtubule dynamics. In this context, the unique capacity of Kidins220/ARMS to interact with MAP1a/b and the members of the stathmin family and its ability to modify their activity places this protein in a privileged position to regulate or “fine tune” their regulatory role on microtubule dynamics during neuronal polarization and development. In accordance with this hypothesis, the Kidins220/ARMS loss of function phenotype would correlate with a moderate shift in the affinity/activity of the axonal relevant MAP1 toward more stable or less dynamic axonal microtubules while modulating the affinity/activity of dendritic relevant microtubule-regulating proteins that would result in an aberrant dendritic arbor.
We find that Kidins220/ARMS knockdown significantly increases total levels of MAP1b HC and reduces its phosphorylation (as detected by the SMI-31 antibody). This phosphorylation regulates MAP1b ability to maintain a more dynamically unstable microtubule population in the distal part of the axon when exposed to neurotrophin, netrin, and reelin (88, 93–96). The rise in total MAP1b HC after Kidins220/ARMS knockdown could simply reflect an increase in axonal structural proteins to produce higher number of axons, and the phosphorylation decrease would promote axonal microtubule stabilization and outgrowth. Thus, normally, Kidins220/ARMS would contribute to maintain the dynamicity of distal axonal microtubules by facilitating MAP1b phosphorylation. It is also worth mentioning Kidins220/ARMS association with MAP2 due to Kidins220/ARMS enrichment in dendrites and the severe dendritic phenotype observed after reducing its levels. Based on Kidins220/ARMS regulation of the phosphorylation-dependent activity of other microtubule-associated proteins, the affinity/activity of MAP2 toward dendritic microtubules could be under a similar control.
Phosphorylation of stathmin at Ser-16 or its equivalent site on SCG10 and Sclip (Ser-50) has been associated with their inactivation (51, 52, 55, 56). These factors sequester or release tubulin, depending on their phosphorylation state. Our data showing that Kidins220/ARMS knockdown results in more than a 50% reduction in Ser(P)-16-stathmin indicates that Kidins220/ARMS would normally favor the phosphorylation and inactivation of this protein (and probably of SCG10 and Sclip, as discussed above). In relation to the effect of this modification on neuronal development, it has been reported that Ser(P)-16-stathmin is necessary for axon formation and polarity establishment in hippocampal neurons grown over laminin (38). The fact that reducing Kidins220/ARMS levels, which produces multiple longer axon-like processes, results in decreased Ser(P)-16-stathmin is intriguing because axons are still able to form. It is possible that Ser(P)-16-stathmin levels after Kidins220/ARMS knockdown are still above a threshold that permits axon growth but might affect other functions, such as the dynamic behavior of the distal axon and therefore its pathfinding functions. Finally, stathmin also appears to regulate dendritic length in Purkinje neurons (36). A decrease in its phosphorylation and activity could affect this process in hippocampal neurons, making a good correlation with the phenotype of neurons where Kidins220/ARMS has been knocked down.
In this context, it is important to mention that Kidins220/ARMS associates with SCG10 and Sclip but not with stathmin. However, the lack of interaction of Kidins220/ARMS with stathmin does not rule out the possibility of Kidins220/ARMS regulating stathmin phosphorylation. It could control this step by modulating different signaling pathways.
To date, no study has addressed the effect of the phosphorylation-dependent inactivation of SCG10 or Sclip on the morphogenesis of hippocampal neurons. In other neuronal types, inhibition of SCG10 increases growth cone size and reduces axonal length (66, 68). In contrast, Sclip specifically regulates axonal branching (66) and is also required for dendritic initiation and development in cerebellar Purkinje cells (37). We could not precisely determine the phosphorylation state of SCG10 or Sclip in our neuronal model. However, we have provided evidence, indicating that SCG10 and Sclip are subjected to important decreases in their phosphorylation state after Kidins220/ARMS knockdown. Because SCG10 and Sclip perform non-overlapping functions on neuronal development, any putative modifications could be relevant for the polarity as well as the aberrant dendritic phenotype of neurons with reduced Kidins220/ARMS levels.
One important issue regarding stathmin family proteins is that they are starting to emerge as essential regulators of specific processes of neuronal development acting at particular sites of the neuron to locally modulate microtubule dynamics. Neuronal differentiation requires not only fine tuning of the plasticity of microtubule and actin networks but also a spaciotemporal regulation of polarized traffic of vesicles from the Golgi complex along cytoskeletal tracks toward their final destination (2, 4, 8, 73–75). Kidins220/ARMS binds directly to SCG10 and Sclip, and they all accumulate at the Golgi apparatus and growth cones (64–66). It is feasible that these complexes could coordinate the polarized delivery of cargoes from this organelle by modulating the local reorganization of microtubules.
MAP1, stathmin, SCG10, and Sclip could perform specific, complementary, and coordinated functions during neuronal differentiation. It will be crucial to further study how Kidins220/ARMS locally modulates the specific phosphorylation and activity of these proteins during the different phases of neuronal differentiation and the signaling pathways involved.
The association of Kidins220/ARMS with multiple polarity regulators and its ability to bind and modulate the activity of microtubule-regulating proteins known to control neuronal morphogenesis reinforce the conclusions derived from overexpression and knockdown phenotypes that identify a novel role for Kidins220/ARMS in the regulation of neuronal polarity and development.
Supplementary Material
Acknowledgments
We are grateful to members of all laboratories for helpful comments and discussions. We are also thankful to Dr. A. Sobel (Institut du Fer a Moulin, Paris, France) for anti-stathmin, anti-SCG10, anti-Sclip, and stathmin phosphoantibodies as well as for critical reading of the manuscript; to Dr. F. Hernádez (Centro de Biologia Molecular Severo Ochoa) for anti-MAP2 antibodies; and to Dr. J. Serrador (Centro Nacional de Investicagión Cardiovascular, CNIC, Instituto de Salud Carlos III) for help with nucleofection experiments. We thank D. Megías and Dr. M. Montoya (Centro Nacional de Investicagión Oncológica, CNIO, Instituto de Salud Carlos III) for assistance quantifying fluorescence intensity from confocal microscopy images as well as J. Pérez and R. Uña from the Scientific Imaging Department of the “Instituto de Investigaciones Biomédicas de Madrid, Consejo Superior de Investigaciones Científicas-Universidad Autónoma de Madrid” for assistance in the neuronal morphometric analysis.
This work was supported by “Ministerio de Ciencia e Innovación” Grant SAF2008-01951, “Neurodegmodels” Grant CAM S-SAL-0202-2006-01 from “Comunidad de Madrid,” and CIBERNED from “Instituto de Salud Carlos III” (Spain) (to T. I.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1–4.
- PKD
- protein kinase D
- DIV
- days in vitro
- GFP
- green fluorescent protein
- NFκB
- nuclear factor κB
- shRNA
- short hairpin RNA
- TRITC
- tetramethylrhodamine isothiocyanate.
REFERENCES
- 1.Arimura N., Kaibuchi K. (2007) Nat. Rev. Neurosci. 8, 194–205 [DOI] [PubMed] [Google Scholar]
- 2.da Silva J. S., Dotti C. G. (2002) Nat. Rev. Neurosci. 3, 694–704 [DOI] [PubMed] [Google Scholar]
- 3.Da Silva J. S., Hasegawa T., Miyagi T., Dotti C. G., Abad-Rodriguez J. (2005) Nat. Neurosci. 8, 606–615 [DOI] [PubMed] [Google Scholar]
- 4.Horton A. C., Ehlers M. D. (2003) Neuron 40, 277–295 [DOI] [PubMed] [Google Scholar]
- 5.Bradke F., Dotti C. G. (2000) Curr. Opin. Neurobiol. 10, 574–581 [DOI] [PubMed] [Google Scholar]
- 6.Dotti C. G., Sullivan C. A., Banker G. A. (1988) J. Neurosci. 8, 1454–1468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bradke F., Dotti C. G. (1999) Science 283, 1931–1934 [DOI] [PubMed] [Google Scholar]
- 8.Witte H., Neukirchen D., Bradke F. (2008) J. Cell Biol. 180, 619–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jacobson C., Schnapp B., Banker G. A. (2006) Neuron 49, 797–804 [DOI] [PubMed] [Google Scholar]
- 10.Yin D. M., Huang Y. H., Zhu Y. B., Wang Y. (2008) J. Neurosci. 28, 8832–8843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cabrera-Poch N., Sánchez-Ruiloba L., Rodríguez-Martínez M., Iglesias T. (2004) J. Biol. Chem. 279, 28592–28602 [DOI] [PubMed] [Google Scholar]
- 12.Iglesias T., Cabrera-Poch N., Mitchell M. P., Naven T. J., Rozengurt E., Schiavo G. (2000) J. Biol. Chem. 275, 40048–40056 [DOI] [PubMed] [Google Scholar]
- 13.Kong H., Boulter J., Weber J. L., Lai C., Chao M. V. (2001) J. Neurosci. 21, 176–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ghanekar Y., Lowe M. (2005) Trends Cell Biol. 15, 511–514 [DOI] [PubMed] [Google Scholar]
- 15.Rozengurt E., Rey O., Waldron R. T. (2005) J. Biol. Chem. 280, 13205–13208 [DOI] [PubMed] [Google Scholar]
- 16.Van Lint J., Rykx A., Maeda Y., Vantus T., Sturany S., Malhotra V., Vandenheede J. R., Seufferlein T. (2002) Trends Cell Biol. 12, 193–200 [DOI] [PubMed] [Google Scholar]
- 17.Yeaman C., Ayala M. I., Wright J. R., Bard F., Bossard C., Ang A., Maeda Y., Seufferlein T., Mellman I., Nelson W. J., Malhotra V. (2004) Nat. Cell Biol. 6, 106–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bisbal M., Conde C., Donoso M., Bollati F., Sesma J., Quiroga S., Díaz Añel A., Malhotra V., Marzolo M. P., Cáceres A. (2008) J. Neurosci. 28, 9297–9308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Czöndör K., Ellwanger K., Fuchs Y. F., Lutz S., Gulyás M., Mansuy I. M., Hausser A., Pfizenmaier K., Schlett K. (2009) Mol. Biol. Cell 20, 2108–2120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang E. J., Reichardt L. F. (2001) Annu. Rev. Neurosci. 24, 677–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Klein R. (2004) Curr. Opin. Cell Biol. 16, 580–589 [DOI] [PubMed] [Google Scholar]
- 22.Martinez A., Soriano E. (2005) Brain Res. 49, 211–226 [DOI] [PubMed] [Google Scholar]
- 23.Pasquale E. B. (2005) Nat. Rev. 6, 462–475 [DOI] [PubMed] [Google Scholar]
- 24.Reichardt L. F. (2006) Phil. Trans. R. Soc. Lond. 361, 1545–1564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou F. Q., Snider W. D. (2006) Phil. Trans. R. Soc. Lond. 361, 1575–1592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arévalo J. C., Yano H., Teng K. K., Chao M. V. (2004) EMBO J. 23, 2358–2368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arévalo J. C., Pereira D. B., Yano H., Teng K. K., Chao M. V. (2006) J. Biol. Chem. 281, 1001–1007 [DOI] [PubMed] [Google Scholar]
- 28.Hisata S., Sakisaka T., Baba T., Yamada T., Aoki K., Matsuda M., Takai Y. (2007) J. Cell Biol. 178, 843–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bracale A., Cesca F., Neubrand V. E., Newsome T. P., Way M., Schiavo G. (2007) Mol. Biol. Cell 18, 142–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sánchez-Ruiloba L., Cabrera-Poch N., Rodríguez-Martínez M., López-Menéndez C., Jean-Mairet R. M., Higuero A. M., Iglesias T. (2006) J. Biol. Chem. 281, 18888–18900 [DOI] [PubMed] [Google Scholar]
- 31.Luo S., Chen Y., Lai K. O., Arévalo J. C., Froehner S. C., Adams M. E., Chao M. V., Ip N. Y. (2005) J. Cell Biol. 169, 813–824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.López-Menéndez C., Gascón S., Sobrado M., Vidaurre O. G., Higuero A. M., Rodríguez-Peña A., Iglesias T., Díaz-Guerra M. (2009) J. Cell Sci. 122, 3554–3565 [DOI] [PubMed] [Google Scholar]
- 33.Banker G. (1998) Culturing Nerve Cells, 2nd Ed., MIT Press, Cambridge, MA [Google Scholar]
- 34.Kaech S., Banker G. (2006) Nat. Protoc. 1, 2406–2415 [DOI] [PubMed] [Google Scholar]
- 35.Grenningloh G., Soehrman S., Bondallaz P., Ruchti E., Cadas H. (2004) J. Neurobiol. 58, 60–69 [DOI] [PubMed] [Google Scholar]
- 36.Ohkawa N., Fujitani K., Tokunaga E., Furuya S., Inokuchi K. (2007) J. Cell Sci. 120, 1447–1456 [DOI] [PubMed] [Google Scholar]
- 37.Poulain F. E., Chauvin S., Wehrlé R., Desclaux M., Mallet J., Vodjdani G., Dusart I., Sobel A. (2008) J. Neurosci. 28, 7387–7398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Watabe-Uchida M., John K. A., Janas J. A., Newey S. E., Van Aelst L. (2006) Neuron 51, 727–739 [DOI] [PubMed] [Google Scholar]
- 39.Gonzalez-Billault C., Jimenez-Mateos E. M., Caceres A., Diaz-Nido J., Wandosell F., Avila J. (2004) J Neurobiol 58, 48–59 [DOI] [PubMed] [Google Scholar]
- 40.Riederer B. M. (2007) Brain Res. Bull. 71, 541–558 [DOI] [PubMed] [Google Scholar]
- 41.Poulain F. E., Sobel A. (2009) Mol. Cell Neurosci., in press [Google Scholar]
- 42.Dhamodharan R., Wadsworth P. (1995) J. Cell Sci. 108, 1679–1689 [DOI] [PubMed] [Google Scholar]
- 43.Fukata Y., Itoh T. J., Kimura T., Ménager C., Nishimura T., Shiromizu T., Watanabe H., Inagaki N., Iwamatsu A., Hotani H., Kaibuchi K. (2002) Nat. Cell Biol. 4, 583–591 [DOI] [PubMed] [Google Scholar]
- 44.Gu Y., Ihara Y. (2000) J. Biol. Chem. 275, 17917–17920 [DOI] [PubMed] [Google Scholar]
- 45.Maccioni R. B., Cambiazo V. (1995) Physiol. Rev. 75, 835–864 [DOI] [PubMed] [Google Scholar]
- 46.Sánchez C., Díaz-Nido J., Avila J. (2000) Prog. Neurobiol. 61, 133–168 [DOI] [PubMed] [Google Scholar]
- 47.Yoshimura T., Kawano Y., Arimura N., Kawabata S., Kikuchi A., Kaibuchi K. (2005) Cell 120, 137–149 [DOI] [PubMed] [Google Scholar]
- 48.Baas P. W., Ahmad F. J., Pienkowski T. P., Brown A., Black M. M. (1993) J. Neurosci. 13, 2177–2185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brown A., Slaughter T., Black M. M. (1992) J. Cell Biol. 119, 867–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li Y., Black M. M. (1996) J. Neurosci. 16, 531–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Antonsson B., Kassel D. B., Di Paolo G., Lutjens R., Riederer B. M., Grenningloh G. (1998) J. Biol. Chem. 273, 8439–8446 [DOI] [PubMed] [Google Scholar]
- 52.Gavet O., Ozon S., Manceau V., Lawler S., Curmi P., Sobel A. (1998) J. Cell Sci. 111, 3333–3346 [DOI] [PubMed] [Google Scholar]
- 53.Goold R. G., Owen R., Gordon-Weeks P. R. (1999) J. Cell Sci. 112, 3373–3384 [DOI] [PubMed] [Google Scholar]
- 54.Goold R. G., Gordon-Weeks P. R. (2004) Biochem. Soc. Trans. 32, 809–811 [DOI] [PubMed] [Google Scholar]
- 55.Larsson N., Marklund U., Gradin H. M., Brattsand G., Gullberg M. (1997) Mol. Cell. Biol. 17, 5530–5539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Marklund U., Larsson N., Gradin H. M., Brattsand G., Gullberg M. (1996) EMBO J. 15, 5290–5298 [PMC free article] [PubMed] [Google Scholar]
- 57.Owen R., Gordon-Weeks P. R. (2003) Mol. Cell Neurosci. 23, 626–637 [DOI] [PubMed] [Google Scholar]
- 58.Fischer I., Romano-Clarke G. (1990) J. Neurochem. 55, 328–333 [DOI] [PubMed] [Google Scholar]
- 59.Johnstone M., Goold R. G., Bei D., Fischer I., Gordon-Weeks P. R. (1997) J. Neurochem. 69, 1417–1424 [DOI] [PubMed] [Google Scholar]
- 60.Lichtenberg-Kraag B., Mandelkow E. M., Biernat J., Steiner B., Schröter C., Gustke N., Meyer H. E., Mandelkow E. (1992) Proc. Natl. Acad. Sci. U.S.A. 89, 5384–5388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Trivedi N., Marsh P., Goold R. G., Wood-Kaczmar A., Gordon-Weeks P. R. (2005) J. Cell Sci. 118, 993–1005 [DOI] [PubMed] [Google Scholar]
- 62.Matthews S. A., Rozengurt E., Cantrell D. (1999) J. Biol. Chem. 274, 26543–26549 [DOI] [PubMed] [Google Scholar]
- 63.Horton A. C., Rácz B., Monson E. E., Lin A. L., Weinberg R. J., Ehlers M. D. (2005) Neuron 48, 757–771 [DOI] [PubMed] [Google Scholar]
- 64.Di Paolo G., Lutjens R., Osen-Sand A., Sobel A., Catsicas S., Grenningloh G. (1997) J. Neurosci. Res. 50, 1000–1009 [DOI] [PubMed] [Google Scholar]
- 65.Gavet O., El Messari S., Ozon S., Sobel A. (2002) J. Neurosci. Res. 68, 535–550 [DOI] [PubMed] [Google Scholar]
- 66.Poulain F. E., Sobel A. (2007) Mol. Cell Neurosci. 34, 137–146 [DOI] [PubMed] [Google Scholar]
- 67.Wu S. H., Arevalo J. C., Sarti F., Tessarollo L., Gan W. B., Chao M. V. (2009) Dev. Neurobiol. 69, 547–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Morii H., Shiraishi-Yamaguchi Y., Mori N. (2006) J. Neurobiol. 66, 1101–1114 [DOI] [PubMed] [Google Scholar]
- 69.Eiseler T., Schmid M. A., Topbas F., Pfizenmaier K., Hausser A. (2007) FEBS Lett. 581, 4279–4287 [DOI] [PubMed] [Google Scholar]
- 70.Liljedahl M., Maeda Y., Colanzi A., Ayala I., Van Lint J., Malhotra V. (2001) Cell 104, 409–420 [DOI] [PubMed] [Google Scholar]
- 71.Prigozhina N. L., Waterman-Storer C. M. (2004) Curr. Biol. 14, 88–98 [DOI] [PubMed] [Google Scholar]
- 72.Woods A. J., White D. P., Caswell P. T., Norman J. C. (2004) EMBO J. 23, 2531–2543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Foletti D. L., Prekeris R., Scheller R. H. (1999) Neuron 23, 641–644 [DOI] [PubMed] [Google Scholar]
- 74.Horton A. C., Ehlers M. D. (2004) Nat. Cell Biol. 6, 585–591 [DOI] [PubMed] [Google Scholar]
- 75.Ye B., Zhang Y. W., Jan L. Y., Jan Y. N. (2006) J. Neurosci. 26, 10631–10632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Barnes A. P., Lilley B. N., Pan Y. A., Plummer L. J., Powell A. W., Raines A. N., Sanes J. R., Polleux F. (2007) Cell 129, 549–563 [DOI] [PubMed] [Google Scholar]
- 77.Bondallaz P., Barbier A., Soehrman S., Grenningloh G., Riederer B. M. (2006) Cell Motil. Cytoskeleton 63, 681–695 [DOI] [PubMed] [Google Scholar]
- 78.Black M. M., Slaughter T., Fischer I. (1994) J. Neurosci. 14, 857–870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Calvert R., Anderton B. H. (1985) EMBO J. 4, 1171–1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.DiTella M. C., Feiguin F., Carri N., Kosik K. S., Cáceres A. (1996) J. Cell Sci. 109, 467–477 [DOI] [PubMed] [Google Scholar]
- 81.Garner C. C., Garner A., Huber G., Kozak C., Matus A. (1990) J. Neurochem. 55, 146–154 [DOI] [PubMed] [Google Scholar]
- 82.Gordon-Weeks P. R., Fischer I. (2000) Microsc. Res. Tech. 48, 63–74 [DOI] [PubMed] [Google Scholar]
- 83.Bloom G. S., Luca F. C., Vallee R. B. (1985) Proc. Natl. Acad. Sci. U.S.A. 82, 5404–5408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Edelmann W., Zervas M., Costello P., Roback L., Fischer I., Hammarback J. A., Cowan N., Davies P., Wainer B., Kucherlapati R. (1996) Proc. Natl. Acad. Sci. U.S.A. 93, 1270–1275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.González-Billault C., Demandt E., Wandosell F., Torres M., Bonaldo P., Stoykova A., Chowdhury K., Gruss P., Avila J., Sánchez M. P. (2000) Mol. Cell Neurosci. 16, 408–421 [DOI] [PubMed] [Google Scholar]
- 86.Gonzalez-Billault C., Avila J., Cáceres A. (2001) Mol. Biol. Cell 12, 2087–2098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.González-Billault C., Engelke M., Jiménez-Mateos E. M., Wandosell F., Cáceres A., Avila J. (2002) J. Neurosci. Res. 67, 713–719 [DOI] [PubMed] [Google Scholar]
- 88.González-Billault C., Del Río J. A., Ureña J. M., Jiménez-Mateos E. M., Barallobre M. J., Pascual M., Pujadas L., Simó S., Torre A. L., Gavin R., Wandosell F., Soriano E., Avila J. (2005) Cereb. Cortex 15, 1134–1145 [DOI] [PubMed] [Google Scholar]
- 89.Meixner A., Haverkamp S., Wässle H., Führer S., Thalhammer J., Kropf N., Bittner R. E., Lassmann H., Wiche G., Propst F. (2000) J. Cell Biol. 151, 1169–1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Riederer B., Cohen R., Matus A. (1986) J. Neurocytol. 15, 763–775 [DOI] [PubMed] [Google Scholar]
- 91.Takei Y., Kondo S., Harada A., Inomata S., Noda T., Hirokawa N. (1997) J. Cell Biol. 137, 1615–1626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ozon S., Maucuer A., Sobel A. (1997) Eur. J. Biochem. 248, 794–806 [DOI] [PubMed] [Google Scholar]
- 93.Del Río J. A., González-Billault C., Ureña J. M., Jiménez E. M., Barallobre M. J., Pascual M., Pujadas L., Simó S., La Torre A., Wandosell F., Avila J., Soriano E. (2004) Curr. Biol. 14, 840–850 [DOI] [PubMed] [Google Scholar]
- 94.Goold R. G., Gordon-Weeks P. R. (2001) J. Cell Sci. 114, 4273–4284 [DOI] [PubMed] [Google Scholar]
- 95.Goold R. G., Gordon-Weeks P. R. (2003) J. Neurochem. 87, 935–946 [DOI] [PubMed] [Google Scholar]
- 96.Goold R. G., Gordon-Weeks P. R. (2005) Mol. Cell Neurosci. 28, 524–534 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.